

Abstract
For any dynamical system, like living organisms, an attractor state is a set of variables or mechanisms that converge towards a stable system behavior despite a wide variety of initial conditions. Here, using multi-dimensional statistics, we investigate the global gene expression attractor mechanisms shaping anaerobic to aerobic state transition (AAT) of Escherichia coli in a bioreactor at early times. Out of 3,389 RNA-Seq expression changes over time, we identified 100 sharply changing genes that are key for guiding 1700 genes into the AAT attractor basin. Collectively, these genes were named as attractor genes constituting of 6 dynamic clusters. Apart from the expected anaerobic (glycolysis), aerobic (TCA cycle) and fermentation (succinate pathways) processes, sulphur metabolism, ribosome assembly and amino acid transport mechanisms together with 332 uncharacterised genes are also key for AAT. Overall, our work highlights the importance of multi-dimensional statistical analyses for revealing novel processes shaping AAT.
Subject terms: Computational biology and bioinformatics, Systems biology
Introduction
Microorganisms are able to adapt to diverse environmental changes, making them the longest surviving living systems on this planet. The well-studied bacterium Escherichia coli is able to switch between 2 stable attractor states, aerobic and anaerobic conditions, based on oxygen availability1. E. coli grows well on both conditions, albeit at a lower rate anaerobically. Over the last few decades, numerous studies have investigated the transition of E. coli between the states from a traditional and reductionist standpoint2,3.
The current understanding of E. coli metabolism during aerobic condition is that the glucose flux moves through the glycolysis pathway, and channelled to the TCA cycle via pyruvate dehydrogenase complex. This mode of respiration yields higher ATP levels, thereby, generating more energy compared to anaerobic respiration. In the absence of oxygen, depending on the availabilities of electron donors or acceptors, pyruvate formate-lyase, nowadays called formate C-acetyltransferase, catalyses pyruvate and coenzyme-A into formate and acetyl-CoA, a reversible conversion. In addition, lactate dehydrogenase also acts on pyruvate to produce lactate, and still others include succinate, acetate and ethanol production. These processes, collectively, suppress the metabolic fluxes channelling to the TCA cycle. Thus, major metabolic switching mechanisms occur during aerobiosis or AAT, and numerous recent studies have focused on deciphering other key novel mechanisms that are also concerted. For example, Green and colleagues used microarray transcript profiling to reveal peroxide stress response and methionine biosynthesis as novel processes induced during aerobiosis4. Although these works have shed light into the novel processes guiding E. coli AAT, it is important to note that living cells are dynamical systems that involve large interplay of cellular networks.
For understanding complex cellular behaviours such as immune response or growth, numerous studies have employed computational models utilizing linear and non-linear differential equations to monitor intracellular as well as extracellular molecular species, such as proteins or metabolites turnover, over time5. In addition, dynamical systems and chaos theories have also been used to study the complex self-organizing (e.g. skin pattern formation) and stochastic or chaotic behaviors (e.g. multiple lineage cell differentiation from single cell origin) of living systems6. Here, the models developed are qualitative, rather than quantitative, in defining the dynamical system, and are often used to understand the sensitivity of the initial conditions, or perturbations, to their longer term steady or periodic states7.
With the recent advent of high-throughput technologies, we are now able to observe transcriptome-wide gene expression changes over time as opposed to only a few proteins or metabolites monitored traditionally. We also know that not individual genes, but rather the complex networks of genes drive key cellular processes. Therefore, genes can be considered to be a set of points in state space8,9. The state refers to the expression vector of a large set of co-regulated genes of interest. In other words, the alteration of expression levels of specific sets of genes can be represented by a continuous trajectory in their expression state space10. Some of these trajectories are attractive, that is, they converge towards a fixed point if the system is perturbed from a nearby state (Fig. 1).
Figure 1.

Schematic illustration of attractor landscape and cellular trajectory. (A,B) Cellular gene expression profile represented by (A) matrix and (B) heat map of gene expression level. Each element represents the expression level of a gene (rows) in a cellular state (columns). (C) Schematic representation of cell trajectory convergence on principal components 1 and 2 (denoted as PC1 and PC2) space. Each point represents a sample’s entire gene expression profile within one of the two transitioning processes caused by two distinct perturbations13. (D) The landscape is a schematic 3-D projection of N (total number of genes) to a two-dimensional state space. In the attractor landscape, many stationary attractors (represented by the local minima), which correspond to the natural cellular phenotypes such as cell fate, might co-exist. Each attractor associates with a unique cellular signature profile (or gene expression profile in this study). The transitioning processes (dashed blue line) guide the cell from one stable attractor to another57. (E) Gene expression attractor landscapes generated by the superimposition of probability distribution of Pearson and mutual information correlation mertics to create a 3-D space18. Existence of stable attractor coinciding the convergence of cellular trajectories is indicated by the local minima.
Each cellular phenotype can correspond to an attractor in the gene expression state space, where they represent stable state, either peaks or valleys in the landscape, to which the system returns given a small biological perturbation11. In other words, the dynamical pattern of gene expressions developing over time reflect the concerted regulation of the transcriptome and can lead to convergence or equilibrium of the cell state, often referred to as attractor state12,13.
The presence of multi-dimensional attractor states based on high-throughput gene expression data has been experimentally implicated in recent times14,15. It has been shown that dynamic gene expression trajectories, using principal components and correlation metrics, reveals subset of the state space convergence in time, a hallmark of attractor, coinciding with the end of cell fate completion16–18 (Fig. 1C,D).
Despite the progresses made for E. coli AAT, as mentioned earlier, detailed gene expression response using multi-dimensional statistical approaches and a global transcriptome-wide scale analyses using dynamical system approach still remain largely underexplored. Most studies investigate only the most differentially expressed genes over time using arbitrary expression fold, e.g. 2-fold, cut-off without investigating the underlying statistical structure or undertake dynamical systems view. However, during aerobiosis or cell state changes, the transcriptomic network invokes a progressive directional change of thousands of gene expressions in time, through which the cell state expression pattern is adopted19–21.
With the recent advances in understanding how transcriptomic networks govern cell fate decisions, it is now possible to explore biological patterns and order using multi-dimensional statistics22–25. Here we, therefore, extended the concept, using multi-dimensional statistical methods on RNA-Seq data, to investigate the anaerobic to aerobic transition of E. coli grown in 3-liter bioreactors. Sampling of cells for transcriptome analysis were obtained rapidly, with 6 time points in the first 10 minutes. Using distribution fitting, linear and non-linear correlations (to develop attractor landscape), PCA (to observe attractor trajectory), hierarchical clustering and gene ontology, we report the gene expression patterns and functions during early aerobiosis.
Results
E. coli transcriptome-wide expressions follow lognormal distribution
The original experiments were performed by Feuer and colleagues26. Briefly, E. coli K-12 strain W3110 cells were grown anaerobically in a 3-liter continuously stirred tank bioreactor at pH7 and 37°C, and stirred at 500 rpm with a Rushton turbine. The first sample was drawn (t = 0) when OD of 3 at 600 nm was achieved, and air supply of 1L/min was then initiated. Subsequent samples were taken at t = 0.5, 1, 2, 5 and 10 min, with 3 biological replicates. The resultant RNA-Seq data, in read counts, for all samples were deposited and available with GEO accession number GSE71562.
The RNA-Seq data needed to be checked and reduced for gene expressions that can be considered noisy, especially the lowly expressed ones. Previously, we had used statistical distributions on normalized expressions as a means to remove unreliable or noisy genes17,23,24,27. Therefore, Transcripts Per Kilobase Million or Transcripts Per Million (TPM) normalization of the read counts of all samples were performed and checked for statistical distribution of all (4240 non-zero) transcripts for all time points (Figs. 2A and S1A). As previously seen for other cell types, we observed theoretical Pareto (power-law) and lognormal distributions best followed the experimental transcript distributions above a threshold of 5 TPM. Next, to check how close the gene expressions match the Pareto and lognormal distributions, we performed a Quantile-Quantile plot (Figs. 2B and S1B). The data show that lognormal has major advantage over Pareto, noticeable for the higher expressions (TPM > 800). The quality of statistical distribution fit was finally confirmed using the Akaike information criterion (Table S1). Notably, lognormal distribution for gene expressions has also been recently implicated for several other cell types27,28. Hence, we concur that our E. coli gene expressions follow lognormal distribution across all replicates and time points (Fig. S1A,B) and retained genes with transcript levels above the 5 TPM threshold for further analysis (N = 3391).
Figure 2.

E. coli transcriptome-wide statistical properties. (A,B) Comparison of transcriptome-wide data with statistical distributions: (A) Cumulative distribution functions versus TPM values in logscale, and (B) Quantile-quantile plot between transcriptome data (experimental data – black colour) and lognormal (red), Pareto (yellow), Burr (cyan), loglogistic (blue), Weibull (purple), and gamma (grey) statistical distributions (Methods). Figure is one representative at t = 10 min for replicate a. See Fig. S1 for other time points and replicates. Transcriptome-wide correlation in time using: (C) Pearson correlation, and (D) Mutual Information-based correlation metrics (Methods) between time t0 (0 min) and ti (0, 0.5, 1, 2, 5, 10 min) for all 3 replicates (replicate a – red, replicate b – green, replicate c - blue) across 3391 genes with expression above 5 TPM (left panels) and 3389 genes upon removal of two highest expressed genes rnpB and lpp (right panels).
Correlation analysis shows gradual transcriptome-wide deviation from origin
Temporal correlation metrics are often used to check how a dataset deviates from their initial condition, time or perturbation17,18,22. Here, to investigate the transcriptome-wide deviation from t = 0, we adopted 4 correlation metrics (Pearson, Spearman, Biweight midcorrelation or bicor29, and Mutual Information-based correlation30 MIc, see Methods). We used these metrics to investigate parametric (Pearson), and non-parametric (Spearman, bicor and MIc) correlations. All metrics pointed to a gradual decay in the transcriptome-wide correlation with time for all 3 replicates (Fig. 2C,D – left panel and Fig. S2). These data suggest that there is a constant and gradual global transcriptomic “movement” in time31.
Looking closer, the Pearson correlation, which measures the linear association between 2 vectors, showed a sharp initial decay (t = 0.5 and 1 min) and a recovery before gradual decay for 2 replicates. This “discrepancy” is absent in the other 3 non-parametric correlations which could be less outlier-sensitive. In addition, the Q-Q plots show that the highly expressed genes deviated from the global statistical distribution (Figs. 2B and S1B). Thus, we removed genes one by one from the highest expression and checked the transcriptome-wide Pearson correlation. Consequently, the removal of the top 2 highest expressed genes (rnpB and lpp) was sufficient to result in smooth correlation decay in all metrics used (Fig. 2C,D – right panel and Fig. S2). Thus, we kept the remaining genes (N = 3389) for further analysis. Next, we investigate which portions of genes are key for guiding the anaerobic to aerobic state transition. To do this, we need to consider the concepts of attractors often used in physics and mathematics18,32.
Defining attractor region and transcriptomic elements
State space for a dynamical system is the set of all possible states, where each state of the system corresponds to a unique point in the state space13,14,20. As it is difficult to define explicit expressions for representing transcriptome-wide dynamics, the analysis of state space as set of all possible pairs of linear and non-linear gene expression correlation dynamics (based on Rv and MIv, see below) provides a useful way for understanding the qualitative features of attractor localizations or visualizations18,22. Note that the modified mutual information (MIv) and Pearson correlation (Rv) used here are slightly different from those used in the previous section (Method).
The fractal nature of transcriptomic response in cell fate determination has been explored in previous studies18,33,34. Basically, discrete subsets of transcriptome are responsible for guiding the transition of gene expressions from one “equilibrium” attractor state to another. The attractor state is the result of the convergence of gene expression dynamics across the transitioning time. Thus, in order to identify the genetic drivers of AAT, we divided the transcriptome into discrete fractions, namely transcriptomic elements18, and compared their individual trajectories against attractor region (Fig. 3A). Any element falling within the attractor boundary is attributed to AAT response.
Figure 3.

Attractor landscape by probability density distributions of correlations, transcriptomic elements and attractor genes. (A) Schematic trajectories for transcriptomic elements falling into attractor (red) and not falling into attractor (blue). (B) Distribution and (C) Standard deviation of Rv (top panel) and MIv (bottom panel) with different transcriptomic element size (denoted as n) of replicate a at 0.5 minutes. Distribution of Rv and MIv for ensembles of n randomly chosen genes (n = 25, 50, 100, 200, 400, 600, 800, 1000) were generated with 100 repeats. Standard deviation of Rv and MIv decreases as n increases (except for when n = 25 for Rv), and follows + c law. See Fig. S3A,B for other time points. (D) 3D plot of the superimposition of the probability distribution (SPD) of Rv and MIv over all time points for the whole transcriptome. SPDs were estimated by getting Rv and MIv values of 100 randomly chosen genes for 100 times, using two-dimensional kernel density estimation. (E) Trajectory of whole genome (3389 genes) falling within attractor boundary (solid contour line) overlaid on SPD of whole transcriptome Rv and MIv. The trajectory was generated by averaging 100 trajectories of 100 randomly chosen genes from the pool of 3389 genes. (F) Trajectories of cumulative attractor (1800), and non-attractor (1589) genes overlaid on SPD of Rv and MIv for whole transcriptome. (G) Distribution of expression level for attractor (red), and non-attractor (blue) genes at representative t = 0. See Fig. S3D for other time points.
A transcriptomic element is a minimum set of genes that possess enough correlation information (based on Rv and MIv) to show whether they fall into a cell attractor state18. To identify the size of a transcriptomic element, n genes were randomly chosen from the whole genome (n = 25, 50, 100, 200, 400, 600, 800, 1000), and their Rv and MIv were evaluated for 100 repeats at all time points. The standard deviation of the Rv and MIv were then plotted for the different n sizes and compared with the law of large numbers or LLN18,35. Both Rv and MIv distributions of the transcriptomic elements converged to specific values for n > 50 (Figs. 3B and S3A). Furthermore, the result shows that the standard deviations of Rv and MIv distributions at each time point decrease with increasing n, approximately following the law of large number (Figs. 3C and S3B). To investigate ensemble of genes that fall into the AAT attractor basin we, therefore, chose n = 100 as a compromise between density distribution and to retain a reasonable number of transcriptomic elements since higher n reduces the total number of elements.
To check the existence of cell attractor state in AAT, we analysed the superimposition of probability density (SPD) distribution for modified Pearson correlations (Rv) and modified mutual information (MIv) of gene expression deviations from t = 0 to 10 minutes18 (Method). We found 2 distinct peaks (Fig. 3D,E), with the major peak coinciding with the state transition end time of the experiments. This implies the existence of cell attractor by the localizations of Rv and MIv on the correlation space. As previously, we defined the attractor region boundary or basin as the inflection plane of the major peak curves18, and observed the transcriptome-wide trajectory also fall within this attractor region (Fig. 3E). Note that there are usually several meta-stable attractor states before the final state transition occurs. Here, we are evaluating the early meta-stable attractor states crucial for AAT.
Identification of genes falling into attractor basin
To identify the number of genes that fall into the attractor basin and their names, we first ranked all genes according to their expression variability, measured by standard deviation (SD) across time18. Secondly, we assembled the ranked genes into transcriptomic elements (n = 100) and checked their Rv-MIv trajectories in time across the coordinates of the attractor region (Fig. S4A). We observe that one element, with the highest SD, fall above the attractor basin, 4 elements (3, 4, 5 and 12) fall into the basin and the rest fall out and remain below the basin (Fig. S4A).
The Euclidean distance of each element compared with the whole transcriptome trajectory is shown in Fig. S3C. Notably, the elements that fall above and into the basin shows the closest distance (less than the mean values) with whole transcriptome trajectory. Thus, we concur that these 5 transcriptomic elements are key in shaping the AAT response, and checked their combined effect. As anticipated, merging the elements into a larger sub-transcriptome resulted in them falling into the attractor region (Fig. S3C, black dotted). Hence, we named them as attractor genes.
Among the remaining 29 transcriptomic elements, 13 show close Euclidian distance to the whole transcriptome trajectory (Fig. S3D, empty circle symbol), while 16 elements have large deviation (Fig. S3D. empty square symbol). We considered them as pseudo-attractor and non-attractor elements as their collective or combined Rv-MIv trajectories fall into and outside the attractor basin, respectively (Fig. S3D, green and blue dotted).
Finally, we combined the attractor (500) and pseudo-attractor (1300) genes and tracked their overall trajectories which resulted in them falling into the attractor basin (Fig. 3F). Since the 1,800 genes collectively enter into the attractor basin, we now re-term them as the attractor genes, while keeping the remaining as the non-attractor genes (Fig. 3F). In other words, our attractor analyses reveal 53% or about half of the transcriptome, spreading across a wide spectrum of expression levels (Figs. 3G and S3D), is responsible for shaping the E. coli AAT. Next, we also conducted a similar attractor study, sorting elements according to fold-changes instead (Fig. S4B). Here, we obtained 65% of the transcriptome fall into the attractor basin.
Overall, contrary to the general impression that only a small number of highly expressed genes shape AAT, our data suggest that gene expression levels are not indicative of AAT, and an order of about half the transcriptome is crucial for the biological state transition.
Principal component analysis as a test for attractor genes
Previous works had utilised principal components (PC) trajectories to investigate the dynamic global response of cell differentiation13,16–18,36. Here, we performed similar analysis for whole, attractor and non-attractor genes. PCs 1 and 2 constituted over 70% variance at t = 0 for all genes (Fig. S5A) and, hence, we tracked their values at each time point (Fig. 4A). As anticipated, our results show that the attractor genes tracked similar whole genome PC trajectories in time.
Figure 4.

Principal component (PC) analysis and auto-correlations of whole transcriptome attractor and non-attractor genes. (A) Gene expression trajectory of whole transcriptome (black), attractor (red), non-attractor (blue), and no response genes (brown), obtained by taking the average trajectories of 3 replicates, presented on first 2 principal components space. Right panel indicates non-attractor trajectory on a larger scale. (B) Temporal correlation of whole transcriptome (black), attractor (red), and non-attractor (blue) genes using Pearson (top left panel), Spearman (top right panel), Biweight midcorrelation (bottom left panel) and Mutual Information-based (bottom right panel) correlation metrics.
Next checking correlations, the attractor genes produce the most variable response (Fig. 4B). Nevertheless, the non-attractor genes also show monotonic or gradual variation in time, suggesting that even these genes can support AAT response, albeit with lower kinetics. Therefore, in this remaining 1,589 genes, we removed 68 genes that had below 1.12 fold changes at any time point, and checked their PCA and correlation plots (Figs. 4A and S5B). Our data show that removing the 68 genes did not noticeably affect the PCA or correlation response. Although the 1521 genes did not enter the attractor basin, they still show gradual response in time. Thus, these were named as collective non-attractor genes.
To summarize, our analysis indicate that 1,800 attractor genes shape the global AAT response, while 1521 genes follow weak but collective global response with remaining 68 genes not having any response. (Note that these 68 genes are additional to the 851 genes already removed during the low and high expression filtering using lognormal distribution fitting.)
Temporal groups of attractor genes and characterization of major biological processes
The previous section highlights the significance of the attractor genes in shaping the global response of E. coli in aerobiosis. To scrutinize the biological functions of attractor genes, hierarchical clustering was utilised, resulting in 13 initial clusters (Fig. 5A). From these clusters, we further refined the gene groupings by setting a Pearson correlation of above 0.7 between each gene’s temporal expression with the average profile for that group. Those that were below the threshold were re-evaluated within subgroups and subsequently re-grouped. As a result, we obtained 6 temporal groups of patterns for the attractor genes (Fig. 5B).
Figure 5.

Major gene expression patterns of attractor genes. (A) Hierarchical clustering of attractor genes reveals 13 clusters of temporal expression profiles. (B) Six temporal average expression profile constructed by regrouping the 13 clusters: Group A: Gradual decay: Group B: Gradual activation; Group C: Fast activation, followed by decay and re-activation; Group D: Early activation, followed by decay; Group E: Early activation followed by plateau; Group F: Early decay, followed by plateau.
To elucidate the major biological processes that are regulated in each of the temporal groups, we performed Gene Ontology enrichment analysis using clusterProfiler R package37 with Entrez Gene database38 (both False Discovery Rate and adjusted p-value cut-off were set at 0.05). Enrichment analysis for the 6 groups of attractor genes resulted in (Fig. 6, Table S2): Group A: Gradual decay, mainly enriched in glycolysis, fermentation, anaerobic respiration, phosphorylation, amino acid metabolism, and locomotion, Group B: Gradual activation, enriched in TCA cycle, aerobic respiration, sulphur compound metabolism, protein folding and response to stress, Group C: Fast activation, followed by decay and re-activation, enriched in ribosome biogenesis, RNA processing, gene expression and response to copper ion, Group D: Early activation, followed by decay, mostly enriched in amino acid metabolism, cation transport, and organelle organization, group E: Early activation followed by plateau, enriched in aerobic respiration, several types of transporters, and ion homeostasis, and group F: Early decay, followed by plateau, enriched in biosynthesis of lipid, organonitrogen and nucleobase-containing compounds, and negative regulation of cellular processes.
Figure 6.

Selected enriched biological processes (coloured hubs) with their associated genes (grey dots) in the 6 major expression patterns of attractor genes. Full list of enriched processes is available in Table S2.
Focusing on the names and functions of each attractor gene (using UniProt39 and EcoCyc40 databases), we observe several novel or uncharacterised genes for all groups (Tables S2–S4). Group A possesses several genes coding for flagellum protein, ligase, transcriptional regulator, hydrogenase/dehydrogenase functions, together with 71 uncharacterized genes. Group B contains ion and small molecular binding, and oxidoreductase genes together with 60 uncharacterized genes. Notably, a large portion of group C are tRNA genes (approximately 20% of the genes in group C) and ribosomal protein genes, with 76 uncharacterized genes. Group D consists of several ribosomal proteins (distinct from group C) and transporters, with 19 uncharacterized genes. Group E comprises mainly of oxidoreductase, ion transferase and binding proteins, with 33 uncharacterized genes. Finally, group F shows many rRNA coding genes, along with other protein binding genes and 73 uncharacterized genes (note that tRNAs and rRNAs were not mapped to any gene ontology term in the Entrez Gene database, they were found in EcoCyc database instead).
To narrow down the pool of the attractor genes and to investigate their individual function, a more stringent cut-off of expression levels (TPM) greater than 500 units at any time point, and a 3-fold change between any 2 time points unveiled 94 genes (individual function of the genes are listed in Table S5). As expected, the majority of the refined Group A (23 genes) belongs or is connected to glycolysis and fermentation. In addition, genes of aldehyde-alcohol dehydrogenase (adhE), transcriptional regulator (gadE), ferritin (ftn), formate hydrogenlyase regulatory protein (hycA) and fumarate reductase (frdB, frdC) are also present in the group.
Refined Group B contains 27 genes and mainly relates to TCA and sulphur metabolism (Table S5). Notably, it contains still several novel or unknown functional genes (gpmA, osmY, ybaY, yfhJ, ytjA). Other genes include those of entericidin B (ecnB), thiol peroxidase (tpx), chaperon protein (hscA) and co-chaperon protein (hscB), stress resistant protein (ycfR), and transcriptional regulators (iscR and soxS) among others.
Group C genes were not retained with the cut-off threshold, and refined Group D consists of 5 genes with various functions. Refined group E contains mostly electron transport and aerobic respiration genes, along with cytochrome bo oxidase genes (cyoA, cyoB, cyoC, cyoD, cyoE), biopolymer transporters (exbB, exbD), ornithine carbamoyltransferase (argF, argI), sigma factor binding protein (crl), peptide methionine sulfoxide reductase (yeaA), and alpha-ketoglutarate permease (kgtP). Finally, refined Group F contains 12 genes, 9 of which specifically code for ribosomal RNA (rrlC, rrlA, rrlD, rrsH, rrlH, rrsG, rrsC, rrsE, rrfA) (Table S5).
Finally, we compared our attractor analysis with the common method of choosing genes with more than 2-fold expression change between any 2 time points. This resulted in 631 genes clustered in 5 temporal groups (Fig. S6A,B). Venn diagram comparative analysis reveals 522 common genes (Fig. 7), with 48 unique novel or uncharacterized genes compared with 219 unique in the attractor set and 113 that are common. The notable novel genes for the 2-fold analysis include uracil permease (yfbP), plasmid stabilization mediating proteins (mokC) and a few tRNA genes (Table S6). Gene expression distribution of the attractor and 2-fold genes showed that the latter possess higher proportion of lowly expressed genes (Fig. S6B).
Figure 7.
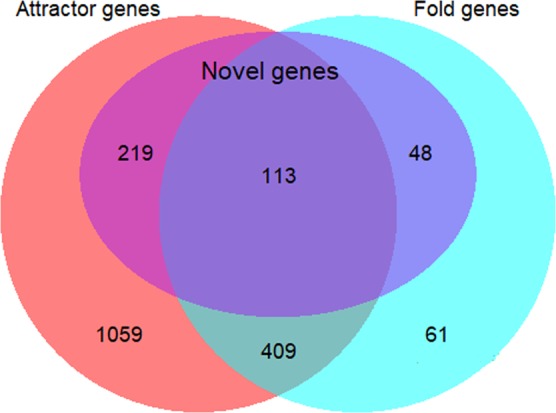
Number of attractor genes (red), 2-fold change genes (cyan), and novel genes (purple).
Discussion
E. Coli aerobiosis is a highly investigated area of research. Although numerous works have been performed, the early dynamic transcriptome-wide response is still poorly understood. Here we investigated RNA-Seq data, consisting of 4,240 non-zero expressions, of anaerobic E. Coli in a bioreactor where readings were taken at 0, 0.5, 1, 2, 5 and 10 min after air supply was initiated. Unlike other approaches that typically investigated arbitrarily selected differential fold changing genes, and performed gene ontology enrichment analysis41–43, here we undertook multi-dimensional statistical approaches, in view of a dynamical system approach, where we queried the portion of transcriptome that are able to track the global transcriptome-wide attractor response. In other words, we investigated the spectrum of genes that are responsible for shaping the AAT.
After incorporating statistical distribution fitting (Fig. 2A,B), we retained 3,389 genes for analysis. We also included the concept of dynamical systems approach into analysing the remaining gene expressions, introducing the state space visualizations through modified linear (Pearson, Rv) and non-linear (mutual information, MIv) correlation density distributions. From these, the attractor regions were generated where a subset of genes (transcriptomic elements) were shown to fall into (Fig. 3). We obtained 1800 genes that fall into the attractor basin (attractor genes), and another 1589 genes fall away (non-attractor genes). Notably, the attractor genes track the global transcriptome-wide trajectory on the PC space (Fig. 4A), and their temporal correlation analysis showed significant variation in time (Fig. 4B). Among the 1589 non-attractor genes, 1521 genes showed small-scaled but clear trajectory on PC space, indicating weak but collective global response, while the remaining 68 did not show any response during the AAT (Fig. S5B).
Hierarchical clustering of the attractor genes showed 6 temporally regulated groups (Fig. 5). Functional enrichment analysis to characterise the function of these important genes shows, as expected, glycolytic, fermentation, anaerobic respiration and cell motility related genes were gradually deactivated (Group A), whereas TCA cycle, aerobic respiration and sulphur compound metabolism were activated (Group B), inversely to Group A. These data are consistent with our general understanding of E. coli aerobiosis4. There were also several novel or uncharacterized genes significantly induced (TPM > 500 and 3-fold changes), including osmY, ybaY, yfhJ, and ytjA. Additionally, entericidin B (ecnB), which plays a role in bacteriolysis44, was also found in this group.
Group C reveals ribosomal biogenesis, translation and gene expression processes. Notably, almost 20% of the genes in this group are tRNA, along with several unknown functional genes (yciG, bdm, YciG, YmdF, YobH, YciH - Table S3). Group F, on the other hand, constitutes mainly of ribosomal RNA. The data from these 2 groups are interesting in that several recent high-profile articles have highlighted the importance of hibernating ribosomes to conserve respiration energy during anaerobic conditions, and certain enzymes “kick in” to revive the metabolism45–49. Also, tRNAs are key for protein synthesis and they play important roles in cellular growth, stress response and general translational regulation. The data here show several genes coding for ribosomes and tRNA perhaps play key roles in translational process that shapes anaerobic to aerobic transition.
Group D and E elucidates several hydrogenase genes which are known to be produced in anaerobic or stress conditions and participates in the reduction of fumarate and dimethyl sulfoxide (fnr)50. In addition to these, cystathionine gama-synthase (metB), alkyl hydroperoxide reductase (ahpF, ahpC), ornithine carbamoyltransferase (argF, argI), and ferric iron reductase (fhuF) are also revealed. Many of these genes were not previously identified with the anaerobic to aerobic transition.
Overall, our work undertaking several statistical metrics, on a dynamical systems viewpoint, to infer the transcriptome-wide response of E. Coli aerobiosis have revealed, for the first time, the existence of a fractal portion of transcriptome (1,800 attractor genes) that tracks transcriptome-wide response, and is collectively crucial for the adaptive state transition. This shows a much higher resolution than the conventional 2-fold expression changing genes selected between any 2 time points (Fig. 7). Notably, previous works have mainly focused on metabolic regulatory genes, but here we show the significance of other types of genes such as tRNAs and rRNAs which are largely involved in post-transcriptional and -translational processes, in addition to 332 uncharacterised attractor genes. Future work could focus on elucidating the function of these genes that are captured for each temporal group of gene regulation.
Methods
Statistical distributions fitting
Fitting gene expression distributions was performed using the maximum-likelihood estimation method (fitdistplus packge51 for parameter estimation and the mass package52 for log-normal, Pareto, Burr, Loglogistic, Weibull and Burr distributions53).
Pearson correlation
The Pearson correlation coefficient r between two vectors (e.g. transcriptome in two different samples), containing n observations (e.g. gene expression values), is defined by (for large n):
where xi and yi are the ith observation in the vectors X and Y, respectively, μX and μY, the average values of each vector, and σx and σy, the corresponding standard deviations. Pearson correlation measures linear relationship between two vectors, where r = 1 if the two vectors are identical, and r = 0 if there are no linear relationships between the vectors.
Spearman correlation
Spearman rank correlations is defined by
where rx,i and ry,i are the ranks of the ith observation xi and yi, in vectors X and Y, respectively.
Bicor (Biweight midcorrelation)
The biweight midcorrelation29 of two vectors X and Y is given by
where
and
noting the weights for x or y, represented by general term p:
and the identity function:
Mutual information-based correlation MIc
Nonlinear dependency between two vectors X and Y can be checked by mutual information:
where54 the joint probability distribution function p(x, y), and marginal probability distribution functions, and are estimated by means of an histogram-based approach by discretizing the rank-transformed gene expression into K = 10 bins18,54,55 for ti (i = 0,1,2..,M, where and M = 10 min). Note that systematic error ε occurs during the discretization, which is then subtracted from the raw MI value. ε is defined as minimum MI for 100 random permutation of the rank-transformed gene expression vector17. The MI-based correlation between X and Y is expressed via30:
Modified Pearson correlation Rv
The dynamic gene expression at time ti can be defined as a N-dimensional vector X(ti) = (x1(ti), x2(ti), …, xN(ti)) with xj(ti) being expression value of the jth gene at ti. The deviation-from-average expression vector at time ti is defined as V(ti) = (v1(ti), v2(ti), …, vN(ti)) where (where is the average expression of jth gene over M + 1 time points)
The modified Pearson correlation is defined as
This index thus measures the temporal correlation of genome-wide expression deviations from their average values so as to allow discriminating gene expressions with different amplification but similar temporal profiles18.
Modified mutual information MIv
Mutual information between vectors V(ti) and V(t0) is defined similar to formula (1) with V(ti) replacing X and V(t0) replacing Y. The probability functions p(x), p(y) and p(x,y) are estimated based on discretized gene deviation data into 10 bins using histogram-based approach. Systematic error ε is defined as minimum MI for 100 random permutation of gene deviation vectors V(ti)18. Finally, to compare MI among different replicates, we used the normalized value:
Ranking genome elements
The whole transcriptome (3389 genes) was sorted according to their standard deviation across 6 time points, from the highest to the lowest: , with being the standard deviation of gene jth expression across 6 time points. After that, we divided the ranked standard deviation vector into p groups, each group with n genes (n = 25, 50, 100, 200, 400, 600, 800, 1000). Note that we choose p = ⌈⌉, the pth group contained n genes which can be overlapped with the (p − 1)th group. Next, we examined the trajectory on MIv − Rv space for each individual group of genes to check whether it fall into attractor region.
Determination of attractor region on Rv-MIv space
Attractor boundary was defined on the superimposed probability density (SPD) distribution of modified Pearson correlation Rv and modified mutual information MIv for whole genome (3389 genes). Distribution of whole genome Rv and MIv was generated by randomly choosing n = 100 genes for 100 times from the pool of 3389 genes, and SPD of these 100 repeats was estimated on discretised lattice by 2D kernel density estimation using the mass library in R programming52.
Attractor boundary was determined by the inflection points on the SPD of whole genome Rv and MIv, where the inflection points18 were determined as highest gradients in vertical and horizontal directions from the local points on the lattice. Averaging the z-coordinates of the vertical and horizontal inflection points determine the z-coordinate of inflection curve, or attractor boundary contour.
Hierarchical clustering
Hierarchical clustering was performed on normalized expressions of attractor and pseudo-attractor genes using Ward clustering method56. Normalized expression of the jth gene at time ti is defined as17 where is expression of the jth gene at time ti, is the mean expression across all time points, and is the standard deviation. As a result, 9 clusters were obtained, which were further regrouped according to 5 distinct temporal average expression patterns for attractor genes, and 4 distinct temporal average expression patterns for pseudo-attractor genes.
Supplementary information
Acknowledgements
This work was supported by IAF-PP research fund of the Biotransformation Innovation Platform (BioTrans), Agency for Science, Technology & Research (A*STAR), Singapore.
Author contributions
T.T.B. developed the R codes for transcriptomics analysis. K.S. conceptualized the idea, and supervised the entire work. T.T.B. and K.S. analysed the data and wrote the paper. All authors have read and approved the final documents.
Data availability
The R-codes for transcriptomics analysis are available from the authors upon request. The E. coli data is obtained using GEO accession number GSE71562.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-62804-3.
References
- 1.Bettenbrock K, et al. Towards a Systems Level Understanding of the Oxygen Response of Escherichia coli. Adv. Microb. Physiol. 2014;64:65–114. doi: 10.1016/B978-0-12-800143-1.00002-6. [DOI] [PubMed] [Google Scholar]
- 2.Regenmortel MHVV. Reductionism and complexity in molecular biology. EMBO Rep. 2004;5:1016–1020. doi: 10.1038/sj.embor.7400284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn AC, Tewari M, Poon C-S, Phillips RS. The Limits of Reductionism in Medicine: Could Systems Biology Offer an Alternative? PLoS Med. 2006;3:e208. doi: 10.1371/journal.pmed.0030208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Partridge JD, Scott C, Tang Y, Poole RK, Green J. Escherichia coli transcriptome dynamics during the transition from anaerobic to aerobic conditions. J. Biol. Chem. 2006;281:27806–27815. doi: 10.1074/jbc.M603450200. [DOI] [PubMed] [Google Scholar]
- 5.Kim OD, Rocha M, Maia P. A Review of Dynamic Modeling Approaches and Their Application in Computational Strain Optimization for Metabolic Engineering. Front. Microbiol. 2018;9:1690. doi: 10.3389/fmicb.2018.01690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selvarajoo, K. Complexity of biochemical and genetic responses reduced using simple theoretical models. In Methods in Molecular Biology1702, 171–201 (Humana Press Inc., 2018). [DOI] [PubMed]
- 7.Selvarajoo K. Understanding multimodal biological decisions from single cell and population dynamics. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012;4:385–399. doi: 10.1002/wsbm.1175. [DOI] [PubMed] [Google Scholar]
- 8.Kauffman S. A proposal for using the ensemble approach to understand genetic regulatory networks. J. Theor. Biol. 2004;230:581–590. doi: 10.1016/j.jtbi.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 9.Huang S, Ingber DE. A non-genetic basis for cancer progression and metastasis: Self-organizing attractors in cell regulatory networks. Breast Dis. 2006;26:27–54. doi: 10.3233/BD-2007-26104. [DOI] [PubMed] [Google Scholar]
- 10.Mar JC, Quackenbush J. Decomposition of Gene Expression State Space Trajectories. PLoS Comput. Biol. 2009;5:e1000626. doi: 10.1371/journal.pcbi.1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kauffman, S. At Home in the Universe. October (Oxford University Press, 1995).
- 12.Ghaffarizadeh A, Podgorski GJ, Flann NS. Applying attractor dynamics to infer gene regulatory interactions involved in cellular differentiation. BioSystems. 2017;155:29–41. doi: 10.1016/j.biosystems.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Huang S, Eichler G, Bar-Yam Y, Ingber DE. Cell Fates as High-Dimensional Attractor States of a Complex Gene Regulatory Network. Phys. Rev. Lett. 2005;94:128701. doi: 10.1103/PhysRevLett.94.128701. [DOI] [PubMed] [Google Scholar]
- 14.Antolović, V., Lenn, T., Miermont, A. & Chubb, J. R. Transition state dynamics during a stochastic fate choice. Dev. 146 (2019). [DOI] [PMC free article] [PubMed]
- 15.Bargaje R, et al. Cell population structure prior to bifurcation predicts efficiency of directed differentiation in human induced pluripotent cells. Proc. Natl. Acad. Sci. 2017;114:2271–2276. doi: 10.1073/pnas.1621412114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mojtahedi M, et al. Cell Fate Decision as High-Dimensional Critical State Transition. PLOS Biol. 2016;14:e2000640. doi: 10.1371/journal.pbio.2000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simeoni O, Piras V, Tomita M, Selvarajoo K. Tracking global gene expression responses in T cell differentiation. Gene. 2015;569:259–266. doi: 10.1016/j.gene.2015.05.061. [DOI] [PubMed] [Google Scholar]
- 18.Tsuchiya, M., Piras, V., Giuliani, A., Tomita, M. & Selvarajoo, K. Collective dynamics of specific gene ensembles crucial for neutrophil differentiation: The existence of genome vehicles revealed. PLoS One5 (2010). [DOI] [PMC free article] [PubMed]
- 19.Marr C, Zhou JX, Huang S. Single-cell gene expression profiling and cell state dynamics: Collecting data, correlating data points and connecting the dots. Current Opinion in Biotechnology. 2016;39:207–214. doi: 10.1016/j.copbio.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felli N, et al. Hematopoietic differentiation: A coordinated dynamical process towards attractor stable states. BMC Syst. Biol. 2010;4:85. doi: 10.1186/1752-0509-4-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selvarajoo K, Giuliani A. Finding self-organization from the dynamic gene expressions of innate immune responses. Front. Physiol. 2012;3:192. doi: 10.3389/fphys.2012.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuchiya M, et al. Emergent Genome-Wide Control in Wildtype and Genetically Mutated Lipopolysaccarides-Stimulated Macrophages. PLoS One. 2009;4:e4905. doi: 10.1371/journal.pone.0004905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piras V, Chiow A, Selvarajoo K. Long-range order and short-range disorder in Saccharomyces cerevisiae biofilm. Eng. Biol. 2019;3:12–19. doi: 10.1049/enb.2018.5008. [DOI] [Google Scholar]
- 24.Piras V, Tomita M, Selvarajoo K. Transcriptome-wide variability in single embryonic development cells. Sci. Rep. 2014;4:1–9. doi: 10.1038/srep07137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selvarajoo, K. Large Scale-Free Network Organization is Likely Key for Biofilm Phase Transition. Eng. Biol. In Press, (2019).
- 26.von Wulffen, J., Ulmer, A., Jäger, G., Sawodny, O. & Feuer, R. Rapid Sampling of Escherichia coli After Changing Oxygen Conditions Reveals Transcriptional Dynamics. Genes (Basel). 8 (2017). [DOI] [PMC free article] [PubMed]
- 27.Tien BT, Giuliani A, Selvarajoo K. Statistical Distribution as a Way for Lower Gene Expressions Threshold Cutoff. Org. J. Biol. Sci. 2018;2:55–58. [Google Scholar]
- 28.Beal J. Biochemical complexity drives log-normal variation in genetic expression. Eng. Biol. 2017;1:55–60. doi: 10.1049/enb.2017.0004. [DOI] [Google Scholar]
- 29.Zheng C-H, Yuan L, Sha W, Sun Z-L. Gene differential coexpression analysis based on biweight correlation and maximum clique. BMC Bioinformatics. 2014;15:S3. doi: 10.1186/1471-2105-15-S15-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joe H. Multivariate dependence measures and data analysis. Comput. Stat. Data Anal. 1993;16:279–297. doi: 10.1016/0167-9473(93)90130-L. [DOI] [Google Scholar]
- 31.Giuliani A. Collective motions and specific effectors: a statistical mechanics perspective on biological regulation. BMC Genomics. 2010;11(Suppl 1):S2. doi: 10.1186/1471-2164-11-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milnor J. On the concept of attractor. Commun. Math. Phys. 1985;99:177–195. doi: 10.1007/BF01212280. [DOI] [Google Scholar]
- 33.Ghorbani M, Jonckheere EA, Bogdan P. Gene expression is not random: Scaling, long-range cross-dependence, and fractal characteristics of gene regulatory networks. Front. Physiol. 2018;9:1446. doi: 10.3389/fphys.2018.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinicola S, et al. A systems biology approach to cancer: Fractals, attractors, and nonlinear dynamics. OMICS A Journal of Integrative Biology. 2011;15:93–104. doi: 10.1089/omi.2010.0091. [DOI] [PubMed] [Google Scholar]
- 35.Piras V, Selvarajoo K. The reduction of gene expression variability from single cells to populations follows simple statistical laws. Genomics. 2015;105:137–144. doi: 10.1016/j.ygeno.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Tsuchyia M, et al. Gene expression waves. Cell cycle independent collective dynamics in cultured cells. FEBS J. 2007;274:2878–2886. doi: 10.1111/j.1742-4658.2007.05822.x. [DOI] [PubMed] [Google Scholar]
- 37.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39:D52-7. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bateman A, et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karp, P. D. et al. The EcoCyc Database. EcoSal Plus6 (2014). [DOI] [PMC free article] [PubMed]
- 41.Maximova SN, et al. Genome-wide analysis reveals divergent patterns of gene expression during zygotic and somatic embryo maturation of Theobroma cacao L., the chocolate tree. BMC Plant Biol. 2014;14:185. doi: 10.1186/1471-2229-14-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He F, Ai B, Tian L. Identification of genes and pathways in esophageal adenocarcinoma using bioinformatics analysis. Biomed. Reports. 2018;9:305–312. doi: 10.3892/br.2018.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu P, Jiang L, Wu L. Identify differential gene expressions in fatty infiltration process in rotator cuff. J. Orthop. Surg. Res. 2019;14:158. doi: 10.1186/s13018-019-1182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bishop RE, Leskiw BK, Hodges RS, Kay CM, Weiner JH. The entericidin locus of Escherichia coli and its implications for programmed bacterial cell death. J. Mol. Biol. 1998;280:583–596. doi: 10.1006/jmbi.1998.1894. [DOI] [PubMed] [Google Scholar]
- 45.Iyer S, Le D, Park BR, Kim M. Distinct mechanisms coordinate transcription and translation under carbon and nitrogen starvation in Escherichia coli. Nat. Microbiol. 2018;3:741–748. doi: 10.1038/s41564-018-0161-3. [DOI] [PubMed] [Google Scholar]
- 46.Li SH-J, et al. Escherichia coli translation strategies differ across carbon, nitrogen and phosphorus limitation conditions. Nat. Microbiol. 2018;3:939–947. doi: 10.1038/s41564-018-0199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beckert B, et al. Structure of a hibernating 100S ribosome reveals an inactive conformation of the ribosomal protein S1. Nature. Microbiology. 2018;3:1115–1121. doi: 10.1038/s41564-018-0237-0. [DOI] [PubMed] [Google Scholar]
- 48.Schmied WH, et al. Controlling orthogonal ribosome subunit interactions enables evolution of new function. Nature. 2018;564:444–448. doi: 10.1038/s41586-018-0773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prossliner T, Skovbo Winther K, Sørensen MA, Gerdes K. Ribosome Hibernation. Annu. Rev. Genet. 2018;52:321–348. doi: 10.1146/annurev-genet-120215-035130. [DOI] [PubMed] [Google Scholar]
- 50.Kovács, Á. T. et al. Anaerobic regulation of hydrogenase transcription in different bacteria. in Biochemical Society Transactions33, 36–38 (Portland Press Limited, 2005). [DOI] [PubMed]
- 51.Delignette-Muller ML, Dutang C. fitdistrplus: An R package for fitting distributions. J. Stat. Softw. 2015;64:1–34. doi: 10.18637/jss.v064.i04. [DOI] [Google Scholar]
- 52.Venables, W. N. & Ripley, B. D. Modern Applied Statistics with S. (Springer, 2002).
- 53.Johnson LD, Kotz NL, Balakrishnan S, Continuous Univariate N. Distributions. J. Am. Stat. Assoc. 1996;91:915. [Google Scholar]
- 54.Kurths J, Daub CO, Weise J, Selbig J, Steuer. The mutual information: detecting and evaluating dependencies between variables. Bioinformatics. 2002;18(Suppl 2):S231–40. doi: 10.1093/bioinformatics/18.suppl_2.S231. [DOI] [PubMed] [Google Scholar]
- 55.Speed TP, Bolstad BM, Irizarry RA, Astrand M. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 56.Wilks, D. S. Cluster Analysis. International Geophysics100, (Wiley, 2011).
- 57.Huang S, Ernberg I, Kauffman S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Seminars in Cell and Developmental Biology. 2009;20:869–876. doi: 10.1016/j.semcdb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R-codes for transcriptomics analysis are available from the authors upon request. The E. coli data is obtained using GEO accession number GSE71562.