

Abstract
LSD1 is a lysine demethylase highly involved in initiation and development of cancer. To design highly effective covalent inhibitors, a strategy is to fill its large catalytic cleft by designing tranylcypromine (TCP) analogs decorated with long, hindered substituents. We prepared three series of TCP analogs, carrying aroyl- and arylacetylamino (1a-h), Z-amino acylamino (2a-o), or double-substituted benzamide (3a-n) residues at the C4 or C3 position of the phenyl ring. Further fragments obtained by chemical manipulation applied on the TCP scaffold (compounds 4a-i) were also prepared. When tested against LSD1, most of 1 and 3 exhibited IC50 values in the low nanomolar range, with 1e and 3a,d,f,g being also the most selective respect to monoamine oxidases. In MV4–11 AML and NB4 APL cells compounds 3 were the most potent, displaying up to submicromolar cell growth inhibition against both cell lines (3a) or against NB4 cells (3c). The most potent compounds in cellular assays were also able to induce the expression of LSD1 target genes, such as GFI-1b, ITGAM, and KCTD12, as functional read-out for LSD1 inhibition. Mouse and human intrinsic clearance data highlighted the high metabolic stability of compounds 3a, 3d and 3g. Further studies will be performed on the new compounds 3a and 3c to assess their anticancer potential in different cancer contexts.
Keywords: histone demethylases, lysine specific demethylase 1, drug discovery, structure–activity relationships, antiproliferative activity
GRAPHICAL ABSTRACT
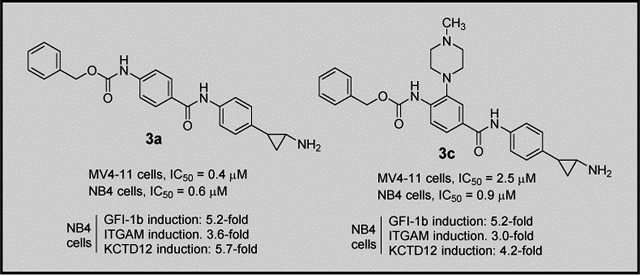
LSD1 is a lysine demethylase highly involved in cancer initiation and progression. Through a X-ray-driven drug discovery process, we identified some tranylcypromine-based compounds 1-4. Among them, 1-3 were active against LSD1 at the submicromolar/low nanomolar range. When tested in MV4–11 AML and NB4 APL cells, 3a and 3c were the most potent to arrest proliferation and induced target genes under the control of LSD1 in cells. These compounds will be further validated in different cancer contexts.
Introduction
Epigenetic methylation of histone tails is regulated by the activity of two families of enzymes: histone methyltransferases (HMTs), that add methyl groups at either lysine or arginine residues (lysine or protein arginine methyltransferases (KMTs or PRMTs), respectively), and lysine demethylases (KDMs), that erase the methyl marks from the ε-amino group of lysine residues using either a flavin adenine dinucleotide (FAD)-dependent (KDM1A and KDM1B) or a 2-oxoglutarate/Fe(II)-dependent (Jumonji-containing enzymes, KDM2–7) mechanism.[1] Lysine specific demethylase 1 (LSD1 or KDM1A) removes the methyl group from mono- and di-methylated lysine residues at the lysine 4 of histone H3 (H3K4me1/2) and, in particular conditions, at H3K9me1/2 (androgen-positive prostate cancer cells).[2] The H3K4me1/2 is an activation mark for transcription while the H3K9me1/2 has a gene silencing role, thus LSD1 can act as transcriptional repressor or activator depending on the substrate used and the cellular context.[3] LSD1 forms complexes with other repressor proteins (CoREST),[4] transcriptional silencers (HDAC1/2), nuclear receptors, and other transcription factors to epigenetically regulate the expression of genes involved in various cellular events.[5] LSD1 is an essential gene in mammalian biology and is involved in many different cellular events such as maintenance of pluripotency in embryonic stem cells, embryonic development, hematopoiesis, neural stem cell proliferation and neuronal development.[6] Pathologically, LSD1 is implicated in tumorigenesis, metastasis, and poor prognosis of cancer,[7] and is overexpressed in several cancer cells, such as breast cancer,[8] neuroblastoma,[9] and glioma[10] cells.
Several irreversible and reversible LSD1 inhibitors have been developed so far and validated in various types of cancers including leukemias, and some of them are in clinical trials (tranylcypromine (TCP), IMG-7289, INCB059872, GSK2879552, seclidemstat, and CC-90011) for the treatment of hematological malignancies or solid tumors (Figure 1).[11]
Figure 1.
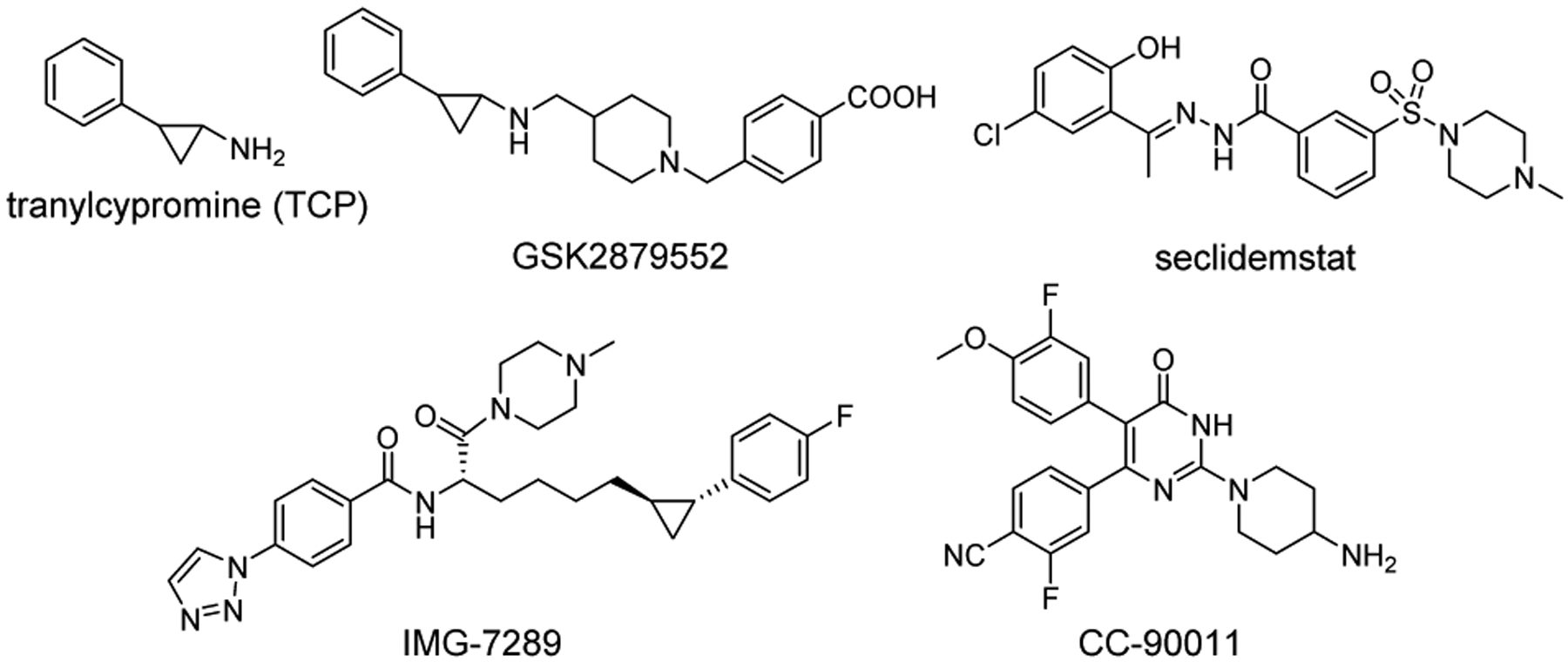
Structures of irreversible and reversible LSD1 inhibitors in clinical trials for treatment of cancer.
We and others pioneered the studies on covalent LSD1 inhibitors starting from mechanistic and crystallographic analyses on the stereoisomers of TCP and their 4-bromo derivatives.[12] Such crystal structures confirmed the covalent bond between TCP and the FAD cofactor and highlighted the presence of an open cleft in the enzyme active site, suggesting the introduction of large substituents at the C4 position of the TCP phenyl ring to design new molecules with improved inhibitory activities.[12] Thus, we prepared a first series of 4-benzamide analogues of TCP, by inserting aroylamino and arylacetylamino moieties with different sizes and shapes at the C4 position of the TCP phenyl ring (1a-h), and a second series of compounds in which we inserted at the same position a N-benzyloxycarbonyl (Z) amino acid residue linked through an amide bond (2a-o) (Figure 2).[12] These more hindered, branched derivatives were designed to gain more information on steric requirements for the occupancy of the LSD1 open cleft and to enhance selectivity towards LSD1 with respect to other flavoenzymes such as monoamine oxidases (MAOs). From these studies two prototypes emerged, 1a and 2a, which were further characterized through chemical investigations of their regio/stereoisomers.[13] Afterwards, to reduce the number of chiral centers, we designed new analogues of 1a by inserting two additional substituents at its benzamide portion (3a-n) (Figure 2). Finally, we explored chemical variations of the TCP motif to find new scaffolds for LSD1 inhibitor development (4a-h) (Figure 2).
Figure 2.

TCP-based LSD1 inhibitors described in this paper.
Here we report for all the derivatives 1–4 the LSD1 inhibitory activities, the effects on cell proliferation in MV4–11 and NB4 cell lines, two prototypes of acute myeloid leukemia (AML) and acute promyelocytic leukemia (APL) cell lines, respectively. We further report on the inhibitors’ ability to reactivate the expression of genes related to silencing by LSD1, as functional read-out of LSD1 inhibition in cells.
Results and Discussion
Design, synthesis and enzyme inhibition data
The chemical syntheses of 1a-h and 2a,c-o were already described by us.[12–13] For the preparation of 2b, the meta isomer of 2a, the known tert-butyl [2-(3-aminophenyl)cyclopropyl]carbamate 5[13a] was coupled with Z-phenylalanine in the presence of (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), triethylamine, and dry N,N-dimethylformamide (DMF) in N2 atmosphere to furnish the tert-butyl carbamate 6. Subsequent acidic deprotection of 6 with 4M HCl in 1.4-dioxane afforded the desired 2b (Scheme 1).
Scheme 1.
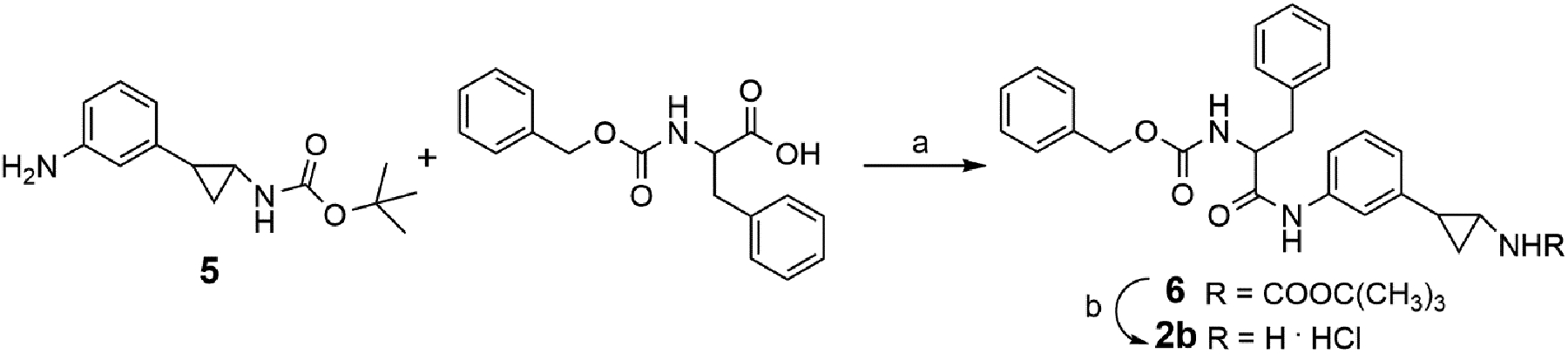
a) PyBOP, triethylamine, dry DMF, N2; b) 4M HCl in 1,4-dioxane, dry THF.
Since 2a-o contain three chiral centers, i.e. two on the cyclopropyl ring and the third on the Z-amino acid chain, we designed compounds 3a-n lacking the third stereogenic center and retaining a branched structure. We decorated the benzamide portion of 1a with two substituents, the N-benzyloxycarbonylamino moiety present also in 2a-o, added to the ortho, meta, or para position, and a second substituent, represented by a cyclic amine as well as a phenyl, phenoxy, or phenylthiol group (3a-l). Additionally, two further analogues were prepared carrying a C2-oxyacetamide function and a C5-benzamide moiety on the benzamide portion of 1a (3m,n).
Among compounds 3, the syntheses of 3b,d-g were reported previously by us.[14] The synthesis of 3a was accomplished by coupling the commercially available 4-{[(benzyloxy)carbonyl]amino}benzoic acid 7 with the tert-butyl [2-(4-aminophenyl)cyclopropyl]carbamate 8[12] in the presence of N-ethyl-N’-(3-N,N-dimethylaminopropyl)carbodiimide (EDC) hydrochloride in dichloromethane (DCM), followed by cleavage of the tert-butoxycarbonyl (Boc)-protected intermediate 9 with 4M HCl solution in 1,4-dioxane (Scheme 2A).
Scheme 2.
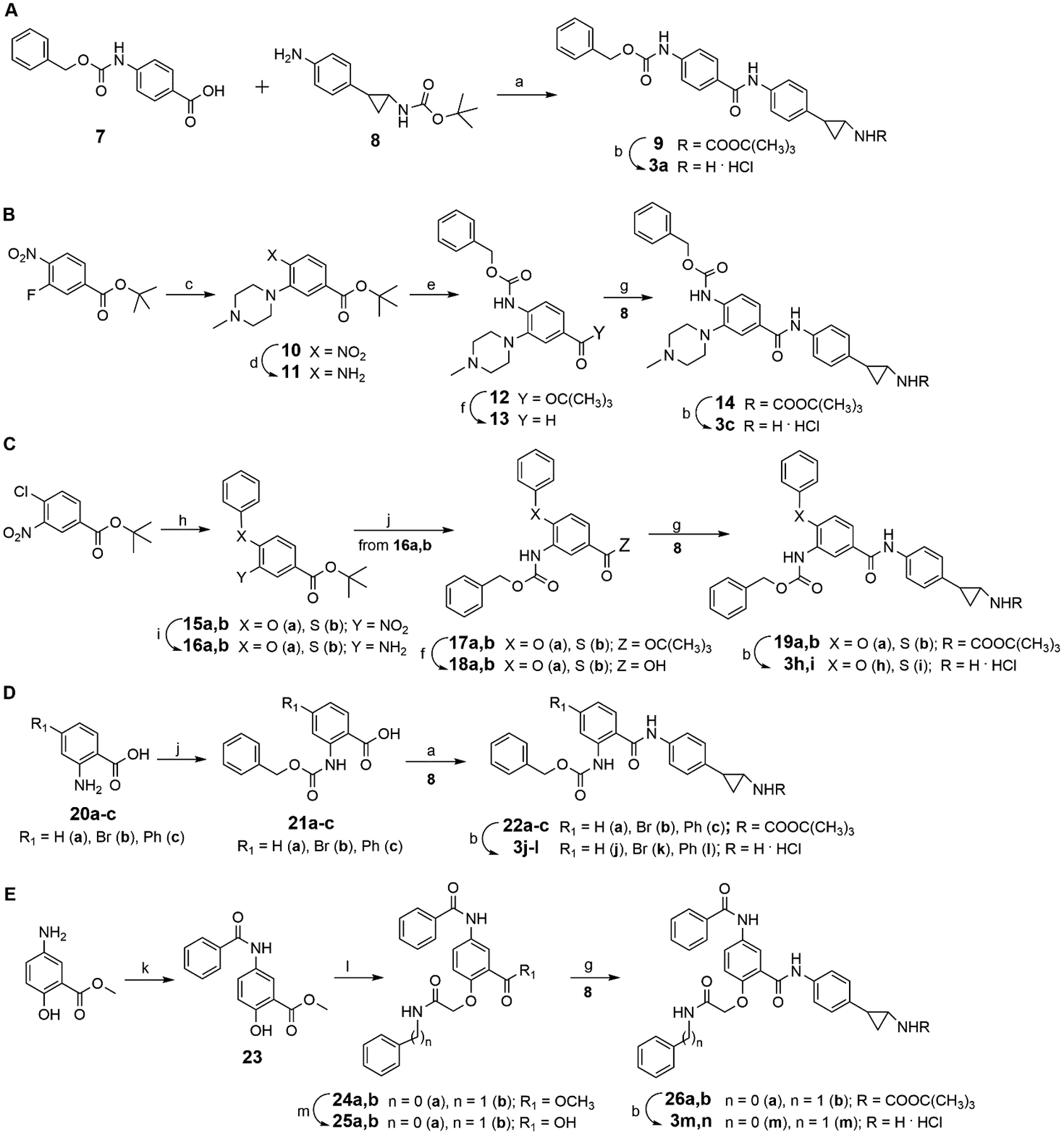
Route A: a) EDC hydrochloride, triethylamine, dry DCM; b) 4M HCl in dioxane, dry THF. Route B: c) N-methylpiperazine, dry potassium carbonate, dry DMF, 100 °C; d) H2, 10% Pd/C, MeOH; e) benzyl chloroformate, triethylamine, dry THF, 0 °C; f) trifluoracetic acid, dry DCM; g) PyBOP, triethylamine, dry DMF, N2; b) 4M HCl in dioxane, dry THF. Route C: h) phenol or thiophenol, dry potassium carbonate, dry DMF, 100 °C; i) H2, 10% Pd/C, EtOH; j) benzyl chloroformate, dry potassium carbonate, THF/H2O = 1:1, 0 °C; f) trifluoracetic acid, dry DCM; g) PyBOP, triethylamine, dry DMF, N2; b) 4M HCl in 1,4-dioxane, dry THF. Route D: j) benzyl chloroformate, dry potassium carbonate, THF/H2O = 1:1, 0 °C; a) EDC hydrochloride, triethylamine, dry DCM; b) 4M HCl in 1,4-dioxane, dry THF. Route E: k) benzoyl chloride, triethylamine, dry DCM; l) N-phenyl- or N-benzyl-2-bromoacetamide, dry potassium carbonate, dry CH3CN, 70 °C; m) 2N LiOH, dry THF, g) PyBOP, triethylamine, dry DMF, N2; b) 4M HCl in 1,4-dioxane, dry THF..
The synthesis of 3c was performed starting from tert-butyl 3-fluoro-4-nitrobenzoate, which was treated with N-methylpiperazine to afford the nitro ester 10. Further palladium-catalyzed reduction of the nitro group with hydrogen and subsequent reaction of the primary amino group (compound 11) with benzyl chloroformate in dry THF furnished the tert-butyl 4-Z-aminobenzoate 12. Such intermediate, after transformation into the corresponding carboxylic acid 13 with trifluoroacetic acid in DCM, was coupled with 8[12] in the presence of PyBOP and triethylamine in dry DMF to give the tert-butyl carbamate 14 which was converted by acidic treatment into the hydrochloride salt of the corresponding free amine 3c (Scheme 2B).
The reaction of the commercially available tert-butyl 4-chloro-3-nitrobenzoate with phenol or thiophenol in the presence of potassium carbonate in dry DMF at 100 °C furnished the nitro intermediates 15a,b, then converted in the corresponding amines 16a,b by palladium-catalyzed hydrogenation. Subsequent reaction of 16a,b with benzyl chloroformate in presence of potassium carbonate in a mixture of THF/water 1:1 furnished the tert-butyl 3-{[(benzyloxy)carbonyl]amino-4-phenoxy- and -phenylthiobenzoates 17a,b, which were converted into the corresponding carboxylic acids 18a,b by treatment with trifluoroacetic acid in dry DCM. The acidic intermediates 18a,b were then coupled with the amine 8[12] using PyBOP as activating agent in presence of triethylamine in dry DMF under nitrogen atmosphere. The obtained Boc-protected derivatives 19a,b were finally treated with 4M HCl in 1,4-dioxane and dry THF to give the desired compounds 3h,i (Scheme 2C).
The synthesis of 3j-l was undertaken by treating the commercially available amino acids 20a-c with benzyl chloroformate. The resulting 2-Z-aminobenzoic acids 21a-c were then coupled with 8[12] to furnish the Boc-protected derivatives 22a-c, which after acidic cleavage provided the desired 3j-l (Scheme 2D).
Reaction of methyl 2-hydroxy-5-aminobenzoate with benzoyl chloride gave the intermediate 23, which was treated with N-phenyl- or -benzyl-2-bromoacetamide to furnish the methyl esters 24a,b. Such intermediates were hydrolyzed with lithium hydroxide to afford the corresponding carboxylic acids 25a,b which, after coupling with 8[12] and removal of the Boc protection (4M HCl in 1,4-dioxane) from the obtained intermediates 26a,b gave the desired compounds 3m,n (Scheme 2E).
Afterwards, we applied some chemical modifications to the TCP structure by preparing compounds 4a-h, in which the primary amine group of TCP has been changed into secondary (N-methyl-TCP, 4a)[15] or tertiary (N,N-dimethyl-TCP, 4b)[16] amine, or transformed into carboxamide (4c)[15] or carbohydrazide (4d). The isomeric 1-phenylcyclopropan-1-amine (4e) and the related benzyl {1-[(4-(1-aminocyclopropyl)phenyl)amino]-1-oxo-3-phenylpropan-2-yl}carbamate hydrochloride (4f, a 2a isomer) were also prepared, together with the related 1-phenylcyclopropane-1-carboxamide (4g),[17] (1-phenylcyclopropyl)methanamine (4h),[17] and 2-phenylaziridin-1-amine (4i).[18] Compounds 4a,b,h,i were prepared according to the reported procedures, while 4c,d were obtained by treating the commercially available 2-phenylcyclopropane-1-carboxylic acid with PyBOP, 33% ammonia and triethylamine (for 4c), or its methyl ester with hydrazine hydrate (for 4d) (Scheme 3A).
Scheme 3.

Route A: a) PyBOP, 33% NH3, triethylamine, dry DCM/DMF 4/1; b) hydrazine hydrate, methanol. Route B: c) diphenylphosphoryl azide, triethylamine, dry tert-butanol, dry benzene, 105 °C, N2; d) 4M HCl in 1,4-dioxane, dry THF; a) PyBOP, 33% NH3, triethylamine, dry DCM/DMF 4/1. Route C: e) 50 % sodium hydroxide, tetrabutylammonium bromide, dry CH3CN, 40 °C; f) 96% sulfuric acid, reflux; c) diphenylphosphoryl azide, triethylamine, dry tert-butanol, dry benzene, 105 °C, N2; g) Pd/C, H2 30 psi, methanol; h) Z-Phe-OH, EDC hydrochloride, triethylamine, hydroxybenzotriazole, dry DCM; d) 4M HCl in 1,4-dioxane, dry THF.
Curtius reaction performed on 1-phenylcyclopropane-1-carboxylic acid using diphenylphosphoryl azide, triethylamine, and dry tert-butanol in dry benzene at 105 °C under N2 atmosphere furnished 4e after acidic hydrolysis of the tert-butyl carbamate 27, while treatment of the same carboxylic acid with PyBOP, 33% ammonia and triethylamine afforded the 1-phenylcyclopropane-1-carboxamide 4g (Scheme 3B). For the synthesis of 4f, first the commercially available 4-nitrophenylacetonitrile was converted into the corresponding cyclopropane intermediate 28 using 1,2-dibromoethane, tetrabutylammonium bromide and 50% sodium hydroxide, and then hydrolyzed under strongly acidic conditions to afford the carboxylic acid 29. Such intermediate was then converted through Curtius reaction into the corresponding nitro carbamate 30, which was in turn reduced to the amino intermediate 31 by palladium-catalyzed hydrogenation. Final coupling of 31 with Z-phenylalanine in the presence of EDC hydrochloride, triethylamine and hydroxybenzotriazole in dry DCM furnished the protected tert-butyl carbamate 32, which after acidic treatment provided 4f as a hydrochloride salt (Scheme 3C).
Compounds 1–4 were incubated with the human recombinant LSD1/CoREST enzymatic complex as well as with human MAO A and MAO B proteins to assess their potency and selectivity, and the results are expressed as IC50 values (for LSD1) or percentage of inhibition at 100 μM (for MAOs) and summarized in Table 1.
Table 1.
Biochemical profile of compounds 1–4[a].
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cpd | R | R1 | R2 | R3 | R4 | R5 | R6 | LSD1, IC50 (μM) | % inhibition at 100 μM | |
| MAO A | MAO B | |||||||||
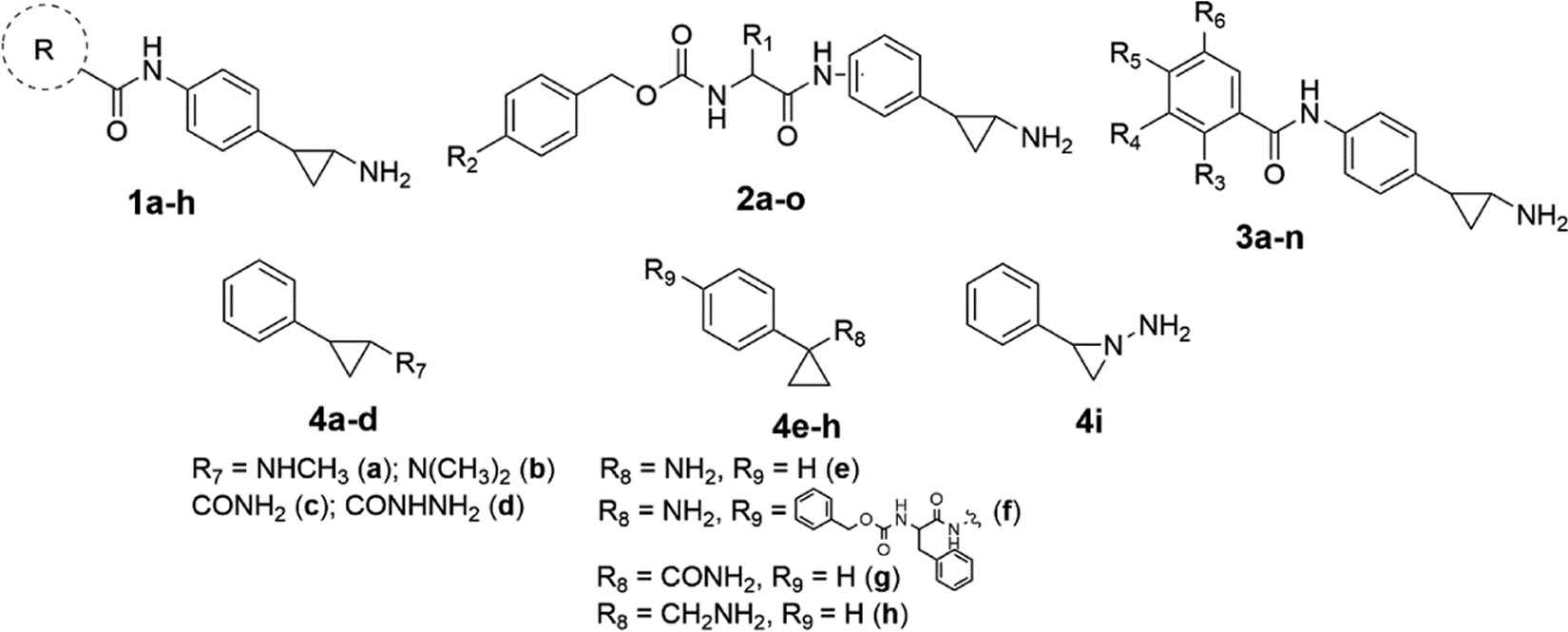
| 1a[b] |  |
0.019 ± 0.004 | 97% | NI[c] | ||||||
| 1b |  |
0.106 ± 0.020 | 96% | 37% | ||||||
| 1c |  |
0.024 ± 0.004 | 96% | 50% | ||||||
| 1d | 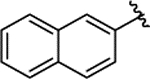 |
0.029 ± 0.005 | 95% | NI | ||||||
| 1e | 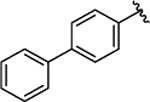 |
0.051 ± 0.008 | 2% | 8% | ||||||
| 1f | 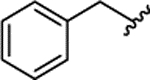 |
0.054 ± 0.010 | 96% | 33% | ||||||
| 1g |  |
0.038 ± 0.009 | 98% | 48% | ||||||
| 1h | 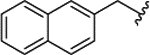 |
0.043 ± 0.010 | 89% | 17% | ||||||
| 2a[d] | 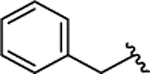 |
H | 0.149 ± 0.028 | 94% | 1% | |||||
| 2b[e] |
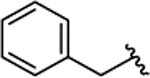 meta |
H | 18.1 ± 2.9 | |||||||
| 2c |  |
H | 0.459 ± 0.090 | |||||||
| 2d | 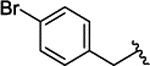 |
H | 0.230 ± 0.042 | |||||||
| 2e | 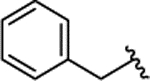 |
Br | 0.080 ± 0.026 | 71% | 29% | |||||
| 2f |  |
H | 0.236 ± 0.039 | |||||||
| 2g |  |
H | 0.130 ± 0.025 | 87% | 42% | |||||
| 2h | 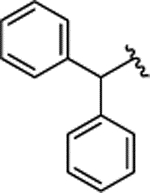 |
H | 0.280 ± 0.052 | |||||||
| 2i | 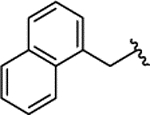 |
H | 0.173 ± 0.028 | |||||||
| 2j | 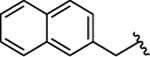 |
H | 0.767 ± 0.104 | |||||||
| 2k |  |
H | 0.190 ± 0.030 | |||||||
| 2l | 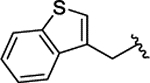 |
H | 0.083 ± 0.012 | |||||||
| 2m | 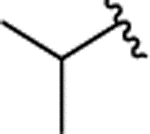 |
H | 0.400 ± 0.072 | |||||||
| 2n | 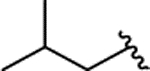 |
H | 0.236 ± 0.040 | |||||||
| 2o | 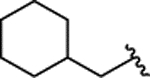 |
H | 0.581 ± 0.071 | |||||||
| 3a | H | H |  |
H | 0.090 ± 0.012 | 37% | 8% | |||
| 3b[f] | H | 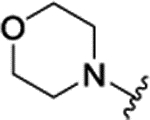 |
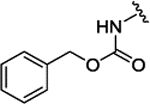 |
H | 0.075 ± 0.011 | 76% | 78% | |||
| 3c | H | 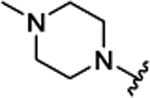 |
 |
H | 0.089 ± 0.010 | 69% | 55% | |||
| 3d[f] | H | 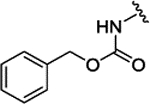 |
H | H | 0.043 ± 0.010 | 46% | NI | |||
| 3e[f] | H |  |
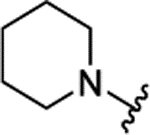 |
H | 0.307 ± 0.069 | |||||
| 3f[f] | H | 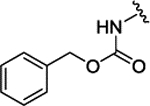 |
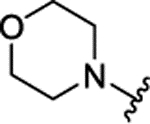 |
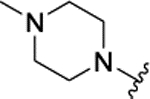
H | 0.061 ± 0.011 | 21% | 17% | |||
| 3g[f] | H |  |
 |
H | 0.089 ± 0.020 | 5% | 13% | |||
| 3h | H |  |
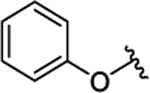 |
H | 0.143 ± 0.028 | |||||
| 3i | H | 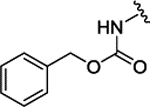 |
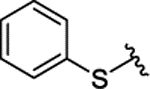 |
H | 0.073 ± 0.011 | |||||
| 3j |  |
H | H | H | 0.146 ± 0.022 | 72% | 17% | |||
| 3k | 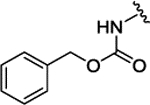 |
H | Br | H | 0.159 ± 0.034 | 94% | NI | |||
| 3l | 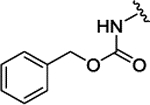 |
H | 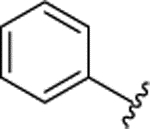 |
H | 0.135 ± 0.029 | |||||
| 3m |  |
H | H | 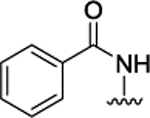 |
3.9 ± 0.2 | |||||
| 3n | 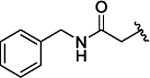 |
H | H | 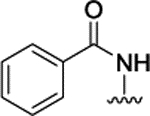 |
1.7 ± 0.05 | |||||
| 4a | 10.8 ± 1.1 | |||||||||
| 4b | 56.2 ± 7.8 | |||||||||
| 4c | NI | |||||||||
| 4d | NI | |||||||||
| 4e | NI | |||||||||
| 4f | NI | |||||||||
| 4g | NI | |||||||||
| 4h | NI | |||||||||
| 4i | NI | |||||||||
| TCP | 11.2 ± 1.8 | 79% | 85% | |||||||
Data represent mean values of at least two separate experiments in duplicate.
Ref. [13a], added for comparison.
NI, no inhibition.
Reff. [13b, 19], added for comparison; compounds 2a,c-o have the Z-aminoacylamino substituent inserted at the C4 position of the TCP phenyl ring.
C3-substituted regioisomer of 2a.
Ref. [14], added for comparison.
As regards to compounds 1, we already reported the high potency of 1a at LSD1 enzyme level.[12–13] The introduction of 1- or 2-naphthoylamine (1c,d) moiety at the C4-position of the TCP phenyl ring furnished low nanomolar potency for LSD1 inhibition, and the insertion at the amide function of longer substituents such as 4-biphenyl, benzyl, 1- or 2-naphthylmethyl moieties led to slightly less but still potent derivatives (1e-h) (Table 1). The benzyl [4-(2-aminocyclopropyl)phenyl]carbamate 1b displayed the lowest potency among the compounds of the series, with an IC50 7-fold higher than 1a. About selectivity, only the 4-biphenyl analogue 1e displayed no inhibition of both MAO-A and MAO-B at 100 μM; all the other compounds strongly inhibited MAO-A, while MAO-B was in general moderately or scarcely affected.
Among the TCP analogues bearing a Z-amino acylamine moiety in C4, we selected the Z-PheCONH derivative 2a (also called MC2580) as a scaffold for further derivatization[12, 13b] (Table 1). Similar to what previously observed with 1a and its benzamide moiety, the shift of the 2a amino acylamine chain from C4 to C3 (see the meta regioisomer 2b) decreased >120-fold the anti-LSD1potency. Replacement of the 2a branched benzyl group with phenyl (2c), 2-naphthylmethyl (2j), 2-propyl (2m), or cyclohexylmethyl (2o) moiety furnished derivatives up to 5-fold less potent than 2a, while 4-bromo- (2d) and 4-methoxybenzyl (2f), 2-phenylethyl (2g), diphenylmethyl (2h), 1-naphthylmethyl (2i), 1H-indol-3-yl (2k), or 2-methyl-1-propyl (2n) substituent had only slight modulatory effects on the inhibitory potency respect to the prototype. The chemical changes affording about 2-fold increase of potency against LSD1 were the introduction of the bromo atom at the C4 position of the benzyl carbamate (2e) and the replacement of the benzyl at Cα with the benzo[b]thiophen-3-yl group (2l). When some of compounds 2 were tested against MAOs, in general appeared more LSD1-selective than compounds 1, due to a slight drop of inhibiting activity against MAO-A.
The insertion of the Z-amino group to the benzamide portion of 1a was devised to generate a sort of chimeras between 1 and 2 (compounds 3), without the disadvantage of the third chiral center of 2. This modification led to highly active compounds, with the meta substituted (3d,f,g) slightly more potent than the corresponding para analogues (3a-c), and the ortho regiosomers (3j-l) definitively less effective (Table 1). The addition of a second substituent generally did not improve the inhibitory potency of the derivatives, and the insertion of a piperidine ring to the meta-substituted 3d (compound 3e) caused a 10-fold drop of potency respect to 3d (Table 1). Further design and synthesis of the related compounds 3m,n furnished LSD1 inhibitors with low micromolar potency (Table 1). When tested against MAOs, compounds 3 seemed to be in general more selective towards LSD1 than 1 and 2, as they mostly displayed scarce inhibition of both the amino oxidases (Table 1).
Final exploration of chemical modifications applied on TCP furnished fragments 4, totally devoid of LSD1/CoREST inhibitory activity. The only exceptions are the N-methyl- and N,N-dimethyl-TCP (4a and 4b, respectively), the first showing the same potency of TCP, the latter being 5-fold less effective (Table 1).
Antiproliferative activities of 1–3 on MV4–11 AML and NB4 APL cell lines
Previous studies described LSD1 playing a relevant role in human leukemias.[12–14, 20] Thus, 1–3 were tested in MV4–11 AML and in NB4 APL cell lines to determine their effects on cell proliferation. The relative IC50 values for antiproliferative activities by 1–3 are reported in Table 2.
Table 2.
Antiproliferative activities of 1–3 on MV4–11 AML and NB4 APL cell lines.
| Compd | IC50 (μM)[a] | |
|---|---|---|
| MV4–11 | NB4 | |
| 1a | >100 | >100 |
| 1b | 22.4 ± 3.8 | 25 ± 3.2 |
| 1c | 8.7 ± 1.9 | 14.4 ± 2.2 |
| 1d | 6.5 ± 0.7 | 9.4 ± 0.9 |
| 1e | 2.3 ± 0.04 | 7.5 ± 0.8 |
| 1f | 49.8 ± 7.7 | 42.4 ± 6.2 |
| 1g | 21.8 ± 3.1 | 17.8 ± 2.0 |
| 1h | 7.8 ± 1.0 | 16.4 ± 3.3 |
| 2a[b] | 10.2 ± 1.2 | 17.4 ± 2.9 |
| 2b | 24.2 ± 3.1 | 42.7 ± 5.8 |
| 2c | 52.3 ± 8.9 | 62.2 ± 9.2 |
| 2d | 4.6 ± 0.5 | 5.8 ± 0.8 |
| 2e | 5.3 ± 0.4 | 7.3 ± 0.6 |
| 2f | 5.0 ± 0.7 | 6.2 ± 0.5 |
| 2g | 8.7 ± 0.9 | 16.5 ± 2.6 |
| 2h | 5.0 ± 0.8 | 8.7 ± 1.0 |
| 2i | 7.6 ± 0.9 | 7.8 ± 0.8 |
| 2j | 15.4 ± 2.7 | 23.2 ± 4.2 |
| 2k | 16.0 ± 3.3 | 24.4 ± 3.9 |
| 2l | 6.8 ± 0.8 | 3.8 ± 0.04 |
| 2m | >100 | 70.4 ± 9.8 |
| 2n | 16.9 ± 2.5 | 46.7 ± 6.2 |
| 2o | 13.2 ± 2.2 | 30.3 ± 3.9 |
| 3a | 0.4 ± 0.01 | 0.6 ± 0.02 |
| 3b | 4.4 ± 0.3 | 7.4 ± 0.6 |
| 3c | 2.5 ± 0.1 | 0.9 ± 0.06 |
| 3d | 1.7 ± 0.02 | 1.3 ± 0.01 |
| 3e | 4.3 ± 0.3 | 5.2 ± 0.4 |
| 3f | 1.9 ± 0.08 | 1.5 ± 0.09 |
| 3g | 1.4 ± 0.06 | 3.2 ± 0.2 |
| 3h | 3.3 ± 0.3 | 5.4 ± 0.6 |
| 3i | 21.4 ± 3.3 | 23.2 ± 3.8 |
| 3j | 8.2 ± 1.2 | 8.1 ± 0.9 |
| 3k | 6.2 ± 0.7 | 9.3 ± 1.0 |
| 3l | 8.1 ± 1.0 | 7.6 ± 0.8 |
| 3m | 5.6 ± 1.1 | 5.5 ± 1.4 |
| 3n | 3.3 ± 0.6 | 3.8 ± 0.8 |
| TCP | >100 | >100 |
Data represent mean values of at least two separate experiments in duplicate.
Ref. [19], added for comparison.
Among 1a-h, all compounds displayed antiproliferative activities against the two leukemia cell lines higher than the prototype 1a. Derivatives bearing bulkier substituents such as 1- and 2-naphthyl (1c and 1d, respectively), 2-naphthylmethyl (1h) and mainly 4-biphenyl (1e) arrested MV4–11 cell growth at single-digit micromolar concentration (in the range 2.3 to 8.7 μM), while only 1d and 1e displayed similar level of potency against NB4 cells (Table 2). Among 2a-o, the shift of the Z-Phe substituent from para to meta position (2b), or the shortening of one of the two arms by replacing the Z-Phe with a Z-phenylglycine (2c) reduced the antiproliferative effect. By contrast, the insertion at the Cα of the Z-aminoacid unit of a 4-substituted benzyl group (2d,f) or a bulkier diphenyl (2h), 1-naphthylmethyl (2i) or benzo[b]thiophen-3-yl (2l) group, or introduction of a bromine into the Z group of 2a (2e) increased the inhibition of proliferation of the two tested leukemia cell lines in the range 3.8 to 8.7 μM [IC50 values for 2a: 10.2 (MV4–11) and 17.4 (NB4) μM] (Table 2). As regards to 3a-n, the introduction of substituent(s) at the C4-benzamide moiety of 1a afforded the most potent antiproliferative agents against MV4–11 and NB4 cells. Indeed, all the compounds 3a-n except for the 3-Z-amino-4-phenylthio derivative 3i exhibited IC50 values for both cell lines in the single-digit micromolar range. The most effective of this series were the 4- and 3-Z-amino substituted compounds 3a, 3c, 3d, 3f and 3g, with 3a and 3c being potent at submicromolar level against MV4–11 (3a) and NB4 (3a and 3c) cells. This generally agreed with their trend for LSD1 inhibition, with some exceptions for 3b and 3i, which although showing potent inhibition of LSD1 displayed less antiproliferative activity, probably due to scarce cell penetration and/or distribution.
LSD1 target modulation: effects of selected 1–3 derivatives on GFI-1b, ITGAM, and KCTD12 gene induction
To confirm that the most potent compounds in enzyme assay as well as in leukemia cells were effectively working through LSD1 inhibition in cells, we detected the levels of transcripts of three genes, GFI-1b, ITGAM/CD11b, and KCTD12, normally silenced by LSD1, in NB4 cells after treatment with selected 1–3 compounds. In particular, GFI-1b physically interacts with LSD1 in a complex acting as a transcriptional repressor and critical regulator of hematopoietic cell lineage development and differentiation,[5a, 21] ITGAM is a differentiation marker in leukemia and its gene target of LSD1,[13, 21b, 22] and KCTD12 is another gene reported to be under the control of LSD1.[20c, 22a]
The inhibitors were added at a concentration equal to their respective biochemical IC50 values, and after 24 h the GFI-1b, ITGAM and KCTD12 mRNA expression levels were measured by quantitative RT-PCR and graphed as fold-induction with respect to the control (DMSO). Data depicted in Figure 3 show that all the selected compounds 1d,e, 2d-f,h,i,l, and 3a-h,j-l increased the expression of the three gene targets in NB4 cells, demonstrating an inhibitory effect on LSD1 in cells.
Figure 3.

GFI-1b, ITGAM and KCTD12 gene expression induction in NB4 cells by selected 1–3 derivatives. The inhibitors were tested at their biochemical anti-LSD1 IC50 values (see Table 1). Fold-inductions were calculated with respect to DMSO, which was used as a control. Data for GFI-1b and ITGAM gene induction by 3b, 3f and 3g have been reported in ref. [14] and added here for comparison.
Preliminary metabolic stability evaluation of selected compounds 3a, 3d and 3g
Finally, the metabolic stability of selected compounds 3a, 3d and 3g was assessed using both human and mouse liver microsomes, to estimate their stability to phase I oxidative metabolism.
As reported in Table 3, all the compounds, according to the well stirred model,[23] showed a low/medium intrinsic clearance, both in mouse and human, suggestive of an high stability to phase I oxidative metabolism.
Table 3.
Antiproliferative activities of 1–3 on MV4–11 AML and NB4 APL cell lines.
| Compd | Metabolic stability data[a] | |
|---|---|---|
| Mouse Clint (μL/min/mg protein) | Human Clint (μL/min/mg protein) | |
| 3a | 7.1 ± 2.2 | 6.7 ± 1.1 |
| 3d | 4.3 ± 0.7 | 12.2 ± 1.7 |
| 3g | 11.2 ± 1.8 | lower than 3 |
Data are expressed as the mean of at least two determinations ± standard deviation.
Conclusion
LSD1 is a KDM epigenetic enzyme highly involved in initiation and development of hematological malignancies as well as solid tumor. In particular, AML cell lines harboring MLL and AML-ETO translocations[20c, 24] displayed high sensitivity to LSD1 inhibitors, and their combination with all-trans retinoic acid (ATRA) was proposed for the use in AML.[25] The analysis of crystal structures of the four stereoisomers of TCP and 4-bromo-TCP complexed with the enzyme revealed that strong inhibitory potency requires the large catalytic cleft of LSD1 to be filled through insertion of long, hindered substituent(s) at the C4 position of the phenyl ring of TCP.[12] Thus, we prepared three different series of TCP derivatives, bearing aroyl- and arylacetylamino (1a-h), Z-amino acylamino (2a-o), or double-substituted benzamide (3a-n) residues at C4 or C3 position of TCP as covalent LSD1 inhibitors. In addition, we applied some chemical changes on the TCP scaffold (compounds 4a-i) to find novel chemical entities suitable for designing novel LSD1 inhibitors. Compounds 1–4 were tested against LSD1, and some of them against MAO-A and -B, to assess their potency and selectivity. Most compounds 1 and 3 displayed IC50 values against LSD1 in the low nanomolar range, while compounds 2 were in general less effective (IC50 values in the submicromolar range) and compounds 4 were totally inactive. The different substituents in 1, 2 and 3 had mostly modulatory effects on the inhibitory potency of the compounds. Only compounds 1e, 3a, 3d, 3f and 3g displayed selectivity towards LSD1 with respect to MAOs, the others exhibiting typically low inhibition for MAO-B and high inhibition for MAO-A. Compounds 1–3 were tested in MV4–11 AML and NB4 APL cells, to determine their effects on cell proliferation. In general, MV4–11 cells were more sensitive than NB4 cells to these inhibitors, and only two compounds of the series 1 (1d,e) and six compounds of the series 2 (2d-f,h,i,l) showed single digit micromolar IC50 values against both the cell lines. Compounds 3, characterized by high potency and selectivity against LSD1 at enzyme level, were also the most potent in these assays: all these derivatives displayed single digit micromolar cell growth inhibition against both the cell lines, with the only exception of 3i, which was less effective. Among them, 3a exhibited submicromolar potency against both cell lines, and 3c against NB4 cells. Target modulation experiments in NB4 cells revealed that the most potent compounds at cellular levels were also able to increase the expression of three genes under the control of LSD1, thus demonstrating that the observed effects were actually due to LSD1 inhibition. Selected compounds 3a, 3d and 3g were profiled for mouse and human microsomal stability, and all of them showed low/medium intrinsic clearance, either in mouse or human microsomes, highlighting their good metabolic stability. Further studies will be performed on the new hit compounds 3a and 3c to assess their anticancer potential in different cancer contexts including cancer stem cells.[26]
Experimental Section
Chemistry
General:
Melting points were determined on a Buchi 530 melting point apparatus and are uncorrected. 1H-NMR spectra were recorded at 400 MHz on a Bruker AC 400 spectrometer; chemical shifts are reported in δ (ppm) units relative to the internal reference tetramethylsilane (Me4Si). Microwave-assisted reactions were performed with a Biotage Initiator (Uppsala, Sweden) high frequency microwave synthesizer working at 2.45 GHz, fitted with magnetic stirrer and sample processor; reaction vessels were Biotage microwave glass vials sealed with applicable cap; temperature was controlled through the internal IR sensor of the microwave apparatus. Low resolution mass spectra of final and intermediate compounds were recorded on an API-TOF Mariner by Perspective Biosystem (Stratford, Texas, USA), samples were injected by a Harvard pump using a flow rate of 5–10 μL/min, infused in the Electrospray system. All compounds were routinely checked by TLC and 1H-NMR. TLC was performed on aluminum backed silica gel plates (Merck DC, Alufolien Kieselgel 60 F254) with spots visualized by UV light. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure of ca. 20 Torr. Organic solutions were dried over anhydrous sodium sulfate. Elemental analysis has been used to determine purity of the described unknown final compounds, that is >95%. Analytical results are within ±0.40% of the theoretical values (Table S1 in Supporting Information). All chemicals were purchased from Sigma Aldrich s.r.l, Milan (Italy), or from TCI Europe N.V., Zwijndrecht (Belgium), and were of the highest purity.
General procedure for the synthesis of the tert-butyl carbamates 6, 14, 19a,b, and 26a,b. Example: Tert-butyl (2-(4-(4-benzamido-2-(2-oxo-2-(phenylamino)ethoxy)benzamido)phenyl) cyclopropyl)carbamate (26a).
Triethylamine (0.06 mL, 0.52 mmol) and PyBop (0.08 g, 0.16 mmol) were added to a solution of 25a (0.05 g, 0.13 mmol) in dry DMF under nitrogen atmosphere. The resulting solution was stirred at room temperature for 45 min, and then 8[12] (0.03 g, 0.13 mmol) was added under nitrogen atmosphere. After 2 h, the reaction was quenched with a saturated sodium chloride solution and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The product was purified by silica gel chromatography eluting with ethyl acetate/n-hexane 1.8/1 to obtain the pure intermediate 26a. Yield = 40 %; mp = 220–222 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, CDCl3) δ 1.42 (s, 9H, NHCOOC(CH3)3), 1.42–1.50 (m, 1H, cyclopropane CHH), 1.63–1.71 (m, 1H, cyclopropane CHH), 2.39–2.46 (m, 1H, cyclopropane CHNHBoc), 3.59–3.66 (m, 1H, cyclopropane PhCH), 4.74 (s, 2H, CH2CONHPh), 5.82–5.84 (d, 1H, NHCOOC(CH3)3), 7.04–7.10 (m, 1H, benzene proton), 7.15–7.17 (d, 1H, benzene proton), 7.19–7.23 (m, 2H, benzene protons), 7.32–7.38 (m, 2H, benzene protons), 7.42–7.59 (m, 7H, benzene protons), 7.68–7.72 (dd, 1H, benzene proton), 7.93–7.97 (m, 2H, benzene protons), 8.13–8.15 (d, 1H, aromatc proton), 9.23 (s, 1H, CONHPh-cyclopropyl), 9.45 (s, 1H, NHCOPh), 9.58 (s, 1H, CH2CONHPh) ppm. MS (ESI) m/z: 621 [M + H]+.
Benzyl (1-((3-(2-((tert-butoxycarbonyl)amino)cyclopropyl)phenyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (6):
Synthesised as described as for 26a. Yield = 66 %; mp = 151–153 °C; solvent = recrystallization toluene; 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 9H, NHCOOC(CH3)3), 1.46–1.53 (m, 1H, cyclopropane CHH), 1.66–1.73 (m, 1H, cyclopropane CHH), 2.42–2.48 (m, 1H, cyclopropane CHNHBoc), 2.96–3.09 (m, 2H, CHCH2Ph), 3.64–3.71 (m, 1H, cyclopropane PhCH), 4.50–4.57 (m, 1H, CHCH2Ph), 5.08 (s, 2H, NHCOOCH2Ph), 5.82–5.84 (d, 1H, NHCOOC(CH3)3), 6.29–6.31 (d, 1H, NHCOOCH2Ph), 7.12–7.16 (m, 1H, benzene proton), 7.19–7.36 (m, 12H, benzene protons), 7.51–7.55 (m, 1H, benzene proton), 9.56 (s, 1H, CONHPh) ppm. MS (ESI) m/z: 530 [M + H]+.
Benzyl (4-((4-(2-((tert-butoxycarbonyl)amino)cyclopropyl)phenyl)carbamoyl)-2-(4-methylpiperazin-1-yl)phenyl)carbamate (14):
Synthesised as described as for 26a. Yield = 74 %; mp = 117–119 °C; recrystallization solvent = cyclohexane; 1H NMR (400 MHz, CDCl3): δ 1.14–1.18 (m, 1H, cyclopropane CHH), 1.34–1.37 (m, 1H, cyclopropane CHH), 1.5 (s, 9H, NHCOOC(CH3)3), 2.25–2.31 (m, 1H, CHNHBoc), 2.76–2.83 (m, 4H, CH3N(CH2)2 piperazine-C3,C5), 3.12–3.17 (m, 4H, PhN(CH2)2 piperazine-C2,C6), 3.32–3.38 (m, 4H, NCH3 and PhCH), 4.89 (bs, 1H, NHCOOC(CH3)3), 5.23 (s, 2H, NHCOOCH2Ph), 7.13–7.16 (m, 2H, benzene protons), 7.34–7.48 (m, 5H, benzene protons), 7.70–7.72 (m, 2H, benzene protons), 7.79–7.86 (m, 2H, benzene protons), 8.01–8.03 (m, 1H, benzene proton), 8.74 (bs, 1H, NHCOOCH2Ph), 10.27 (bs, 1H, PhCONH) ppm. MS (ESI) m/z: 600 [M + H]+.
Benzyl (5-((4-(2-((tert-butoxycarbonyl)amino)cyclopropyl)phenyl)carbamoyl)-(2-phenoxyphenyl)carbamate (19a):
Synthesised from the carboxylic acid 18a as described as for 26a. Yield = 73 %; mp = 203–205 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, CDCl3) δ 1.42 (s, 9H, NHCOOC(CH3)3), 1.43–1.50 (m, 1H, cyclopropane CHH), 1.63–1.71 (m, 1H, cyclopropane CHH), 2.39–2.45 (m, 1H, cyclopropane CHNHBoc), 3.59–3.67 (m, 1H, cyclopropane PhCH), 5.18 (s, 2H, NHCOOCH2Ph), 5.82–5.84 (d, 1H, NHCOOC(CH3)3), 7.01–7.05 (m, 2H, benzene protons), 7.08–7.13 (m, 2H, benzene protons), 7.21–7.24 (m, 2H, benzene protons), 7.26–7.31 (m, 1H, benzene proton), 7.32–7.38 (m, 7H, benzene protons, NHCOOCH2Ph), 7.54–7.58 (m, 2H, benzene protons), 7.90–7.94 (dd, 1H, benzene proton), 8.38–8.40 (d, 1H, benzene proton), 8.55 (s, 1H, PhCONH) ppm. MS (ESI) m/z: 594 [M + H]+.
Benzyl (5-((4-(2-(tert-butoxycarbonylamino)cyclopropylphenyl)carbamoyl)-2-(phenylthio)phenyl)carbamate (19b):
Synthesised from the carboxylic acid 18b as described as for 26a. Yield = 77 %; mp = 189–191 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 9H, NHCOOC(CH3)3), 1.45–1.52 (m, 1H, cyclopropane CHH), 1.65–1.72 (m, 1H, cyclopropane CHH), 2.39–2.46 (m, 1H, cyclopropane CHNHBoc), 3.59–3.68 (m, 1H, cyclopropane PhCH), 5.16 (s, 2H, NHCOOCH2Ph), 5.84–5.86 (d, 1H, NHCOOC(CH3)3), 7.20–7.26 (m, 3H, benzene protons), 7.28–7.32 (m, 3H, benzene protons), 7.33–7.36 (m, 7H, benzene protons, NHCOOCH2Ph), 7.49–7.51 (d, 1H, benzene proton), 7.54–7.58 (m, 2H, benzene protons), 7.89–7.93 (dd, 1H, benzene proton), 8.07–8.09 (d, 1H, benzene proton), 8.57 (s, 1H, PhCONH) ppm. MS (ESI) m/z: 610 [M + H]+.
Tert-butyl (2-(4-(5-benzamido-2-(2-(benzylamino)-2-oxoethoxy)benzamido)phenyl) cyclopropyl)carbamate (26b):
Synthesised from the carboxylic acid 25b as described as for 26a. Yield = 30 %; mp = 215–218 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, CDCl3) δ 1.40 (s, 9H, NHCOOC(CH3)3), 1.44–1.52 (m, 1H, cyclopropane CHH), 1.64–1.73 (m, 1H, cyclopropane CHH), 2.34–2.47 (m, 1H, cyclopropane CHNHBoc), 3.58–3.67 (m, 1H, cyclopropane PhCH), 4.38–4.41 (m, 2H, CH2CONHCH2Ph), 4.54 (s, 2H, CH2CONHCH2Ph), 5.82–5.84 (d, 1H, NHCOOC(CH3)3), 7.14–7.16 (d, 1H, benzene proton), 7.19–7.35 (m, 7H, benzene protons), 7.42–7.59 (m, 6H, benzene protons, CH2CONHCH2Ph), 7.68–7.72 (dd, 1H, benzene proton), 7.93–7.97 (m, 2H, benzene protons), 8.13–8.15 (d, 1H, benzene proton), 9.25 (s, 1H, CONHPhcyclopropyl), 9.45 (s, 1H, NHCOPh), ppm. MS (ESI) m/z: 635 [M + H]+.
General procedure for the synthesis of the cyclopropylamines hydrochlorides 2b, 3a,c,h-n, 4f. Example: Benzyl (2-((4-(2-aminocyclopropyl)phenyl)carbamoyl)phenyl)carbamate hydrochloride (3j).
A 4M HCl solution in 1,4-dioxane (1.0 mL, 4.20 mmol) was added to a solution of 22a (0.06 g, 0.12 mmol) in THF (2 mL). The reaction was stirred at room temperature for 5 h; then the solvent was removed in vacuo and the product was triturated with a mixture of diethyl ether/petroleum ether and was filtered to obtain 3j as a pure colourless solid. Yield = 75 %; mp = 150–152 °C; recrystallization solvent = toluene; 1H NMR (400 MHz, DMSO-d6) δ 1.21–1.30 (m, 1H, cyclopropane CHH), 1.57–1.65 (m, 1H, cyclopropane CHH), 2.11–2.20 (m, 1H, cyclopropane CHNH3+), 2.90–2.98 (m, 1H, cyclopropane PhCH), 5.15 (s, 2H, NHCOOCH2Ph), 7.14–7.16 (d, 2H, benzene protons), 7.16–7.21 (t, 1H, benzene proton), 7.34–7.40 (m, 5H, benzene protons), 7.54–7.58 (t, 1H, benzene proton), 7.63–7.65 (d, 2H, benzene protons), 7.81–7.83 (d, 1H, benzene proton), 8.07–7.09 (d, 1H, benzene proton), 8.36 (bs, 3H, NH3+), 10.30 (s, 1H, PhCONH), 10.44 (s, 1H, -NHCOOCH2Ph) ppm. MS (ESI), m/z: 402 [M + H]+.
Benzyl (1-((3-(2-aminocyclopropyl)phenyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate hydrochloride (2b):
Synthesised from the tert-butyl carbamate 6 as described as for 3j. Yield = 74 %; mp = 210–212 °C; solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6) δ 1.26–1.34 (m, 1H, cyclopropane CHH), 1.61–1.69 (m, 1H, cyclopropane CHH), 2.09–2.20 (m, 1H, cyclopropane CHNH3+), 2.92–3.06 (m, 3H, cyclopropane PhCH, CHCH2Ph), 4.41–4.48 (m, 1H, CHCH2Ph), 5.05 (s, 2H, NHCOOCH2Ph), 7.02–7.08 (m, 1H, benzene proton), 7.13–7.17 (m, 8H, benzene protons), 7.21-.7.27 (m, 3H, benzene protons), 7.76–7.83 (m, 2H, benzene protons), 8.57 (bs, 3H, NH3+), 9.86 (s, 1H, CONHPh), 10.07 (s, 1H, NHCOOCH2Ph) ppm. MS (ESI), m/z: 430 [M + H]+.
Benzyl (4-((4-(2-aminocyclopropyl)phenyl)carbamoyl)phenyl)carbamate hydrochloride (3a):
Synthesised from the tert-butyl carbamate 9 as described as for 3j. Yield = 68 %; mp = 238–240 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6) δ 1.22–1.29 (m, 1H, cyclopropane CHH), 1.58–1.65 (m, 1H, cyclopropane CHH), 2.11–2.20 (m, 1H, cyclopropane CHNH3+), 2.90–2.98 (m, 1H, cyclopropane PhCH), 5.19 (s, 2H, NHCOOCH2Ph), 7.12–7.14 (d, 2H, benzene protons), 7.35–7.46 (m, 5H, benzene protons), 7.59–7.61 (d, 2H, benzene protons), 7.68–7.70 (d, 2H, benzene protons), 7.91 (d, 2H, benzene protons), 8.24 (bs, 3H, NH3+), 10.07 (s, 1H, PhCONH), 10.11 (s, 1H, NHCOOCH2Ph) ppm. MS (ESI), m/z: 402 [M + H]+.
Benzyl 4-(4-(2-aminocyclopropyl)phenylcarbamoyl)-2-(4-methylpiperazin-1-yl)phenylcarbamate hydrochloride (3c):
Synthesised from the tert-butyl carbamate 14 as described as for 3j. Yield = 60 %; mp = 190–192 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6) δ 1.21–1.29 (m, 1H, cyclopropane CHH), 1.58–1.65 (m, 1H, cyclopropane CHH), 2.11–2.20 (m, 1H, cyclopropane CHNH3+), 2.31 (s, 3H, NCH3), 2.69–2.79 (m, 4H, CH3N(CH2)2 at piperazine-C3,C5), 2.91–2.97 (m, 1H, cyclopropane PhCH), 3.16–3.30 (m, 4H, PhN(CH2)2 at piperazine-C2,C6), 5.23 (s, 2H, NHCOOCH2Ph), 7.13–7.16 (d, 2H, benzene protons), 7.34–7.48 (m, 5H, benzene protons), 7.70–7.72 (d, 2H, benzene protons), 7.79–7.86 (m, 2H, benzene protons), 8.01–8.03 (d, 1H, benzene proton), 8.56 (bs, 3H, NH3+), 8.74 (bs, 1H, PhCONH), 10.27 (bs, 1H, NHCOOCH2Ph) ppm; MS (ESI) m/z: 500 [M + H]+.
Benzyl (5-((4-(2-aminocyclopropyl)phenyl)carbamoyl)-2-phenoxyphenyl)carbamate hydrochloride (3h):
Synthesised from the tert-butyl carbamate 19a as described as for 3j. Yield = 68 %; mp = 198–200 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6) δ 1.21–1.30 (m, 1H, cyclopropane CHH), 1.58–1.66 (m, 1H, cyclopropane CHH), 2.10–2.20 (m, 1H, cyclopropane CHNH3+), 2.90–2.98 (m, 1H, cyclopropane PhCH), 5.13 (s, 2H, NHCOOCH2Ph), 6.94–6.96 (d, 2H, benzene protons), 7.03–7.05 (d, 2H, benzene protons), 7.12–7.16 (m, 3H, benzene protons), 7.39 (m, 7H, benzene protons), 7.70 (m, 3H, benzene protons), 8.30 (bs, 3H, NH3+), 9.33 (s, 1H, PhCONH), 10.19 (s, 1H, NHCOOCH2Ph) ppm. MS (ESI), m/z: 494 [M + H]+.
Benzyl (5-((4-(2-aminocyclopropyl)phenyl)carbamoyl)-2-(phenylthio)phenyl)carbamate hydrochloride (3i):
Synthesised from the tert-butyl carbamate 19b as described as for 3j. Yield = 68 %; mp = 227–229 °C; recrystallization solvent = methanol; 1H NMR (400 MHz, DMSO-d6) δ 1.22–1.29 (m, 1H, cyclopropane CHH), 1.57–1.66 (m, 1H, cyclopropane CHH), 2.11–2.20 (m, 1H, cyclopropane CHNH3+), 2.90–2.99 (m, 1H, cyclopropane PhCH), 5.18 (s, 2H, NHCOOCH2Ph), 6.94–6.96 (d, 2H, benzene protons), 7.03–7.05 (d, 2H, benzene protons), 7.12–7.21 (m, 3H, benzene protons), 7.34–7.40 (m, 7H, benzene protons), 7.64–7.72 (m, 3H, benzene protons), 8.30 (bs, 3H, NH3+), 9.79 (s, 1H, PhCONH), 10.56 (s, 1H, NHCOOCH2Ph) ppm. MS (ESI), m/z: 510 [M + H]+.
Benzyl (2-((4-(2-aminocyclopropyl)phenyl)carbamoyl)-5-bromophenyl)carbamate hydrochloridrate (3k):
Synthesised from the tert-butyl carbamate 22b as described as for 3j. Yield = 65 %; mp = 206–208 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6,) δ 1.22–1.29 (m, 1H, cyclopropane CHH), 1.58–1.66 (m, 1H, cyclopropane CHH), 2.11–2.20 (m, 1H, cyclopropane CHNH3+), 2.90–2.98 (m, 1H, cyclopropane PhCH), 5.23 (s, 2H, NHCOOCH2Ph), 7.16–7.24 (m, 1H, benzene proton), 7.33–7.42 (m, 4H, benzene protons), 7.45–7.49 (dd, 1H, benzene proton), 7.75–7.77 (d, 1H, benzene proton), 8.14–8.16 (d, 1H, benzene proton), 8.22–8.24 (d, 1H, PhCONH), 8.77 (bs, 3H, NH3+), 10.30 (s, 1H, NHCOOCH2Ph), ppm. MS (ESI), m/z: 480 [M + H]+.
Benzyl (4-((4-(2-aminocyclopropyl)phenyl)carbamoyl)-[1,1’-biphenyl]-3-yl)carbamate hydrochloride (3l):
Synthesised from the tert-butyl carbamate 22c as described as for 3j. Yield = 65 %; mp = 200–202 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6,) δ 1.22–1.30 (m, 1H, cyclopropane CHH), 1.57–1.63 (m, 1H, cyclopropane CHH), 2.11–2.20 (m, 1H, cyclopropane CHNH3+), 2.90–2.99 (m, 1H, cyclopropane PhCH), 5.15 (s, 2H, NHCOOCH2Ph), 7.16–2.21 (m, 2H, benzene protons), 7.23–2.28 (m, 1H, benzene proton), 7.32–7.39 (m, 4H, benzene protons), 7.41–7.43 (m, 1H, benzene proton), 7.46–7.51 (m, 2H, benzene protons), 7.56–7.60 (m, 4H, benzene protons), 7.65–7.78 (m, 1H, benzene proton), 7.98–8.00 (d, 1H, benzene proton), 8.21–8.23 (d, 1H, benzene proton), 8.59 (bs, 3H, NH3+), 9.91 (s, 1H, PhCONH), 10.17 (s, 1H, NHCOOCH2Ph) ppm. MS (ESI), m/z: 478 [M + H]+.
N-(4-(2-aminocyclopropyl)phenyl)-5-benzamido-2-(2-oxo-2-(phenylamino)ethoxy)benzamide hydrochloride (3m):
Synthesised from the tert-butyl carbamate 26a as described as for 3j. Yield = 70 %; mp = 190–192 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6) δ 1.22–1.29 (m, 1H, cyclopropane CHH), 1.58–1.66 (m, 1H, cyclopropane CHH), 2.11–2.20 (m, 1H, cyclopropane CHNH3+), 2.90–2.99 (m, 1H, cyclopropane PhCH), 4.74 (s, 2H, CH2CONHPh), 7.03–7.09 (m, 1H, benzene proton), 7.16–7.22 (m, 3H, benzene protons), 7.31–7.37 (m, 2H, benzene protons), 7.52–7.63 (m, 7H, benzene protons), 7.71–7.75 (dd, 1H, benzene proton), 7.93–7.99 (m, 2H, benzene protons), 8.19–7.21 (d, 1H, benzene proton), 8.94 (bs, 3H, NH3+), 9.60 (s, 1H, CONHPh-cyclopropyl), 9.93 (s, 1H, CH2CONHPh), 10.11 (s, 1H, NHCOPh) ppm. MS (ESI), m/z: 521 [M + H]+.
N-(4-(2-aminocyclopropyl)phenyl)-5-benzamido-2-(2-(benzylamino)-2-oxoethoxy)benzamide (3n):
Synthesized from the tert-butyl carbamate 26b as described as for 3j. Yield = 68%; mp = 218–220 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6) δ 1.21–1.28 (m, 1H, cyclopropane CHH), 1.57–1.66 (m, 1H, cyclopropane CHH), 2.10–2.20 (m, 1H, cyclopropane CHNH3+), 2.90–2.99 (m, 1H, cyclopropane PhCH), 4.34–4.39 (m, 2H, CH2CONHCH2Ph), 4.75 (s, 2H, CH2CONHCH2Ph), 7.16–7.19 (m, 3H, benzene protons), 7.25–7.31 (m, 5H, benzene protons), 7.51–7.63 (m, 5H, benzene protons), 7.74–7.78 (dd, 1H, benzene proton), 7.92–7.98 (m, 2H, benzene protons), 8.15–8.19 (m, 2H, benzene protons), 8.29 (m, 1H, CONHPh-cyclopropyl) 8.54 (bs, 3H, NH3+), 9.97 (s, 1H, CH2CONHCH2Ph), 10.18 (s, 1H, NHCOPh). ppm. MS (ESI), m/z: 535 [M + H]+.
Benzyl (1-((4-(1-aminocyclopropyl)-phenyl)-amino)-1-oxo-3-phenylpropan-2-yl)-carbamate hydrochloride (4f):
Synthesized from the tert-butyl carbamate 32 as described as for 3j. Yield = 93 %, mp > 250 °C; recrystallization solvent = methanol/THF; 1H-NMR (400 MHz, DMSO-d6): δ 1.16–1.18 (t, 2H, CH2CH2 cyclopropane), 1.32–1.35 (t, 2H, CH2CH2 cyclopropane), 2.83–2.89 (m, 1H, PhCHHCH(NH)CO), 3.01–3.06 (m, 1H, PhCHHCH(NH)CO), 4.40–4.45 (m, 1H, PhCH2CH(NH)CO), 4.97 (s, 2H, PhCH2O), 7.19–7.23 (t, 2H, benzene protons), 7.26–7.32 (t, 4H, benzene protons), 7.32–7.35 (t, 4H, benzene protons), 7.37–7.39 (d, 2H, benzene protons), 7.62–7.64 (d, 2H, benzene protons), 7.73–7.75 (d, 1H, PhCH2OCONH), 8.73 (br s, 3H, NH3+), 10.31 (s, 1H, CONHPh) ppm. MS (ESI), m/z: 466 [M + H]+.
General procedure for the synthesis of the tert-butyl carbamates 9, 22a-c. Example: Benzyl (2-((4-(2-((tert-butoxycarbonyl)amino)cyclopropyl)phenyl)carbamoyl)phenyl)carbamate (22a):
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC × HCl) (0.06 g, 0.32 mmol) and triethylamine (0.05 mL, 0.80 mmol) were added to a solution of 2-(((benzyloxy)carbonyl)amino)benzoic acid 21a (0.05 g, 0.20 mmol) and tert-butyl (2-(4-aminophenyl)cyclopropyl)carbamate 8[12] (0.07 g, 0.28 mmol) in dry DCM (3 mL). After 1 h the reaction was quenched with water and extracted with DCM (3 × 5 mL). Combined organic layers were washed with saturated sodium chloride solution (3 × 5 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The product was purified by silica gel chromatography eluting with ethyl acetate/n-hexane 1/3 to obtain the pure tert-butyl carbamate 22a. Yield = 85 %; mp = 212–215 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, CDCl3) δ 1.42 (s, 9H, NHCOOC(CH3)3), 1.44–1.51 (m, 1H, cyclopropane CHH), 1.64–1.73 (m, 1H, cyclopropane CHH), 2.39–2.45 (m, 1H, cyclopropane CHNHCOOC(CH3)3), 3.58–3.67 (m, 1H, cyclopropane PhCH), 5.18 (s, 2H, NHCOOCH2Ph), 5.81–5.83 (d, 1H, NHCOOC(CH3)3), 7.13–7.24 (m, 3H, benzene protons), 7.26–7.31 (m, 1H, benzene proton), 7.32–7.36 (m, 4H, benzene protons), 7.45–7.51 (m, 1H, benzene proton), 7.53–7.58 (m, 2H, benzene protons), 7.74–7.89 (m, 1H, benzene proton), 8.27–8.31 (dd, 1H, benzene proton), 8.82 (s, 1H, PhCONH), 9.35 (s, 1H, NHCOOCH2Ph) ppm. MS (ESI) m/z: 502 [M + H]+.
Benzyl (4-((4-(2-((tert-butoxycarbonyl)amino)cyclopropyl)phenyl)carbamoyl)phenyl) carbamate (9):
Synthesised from the carboxylic acid 7 as described as for 22a. Yield = 80 %; mp = 203–205 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, CDCl3) δ 1.42 (s, 9H, NHCOOC(CH3)3), 1.45–1.52 (m, 1H, cyclopropane CHH), 1.65–1.72 (m, 1H, cyclopropane CHH), 2.40–2.46 (m, 1H, cyclopropane CHNHCOOC(CH3)3), 3.49–3.66 (m, 1H, cyclopropane PhCH), 5.16 (s, 2H, NHCOOCH2Ph), 5.82–5.84 (d, 1H, NHCOOC(CH3)3), 7.20–7.24 (m, 2H, benzene protons), 7.26–7.32 (m, 1H, benzene proton), 7.33–7.36 (m, 4H, benzene protons), 7.55–7.58 (m, 2H, benzene protons); 7.61–7.64 (m, 2H, benzene protons), 7.96–7.99 (m, 2H, benzene protons), 8.70 (s, 1H, NHCOOCH2Ph), 9.06 (s, 1H, PhCONH) ppm. MS (ESI) m/z: 502 [M + H]+.
Benzyl (5-bromo-2-((4-(2-((tert-butoxycarbonyl)amino)cyclopropyl)phenyl)carbamoyl)phenyl) carbamate (22b):
Synthesised from the carboxylic acid 21b as described as for 22a. Yield = 80 %; mp = 222–223 °C; recrystallization solvent = methanol; 1H NMR (400 MHz, CDCl3) δ 1.42 (s, 9H, -NHCOOC(CH3)3), 1.44–1.52 (m, 1H, cyclopropane CHH), 1.65–1.73 (m, 1H, cyclopropane CHH), 2.39–2.46 (m, 1H, cyclopropane CHNHCOOC(CH3)3), 3.58–3.66 (m, 1H, cyclopropane PhCH), 5.16 (s, 2H, NHCOOCH2Ph), 5.83–5.85 (d, 1H, NHCOOC(CH3)3), 7.19–7.24 (m, 2H, benzene protons), 7.26–7.31 (m, 1H, benzene proton), 7.33–7.36 (m, 4H, benzene protons), 7.46–7.50 (dd, 1H, benzene proton), 7.54–7.58 (m, 2H, benzene protons), 7.84–7.86 (d, 1H, benzene proton), 8.14–8.16 (d, 1H, benzene proton), 9.14 (s, 1H, PhCONH), 9.44 (s, 1H, NHCOOCH2Ph) ppm. MS (ESI) m/z: 580 [M + H]+.
Benzyl (4-((4-(2-((tert-butoxycarbonyl)amino)cyclopropyl)phenyl)carbamoyl)-[1,1’-biphenyl]-3-yl)carbamate (22c):
Synthesised from the carboxylic acid 21c as described as for 22a. Yield = 82 %; mp = 217–219 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, CDCl3) δ 1.40 (s, 9H, NHCOOC(CH3)3), 1.44–1.53 (m, 1H, cyclopropane CHH), 1.64–1.72 (m, 1H, cyclopropane CHH), 2.39–2.46 (m, 1H, cyclopropane CHNHBoc), 3.58–3.65 (m, 1H, cyclopropane PhCH), 5.18 (s, 2H, NHCOOCH2Ph), 5.84–5.86 (d, 1H, NHCOOC(CH3)3), 7.20–7.24 (m, 2H, benzene protons), 7.26–7.31 (m, 1H, benzene proton), 7.32–7.40 (m, 5H, benzene protons), 7.42–7.49 (m, 2H, benzene protons), 7.53–7.59 (m, 4H, benzene protons), 7.61–7.65 (m, 1H, benzene proton), 7.96–7.98 (d, 1H, benzene proton), 8.00–8.02 (d, 1H, benzene proton), 9.11 (s, 1H, -PhCONH), 9.36 (s, 1H, NHCOOCH2Ph) ppm. MS (ESI) m/z: 578 [M + H]+.
Tert-butyl 3-(4-methylpiperazin-1-yl)-4-nitrobenzoate (10):
A suspension of tert-butyl 3-fluoro-4-nitrobenzoate (7.76 mmol, 2.00 g), dry potassium carbonate (23.28 mmol, 3.22 g) and N-methylpiperazine (23.28 mmol, 2.58 mL) was stirred in dry DMF (10 mL) at 90 °C for 5 h in a sealed tube. After this time, the reaction was quenched by water (50 mL) and extracted with ethyl acetate (3 × 30 mL), washed with brined (2 × 50 mL) and dried with sodium sulfate. The collected organic phases were concentrated, and the residue was purified by chromatography on silica gel 60 eluting with ethyl acetate to obtain pure 10 as a yellow solid. Yield = 76 %; mp = 153–155 °C; recrystallization solvent = toluene; 1H NMR (400 MHz, CDCl3) 1.60 (s, 9H, COOC(CH3)3), 2.37 (s, 3H, NCH3), 2.57–2.60 (t, 4H, CH3N(CH2)2 piperazine-C3,C5), 3.18–8.21 (t, 4H, PhN(CH2)2 piperazine-C2,C6), 7.07–7.09 (d, 1H, benzene proton), 8.03–8.05 (d, 1H, benzene proton), 8.37 (s, 1H, benzene proton) ppm. MS (ESI) m/z: 322 [M + H]+.
Tert-butyl 4-amino-3-(4-methylpiperazin-1-yl)benzoate (11):
A suspension of tert-butyl 3-(4-methylpiperazin-1-yl)-4-nitrobenzoate 10 (2.49 mmol, 0.8 g) in methanol (30 mL) and 10% palladium on charcoal (0.12 mmol, 0.13 g) was placed in a Parr apparatus and hydrogenated at 50 psi and 25 °C for 5 h. Afterwards, palladium was filtered and methanol was evaporated to afford an oily residue that was chromatographed over silica gel by eluting with chloroform:methanol 10:1 to provide the amine ester derivative 11 as a yellow solid. Yield = 77 %; mp = 128–130 °C; recrystallization solvent = cyclohexane; 1H NMR (400 MHz, CDCl3): δ 1.58 (s, 9H, COOC(CH3)3), 2.39 (s, 3H, NCH3), 2.56–2.61 (t, 4H, CH3N(CH2)2 piperazine-C3,C5), 2.95–2.99 (t, 4H, PhN(CH2)2 piperazine C2,C6), 4.35 (bs, 2H, PhNH2), 6.68–2.70 (d, 1H, benzene proton), 7.62–7.64 (d, 1H, benzene proton), 7.71 (s, 1H, benzene proton) ppm. MS (ESI) m/z: 292 [M + H]+.
Tert-butyl 4-(benzyloxycarbonylamino)-3-(4-methylpiperazin-1-yl)benzoate (12):
Benzyl chloroformate (2.1 mmol, 0.3 mL) was slowly added to a solution of tert-butyl 4-amino-3-(4-methylpiperazin-1-yl)benzoate 11 (1.72 mmol; 0.50 g) in dry THF (10 mL) and triethylamine (2.1 mmol, 0.29 mL) at 0 °C. The resulting mixture was stirred at room temperature for 1.5 h, then was quenched with water (20 mL) and extracted with DCM (3 × 20 mL). The organic phases were washed with brine (2 × 50 mL), dried by sodium sulfate and concentrated to afford a residue that was chromatographed on silica gel eluting with ethyl acetate/chloroform 1/1 to provide pure 12 as an oil. Yield = 72 %; 1H NMR (400 MHz, CDCl3): δ 1.60 (s, 9H, COOC(CH3)3), 2.39 (s, 3H, NCH3), 2.60–2.64 (t, 4H, CH3N(CH2)2 piperazine-C3,C5), 2.89–2.93 (t, 4H, PhN(CH2)2 piperazine C2,C6), 5.28 (s, 2H, NHCOOCH2Ph), 7.15–7.17 (d, 1H, benzene proton), 7.37–7.47 (m, 5H, benzene protons), 7.69–7.72 (m, 2H, benzene protons), 8.69 (bs, 1H, NHCOOCH2Ph) ppm. MS (ESI) m/z: 426 [M + H]+.
General procedure for the synthesis of the benzoic acids 13 and 18a,b. Example: 4-(Benzyloxycarbonylamino)-3-(4-methylpiperazin-1-yl)benzoic acid (13):
A solution of tert-butyl 4-(benzyloxycarbonylamino)-3-(4-methylpiperazin-1-yl)benzoate 12 (0.47 mmol, 0.2 g) and trifluoroacetic acid (9.4 mmol, 0.72 mL) in dry DCM (5 mL) was stirred at room temperature overnight. The solvent was removed, and the crude residue was purified on silica gel chromatography eluting with ethyl acetate/chloroform 1/1 to furnish the pure 13 as a colorless solid. Yield = 83%; mp = 191–193 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6) δ 2.87 (s, 3H, NCH3), 2.99–3.12 (m, 4H, CH3N(CH2)2 piperazine-C3,C5), 3.44–3.48 (t, 4H, PhN(CH2)2 piperazine-C2,C6), 5.23 (s, 2H, NHCOOCH2Ph), 7.36–7.47 (m, 5H, benzene protons), 7.70 (s, 1H, benzene proton), 7.75–7.77 (d, 1H, benzene proton), 8.04–8.06 (d, 1H, benzene proton), 8.76 (bs, 1H, NHCOOCH2Ph), 9.72 (bs, 1H, NH+), 12.88 (bs, 1H, COOH) ppm. MS (ESI) m/z: 368 [M - H]−.
3-(((Benzyloxy)carbonyl)amino)-4-phenoxybenzoic acid (18a):
Synthesized from 17a as described as for 13. Yield = 71 %; mp = 235–237 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6): δ 5.15 (s, 2H, NHCOOCH2Ph), 7.01–7.03 (d, 1H, benzene proton), 7.05–7.13 (m, 3H, benzene protons), 7.25–7.31 (m, 1H, benzene proton), 7.31–7.39 (m, 6H, benzene protons), 7.77–7.81 (dd, 1H, benzene proton), 8.44 (s, 1H, NHCOOCH2Ph), 8.44–8.46 (d, 1H, benzene proton), 12.41 (bs, 1H, COOH) ppm. MS (ESI) m/z: 362 [M - H]−.
3-(((benzyloxy)carbonyl)amino)-4-(phenylthio)benzoic acid (18b):
Synthesized from 17b as described as for 13. Yield = 68 %; mp = 221–223 °C; recrystallization solvent = acetonitrile/methanol; 1H NMR (400 MHz, DMSO-d6): δ 5.14 (s, 2H, NHCOOCH2Ph), 7.25–7.37 (m, 10H, benzene protons), 7.41–7.43 (d, 1H, benzene proton), 7.80–7.85 (dd, 1H, benzene proton), 8.46–8.48 (d, 1H, aomatic proton), 9.44 (s, 1H, NHCOOCH2Ph), 12.65 (bs, 1H, COOH) ppm. MS (ESI) m/z: 378 [M - H]−.
General procedure for the synthesis of tert-butyl 3-nitro-4-phenoxybenzoate (15a) and tert-butyl 3-nitro-4-(phenylthio)benzoate (15b). Example: Tert-butyl 3-nitro-4-(phenylthio)benzoate (15b):
Benzenethiol (0.15 g, 1.40 mmol) and anhydrous potassium carbonate (0.48 g, 3.49 mmol) were added to a solution of tert-butyl 4-chloro-3-nitrobenzoate (0.30 g, 1.16 mmol) in dry DMF. The resulting mixture was stirred at 100 °C for 5 h, then the reaction was quenched with water and extracted with ethyl acetate (3 × 10 mL). Combined organic layers were washed with saturated solution of sodium chloride (2 × 10 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The crude residue was purified on silica gel chromatography eluting with ethyl acetate/n-hexane 1/3 to furnish the product 15b. Yield = 70%; mp = 124–126 °C; recrystallization solvent = cyclohexane; 1H NMR (400 MHz, CDCl3): δ 1.57 (s, 9H, COOC(CH3)3), 7.20–7.26 (m, 1H, benzene proton), 7.30–7.37 (m, 4H, benzene protons), 7.49–7.51 (d, 1H, benzene proton), 8.17–8.21 (dd, 1H, benzene proton), 8.60–8.62 (d, 1H, benzene proton) ppm. MS (ESI) m/z: 332 [M + H]+.
Tert-butyl 3-nitro-4-phenoxybenzoate (15a):
Synthesized as described for 15b. Yield = 72 %; mp = 135–137 °C; recrystallization solvent = cyclohexane/toluene; 1H NMR (400 MHz, CDCl3): δ 1.55 (s, 9H, COOC(CH3)3), 6.99–7.03 (m, 2H, benzene protons), 7.07–7.13 (m, 1H, benzene proton), 7.16–7.18 (d, 1H, benzene proton), 7.32–7.37 (m, 2H, benzene protons), 8.17–8.21 (dd, 1H, benzene proton), 8.50–8.52 (d, 1H, benzene proton) ppm. MS (ESI) m/z: 316 [M + H]+.
General procedure for the synthesis of tert-butyl 3-amino-4-phenoxybenzoate (16a) and tert-butyl 3-amino-4-(phenylthio)benzoate (16b). Example: Tert-butyl 3-amino-4-phenoxybenzoate (16a):
10% Palladium on charcoal (0.03 g, 0.03 mmol) was added under nitrogen atmosphere to a solution of 15a (0.20 g, 0.63 mmol) in dry ethanol (50 mL). The resulting mixture was degassed with nitrogen for 3 min, followed by hydrogenation by using a Parr apparatus at 30 psi for 1 h. After this time, the mixture was filtered and concentrated in vacuo to obtain the pure colourless solid 16a. Yield = 95 %; mp = 122–124 °C; recrystallization solvent = cyclohexane; 1H NMR (400 MHz, DMSO-d6): δ 1.56 (s, 9H, COOC(CH3)3), 4.49 (s, 2H, NH2), 6.68–6.70 (d, 1H, benzene proton), 7.00–7,04 (m, 2H, benzene protons), 7.08–7.13 (m, 1H, benzene proton), 7.32–7.38 (m, 2H, benzene protons), 7.44–7.46 (d, 1H, benzene proton), 7.50–7.53 (m, 1H, benzene proton) ppm. MS (ESI) m/z: 286 [M + H]+.
Tert-butyl 3-amino-4-(phenylthio)benzoate (16b):
Synthesized from 15b as described for 16a. Yield = 72 %; mp = 112–114 °C; recrystallization solvent = cyclohexane; 1H NMR (400 MHz, DMSO-d6): δ 1.55 (s, 9H, COOC(CH3)3), 4.78 (s, 2H, NH2), 7.21–7.26 (m, 1H, benzene proton), 7.28–7.36 (m, 4H, benzene protons), 7.38–7.40 (d, 1H, benzene proton), 7.46–7.48 (d, 1H, benzene proton), 7.78–7.82 (dd, 1H, benzene proton) ppm. MS (ESI) m/z: 302 [M + H]+.
General procedure for the synthesis of benzyl carbamates 17a,b and 21a-c. Example: Tert-butyl 3-(((benzyloxy)carbonyl)amino)-4-(phenylthio)benzoate (17b):
Dry potassium carbonate (0.10 g, 0.70 mmol) was added to a solution of 16b (0.15 g, 0.50 mmol) in dry THF (5 mL). The resulted mixture was stirred at room temperature for 15 min, then benzyl chloroformate (0.08 mL, 0.60 mmol) was added dropwise at 0 °C to the mixture. After 2 h, the reaction was quenched with water and extracted with ethyl acetate (3 × 10 mL). Combined organic layers were washed with saturated sodium chloride solution (3 × 20 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The product was triturated in petroleum ether and the colourless solid 17b was filtered. Yield = 63 %; mp = 123–125 °C; recrystallization solvent = cyclohexane; 1H NMR (400 MHz, CDCl3): δ 1.55 (s, 9H, COOC(CH3)3), 5.16 (s, 2H, NHCOOCH2Ph), 7.21–7.26 (m, 1H, benzene proton), 7.27–7.30 (m, 1H, benzene proton), 7.30–7.32 (m, 2H, benzene protons), 7.33–7.36 (m, 6H, benzene protons), 7.47–7.49 (d, 1H, benzene proton), 7.85–7.89 (dd, 1H, benzene proton), 8.33 (s, 1H, NHCOOCH2Ph), 8.35–8.37 (d, 1H, benzene proton) ppm. MS (ESI) m/z: 436 [M + H]+.
Tert-butyl 3-(((benzyloxy)carbonyl)amino)-4-phenoxybenzoate (17a):
Synthesized from 16a as described as for 17b. Yield = 67 %; mp = 132–134 °C; recrystallization solvent = cyclohexane; 1H NMR (400 MHz, CDCl3): δ 1.54 (s, 9H, COOC(CH3)3), 5.14 (s, 2H, NHCOOCH2Ph), 7.00–7.04 (m, 2H, benzene proton), 7.06–7.13 (m, 2H, benzene protons), 726–7.31 (m, 1H, benzene proton), 7.31–7.37 (m, 6H, benzene protons), 7.64–7.68 (dd, 1H, benzene proton), 8.35 (s, 1H, NHCOOCH2Ph), 8.44–8.46 (d, 1H, benzene proton) ppm. MS (ESI) m/z: 420 [M + H]+.
2-(((benzyloxy)carbonyl)amino)benzoic acid (21a):
Synthesized from 20a as described as for 17b. Yield = 70 %; mp = 205–208 °C; recrystallization solvent = acetonitile/methanol; 1H NMR (400 MHz, DMSO-d6): δ 5.16 (s, 2H, NHCOOCH2Ph), 7.14–7.20 (m, 1H, benzene proton), 7.25–7.31 (m, 1H, benzene proton), 7.32–7.37 (m, 4H, benzene protons), 7.52–7.58 (m, 1H, benzene proton), 7.98–8.03 (dd, 1H, benzene proton), 8.06–8.09 (dd, 1H, benzene proton), 10.27 (s, 1H, NHCOOCH2Ph), 13.29 (bs, 1H, COOH) ppm. MS (ESI) m/z: 270 [M - H]−.
2-(((Benzyloxy)carbonyl)amino)-4-bromobenzoic acid (21b):
Synthesized from 20b as described as for 17b. Yield = 60 %; mp = >250 °C; recrystallization solvent = methanol; 1H NMR (400 MHz, DMSO-d6): δ 5.14 (s, 2H, NHCOOCH2Ph), 7.25–7.31 (m, 1H, benzene proton), 7.32–7.36 (m, 4H, benzene protons), 7.39–7.44 (dd, 1H, benzene proton), 7.91–7.93 (d, 1H, benzene proton), 8.14–8.16 (d, 1H, aomatic proton), 10.37 (s, 1H, NHCOOCH2Ph), 13.22 (bs, 1H, COOH) ppm. MS (ESI) m/z: 348 [M - H]−.
3-(((benzyloxy)carbonyl)amino)-[1,1’-biphenyl]-4-carboxylic acid (21c):
Synthesized from 20c as described as for 17b. Yield = 77 %; m.p. = >250 °C; recrystallization solvent = methanol; 1H NMR (400 MHz, DMSO-d6): δ 5.15 (s, 2H, NHCOOCH2Ph), 7.25–7.31 (m, 1H, benzene proton), 7.32–7.42 (m, 5H, benzene protons), 7.43–7.49 (m, 2H, benzene protons), 7.56–7.60 (m, 2H, benzene protons), 7.63–7.67 (m, 1H, benzene protons), 7.79–7.81 (d, 1H, benzene proton), 8.19–8.21 (d, 1H, benzene proton), 10.33 (s, 1H, NHCOOCH2Ph), 13.25 (bs, 1H, COOH) ppm. MS (ESI) m/z: 346 [M - H]−.
Methyl 5-benzamido-2-hydroxybenzoate (23):
A solution of methyl 5-amino-2-hydroxybenzoate (0.20 g, 1.46 mmol) and triethylamine (0.22 mL, 1.60 mmol) in dry DCM (4.0 mL) was stirred at room temperature for 15 min, then the solution was cooled to 0 °C and benzoyl chloride (0.17 mL, 1.46 mmol) was added dropwise. After 2 h, the reaction was quenched with water and extracted with DCM (3 × 10 mL). The combined organic layers were washed with a saturated solution of sodium chloride (2 × 20 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The extract was triturated in diethyl ether and the product 23, precipitated as colorless solid, was filtered and rinsed with diethyl ether. Yield = 81 %; mp = 162–164 °C; recrystallization solvent = toluene; 1H NMR (400 MHz, CDCl3) δ 3.86 (s, 3H, COOCH3), 7.01–7.03 (d, 1H, benzene proton), 7.42–7.55 (m, 4H benzene protons), 7.93–7.97 (m, 2H, benzene portons), 8.11–8.13 (d, 1H, benzene proton), 9.76 (s, 1H, NHCOPh), 10.69 (s, 1H, OH) ppm. MS (ESI) m/z: 272 [M +H]+.
General procedure for the synthesis of methyl 5-benzamido-2-(2-oxo-2-(phenylamino)ethoxy)benzoate (24a) and methyl 5-benzamido-2-(2-(benzylamino)-2-oxoethoxy)benzoate (24b). Example: Methyl 5-benzamido-2-(2-oxo-2-(phenylamino)ethoxy)benzoate (24a):
Dry potassium carbonate (0.12 g, 0.88 mmol) and 2-bromo-N-phenylacetamide (0.16 g, 0.74 mmol) were added to a solution of 23 (0.20 g, 0.74 mmol) in dry acetonitrile (5 mL). The reaction was stirred at 70 °C overnight then was quenched with water (30 mL), and the precipitated product was filtered and dried. The solid was purified on silica gel chromatography eluting with ethyl acetate/n-hexane 1/2 to furnish pure 24a. Yield = 90 %; mp = 170–173 °C; recrystallization solvent = toluene/acetonitrile; 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H, COOCH3), 4.73 (s, 2H, CH2CONHPh), 7.04–7.10 (m, 1H, benzene proton), 7.15–7.17 (d, 1H, benzene proton), 7.32–7.38 (m, 2H, benzene protons), 7.42–7.58 (m, 5H, benzene portons), 7.64–7.68 (dd, 1H, benzene proton), 7.93–7.98 (m, 2H, benzene protons), 8.15–8.17 (d, 1H, aromatc proton), 9.47 (s, 1H, NHCOPh), 9.57 (s, 1H, CH2CONHPh) ppm. MS (ESI) m/z: 405 [M + H]+.
Methyl 5-benzamido-2-(2-(benzylamino)-2-oxoethoxy)benzoate (24b):
Synthesized from 23 as described as for 24a. Yield = 75 %; mp = 165–168 °C; recrystallization solvent = toluene/acetonitrile; 1H NMR (400 MHz, CDCl3) δ 3.86 (s, 3H, COOCH3), 4.38–4.41 (m, 2H, CH2CONHCH2Ph), 4.55 (s, 2H, CH2CONHCH2Ph), 7.15–7.17 (d, 1H, benzene proton), 7.22–7.35 (m, 5H, benzene protons), 7.42–7.57 (m, 4H, benzene protons), 7.93–7.98 (m, 2H, benzene protons), 8.15–8.17 (d, 1H, benzene proton), 9.49 (s, 1H, NHCOPh) ppm. MS (ESI) m/z: 419 [M + H]+.
General procedure for the synthesis of 5-benzamido-2-(2-oxo-2-(phenylamino)ethoxy)benzoic acid (25a) and 5-benzamido-2-(2-(benzylamino)-2-oxoethoxy)benzoic acid (25b). Example: Procedure for the synthesis of 5-benzamido-2-(2-(benzylamino)-2-oxoethoxy)benzoic acid (25b):
A 2N aqueous solution of lithium hydroxide (0.02 g, 0.54 mmol) was added to a solution of 24b (0.15 g, 0.36 mmol) in THF (5 mL), and the resulting mixture was stirred at room temperature for 12 h. After this time, the THF was evaporated in vacuo and the carboxylate product obtained was titrated with 2N HCl aqueous solution, added at 0 °C dropwise until pH 1. The colorless solid obtained 25b was filtered, washed with water and dried in vacuo. Yield = 82 %; mp = 240–242 °C; recrystallization solvent = methanol; 1H NMR (400 MHz, DMSO-d6) δ 4.35–4.39 (m, 2H, CH2CONHCH2Ph), 4.54 (s, 2H, CH2CONHCH2Ph), 7.08–7.10 (d, 1H, benzene proton), 7.21–7.35 (m, 5H, benzene protons), 7.45–7.57 (m, 2H, benzene protons), 7.76–7.80 (dd, 1H, benzene proton), 7.93–7.98 (m, 2H, benzene protons), 8.07–8.09 (d, 1H, benzene proton), 8.27–8.30 (t, 1H, benzene proton), 10.13 (s, 1H, NHCOPh), 12.70 (bs, 1H, COOH) ppm. MS (ESI) m/z: 403 [M - H]−.
5-benzamido-2-(2-oxo-2-(phenylamino)ethoxy)benzoic acid (25a):
Synthesized from 24a as described as for 25b. Yield = 80 %; mp = > 250 °C; recrystallization solvent = methanol; 1H NMR (400 MHz, DMSO-d6) δ 4.72 (s, 2H, CH2CONHPh), 7.02–7.06 (m, 1H, benzene proton), 7.09–7.11 (d, 1H, benzene proton), 7.28–7.34 (m, 2H, benzene protons), 7.45–7.57 (m, 3H, benzene protons), 7.58–7.63 (m, 2H, benzene protons), 7.76–7.81 (dd, 1H, benzene proton), 7.93–7.98 (m, 2H, benzene protons), 8.07–8.09 (d, 1H, benzene proton), 9.93 (s, 1H, CH2CONHPh), 10.13 (s, 1H, NHCOPh), 12.70 (bs, 1H, COOH) ppm. MS (ESI) m/z: 389 [M - H]−.
General procedure for the synthesis of 2-phenylcyclopropane-1-carboxamide (4c) and 1-phenylcyclopropane-1-carboxamide (4g). Example: 1-Phenylcyclopropane-1-carboxamide (4g):
To a solution of 1-phenylcyclopropane-1-carboxylic acid (1.0 g, 6.17 mmol) in a mixture of dry DCM/DMF 4/1 (20 mL + 5 mL) were added in sequence triethylamine (3.44 mL, 24.66 mmol) and PyBOP (4.01 g, 7.71 mmol), and the resulting mixture was left under stirring at room temperature for 45 min. Afterwards, a 33% ammonia aqueous solution (0.59 mL, 30.83 mmol) was added and the final mixture was left under stirring at room temperature for 24 h. At the end of the reaction, the reaction mixture was concentrated and poured into water (50 mL) and then extracted with ethyl acetate (4 × 50 mL). The organic layers were washed in sequence with 2N HCl (2 × 20 mL), saturated sodium bicarbonate solution (2 × 20 mL), and saturated sodium chloride solution (1 × 20 mL), then were dried over anhydrous sodium sulfate and concentrated under vacuum to furnish a crude residue that was purified by silica gel column chromatography eluting with a mixture ethyl acetate/n-hexane 1/1 to afford 4g as a white powder. Yield = 84 %, mp = 98–100 °C, recrystallization solvent: cyclohexane. 1H-NMR (400 MHz, CDCl3): δ 1.10–1.13 (q, 2H, CHH-CHH cyclopropane), 1.62–1.65 (q, 2H, CHH-CHH cyclopropane), 5.33 (br s, 1H, CONHH), 5.72 (br s, 1H, CONHH), 7.33–7.35 (m, 1H, benzene proton), 7.37–7.41 (m, 2H, benzene protons), 7.45–7.47 (m, 2H, benzene protons) ppm. MS (ESI), m/z: 162 [M + H]+.
2-Phenylcyclopropane-1-carboxamide (4c):
Synthesized from 2-phenylcyclopropane-1-carboxylic acid as described as for 4g. Yield = 89.3 %, mp = 78–80 °C, recrystallization solvent: cyclohexane. 1H-NMR (400 MHz, CDCl3): δ 1.31–1.35 (m, 1H, PhCHCHHCH cyclopropane), 1.64–1.72 (m, 2H, CHCONH2 and PhCHCHHCH cyclopropane), 2.52–2.57 (m, 1H, PhCHCH2CH cyclopropane), 5.36–5.39 (br s, 1H, CONHH), 5.60–5.63 (br s, 1H, CONHH), 7.12–7.14 (m, 2H, benzene protons), 7.21–7.24 (m, 1H, benzene proton), 7.31–7.33 (m, 2H, benzene protons) ppm. MS (ESI), m/z: 162 [M + H]+.
Synthesis of 2-phenylcyclopropane-1-carbohydrazide (4d):
Hydrazine hydrate (0.07 mL, 1.42 mmol) was added to a solution of methyl-2-phenylcyclopropane-1-carboxylate (0.05 g, 0.28 mmol) in methanol (10 mL), and the reaction was stirred at room temperature overnight. After this time, the methanol was removed in vacuo and the solid formed was triturated in diethyl ether, filtered and washed with diethyl ether (3 × 5 mL) to afford pure 4d. Yield: 78 %, mp 125–127 °C, recrystallization solvent: ethanol; 1H NMR (400 MHz, DMSO-d6) δ 1.41 (m, 2H, CH2 cyclopropane), 1.98 (m, 1H, CH cyclopropane), 2.40 (m, 1H, CH cyclopropane), 4.08 (s, 2H, CONHNH2), 7.23 (m, 5H, benzene protons), 9.19 (s, 1H, CONHNH2) ppm. MS (ESI), m/z: 176 [M+H]+
General procedure for the synthesis of tert-butyl (1-phenylcyclopropyl)carbamates 27 and 30. Example: Tert-butyl (1-phenylcyclopropyl)carbamate (27):
Diphenylphosphoryl azide (1.53 g, 5.55 mmol), triethylamine (0.81 mL, 5.79 mmol) and tert-butanol (5.35 g, 6.92 mL, 72.4 mmol) were added to a solution of 1-phenylcyclopropane-1-carboxylic acid (0.78 g, 4.83 mmol) in dry toluene (30 mL), and the mixture was stirred at 105 °C under nitrogen atmosphere for 24 h. At the end of the reaction, the solvent was removed under vacuum and the residue was purified by silica gel column chromatography eluting with a mixture ethyl acetate/n-hexane 1/7 to give pure 27. Yield = 68.4 %, oil. 1H-NMR (400 MHz, CDCl3): δ 1.25–1.30 (br d, 4H, CH2CH2 cyclopropane), 1.47 (br s, 9H, COOC(CH3)3), 5.28 (br s, 1H, NHCOOC(CH3)3), 7.18–7.33 (m, 5H, benzene protons) ppm. MS (ESI), m/z: 234 [M + H]+.
Tert-butyl (1-(4-nitrophenyl)cyclopropyl)carbamate (30):
Yield = 68.3 %, mp = 122–124 °C, recrystallization solvent = benzene. 1H-NMR (400 MHz, CDCl3): δ 1.37 (br m, 4H, CH2CH2 cyclopropane), 1.48 (s, 9H, COOC(CH3)3), 5.31 (br s, 1H, NHCOOC(CH3)3), 7.32–7.34 (d, 2H, benzene protons), 8.16–8.18 (d, 2H, benzene protons) ppm. MS (ESI), m/z: 279 [M + H]+.
1-Phenylcyclopropan-1-amine hydrochloride (4e):
A solution of 27 (0.20 g, 0.86 mmol) in dry THF (5 mL) was treated with a solution of 4N HCl in 1,4-dioxane (18.85 mL, 51.43 mmol) at 0 °C, and the resulting mixture was stirred at room temperature for 24 h. At the end of the reaction, the solution was concentrated under vacuum, and the resulting residue was triturated with dry diethyl ether (10 mL), isolated by filtration and washed over filter with dry diethyl ether to give 3 as a white powder. Yield = 98 %, mp > 250 °C, recrystallization solvent: methanol/THF. 1H-NMR (400 MHz, DMSO-d6): δ 1.19 (m, 2H, CHH-CHH cyclopropane), 1.39 (m, 2H, CHH-CHH cyclopropane), 7.35–7.45 (m, 5H, benzene protons), 8.78 (br s, 3H, NH3+). MS (ESI), m/z: 170 [M + H]+.
1-(4-Nitrophenyl)cyclopropane-1-carbonitrile (28):
50% Sodium hydroxide (7.9 mL, 197.6 mmol) was added dropwise at room temperature to a mixture of 4-nitrophenylacetonitrile (4.0 g, 24.7 mmol), 1,2-dibromoethane (11.6 g, 5.32 mL, 61.7 mmol) and tetrabutylammonium bromide (3.98 g, 12.4 mmol) in dry acetonitrile (40 mL). Then, the resulting mixture was stirred at 40 °C for 8 h. Upon completion, the reaction mixture was poured into water (50 mL) and extracted with ethyl acetate (3 × 30 mL). The organic layers were washed with 2N potassium hydroxide (2 × 20 mL) and saturated sodium chloride solution (1 × 30 mL), dried over anhydrous sodium sulfate and concentrated under vacuum. The crude residue was purified by silica gel column chromatography eluting with a mixture ethyl acetate/n-hexane 1:3 to afford 28. Yield = 85 %, mp = 113–115 °C, recrystallization solvent: benzene.1H-NMR (400 MHz, DMSO-d6): δ 1.69–1.72 (dd, 2H, CHH-CHH cyclopropane), 1.93–1.96 (dd, 2H, CHH-CHH cyclopropane), 7.58–7.60 (d, 2H, benzene protons), 8.22–8.25 (d, 2H, benzene protons). MS (ESI), m/z: 189 [M + H]+.
1-(4-Nitrophenyl)cyclopropane-1-carboxylic acid (29):
Compound 28 (2.6 g, 13.8 mmol) was suspended in water (16 mL), treated with 96% (w/w) sulfuric acid (11 mL) and stirred under reflux overnight. Upon completion, the reaction mixture was cooled and the resultant precipitate was filtered under vacuum, washed over filter with water (30 mL), and dried overnight under vacuum to give pure 29. Yield = 82 %, mp = 130–132 °C, recrystallization solvent: benzene. 1H-NMR (400 MHz, DMSO-d6): δ 1.23–1.26 (dd, 2H, CHH-CHH cyclopropane), 1.52–1.55 (dd, 2H, CHH-CHH cyclopropane), 7.61–7.63 (d, 2H, benzene protons), 8.15–8.17 (d, 2H, benzene protons), 12.48–12.75 (br s, 1H, COOH). MS (ESI), m/z: 206 [M - H]−.
Tert-butyl (1-(4-aminophenyl)-cyclopropyl)-carbamate (31):
10% Palladium on charcoal (0.017 g, 0.017 mmol) was added to a solution of 30 (0.50 g, 1.82 mmol) in methanol (120 mL). The system was degassed (N2), then treated with H2 (30 psi) for 65 min. The mixture was filtered, washed over filter with methanol and concentrated in vacuo. The resulting crude residue was purified by silica gel column chromatography eluting with a mixture ethyl acetate/n-hexane 3/1 to afford the pure 31. Yield = 77 %, oil. 1H-NMR (400 MHz, CDCl3): δ 1.12 (br s, 2H, CHH-CHH cyclopropane), 1.18 (br s, 2H, CHH-CHH cyclopropane), 1.44 (s, 9H, COOC(CH3)3), 3.25–4.04 (br s, 2H, NH2), 4.92–5.28 (br s, 1H, NHCOOC(CH3)3), 6.63–6.65 (d, 2H, benzene protons), 7.10–7.12 (d, 2H, benzene protons) ppm. MS (ESI), m/z: 249 [M + H]+.
Benzyl (1-((4-(1-((tert-butoxycarbonyl)amino)cyclopropyl)phenyl)amino)-1-oxo-3-phenylpropan-2-yl)-carbamate (32):
Triethylamine (0.17 mL, 1.21 mmol) and N-ethyl-N’-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.12 g, 0.60 mmol) were added to a mixture of N-carbobenzyloxy-l-phenylalanine (0.18 g, 0.60 mmol), N-hydroxybenzotriazole (0.09 g, 0.60 mmol) and 31 (0.10 g, 0.40 mmol) in dry DCM (6 mL), and the resulting mixture was stirred for 24 h at room temperature. The reaction was poured into water (50 mL) and extracted with DCM (3 × 10 mL). The organic layers were washed with saturated sodium chloride solution (3 × 10 mL), dried with anhydrous sodium sulfate and concentrated under vacuum. The crude residue was purified by trituration with a mixture of diethyl ether and petroleum ether to afford 32. Yield = 70 %, mp = 116–118 °C, recrystallization solvent: toluene. 1H-NMR (400 MHz, CDCl3): δ 1.18 (br s 2H, CHH-CHH cyclopropane), 1.20 (br s 2H, CHH-CHH cyclopropane), 1.45 (s, 9H, COOC(CH3)3), 3.13–3.15 (m, 1H, Ph-CHH-CH(NH)CO), 3.20–3.22 (m, 1H, Ph-CHH-CH(NH)CO), 4.50 (m, 1H, PhCH2CH(NH)CO), 5.14 (s, 2H, PhCH2OCO), 5.24 (br s, 1H, PhCH2OCONH), 5.40 (br s, 1H, NHCOOC(CH3)3), 7.07 (m, 1H, benzene proton), 7.16–7.18 (m, 3H, benzene protons), 7.25–7.27 (m, 4H, benzene protons), 7.31–7.35 (m, 6H, benzene protons), 7.44 (br s, 1H, CONHPh) ppm. MS (ESI), m/z: 530 [M + H]+.
Biochemistry.
LSD1 enzyme inhibition assay.
The complex of human recombinant LSD1/CoREST protein was produced in E. coli as separate proteins and co-purified following previously reported procedures.[27] The experiments were performed in 96 well half area white plates (cat. 3693, Corning, Corning, NY) using a mono-methylated H3-K4 peptide containing 21 amino acids (custom synthesis done by Thermo Scientific) as substrate in 40 μL volume of 50 mM Tris-HCl, pH 8.0 and 0.05 mg/ml BSA buffer. The peptide purity was >95% as checked by analytical high-pressure liquid chromatography and mass spectrometry. The demethylase activity was estimated under aerobic conditions and at room temperature by measuring the release of H2O2 produced during the catalytic process by the Amplex UltraRed detection system coupled with horseradish peroxidase (HRP). Briefly, 20 nM of LSD1/CoREST complex was incubated at room temperature for 15 min in the absence and/or the presence of various concentrations of the inhibitors, 50 μM Amplex UltraRed (Life Technologies) and 0.023 μM HRP (Sigma) in 50 mM Tris-HCl pH 8.0 and 0.05 mg/ml BSA. The inhibitors were tested twice in duplicates at each concentration. Tranylcypromine (Sigma) was used as control. After preincubation of the enzyme with the inhibitor, the reaction was initiated by addition of 4.5 μM of mono-methylated H3-K4 peptide. The conversion of the Amplex Ultra Red reagent to Amplex UltroxRed was monitored by fluorescence (excitation at 510 nm, emission at 595 nm) for 12 min and by using a microplate reader (Infinite 200, Tecan Group, Switzerland). Arbitrary units were used to measure the level of H2O2 produced in the absence and/or in the presence of inhibition. The maximum demethylase activity of LSD1/CoREST was obtained in the absence of inhibitors and corrected for background fluorescence in the absence of the substrate. The IC50 values were calculated using GraphPad Prism version 4.0 (GraphPad Software, San Diego, CA).
Anti-MAO assays.
Human recombinant MAO A and MAO B were expressed in Pichia pastoris and purified as published.[28] Inhibition was measured by the horseradish peroxidase coupled-assay using a Cary-Eclipse spectrofluorimeter. Assays were performed using kynuramine (MAO A) and benzylamine (MAO B) as substrates in 50 mM Hepes/NaOH pH 7.5, 0.5% (v/v) reduced Triton X-100 after 15-min incubation of the enzyme with the inhibitor.
Biology
Cell growth assays.
The effect of 1–3 on cell proliferation were evaluated against the AML MV4–11 and the APL NB4 cell lines using the CellTiter-Fluor Cell Viability Assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The cells were incubated for 48 h with various inhibitor concentrations. An equivalent of the CellTiter-Fluor reagent was then added, the content was mixed and incubates for at least 90 min at 37 °C to obtain a stable signal. The fluorescence was recorded using an excitation wavelength of 360 nm and an emission at 535 nm. IC50 values were calculated using GraphPad software.
Gene modulation assays.
Human APL NB4 cells were grown in RPMI supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and antibiotics and maintained in a humidified tissue culture incubator at 37 °C in 5% CO2. Cells were treated at the biochemical IC50 or with vehicle (DMSO). After 24 h the cells were collected for RNA analysis. Total RNA was purified using RNeasy Mini Kit (Qiagen, Valencia, CA), quantified and reverse transcribed. mRNA levels were measured by quantitative RT-PCR (Fast SYBR Green Master mix, Applied Biosystems Foster City, CA) using specific primers and normalized against TBP mRNA. Results are presented as fold induction relative to vehicle treated cells (DMSO). Primers used in this study were:
GFI-1b, TCTGGCCTCATGCCCTTA - TCTGGCCTCATGCCCTTA;
ITGAM, AACCCCTGGTTCACCTCCT - CATGACATAAGGTCAAGGCTGT;
KCTD12, CCTGCCCGAGAGATGATTAC - TACTGGAGGGGGAGAATGG
TBP, GCTGGCCCATAGTGATCTTT - CTTCACACGCCAAGAAACAGT.
Microsomal stability in liver microsomes.
Test compounds were dissolved in DMSO to obtain 0.2 mM solutions and pre-incubated, at the final concentration of 1 μM, for 10 min at 37 °C in potassium phosphate buffer 50 mM, pH 7.4, 3 mM MgCl2, with mouse and human liver microsomes (Sigma) at the final concentration of 0.5 mg/mL. After the pre-incubation period, reactions was started by adding the cofactors mixture (NADP+, Glc6P, Glc6P-DH in 2% sodium bicarbonate); samples (25 μL) were taken at time 0, 10, 20, 30 and 60 min added to 150 μL of acetonitrile with verapamil 0.02 μM as Internal Standard (IS) to stop the reaction. After centrifugation the supernatants were analyzed by LC-MS/MS. A control sample without cofactors was added in order to check the stability of test compounds in the matrix after 60 min. Samples were analyzed on UFLC Shimadzu AC20 coupled with a API 3200 Triple Quadrupole or on a 4500 Qtrap ABSciex. Compounds were analyzed either using acidic or basic conditions. The percent of the area of test compound remaining at the various incubation times were calculated in respect to the area of compound at time 0 min.
Intrinsic Clearance: The rate constant, k (min−1) derived for the exponential decay equation (peak area/IS vs time) was used to calculate the rate of intrinsic clearance (Clint) of the compound using the following equation:
Clint (μL/min/mg) = k/microsomal conc. × 1000
where k = rate constant (min−1)
microsomal protein conc. = 0.5 mg protein/mL
Supplementary Material
Acknowledgements
This work was supported by PRIN 2016 (prot. 20152TE5PK) (A.Mattevi, A.Mai), Ricerca Finalizzata 2013 PE-2013-02355271 (A.Mai), AIRC (IG-19162, PI: A.Mai; IG-11342, PI: A.Mattevi), NIH (n. R01GM114306 - A.Mai), Fondazione Regionale Ricerca Bio-medica, Milan, Italy (n. 2015-0042 A. Mattevi) and Progetto di Ateneo “Sapienza” 2016 n. RG116154BED07FE9 (A.Mai) and 2017 n. RM11715C7CA6CE53 (D.R.) funds.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information for this article is given via a link at the end of the document.
Dedicated to the memory of Prof. Maurizio Botta, outstanding scientist and unforgettable friend.
References
- [1].a Helin K, Dhanak D, Nature 2013, 502, 480–488; [DOI] [PubMed] [Google Scholar]; b Kaniskan HU, Martini ML, Jin J, Chem Rev 2018, 118, 989–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rotili D, Mai A, Genes Cancer 2011, 2, 663–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maiques-Diaz A, Somervaille TC, Epigenomics 2016, 8, 1103–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee MG, Wynder C, Cooch N, Shiekhattar R, Nature 2005, 437, 432–435. [DOI] [PubMed] [Google Scholar]
- [5].a Saleque S, Kim J, Rooke HM, Orkin SH, Mol Cell 2007, 27, 562–572; [DOI] [PubMed] [Google Scholar]; b Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, Metzger E, Schule R, Nat Cell Biol 2007, 9, 347–353. [DOI] [PubMed] [Google Scholar]
- [6].Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG, Nature 2007, 446, 882–887. [DOI] [PubMed] [Google Scholar]
- [7].Kosumi K, Baba Y, Sakamoto A, Ishimoto T, Harada K, Nakamura K, Kurashige J, Hiyoshi Y, Iwatsuki M, Iwagami S, Sakamoto Y, Miyamoto Y, Yoshida N, Oki E, Watanabe M, Hino S, Nakao M, Baba H, Int J Cancer 2016, 138, 428–439. [DOI] [PubMed] [Google Scholar]
- [8].Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, Kirfel J, Carcinogenesis 2010, 31, 512–520. [DOI] [PubMed] [Google Scholar]
- [9].Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schule R, Eggert A, Buettner R, Kirfel J, Cancer Res 2009, 69, 2065–2071. [DOI] [PubMed] [Google Scholar]
- [10].Singh MM, Johnson B, Venkatarayan A, Flores ER, Zhang J, Su X, Barton M, Lang F, Chandra J, Neuro Oncol 2015, 17, 1463–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mould DP, McGonagle AE, Wiseman DH, Williams EL, Jordan AM, Med Res Rev 2015, 35, 586–618. [DOI] [PubMed] [Google Scholar]
- [12].Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A, Ciossani G, Botrugno OA, Forneris F, Tardugno M, Edmondson DE, Minucci S, Mattevi A, Mai A, J Am Chem Soc 2010, 132, 6827–6833. [DOI] [PubMed] [Google Scholar]
- [13].a Valente S, Rodriguez, Mercurio, Vianello, Saponara, Cirilli, Ciossani, Labella, Marrocco, Monaldi, Ruoppolo, Tilset, Botrugno, Dessanti, Minucci, Mattevi, Varasi, Mai, Eur J Med Chem 2015, 94, 163–174; [DOI] [PubMed] [Google Scholar]; b Valente S, Rodriguez V, Mercurio C, Vianello P, Saponara, Cirilli R, Ciossani G, Labella D, Marrocco B, Ruoppolo G, Botrugno OA, Dessanti P, Minucci S, Mattevi A, Varasi M, Mai A, ACS Med Chem Lett 2015, 6, 173–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vianello P, Botrugno OA, Cappa A, Dal Zuffo R, Dessanti P, Mai A, Marrocco B, Mattevi A, Meroni G, Minucci S, Stazi G, Thaler F, Trifiro P, Valente S, Villa M, Varasi M, Mercurio C, J Med Chem 2016, 59, 1501–1517. [DOI] [PubMed] [Google Scholar]
- [15].Cho SJ, Jensen NH, Kurome T, Kadari S, Manzano ML, Malberg JE, Caldarone B, Roth BL, Kozikowski AP, J Med Chem 2009, 52, 1885–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mills LR, Barrera Arbelaez LM, Rousseaux SAL, J Am Chem Soc 2017, 139, 11357–11360. [DOI] [PubMed] [Google Scholar]
- [17].Chan KS, Fu HY, Yu JQ, J Am Chem Soc 2015, 137, 2042–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Müller RK, Joos R, Felix D, Schreiber J, Wintner C, Eschenmoser A, Organic Syntheses 1976, 55, 114–114. [Google Scholar]
- [19].Rodriguez V, Valente S, Rovida S, Rotili D, Stazi G, Lucidi A, Ciossani G, Mattevi A, Botrugno OA, Dessanti P, Mercurio C, Vianello P, Minucci S, Varasi M, Mai A, MedChemComm 2015, 6, 665–670. [Google Scholar]
- [20].a Vianello P, Botrugno OA, Cappa A, Ciossani G, Dessanti P, Mai A, Mattevi A, Meroni G, Minucci S, Thaler F, Tortorici M, Trifiro P, Valente S, Villa, Varasi M, Mercurio C, Eur J Med Chem 2014, 86, 352–363; [DOI] [PubMed] [Google Scholar]; b Niebel D, Kirfel J, Janzen V, Holler T, Majores M, Gutgemann I, Blood 2014, 124, 151–152; [DOI] [PubMed] [Google Scholar]; c McGrath JP, Williamson KE, Balasubramanian S, Odate S, Arora, Hatton C, Edwards TM, O’Brien T, Magnuson S, Stokoe D, Daniels DL, Bryant BM, Trojer P, Cancer Res 2016, 76, 1975–1988. [DOI] [PubMed] [Google Scholar]
- [21].a van der Meer LT, Jansen JH, van der Reijden BA, Leukemia 2010, 24, 1834–1843; [DOI] [PubMed] [Google Scholar]; b Ishikawa Y, Gamo K, Yabuki M, Takagi S, Toyoshima K, Nakayama K, Nakayama A, Morimoto M, Miyashita H, Dairiki R, Hikichi Y, Tomita N, Tomita D, Imamura S, Iwatani M, Kamada Y, Matsumoto S, Hara R, Nomura T, Tsuchida K, Nakamura K, Mol Cancer Ther 2017, 16, 273–284; [DOI] [PubMed] [Google Scholar]; c Sugino N, Kawahara M, Tatsumi G, Kanai A, Matsui H, Yamamoto R, Nagai Y, Fujii S, Shimazu Y, Hishizawa M, Inaba T, Andoh A, Suzuki T, Takaori-Kondo A, Leukemia 2017, 31, 2303–2314. [DOI] [PubMed] [Google Scholar]
- [22].a Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, Guibourt N, Perdones A, Lufino MMP, Somervaille TCP, Wiseman DH, Duy C, Melnick A, Willekens C, Ortega A, Martinell M, Valls N, Kurz G, Fyfe M, Castro-Palomino JC, Buesa C, Cancer Cell 2018, 33, 495–511 e412; [DOI] [PubMed] [Google Scholar]; b Fang J, Ying H, Mao T, Fang Y, Lu Y, Wang H, Zang I, Wang Z, Lin Y, Zhao M, Luo X, Wang Z, Zhang Y, Zhang C, Xiao W, Wang Y, Tan W, Chen Z, Lu C, Atadja P, Li E, Zhao K, Liu J, Gu J, Oncotarget 2017, 8, 85085–85101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Houston JB, Biochem Pharmacol 1994, 47, 1469–1479. [DOI] [PubMed] [Google Scholar]
- [24].Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, Miller CJ, Ogilvie DJ, Somervaille TC, Cancer Cell 2012, 21, 473–487. [DOI] [PubMed] [Google Scholar]
- [25].Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, Casero RA Jr., Marton L, Woster P, Minden MD, Dugas M, Wang JC, Dick JE, Muller-Tidow C, Petrie K, Zelent A, Nat Med 2012, 18, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, Liu Y, Ward D, Quan J, Ye T, Zhang H, Cancer Res 2011, 71, 7238–7249; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D, Wu X, Cao Y, Liang W, Liu Y, Sun H, Ye T, Zhang H, Cell Rep 2013, 5, 445–457; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sareddy GR, Viswanadhapalli S, Surapaneni P, Suzuki T, Brenner A, Vadlamudi RK, Oncogene 2017, 36, 2423–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A, J Biol Chem 2007, 282, 20070–20074; [DOI] [PubMed] [Google Scholar]; b Forneris F, Binda C, Battaglioli E, Mattevi A, Trends Biochem Sci 2008, 33, 181–189. [DOI] [PubMed] [Google Scholar]
- [28].Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A, Proc Natl Acad Sci U S A 2003, 100, 9750–9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.