

Abstract
The presence and activity of CD8+ T cells within the tumor microenvironment is essential for the control of tumor growth. Utilizing B16-F10 melanoma tumors that express altered peptide ligands of chicken ovalbumin, OVA257–264, we measured high- and low-affinity OVA-specific responses following adoptive transfer of OT-I CD8+ T cell into mice subsequently challenged with tumors. TCR affinity positively correlated with the frequency of OT-I tumor infiltrating lymphocytes (TILs). Differences in TCR affinity inversely corresponded to in vivo tumor growth rate. Blockade of the PD-1 and CTLA-4 checkpoints preferentially increased the frequency and antitumor function of TIL responding to high-affinity antigens, while failing to enhance the antitumor activity of low-affinity T cells. To determine whether lowering the TCR activation threshold could enhance the breadth and magnitude of the antitumor T cell response, we inhibited Src homology region 2 domain-containing phosphatase 1 (SHP-1) in OT-I T cells prior to tumor antigen exposure. SHP-1 knockdown increased the cytokine producing potential of high- and low-affinity T cells, but failed to enhance control of tumor growth. In contrast, when SHP-1 knockdown of OT-I T cells was combined with immunotherapy, we observed a significant and long-lasting suppression of tumor growth mediated by low-affinity T cells. We conclude that lowering of TCR activation threshold by targeting SHP-1 expands the repertoire of T cells available to respond to conventional checkpoint blockade, leading to enhanced control of tumor growth.
Introduction
Multiple immunotherapeutic approaches are now available to treat melanoma and other cancers, including administration of high-dose cytokines (1–3), checkpoint blockade inhibitors (4–10), adoptive transfer of in vitro expanded tumor-specific T cells, engineering of T cells, expression of genetically modified or chimeric antigen receptors and use of oncolytic viruses (11–13). Although T cell-directed immunotherapies have successfully induced durable antitumor responses in a subset of patients and increased overall survival, many patients continue to be resistant to such approaches. Consequently, efforts are underway to understand mechanisms of resistance and design strategies for expanding both the tumor types and patient pool that can respond to immunotherapy.
T cells limit tumor growth (14,15). The presence or migration of tumor infiltrating lymphocytes (TIL) corresponds to responsiveness to tumor immunotherapies such as checkpoint blockade, as well as overall patient survival for multiple tumor types (16,17). However, even in settings with brisk TIL responses, response to tumor immunotherapy may be variable. Factors that suppress the ability of TIL to eradicate tumor cells may include inefficient T cell activation, dysregulated cytokine signaling, acquisition of exhausted or anergic states and the impact of the immunosuppressive tumor microenvironment (TME) (18). The reasons for failure to generate TIL may also vary. Whereas active immunosuppression may prevent activation or migration of antitumor T cells, an absence of mutated neo-antigens may also limit generation of high-affinity T cell responses. Mutation burden corresponds to response to checkpoint blockade therapies and patient outcome (19–21). The impact of existing checkpoint blockade therapies on activation and function of low-affinity T cells specific for tumor-associated self-antigens or weakly reactive neo-antigens is not fully understood. Enhancing efficacy of checkpoint blockade therapies in patients with ineffective TIL, or lacking TIL altogether, will likely require development of strategies for expanding the repertoire of tumor-reactive T cells.
The role of TCR affinity during an in vivo antitumor response is complex. High-affinity CD8+ T cells may become tolerized once in the TME (22–24). Indeed, continual or prolonged periods of antigen stimulation via the TCR can induce functional exhaustion (25,26). However, T cell function may be rescued and enhanced through antibody blockade of T cell activation checkpoints, most prominently, CTLA-4 and PD-1 (immune checkpoint blockade, ICB) (27). Although T cells in the tumor setting may respond to neo-antigens, T cells also respond robustly across a range of affinities to tumor-associated self-antigens. For example, CD8+ T cells specific for the human melanoma antigen, gp100, exhibited a range of antigen affinities with similar antitumor activity (28). Additionally, two different TCR transgenic T cell lines specific for the tissue-restricted TRP-1 antigen which exhibited disparate affinities displayed no significant differences in their ability to control tumor growth (29). Furthermore, CD8+ T cell specific for the human telomerase reverse transcriptase (hTERT) reacting to a range of hTERT altered peptide ligands (APLs) demonstrated no optimal affinity at which maximum sensitivity and polyfunctionality occur. Thus, low-affinity T cells may demonstrate antitumor activity. These studies suggest existence of a TCR affinity threshold for T cell activation and also that functional differentiation of activated T cells is not dependent on TCR affinity, a concept we have validated in infectious disease model systems (30,31).
Efforts are underway to identify additional checkpoints on T cell activation. However, immunotherapy based on such checkpoints might fail to target previously activated T cells, expand the repertoire of tumor-responsive T cells, or enhance recruitment or increase activation of low-affinity T cells. We hypothesized that targeting the T cell activation threshold could expand the repertoire of T cells available to respond to ICB. Src homology 2 domain-containing protein tyrosine phosphatase-1 (SHP-1) regulates TCR-driven T cell activation threshold and inhibits early events after TCR triggering, including phosphorylation of ZAP-70 (32). SHP-1 is conserved between mice and humans and plays a role in establishment and maintenance of peripheral tolerance (33). The absence of SHP-1 in CD8+ T cells allows them to resist suppression by Treg activity in a T cell intrinsic manner (34), which may be crucial to survival of those T cells once they enter the TME. The relationship between SHP-1 and TCR affinity and the ability of SHP-1 to control TCR signaling within the immune cell is established (11). Although PD-1-directed blockade may indirectly impact TCR signaling via activation of Src homology 2 domain-containing protein tyrosine phosphatase-2 (SHP-2), SHP-1 impacts TCR signaling directly and independently (35). During T cell activation, SHP-1 does not localize with PD-1. Instead, SHP-2 is preferentially activated by PD-1 (36). PD-1-mediated T cell inhibition can occur independently of SHP-2 (37). Thus, modulation of TCR signal strength and activation threshold might improve responses to existing checkpoint blockade immunotherapy.
To address the role of TCR affinity in antitumor recruitment and function, as well as the impact of existing immunotherapies on low-affinity T cells, we utilized a model system in which mouse B16 tumor cells were engineered to express wild-type chicken ovalbumin (OVA), or OVA with point mutations in the immunodominant epitope recognized by the OT-I TCR. Because these point mutations impair TCR binding but not MHC binding, the resulting altered peptide ligands (APLs) can be used to measure OT-I T cell responses across a wide range of TCR affinities (38,39). Using this model system, we found that TCR affinity regulated the antitumor activity of adoptively transferred OT-I T cells. Furthermore, treatment with ICB targeting PD-1 and CTLA-4 selectively enhanced the antitumor activity of high-affinity, but not low-affinity, OT-I. In order to test the impact of strategies aimed at improving the recruitment and activation of low-affinity OT-I, we targeted SHP-1. Although SHP-1 inhibition alone failed to increase the ability of OT-I to control tumor growth expressing either high- or low-affinity OVA variants, it enhanced recruitment and cytokine production by low-affinity T cells. Combined ICB and SHP-1 inhibition enhanced the ability of low-affinity T cells to control tumor growth. Overall, we conclude that targeting SHP-1 expands the repertoire of T cells available to respond to ICB and induces antitumor activity by low-affinity T cells.
Materials and Methods:
Mice.
C57BL/6 (6 to 8 weeks old) mice were purchased from Jackson Laboratories. OT-I transgenic mice (on Thy1.1+ background) were maintained in our colony at the University of Utah. All mouse experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Utah.
Tumor Cell Lines.
Mouse B16-F10 cells were purchased from American Type Culture Collection (Manassas, VA) in 2014 and grown in DMEM with 10% FBS, penicillin and streptomycin at 37° C with 5% CO2, then frozen in liquid nitrogen. For all experiments, cells were recovered from frozen aliquots and cultured for 1–2 weeks prior to inoculation of mice. The B16-F10 parental cell line has not been re-authenticated or subjected to mycoplasma testing in the past year. B16 cells expressing OVA or its variants were all derived from the same parental B16-F10 line. All MigR1 retroviral vectors that expressed OVA200–290, or the indicated single amino acid variants (39), and a GFP or mCherry fluorescent reporter under the control of an IRES were transfected into 293T cells, a virus producing cell line (Polyplus jetPRIME®, Polyplus Transfection, Illkirch-Graffenstaden, France). B16-OVA(APL) cell lines were created by harvesting viral supernatant from retrovirally transfected 293T cells and placing on top of B16-F10 cells in a 6-well plate in the presence of polybrene (1 μg/mL). Transduction of cells with viral supernatant was promoted by spinning at 2500 rpm and 32°C for 90 minutes. 24 hours after transduction, B16-OVA(APL) cell lines were purified via FACS gated on GFP or mCherry fluorescence.
Tumor Experiments.
C57BL/6 mice received 1–2 × 104 OT-I CD8+ T cells (unsorted total splenocytes) i.v., followed by 1 × 106 B16-OVA(APL) cells injected into the hind flank one day later. B16-OVA(APL) cells were cultured in such a way that they were 60–80% confluent in a 175cm2 tissue culture flasks the day of injection. Tumor volume was calculated with the formula V=a(b2)/2, where a and b are tumor length and width (mm), respectively (40). On various days after tumor implantation, mice were euthanized and analysis of T cells in tumors and draining lymphoid organs was performed. For tumor killing assay experiments, the initial tumor cell implantation consisted of a 1:1 mixture of B16-F10 cells (EV) and a B16-OVA(APL) cell line, each expressing a different fluorescent marker (GFP or mCherry).
Checkpoint Blockade.
Mice were treated at 7 and 10 days post tumor implantation. Each mouse in the treatment group received an intraperitoneal injection of anti-PD-1 (250 μg/dose, clone RMP1–14), anti-PD-L1 (250 μg/dose, clone 10F.9G2), and anti-CTLA-4 (100 μg/dose, clone 9H10) (BioXCell, West Lebanon, NH) as previously described (41).
Cell preparations and flow cytometry.
Splenocytes and lymph node cells were mechanically disrupted and single cell suspensions were placed in cell culture media described above. Tumors were excised, weighed and mechanically disrupted before being digested for 45 minutes in 0.25 mg/mL Collagenase IV (Gibco) and 14.5 μg/mL DNaseI (Sigma) with gentle shaking at 37°C. Digested tumor samples were filtered (40μm) and RBCs were lysed to produce single cell suspensions. For cell-surface stains, single cell suspensions were incubated with fluorescently conjugated antibodies diluted in antibody staining buffer (PBS containing 1% FBS) at 4°C for 30–45 minutes. For intracellular cytokine assays, splenocytes were restimulated for 4 hours with 0.1 μg/ml of full-length OVA peptide or OVA APL peptides at 37°C in the presence of Brefeldin A (GolgiPlug 1μl/mL), permeabilized with a kit (BD Biosciences) and stained with fluorescently labeled antibodies specific to the indicated cytokines. Transcription factor analysis was performed using Foxp3 Fixation/Permeabilization Buffer and accompanying protocol (eBioscience, San Diego, CA). Phosphorylated protein detection was performed as previously described (31). Flow cytometry gating strategy is depicted in Supplementary Fig. S5.
Antibodies.
The following fluorophore-conjugated antibodies were used for flow cytometry: anti-CD8 (53–6.7), anti-Thy1.1 (OX-7), anti-Vα2 (B20.1), anti-CD45 (30-F11), anti-PD-1 (29F.1A12), anti-CXCR3 (CXCR3–173), anti-IFNγ (XMG1.2), anti-IL-2 (JES6–5H4), anti-TNFα (MP6-XT22), anti-CD28 (37.51), anti-Tbet (4B10) (Biolegend); anti-CD27 (LG.7F9), anti-LAG3 (C9B7W), anti-granzyme B (Ngzb), anti-Eomes (Dan11mag) (Thermo Fisher); anti-pCD3ζ (pY142) (K25–407.69), anti-CXCR5 (2G8) (BD Biosciences).
SHP-1 retroviral knockdown.
Retroviral vectors (pMig-R1) were used to express shRNA KD constructs specific for SHP-1 (SHP-1 KD) as previously described (42). Vectors that expressed this shRNA construct utilized a human microRNA (mir30) flanking sequence allowing for optimal expression and processing of siRNA (43). KD was confirmed in an EL-4 thymoma cell line. KD in primary OT-I CD8+ T cells was accomplished by transducing OT-I bone marrow with SHP-1 KD or EV retrovirus and transplanting into irradiated Rag1−/− mice. After reconstitution (8–10 weeks later), OT-I CD8+ T cells (GFP+ and GFP−) were then isolated from the spleen and transferred into B6 recipient mice that received B16-OVA(APL) cells one day later (31).
Statistical Analysis
Graphical representation of data along with statistical analysis was performed using Prism (Graphpad v8.2.1) software. Experimental replicates are denoted in figure legends. Statistical analyses were performed using an unpaired two-tailed t test. Significant P values are marked with “*”, signifying *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Results
Tumor antigen affinity affects CD8+ T cell tumor trafficking and controls tumor growth rate
In order to establish a system for tracking high- and low-affinity T cell responses to antigens expressed by tumor cells, we employed the previously characterized altered peptide ligands (APLs) of the immunodominant H-2Kb-restricted epitope of chicken ovalbumin, OVA257–264. APLs of this epitope are recognized by OT-I TCR transgenic T cells across a ~700-fold range of TCR affinities (38,39). Because single amino acid substitutions impact TCR contact but not MHC binding for each peptide, high- and low-affinity OT-I responses to each peptide are directly comparable. Previous work utilizing these APLs found that OT-I T cells respond to very low-affinity APLs in an acute bacterial infection model, although the magnitude and kinetics of the response was altered (39). The OVA257–264 APLs we assessed were, in order of decreasing affinity for the OT-I transgenic TCR: N4 (WT), A2 (~50% lower affinity as compared to N4), Y3 (~75% lower affinity), Q4 (~88% lower affinity), T4 (~98.75% lower affinity) and V4 (~99.8% lower affinity). OVA constructs expressing each of the OVA APLs were individually retrovirally transfected into B16-F10 melanoma cells. Due to the presence of a GFP reporter, antigen expression was normalized by FACS sorting based on fluorescence intensity.
To determine the response of high- and low-affinity T cells to B16 tumor growth, B6 mice received 1 × 104 naïve OT-I CD8+ T cells one day before being subcutaneously inoculated with 1 ×106 B16 cells expressing WT OVA or an OVA APL in the hind flank. Tumors expressing WT OVA were effectively controlled by OT-I T cells beginning at day 7. Control of tumor growth required the presence of OT-I T cells, as mice that did not receive OT-I T cells failed to slow the rate of tumor growth regardless of tumor affinity (Fig. 1A). Although mice receiving OT-I cells controlled the growth of N4 (WT) tumors, B6 mice that were not given OT-I cells demonstrated no observable difference in basal tumorigenicity of N4 (WT) versus V4 (low-affinity) tumors (Fig. 1A), suggesting that endogenous CD8+ T cell responses to OVA or its antigens were not sufficient to delay tumor growth. OT-I affinity for OVA APLs inversely corresponded to the rate of tumor growth starting at day 7, as well as tumor size at day 15 (Fig. 1B). These results show that OT-I affinity for tumor antigen corresponds to the ability of CD8+ T cells to control tumor growth in vivo.
Figure 1:
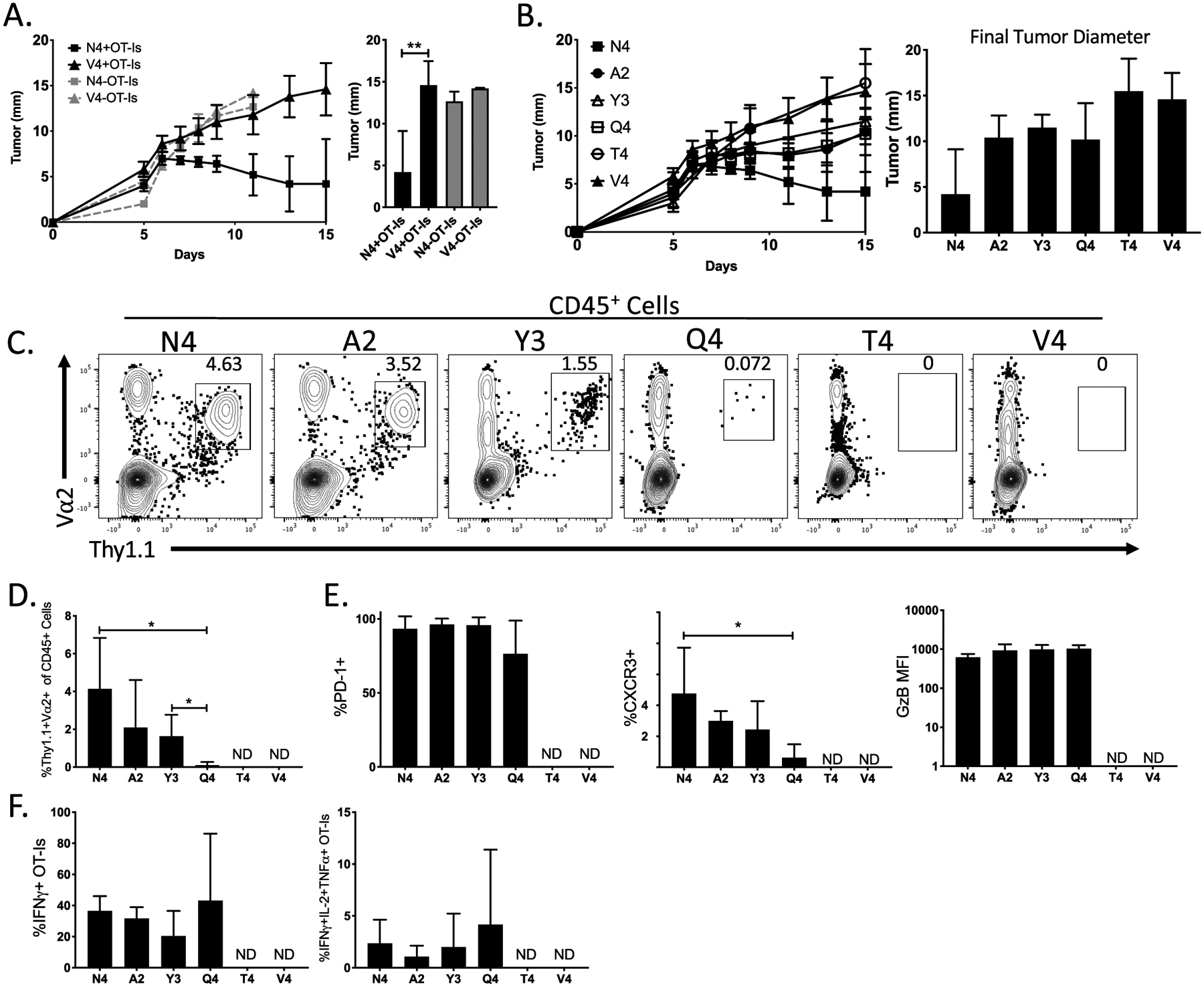
Antitumor responses are dominated by high-affinity CD8+ T cells. (A) Line graph indicates the growth of B16-OVA(N4) or B16-OVA(V4) tumors in the presence or absence of OT-I CD8+ T cells. The bar graph depicts the final tumor diameter at day 15 post implantation. (B) The line graph indicates the growth kinetics of B16-OVA(APL) tumors in B6 mice that received OT-I CD8+ T cells. The bar graph shows the final tumor diameters for all 6 different OVA(APL)s. (C) Representative flow plots show the gating scheme used to determine the frequency of OT-I CD8+ T cells (CD45+Thy1.1+Va2+) within the tumor. (D) Bar graph indicates the frequency of OT-I T cells within the CD45+ cell population in B16-OVA(APL) tumors 14 days after implantation. (E) Bar graphs show the expression of PD-1, CXCR3 and Granzyme B in either frequency or mean fluorescence intensity (MFI) via flow cytometry of OT-I CD8+ T cells within the tumor. (F) Bar graphs indicate the frequency of single or multi-cytokine producing OT-I T cells after ex vivo restimulation with corresponding OVA(APL) peptide. Error bars indicate SD. Statistical significance was determined by an unpaired t test *P < 0.05, **P < 0.01 (n=5–8 mice per group, representative of two independent experiments).
The recruitment of tumor infiltrating lymphocyte (TIL) OT-I T cells also correlated with TCR affinity (Fig. 1C). Although the high-affinity ligands N4 and A2 readily induced OT-I TIL, low-affinity ligands Y3 and Q4 recruited reduced frequencies of OT-I TIL (Fig. 1C–D). Very low-affinity ligands (T4, V4) failed to induce OT-I infiltration into the tumor (Fig. 1C–D). The number of OT-I T cells recruited by the low-affinity antigen, Q4, was low, and the phenotypes displayed by OT-I T cells specific for this APL are uncertain. Determination for positive staining was ascertained using a negative control B16 that did not express OVA or any of its variants. TCR affinity for tumor antigen correlated with surface expression of CXCR3, a chemokine receptor expressed by activated TILs in melanoma (44). Higher affinity interactions resulted in increased expression. Despite differences in infiltration, OT-I T cells did not demonstrate TCR affinity-dependent differences in expression of PD-1 and granzyme B (Fig. 1E) or differences in frequency of OT-I TIL expressing IFNγ, TNFα or IL-2 (Fig. 1F).
To confirm that control of tumor growth was antigen specific, we transduced B16-F10 cells with retroviruses generated using an empty expression vector with a mCherry reporter (B16-EV). We then co-inoculated those cells in a 1:1 ratio with B16-F10 expressing an empty vector (GFP+) or an OVA APL (N4, A2, Y3, Q4) (GFP+) into the hind flank of B6 mice. OT-I T cells selectively controlled the growth of B16-EV/N4 co-implanted tumors beginning after day 6, as compared to B16-EV/EV tumors alone (Supplementary Fig. S1A). By day 7, OT-I T cells only controlled growth of tumors expressing high-affinity antigen (N4). Tumors expressing lower affinity antigens (A2, Y3, Q4) were not controlled by the presence of OT-I (Supplementary Fig. S1B–C). By day 14, however, OT-I T cells were able to delay the growth of B16 cells expressing lower affinity antigens and EV B16 cells made up 90–95% of all tumor cells (Supplementary Fig. S1B). These experiments show targeted tumor cell elimination by OT-I T cells that is TCR affinity dependent and occurs in a temporal manner. Our findings confirm that TCR affinity for tumor antigen guides development of TIL responses during B16 tumor growth.
Immune checkpoint blockade preferentially rescues high-affinity T cells
We next sought to test whether ICB enhances the antitumor activity of high-affinity T cells only or additionally broadens the antitumor T cell response by enhancing the recruitment and effector response of low-affinity T cells. A previous study employed a model of adoptive cell therapy combined with peptide vaccination to conclude that PD-1 blockade could rescue low-affinity T cell responses (45). However, an in-depth analysis of the impact of ICB on low and high-affinity T cells responding de novo without additional manipulation has not been performed. We adoptively transferred OT-I CD8+ T cells into B6 mice that were subcutaneously inoculated with B16 cells expressing WT OVA (N4) or an OVA APL (A2, Y3, Q4, T4, V4) one day later. Although previous treatment approaches have initiated ICB at day 3 post-inoculation, we wished to allow sufficient time for establishment of the effector T cell response prior to treatment. Therefore, we delayed our ICB regimen until seven and ten days after tumor cell transplantation using an antibody cocktail consisting of anti-PD-1, anti-PD-L1 and anti-CTLA-4. Following treatment, only OT-Is responding to WT OVA significantly limited tumor growth, as compared to untreated controls (Fig. 2A). ICB induced a significant increase in the frequency of OT-I TIL in response to tumors expressing WT OVA (N4), but not to tumors expressing lower affinity APLs (A2, Y3, Q4) (Fig. 2B). Furthermore, ICB treatment of mice inoculated with tumors expressing very low-affinity antigens (T4, V4) failed to induce recruitment of OT-I TIL (Fig. 2B).
Figure 2:
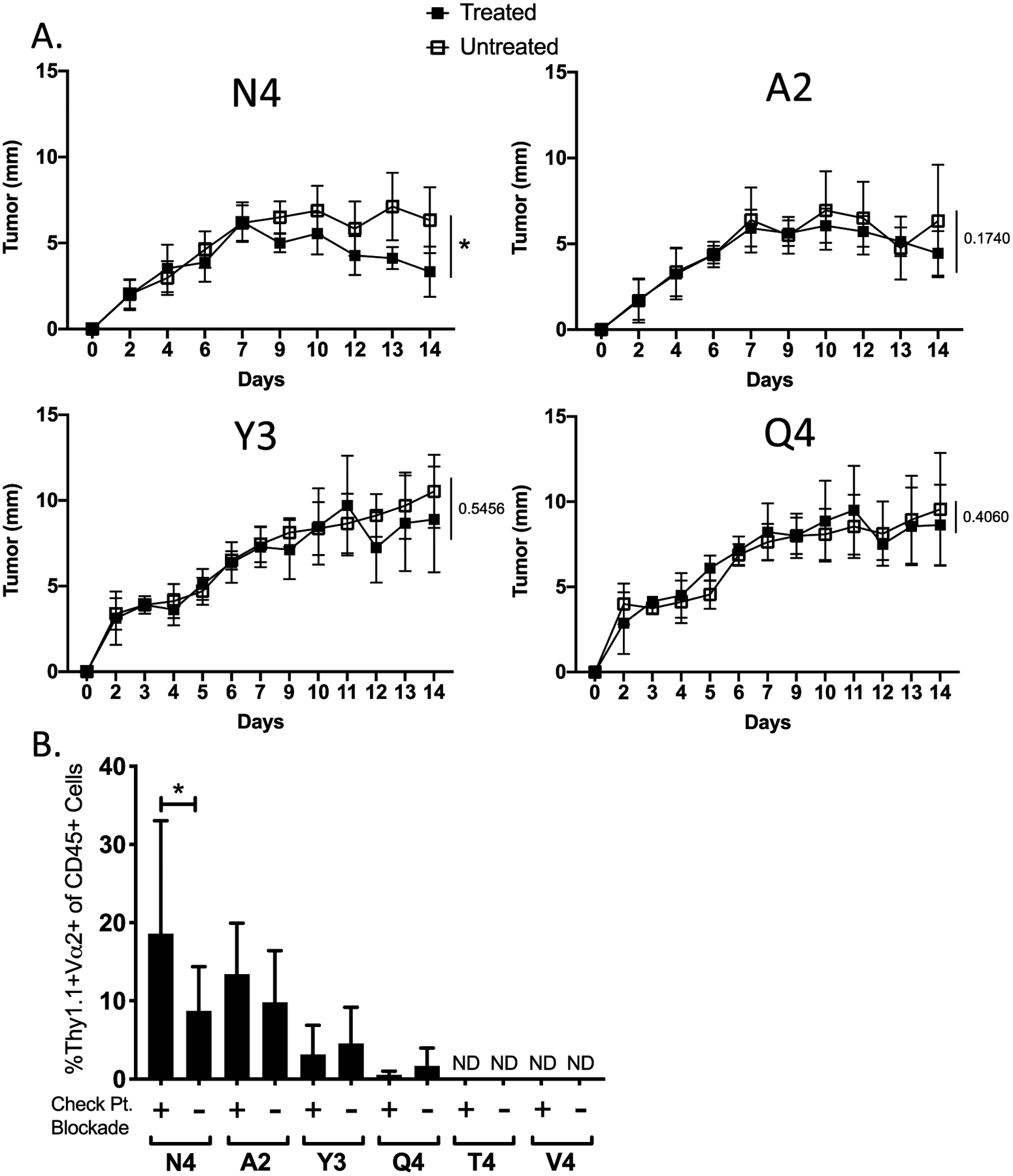
Checkpoint blockade therapy preferentially enhances high-affinity T cell responses. (A) Line graphs show the average growth kinetics of the different B16-OVA(APL) tumors in the presence (filled square) or absence (open square) of checkpoint blockade therapy administered on days 7 and 10 post tumor inoculation. (B) Bar graph indicates the frequency of OT-Is in the CD45+ population within each tumor subset at day 14 post implantation. Error bars indicate SD. Statistical significance was determined by comparing the area under the tumor growth curve (A) or group means (B) using an unpaired t test (n=18 mice per group, representative of two independent experiments). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Despite their inability to efficiently control tumor growth, a number of functional and phenotypic changes to low-affinity T cells were observed following ICB. These included increased expression of CXCR5 by both high- and low-affinity OT-I (Fig. 3A–B), a marker associated with an ICB response. Additionally, low-affinity T cells responded to ICB by increasing expression of Granzyme B, although high-affinity T cells decreased expression of CD27 (Fig. 3A–B), both associated with increased effector differentiation. However, OT-I T cells did not alter their expression of PD-1, and cytokine production following restimulation was unchanged by ICB (Fig. 3B–C). Furthermore, OT-I presence and activity within the draining lymph nodes was not significantly altered upon ICB (Supplementary Fig. S2). From these results we concluded that although ICB has some effects on low-affinity T cells, it only enhanced control of tumor growth in the presence of high-affinity T cells. ICB failed to expand the repertoire of T cells responding to the tumor, as very low-affinity T cells that initially failed to become activated (T4, V4) were non-responsive to ICB.
Figure 3:
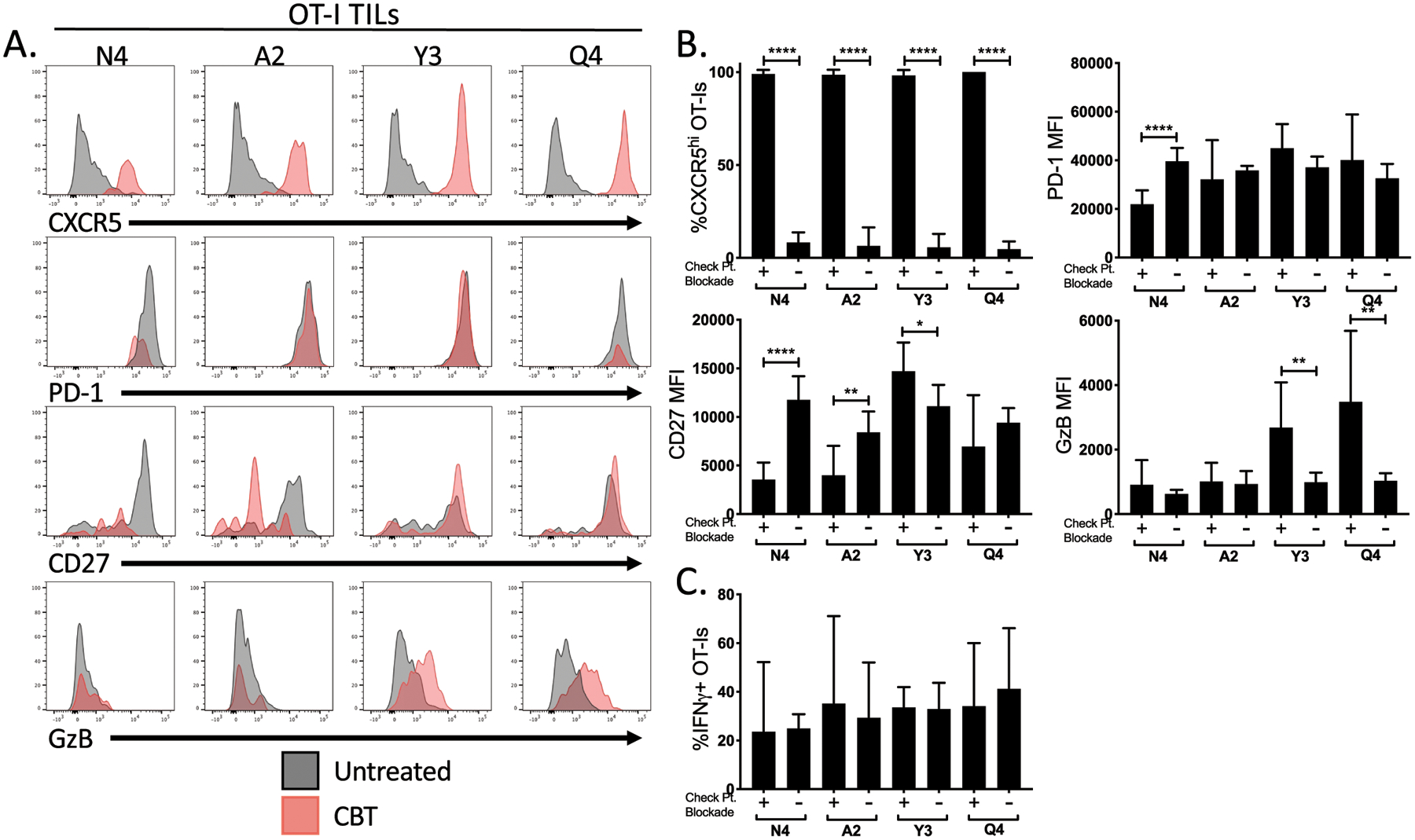
Functional responses to ICB differ based on TCR affinity for tumor antigen. (A) Representative flow plots show the expression of CXCR5, PD-1, CD27 and Granzyme B within OT-I T cells extracted from the CB treated (red) and untreated tumors (grey). (B) Bar graphs indicate the expression of CXCR5, PD-1, LAG-3, CD27, and Granzyme B in either frequency or MFI on OT-I T cells isolated from B16-OVA(APL) tumors from CB treated (+) or untreated (–) mice. (C) Bar graphs show the production of IFNγ by OT-I CD8+ T cells following ex vivo restimulation with corresponding OVA(APL) for each group. Error bars indicate SD, and statistical significance was determined by an unpaired t test (n=8 mice per group, representative of two independent experiments). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
SHP-1 controls antitumor response of low-affinity T cells
Due to their ability to influence T cell activation, differentiation and function, protein tyrosine phosphatases (PTPs) that regulate intracellular signaling within T cells are an attractive target for improving antitumor activity. SHP-1 phosphatase modulates TCR-mediated activation threshold and signal strength. We have previously reported that SHP-1 regulates TCR-dependent effector and memory T cell differentiation (31). Therefore, we hypothesized that modulation of SHP-1 would enhance the ability of low-affinity T cells to join the antitumor response. In support of this hypothesis, SHP-1 inhibition promotes antitumor immunity in some experimental settings (46). However, the mechanisms underlying protection are unclear, and the role of SHP-1 has largely been defined in high-affinity T cells. Complete abrogation of SHP-1 activity through genetic mutation impairs T cell selection in the thymus (47–49). Therefore, we pursued a targeted approach to partially inhibit SHP-1 activity in OT-I T cells. We generated bone marrow chimeras by transducing OT-I bone marrow with SHP-1 shRNA retroviral vectors (SHP-1 KD), or an empty vector (EV) control, and transplanting into irradiated Rag−/− recipients. We have previously used this method to achieve ~70% knockdown (KD) of SHP-1 expression (31). Eight to ten weeks later, GFP+ (transduced) and GFP− (non-transduced) OT-I T cells were isolated from the spleen and adoptively transferred into recipient B6 mice that received B16-OVA(APL) cells one day later. We compared OT-I T cells with decreased SHP-1 (GFP+) to those with normal amounts (GFP−) in the same mouse, as well as OT-I cells expressing an empty vector in separate control mice.
SHP-1 KD OT-I T cells did not limit tumor growth compared to WT counterparts, regardless of the presence of a high- or low-affinity epitope (Fig. 4A). We additionally compared recruitment of SHP-1 KD and WT OT-I to the tumor in the same mouse by measuring their relative ratio as compared to the initial ratio at the time of OT-I transfer. Although the ratio of KD to WT OT-I remained unchanged for high-affinity OT-I responses, the low-affinity response to Q4 favored SHP-1 KD OT-I (Fig. 4B–C). However, SHP-1 KD failed to induce a response to low-affinity antigens (T4, V4), as these tumors failed to recruit either WT or SHP-1 KD OT-I (Fig. 4C).
Figure 4:
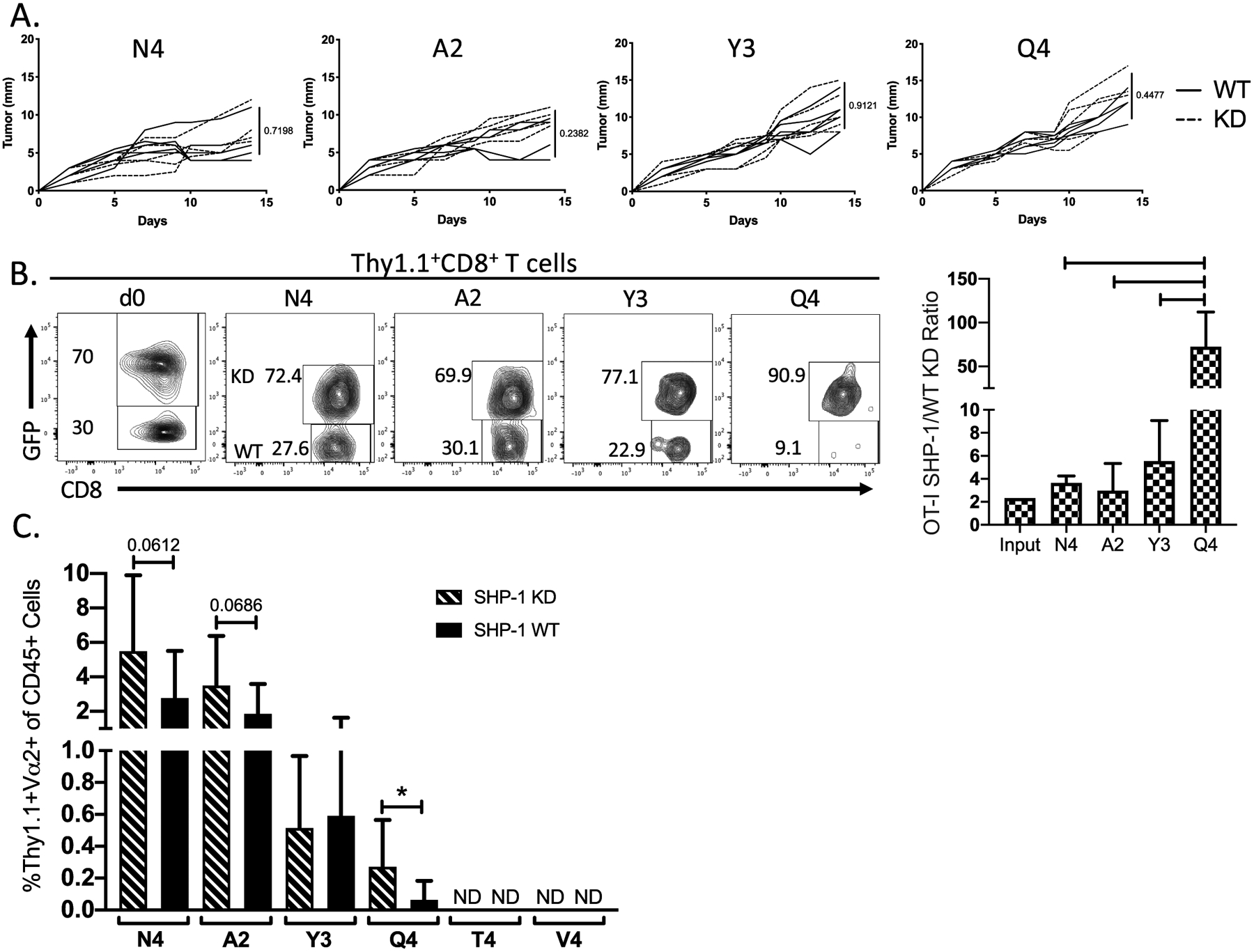
Inhibition of SHP-1 in antitumor CD8+ T cells expands available repertoire for low-affinity tumor antigen. We generated OT-I bone marrow chimeras expressing a SHP-1-specific shRNA, along with a GFP reporter. One day before tumor cell implantation, OT-I T cells were adoptively transferred into naïve B6 recipient mice. GFP+ (SHP-1 KD) and GFP− (non-transduced, SHP-1 WT) OT-I T cells in the tumor were analyzed. (A) Line graphs indicate the growth kinetics of the different B16-OVA(APL) tumors in the presence of SHP-1 KD (dashed) or WT (solid) OT-I CD8+ T cells. (B) Representative flow plots show the frequency of GFP+ (SHP-1 KD) compared to GFP− (SHP-1 WT) OT-I T cells within the OT-I CD8+ T cell population of a single tumor at time of harvest, including the frequency of the OT-I population upon initiation of the experiment (d0). Bar graph indicates the ratio of SHP-1 KD to WT OT-I T cells found in the tumor at day 14 post implantation. (C) Bar graph shows the percent OT-Is within the CD45+ cell population SHP-1 KD OT-I T cells (dashed) compared to WT counterparts (filled) within the same tumor. Frequencies are normalized to input of KD:WT OT-Is at time of adoptive transfer. Error bars indicate SD. Values that are not detectable above background are labeled ND. Statistical significance was determined by comparing the area under the tumor growth curve (A) or group mean (B-C) using an unpaired t test (n=5–8 mice per group, representative of two independent experiments). *P < 0.05, **P < 0.01.
SHP-1 KD resulted in a significant increase in frequency of OT-I that made IFNγ, TNFα and IL-2 following activation by both high-affinity (N4) and low-affinity (A2, Y3, Q4) tumors (Fig. 5A–B). These differences were measured in draining lymph nodes as well but not observed in the empty vector control tumors (Supplementary Fig. S3). Although no differences in CXCR5 or Granzyme B expression were observed, there were significant increases in the markers CD27 and PD-1 (Fig. 5C), which have been linked to increased TCR-stimulated proliferation of T cells in humans (50–52). There was also a significant difference in TCR signal duration observed during in vitro stimulations, as knockdown of SHP-1 caused an increase in CD3ζ phosphorylation over a sustained period of time across a range of TCR affinities (Supplementary Fig. S3). Overall, these findings demonstrate that SHP-1 KD functionally enhances low-affinity T cells, but with limited therapeutic benefit.
Figure 5:
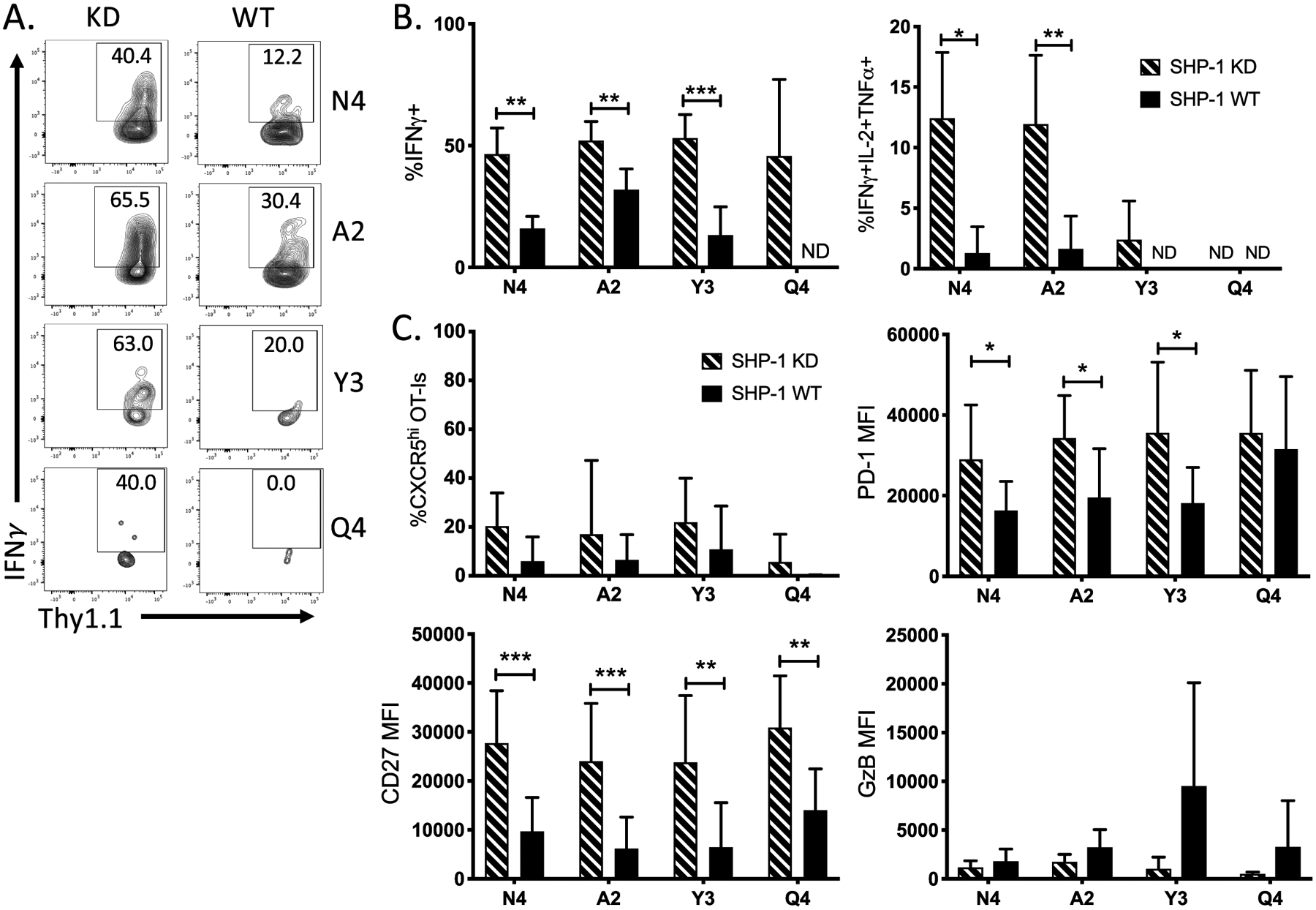
Inhibition of SHP-1 in antitumor CD8+ T cells enhances cytokine production. (A) Representative flow plots show the production of IFNγ by either SHP-1 KD OT-I CD8+ TILs or their WT counterparts following ex vivo restimulation with full-length OVA peptide. (B) Bar graphs indicate the frequency of cytokine producing OT-I SHP-1 KD (dashed) or WT (solid) T cells at d14 post tumor cell implantation. (C) Bar graphs show the frequency of expression or MFI of surface markers CXCR5, PD-1, CD27 and Granzyme B on OT-I T cells. Error bars indicate SD. Statistical significance was determined by comparing the group means using an unpaired t test (n=8–15 mice per group, representative of three independent experiments). *P < 0.05, **P < 0.01, ***P < 0.001.
Combining SHP-1 knockdown with ICB enables control of tumor growth by low-affinity T cells
Because SHP-1 KD increased the recruitment and effector function of low-affinity OT-I, we hypothesized that limiting SHP-1 activity would expand the antitumor function of low-affinity T cells following ICB. Although our previous results found that ICB alone only limited tumor growth in the presence of high-affinity OT-I (N4) (Fig. 2), combined ICB and SHP-1 KD resulted in a significant delay in tumor growth in the presence of low-affinity OT-I (A2, Y3, Q4)(Fig. 6A–B). The delay in tumor growth was rapid and durable, resulting in a significant decrease in tumor mass size in the week following treatment (Fig. 6B) and lasting at least 20 days (Fig. 6A). By day 20, the combination of SHP-1 knockdown and checkpoint blockade therapy resulted in no observable tumor for the wildtype (N4: 6/10) and the lower affinity variants (A2: 5/10; Y3: 4/10; Q4: 3/10). Tumor eradication was not observed following challenge with lower affinity tumors in other treatment settings. The reduced tumor growth could not be fully explained by an increase in OT-I recruitment to the tumor, as combined therapy did not result in an increase in OT-I frequency in response to the lower affinity OVA-APLs (Fig. 7A). However, an increase in CXCR3 expression was measured following combined treatment across all affinities (Fig. 7A), suggesting an increase in T cell activation and antitumor activity of the OT-I cells in response to increased TCR signaling and PD-1 blockade (53). To explore alternative mechanisms of antitumor activity mediated by low-affinity T cells, we measured endogenous CD8+ T cell responses to WT OVA (N4) or OVA APLs (A2, Y3, Q4). For lower antigen affinities (Y3 and Q4), an increase in the endogenous antitumor response was observed, as measured by endogenous CD8+ T cell frequency, CXCR3 expression and Granzyme B production (Fig. 7B). This phenotype was further supported by the frequency of endogenous CD8+ T cells capable of producing IFNγ upon ex vivo restimulation (Fig. 7C). Empty vector control experiments did not result in significant changes to any of these parameters, nor did ICB alone elicit an increase in endogenous IFNγ production (Supplementary Fig. S4). These results suggest that low-affinity OT-I rescued by combined ICB and SHP-1 KD mediate their antitumor effect by enhancing immune activation within the tumor microenvironment rather than by direct tumor killing.
Figure 6:
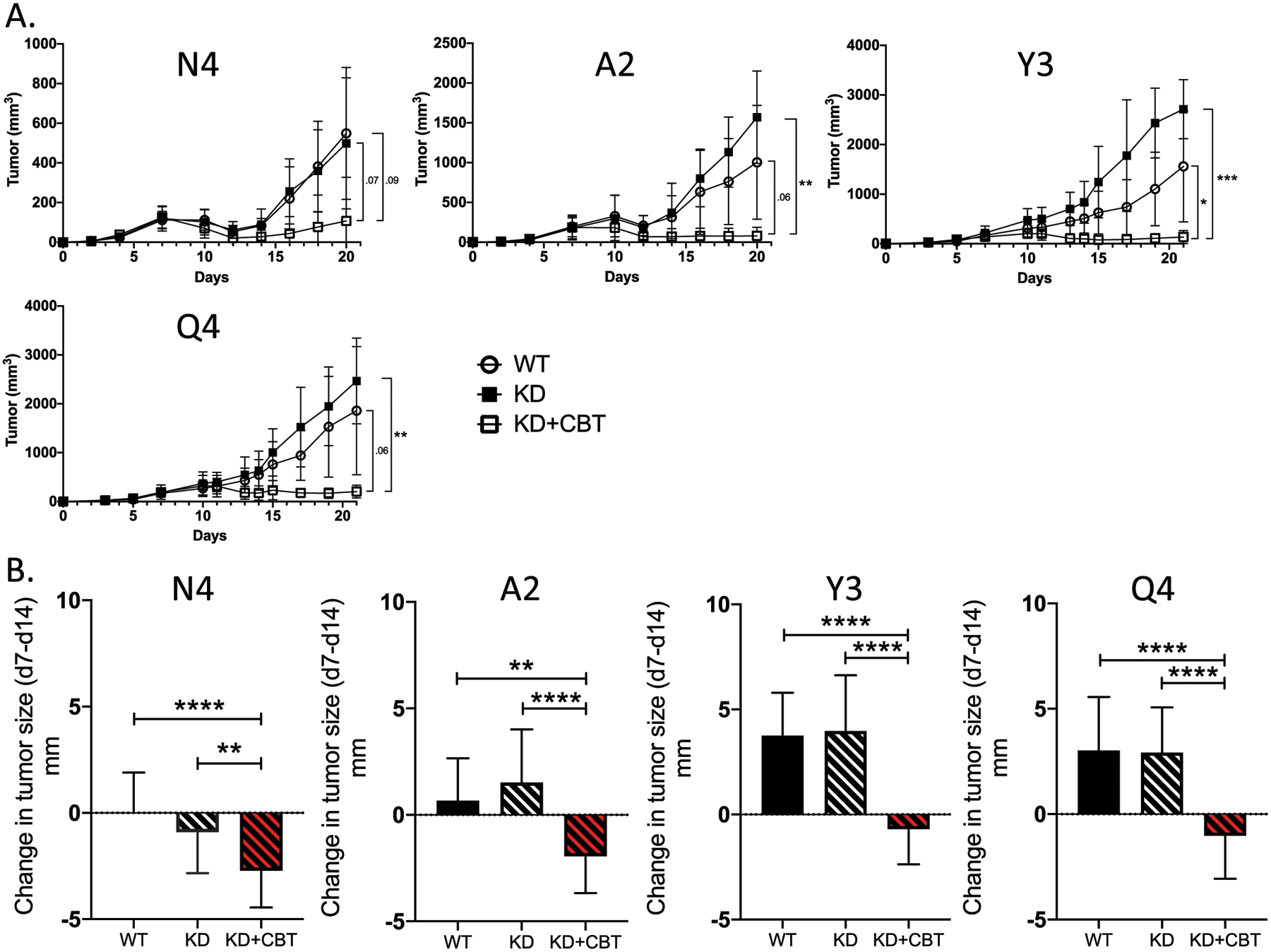
Inhibition of SHP-1, combined with ICB, promotes tumor regression across a wide range of tumor antigen affinities. (A) Line graphs indicate the growth kinetics of tumors in the presence of WT OT-I CD8+ T cells (open circle), SHP-1 KD OT-Is (filled square) and SHP-1 KD OT-Is in the presence of ICB (open square). (B) Bar graphs show the change in tumor diameter between day 7 (pre-treatment) and day 14 (post-treatment). Comparisons between WT (solid), SHP-1 KD (black dashed), and SHP-1 KD with CB treatment (red dashed) are made for each OVA(APL). Error bars indicate SD. Statistical significance was determined by comparing the area under the tumor growth curve (A) or group mean (B) using an unpaired t test (n=10–15 mice per group, representative of two independent experiments). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 7:
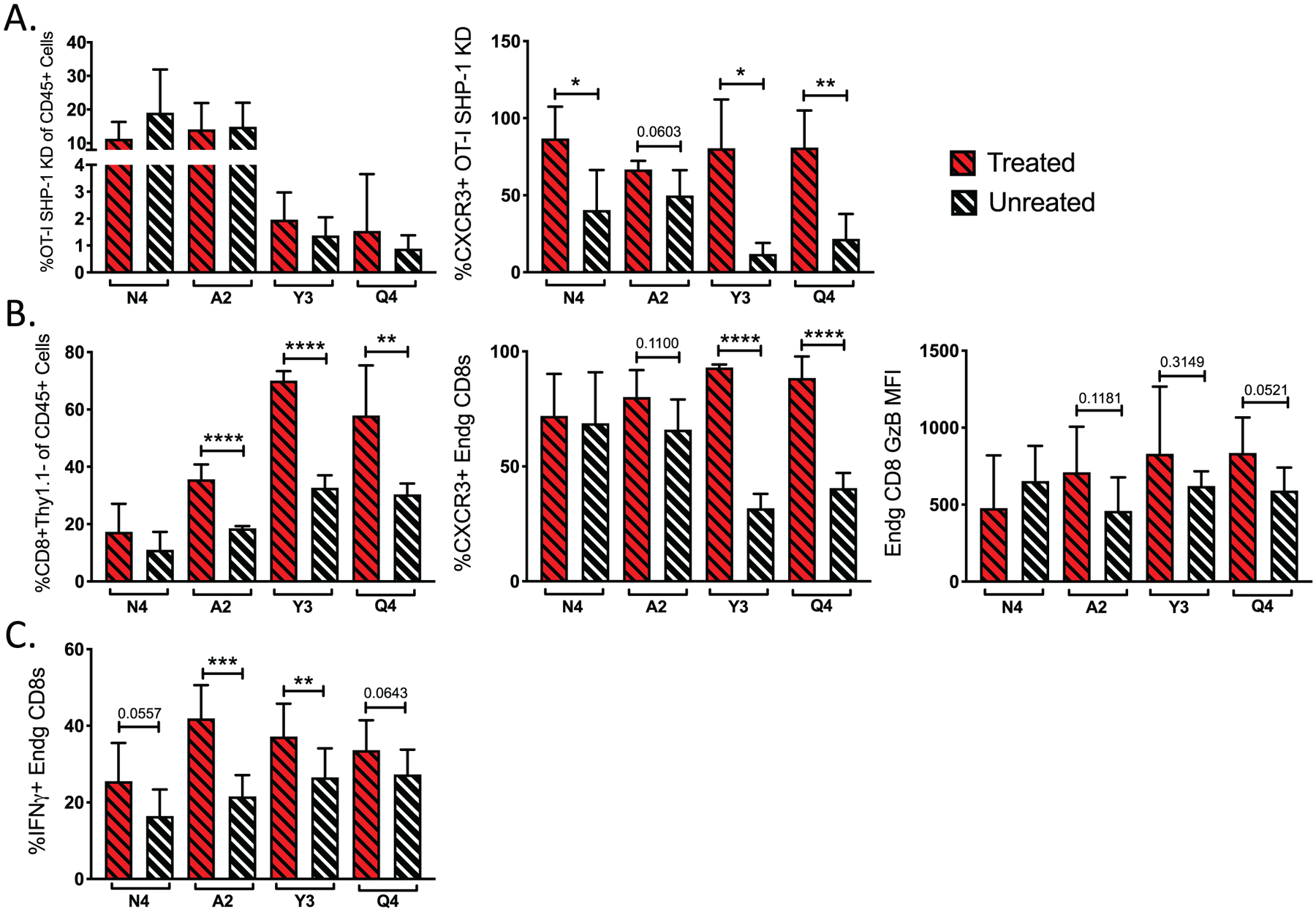
ICB combined with SHP-1 inhibition increases the frequency of IFNγ-producing endogenous antitumor T cells. (A) Bar graphs indicate the frequency of SHP-1 KD OT-Is in the CD45+ population with (red dashed) and without (black dashed) ICB and CXCR3 surface expression on KD OT-Is. (B) Bar graphs show the frequency of endogenous CD8+ T cells (Thy1.1−) within the CD45+ population in the tumor and expression of CXCR3 and Granzyme B on endogenous CD8+ T cells with (red dashed) or without (black dashed) ICB. (C) Bar graphs indicate the frequency of IFNγ producing endogenous CD8+ T cells after ex vivo restimulation with corresponding OVA(APL) peptide. Error bars indicate SD, and statistical significance was determined by an unpaired t test (n=10–15 mice per group, representative of two independent experiments). *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
The results we present in this study demonstrate that inhibition of SHP-1 potentiates the antitumor activity of low-affinity T cells responding to tumor antigen, particularly in combination with blockade of PD-1 and CTLA-4. Our findings clarify the role of TCR affinity in conferring on antitumor CD8+ T cells the ability to traffic to and control growth of solid tumors. Although TCR affinity enforced a threshold for efficient activation of anti-melanoma T cells, as well as their frequency within the tumor, low-affinity T cells demonstrated profound therapeutic potential. Altering the activation threshold of low-affinity T cells via inhibition of SHP-1, combined with ICB, demonstrated that low-affinity T cells can control tumor growth. The mechanism by which SHP-1 knockdown enhances the antitumor function of low-affinity T cells remains a topic of future investigation. Although our results show an increase TCR signal strength following SHP-1 inhibition, we also observe an increase in IFNγ production that may not be related to the role of SHP-1 in regulation of TCR activation threshold. Indeed, a previous study showed that T cell specific deletion of SHP-1 enhanced cytokine production without altering TCR activation threshold (33). SHP-1 may regulate negative selection mediated by low-affinity epitopes in the thymus (54). We use a knockdown approach that results in partial inhibition of SHP-1, rather than germline deletion, which may explain some of the differences between our study and previous approaches. Future studies will be needed to decipher TCR-dependent and TCR-independent roles for SHP-1 in regulation T cell activation and effector function.
Low-affinity T cells may represent a source of antitumor activity during natural tumor growth, as supported by our finding that antitumor responses induced by ICB are dominated by high-affinity T cells. The induction of high-affinity TCRs for either tumor-associated self-antigens or neo-antigens presents the risk of off-target effects that may lead to autoimmune activity. One study that induced tumor antigen expression in off-target organs determined that low-affinity T cell interactions avoided concomitant autoimmunity in an ovarian carcinoma model (55). Other studies observed no differences in the antitumor capability of high-affinity and moderate affinity antitumor TCRs (28,29,56). High-affinity T cells may be more prone to increased checkpoint molecule expression and the development of functional exhaustion (24). In sum, we argue that moderate- to low-affinity TCRs may be induced to acquire antitumor activity. Such cells may be useful in combination with other immunotherapies and may present less danger of off-target toxicity.
Cytolytic antitumor activity is mediated by CD8+ T cells, and the frequency of CD8+ T cells in tumors corresponds to their ability to control tumor growth. Our findings show that ICB effectiveness corresponds to an increase in the frequency of high-affinity T cells in the tumor. In contrast, low-affinity T cells rescued by SHP-1 inhibition may utilize distinct mechanisms for controlling tumor growth. One possibility is that low-affinity TIL re-shape the tumor microenvironment to promote antitumor immunity. Two pieces of evidence in support of that are the increased IFNγ production by TIL after SHP-1 knockdown and the increase in the frequency of endogenous CD8+ T cells responding to OVA after combined ICB and SHP-1 knockdown. These results may relate to a prior study that found that cytotoxic and cytokine-dependent antitumor functions could be uncoupled in a TCR affinity-dependent manner (57). Future studies are needed to elucidate the mechanisms by which SHP-1-deficient T cells mediate their antitumor function.
SHP-1-regulated TCR signal strength plays a role in the functional differentiation of T cells (31). For example, there is an optimal TCR affinity for tumor antigen that generates intense and sustained TCR signals within NY-ESO-1 specific CD8+ T cells, with a role for SHP-1 in determining signal strength (35). Although in vitro studies have shown that the phosphatases SHP-1 and SHP-2 have overlapping specificities, other studies have indicated that they preferentially co-localize with the TCR and PD-1, respectively. Therefore, in vivo SHP-1 and SHP-2 appear to act in distinct and separate pathways (36,37,58,59). SHP-1 and PD-1 inhibit T cell activation independently, with PD-1 (and possibly SHP-2) preferentially inhibiting high-affinity T cells and SHP-1 limiting activation incrementally as TCR affinity increases (11). Deficiency in SHP-1 confers resistance to Treg suppression in both in vitro and in vivo settings (34). Previous work in a leukemic model supports the rationale for targeting SHP-1 activity in antitumor T cells (46).
While we demonstrate that ICB preferentially rescues high-affinity antitumor responses, ICB has been previously shown to increase the antitumor activity of both high- and low-affinity TILs in a B16-OVA melanoma model (45). However, that study utilized a model of therapeutic cell transfer, suggesting that ICB may target low-affinity T cells if T cell activation thresholds are lowered or bypassed. This may correspond to our finding that SHP-1 inhibition, which lowers TCR activation threshold, combines with ICB to enhance the antitumor function of low-affinity T cells. In fact, there are likely a number of activation thresholds that must be overcome in order to efficiently activate and recruit antitumor CD8+ T cells, including thresholds for activation, migration to the tumor and antitumor effector functions. In an adoptive cell therapy model, activation of OT-I T cells before adoptive transfer into a tumor bearing mouse was necessary for antitumor activity (60), supporting a connection between proper T cell activation and effector function within the tumor. Our current study, focused on the role of SHP-1 inhibition in T cells activated by in vivo tumor growth, suggests SHP-1 may prove a viable target in settings of adoptive cell therapy or CAR T cell therapy.
Supplementary Material
Acknowledgments:
The authors declare no competing financial interests.
Dr. Dietmar Zehn kindly provided OVA-APL constructs.
References:
- 1.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999;17(7):2105–16. [DOI] [PubMed] [Google Scholar]
- 2.Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. Journal of the National Cancer Institute 2010;102(7):493–501 doi 10.1093/jnci/djq009. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994;271(12):907–13. [PubMed] [Google Scholar]
- 4.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369(2):134–44 doi 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711–23 doi 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. Journal of leukocyte biology 2013;94(1):25–39 doi 10.1189/jlb.1212621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271(5256):1734–6. [DOI] [PubMed] [Google Scholar]
- 8.Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Current opinion in immunology 2015;33C:23–35 doi 10.1016/j.coi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366(26):2443–54 doi 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369(2):122–33 doi 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, et al. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest 2013;123(3):1044–56 doi 10.1172/JCI65325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nature reviews Immunology 2012;12(4):269–81 doi 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turcotte S, Rosenberg SA. Immunotherapy for metastatic solid cancers. Advances in surgery 2011;45:341–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trapani JA. Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B. Aust N Z J Med 1995;25(6):793–9. [DOI] [PubMed] [Google Scholar]
- 15.Young JD, Hengartner H, Podack ER, Cohn ZA. Purification and Characterization of a Cytolytic Pore-Forming Protein from Granules of Clones Lymphocytes with Natural Killer Activity. Cell 1986;44:849–59. [DOI] [PubMed] [Google Scholar]
- 16.Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol 2014;232(2):199–209 doi 10.1002/path.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell 2016;29(3):285–96 doi 10.1016/j.ccell.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol 2012;33(7):364–72 doi 10.1016/j.it.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther 2017;16(11):2598–608 doi 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348(6230):124–8 doi 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350(6257):207–11 doi 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janicki CN, Jenkinson SR, Williams NA, Morgan DJ. Loss of CTL function among high-avidity tumor-specific CD8+ T cells following tumor infiltration. Cancer Res 2008;68(8):2993–3000 doi 10.1158/0008-5472.CAN-07-5008. [DOI] [PubMed] [Google Scholar]
- 23.Wang RF, Wang HY. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res 2017;27(1):11–37 doi 10.1038/cr.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahm CD, Colluru VT, McNeel DG. Vaccination with High-Affinity Epitopes Impairs Antitumor Efficacy by Increasing PD-1 Expression on CD8(+) T Cells. Cancer Immunol Res 2017;5(8):630–41 doi 10.1158/2326-6066.CIR-16-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. Journal of Virology 2003;77(8):4911–27 doi 10.1128/jvi.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15(8):486–99 doi 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res 2013;73(12):3591–603 doi 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A 2013;110(17):6973–8 doi 10.1073/pnas.1221609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dougan SK, Dougan M, Kim J, Turner JA, Ogata S, Cho HI, et al. Transnuclear TRP1-specific CD8 T cells with high or low affinity TCRs show equivalent antitumor activity. Cancer Immunol Res 2013;1(2):99–111 doi 10.1158/2326-6066.CIR-13-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim C, Wilson T, Fischer KF, Williams MA. Sustained interactions between T cell receptors and antigens promote the differentiation of CD4(+) memory T cells. Immunity 2013;39(3):508–20 doi 10.1016/j.immuni.2013.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Snook JP, Kim C, Williams MA. TCR signal strength controls the differentiation of CD4+ effector and memory T cells. Sci Immunol 2018(3) doi 10.1126/sciimmunol.aas9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plas DR, Johnson R, Pingel JT, Matthews RJ, Dalton M, Roy G, et al. Direct Regulation of ZAP-70 by SHP-1 in T Cell Antigen Receptor Signaling. Science 1996;272(5265):1173–6 doi 10.1126/science.272.5265.1173. [DOI] [PubMed] [Google Scholar]
- 33.Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG. Shp1 regulates T cell homeostasis by limiting IL-4 signals. J Exp Med 2013;210(7):1419–31 doi 10.1084/jem.20122239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mercadante ER, Lorenz UM. T Cells Deficient in the Tyrosine Phosphatase SHP-1 Resist Suppression by Regulatory T Cells. J Immunol 2017;199(1):129–37 doi 10.4049/jimmunol.1602171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Presotto D, Erdes E, Duong MN, Allard M, Regamey PO, Quadroni M, et al. Fine-Tuning of Optimal TCR Signaling in Tumor-Redirected CD8 T Cells by Distinct TCR Affinity-Mediated Mechanisms. Front Immunol 2017;8:1564 doi 10.3389/fimmu.2017.01564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017;355(6332):1428–33 doi 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rota G, Niogret C, Dang A, Barros C, Fonta N, Alfei F, et al. Shp-2 Is Dispensable for Establishing T Cell Exhaustion and for PD-1 Signaling In Vivo. Cell Rep 2018. doi 10.1016/j.celrep.2018.03.026. [DOI] [PubMed] [Google Scholar]
- 38.Krummey SM, Martinez RJ, Andargachew R, Liu D, Wagener M, Kohlmeier JE, et al. Low-Affinity Memory CD8+ T Cells Mediate Robust Heterologous Immunity. J Immunol 2016;196(6):2838–46 doi 10.4049/jimmunol.1500639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature 2009;458(7235):211–4 doi 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bobek V, Kolostova K, Pinterova D, Kacprzak G, Adamiak J, Kolodziej J, et al. A clinically relevant, syngeneic model of spontaneous, highly metastatic B16 mouse melanoma. Anticancer Research 2010;30:4799–804. [PubMed] [Google Scholar]
- 41.Huffaker TB, Lee SH, Tang WW, Wallace JA, Alexander M, Runtsch MC, et al. Antitumor immunity is defective in T cell-specific microRNA-155-deficient mice and is rescued by immune checkpoint blockade. J Biol Chem 2017;292(45):18530–41 doi 10.1074/jbc.M117.808121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahmood S, Kanwar N, Tran J, Zhang ML, Kung SK. SHP-1 phosphatase is a critical regulator in preventing natural killer cell self-killing. PLoS One 2012;7(8):e44244 doi 10.1371/journal.pone.0044244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen R, Belanger S, Frederick MA, Li B, Johnston RJ, Xiao N, et al. In vivo RNA interference screens identify regulators of antiviral CD4(+) and CD8(+) T cell differentiation. Immunity 2014;41(2):325–38 doi 10.1016/j.immuni.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009;69(7):3077–85 doi 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez-Usatorre A, Donda A, Zehn D, Romero P. PD-1 Blockade Unleashes Effector Potential of Both High- and Low-Affinity Tumor-Infiltrating T Cells. J Immunol 2018;201(2):792–803 doi 10.4049/jimmunol.1701644. [DOI] [PubMed] [Google Scholar]
- 46.Stromnes IM, Fowler C, Casamina CC, Georgopolos CM, McAfee MS, Schmitt TM, et al. Abrogation of SRC homology region 2 domain-containing phosphatase 1 in tumor-specific T cells improves efficacy of adoptive immunotherapy by enhancing the effector function and accumulation of short-lived effector T cells in vivo. J Immunol 2012;189(4):1812–25 doi 10.4049/jimmunol.1200552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khaled AR, Butfiloski EJ, Sobel ES, Schiffenbauer J. Functional consequences of the SHP-1 defect in motheaten viable mice: role of NF-kB. Cell Immunol 1998;185:49–58. [DOI] [PubMed] [Google Scholar]
- 48.Schultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol 1997;15:302–7. [DOI] [PubMed] [Google Scholar]
- 49.Tsui HW, Siminovitch KA, de Souza L, Tsui FWL. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet 1993;4:124–9. [DOI] [PubMed] [Google Scholar]
- 50.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014;124(5):2246–59 doi 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inozume T, Hanada K, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother 2010;33(9):956–64 doi 10.1097/CJI.0b013e3181fad2b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powell DJ, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood 2005;105(1):241–50 doi 10.1182/blood-2004-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, et al. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 2019;50(6):1498–512 e5 doi 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez RJ, Morris AB, Neeld DK, Evavold BD. Targeted loss of SHP1 in murine thymocytes dampens TCR signaling late in selection. Eur J Immunol 2016;46(9):2103–10 doi 10.1002/eji.201646475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller AM, Bahmanof M, Zehn D, Cohen EEW, Schoenberger SP. Leveraging TCR Affinity in Adoptive Immunotherapy against Shared Tumor/Self-Antigens. Cancer Immunol Res 2019;7(1):40–9 doi 10.1158/2326-6066.CIR-18-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Irving M, Zoete V, Hebeisen M, Schmid D, Baumgartner P, Guillaume P, et al. Interplay between T cell receptor binding kinetics and the level of cognate peptide presented by major histocompatibility complexes governs CD8+ T cell responsiveness. J Biol Chem 2012;287(27):23068–78 doi 10.1074/jbc.M112.357673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clancy-Thompson E, Devlin CA, Tyler PM, Servos MM, Ali LR, Ventre KS, et al. Altered Binding of Tumor Antigenic Peptides to MHC Class I Affects CD8(+) T Cell-Effector Responses. Cancer Immunol Res 2018;6(12):1524–36 doi 10.1158/2326-6066.CIR-18-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12(4):252–64 doi 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao M, Guo W, Wu Y, Yang C, Zhong L, Deng G, et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade Acta Pharmaceutica Sinica B 2018;9(2):304–15 doi 10.1016/j.apsb.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riquelme E, Carreno LJ, Gonzalez PA, Kalergis AM. The duration of TCR/pMHC interactions regulates CTL effector function and tumor-killing capacity. Eur J Immunol 2009;39(8):2259–69 doi 10.1002/eji.200939341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.