

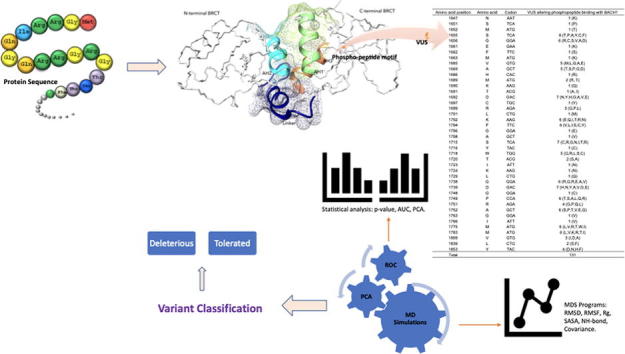
Graphical abstract
Keywords: BRCA1, BRCT, VUS, Unclassified variants, Molecular dynamics simulation, PCA
Abstract
Pathogenic mutation in BRCA1 gene is one of the most penetrant genetic predispositions towards cancer. Identification of the mutation provides important aspect in prevention and treatment of the mutation-caused cancer. Of the large quantity of genetic variants identified in human BRCA1, substantial portion is classified as Variant of Uncertain Significance (VUS) or unclassified variants due to the lack of functional evidence. In this study, we focused on the VUS and unclassified variants in BRCT repeat located at BRCA1 C-terminal. Utilizing the well-determined structure of BRCT repeats, we measured the influence of the variants on the structural conformations of BRCT repeats by using molecular dynamics simulation (MDS) consisting of RMSD (Root-mean-square-deviation), RMSF (Root-mean-square-fluctuations), Rg (Radius of gyration), SASA (Solvent accessible surface area), NH bond (hydrogen bond) and Covariance analysis. Using this approach, we analyzed 131 variants consisting of 89 VUS (Variant of Uncertain Significance) and 42 unclassified variants (unclassifiable by current methods) within BRCT repeats and were able to differentiate them into 78 Deleterious and 53 Tolerated variants. Comparing the results made by the saturation genome editing assay, multiple experimental assays, and BRCA1 reference databases shows that our approach provides high specificity, sensitivity and robust. Our study opens an avenue to classify VUS and unclassified variants in many cancer predisposition genes with known protein structure.
1. Introduction
BRCA1 plays essential roles in maintaining genome integrity [1]. Pathogenic mutation in BRCA1 damages the function of BRCA1 and has been reported as one of the utmost penetrating genetic predispositions towards breast and ovarian cancer [2]. Identification of the mutation carriers for these not yet developed cancers will be critical in preventing cancer whereas for those already developed cancer, it will be a crucial stride towards targeted cancer treatment such as the use of PARP inhibitors. Since the discovery of the relationship between mutation in BRCA1 and cancer, extensive efforts have been made to determine the mutation spectrum of BRCA1, with a large quantity of BRCA1 variants identified mostly in Caucasian population [3], [4]. A widely used five-classification system has been applied to classify the variants into Benign, Likely Benign, Variant of Uncertain Significance (VUS), Likely Pathogenic and Pathogenic [5], [6]. While the variants classified as Pathogenic, Likely Pathogenic, Benign and Likely Benign have clinical significance, the variants classified as VUS are of utmost concern as their clinical significance cannot be determined due to the lack of sufficient evidence to determine if they are pathogenic or benign. Of the over 5000 BRCA1 variants in ClinVar database, 29% are classified as VUS (https://www.ncbi.nlm.nih.gov/clinvar). In addition, a large quantity of variants is grouped as the unclassified variants as these are unclassifiable under the current five-classification system. The presence of VUS and unclassified variants is a serious obstacle for clinical prognosis and treatment of BRCA1-related cancer.
Analyzing variant-induced protein structural change is a widely used approach to study the effects of genetic variation on the affected protein, particularly for these with well-defined protein structure [7]. The two tandem BRCT (BRCA1 C-terminus) repeats are one of functional domains in BRCA1 highly conserved in multiple proteins [8]. BRCA1 BRCT repeats play important roles in tumor suppressor function of BRCA1 by interaction with multiple phospho-proteins including BACH1, CtIP, CCDC98 and RAP80 through partner protein’s phosphorylated peptide motif [9]. As intact tandem BRCT structure is required for BRCA1 function, variation in BRCA1 BRCT repeats effecting the native structure can have severe consequence of impairing BRCA1 function. This is evidenced by enriched missense variation within BRCT repeats in early onset of breast cancer patients [10], [11], [12], decreased BRCA1 BRCT dimerization in the dimer interface in cancer [13], and by oncogenesis effects in deletion of BRCA1 BRCT repeats [14]. Indeed, multiple attempts have been made to classify the VUS in BRCA1 BRCT repeats through computational approaches developed over the years [15], [16], [17], [18]. Majority of these approaches utilized evolution-based sequence conservation approach [19], [20], [21], [22].
We reasoned that the influence of variants in structural stability could be used to evaluate the impact of genetic variants. Although the structure of entire BRCA1 has not been reported, the structure of BRCA1 BRCT repeats is well defined [23]. With ~100 amino acid residues, each BRCA1 BRCT repeat comprises a central, four stranded β sheets, surrounded by two α helices (α1 and α3) on one face and a single helix (α2) on the opposite face of β sheets (Fig. 1A). The two BRCA1 BRCT repeats form an elongated structure, with each BRCT repeat adopting a globular α/β fold [24]. Relative arrangement of α1, α3 and the central β sheet is conserved on aligning the BRCA1 BRCT repeats with other DNA repair proteins, such as XRCC1 repeats, whereas the orientation of α2 is much less conserved than the central β-sheet connecting loops. Folding of key hydrophobic residues (S1655, G1656, K1702) maintains the conservation of α1-α3-β-sheet structure [25]. Hydrophobic residues within these helices pack tightly contributing to its inter-repeat interface required for its phospho-peptide recognition and interaction with phospho-dependent interacting proteins of BACH1, CtIP, CCDC98 and RAP80. The two BRCT repeats stack closely against each other through a large hydrophobic interface, giving rise to a deep surface cleft (Fig. 1B). Sequence and structural analyses revealed that this surface cleft is highly conserved among BRCA1 orthologs across species [26].
Fig. 1.

Structure of BRCT repeats. (A). Features of the structure. Each BRCA1 BRCT repeat shown as alpha carbon trace is comprised of a central, four stranded β sheets, surrounded by two α helices (α1 and α3) on one face and a single helix (α2) on the opposite face of β sheet. The AH2, AH3′ and AH1′ along with the linker helix forming the phosphor-peptide binding motif are shown in ribbon conformation. (B). Amino acid positions within BRCT phosphor-peptide binding motif in the hydrophobic cleft involved in phospho-peptide binding. The up part and the low part are 180-degree horizontal turn to show the variants on both sides of BRCA1 BRCT repeats. The up part shows the variant S1655, D1692, K1702, F1704, A1708, S1715, P1749, M1775, L1839, Y1853, the low part shows the variant D1692, K1702, S1715, W1718, T1720, D1739, R1751, M1783, L1839, and Y1853.
In this study, we analyzed the equilibrium dynamics for variants in phosphoserine recognition pocket (pSer-x-x-Phe) within the NH2-terminal of BRCT repeat (native and mutant structure) through Molecular Dynamics Simulations (MDS) in order to measure the impact of variants on phospho-peptide binding in BRCT, and interaction with other functional domains involved in DNA repair such as BACH1, TP53BP1, DNA ligase IV, and XRCC4 [27], [28]. We analyzed a set of 131 variants in the BRCA1 BRCT repeats consisting of 89 VUS and 42 unclassified variants within the phospho-peptide motif, and successfully differentiated them into the Deleterious and Tolerated variants based on the full agreement of the respective scores from known Pathogenic and Benign controls (We used Deleterious or Tolerated rather than Pathogenic and Benign to describe the results from our study to avoid confusion as Pathogenic and Benign variants have specific clinical implications).
2. Materials and methods
2.1. Modelling BRCT mutant structure
The BRCA1 BRCT repeats comprising of phospho-peptide binding motif spanning aa 1648–1837 was from UniprotKB database (accession no: P38398) [29]. We retrieved the phospho-peptide binding motif from native BRCT repeat structure (PDBID:1JNX) (www.rcsb.org) and utilized it as template to build the BRCT mutants using the Modeller package (version 9.22) to generate structure model of the BRCA1 BRCT repeats comprising phospho-peptide binding motif [30]. The phospho-peptide region comprised of a large hydrophobic interface between BRCT1 and BRCT2 is formed by α2 (from BRCT1) and α′1 and α′3 (from BRCT2), with a linker between the two domains critical for the tumor suppressor function of BRCA1 [31], [32]. Through extensive database/literature searching, a total of 131 variants within BRCA1 BRCT1 and BRCT2 were identified, constituting 89 VUS and 42 unclassified variants, only 42 (32%) were present in dbSNP (version 150) [15], [16], [18], [33], [34], [35], [36], [37]. All of the variants were located within the phospho-peptide binding motif of BRCT repeats. A total of 42 amino acid substitutions at respective amino acid positions for each VUS involved in phospho-peptide binding [38] were introduced through UCSF Chimera [39], including N1647, S1651, M1652, S1655, G1656, E1661, F1662, M1663, V1665, A1669, H1686, M1689, K1690, T1691, D1692, C1697, R1699, L1701, K1702, F1704, G1706, A1708, S1715, Y1716, W1718, T1720, I1723, K1724, L1729, G1738, D1739, G1748, P1749, R1751, A1752, G1763, I1766, M1775, M1783, V1809, L1839, Y1853. Modeller package consisted of multistep processes, in which the input was an alignment of a sequence to be modelled with the template structure, the atomic coordinates and a script file. Four 3D models were generated for submitted query sequence and the one with the lowest energy was selected as the final model. PROCHECK [40] and PROSA [41] programs were used independently to evaluate the modelled mutant structure. Both programs evaluated the number of amino acid residues in favorable or disallowed regions and recognized structural errors within the modelled structure.
2.2. Predicting effects of mutation on BRCT stability by MDS
To delineate the diverse structural characteristics, complexes of the native and mutant BRCT structure were analyzed with MDS [42] composed of RMSD (Root-mean-square-deviation), RMSF (Root-mean-square-fluctuations), Rg (Radius of gyration), SASA (Solvent accessible surface area), and NH bond (hydrogen bond) programs. RMSD is commonly used as an indicator of structural convergence towards an equilibrium state [43]. It measures low values of deviation for native and variant average structures over a period of time. All variants having RMSD value >0.3 were classified as Deleterious and those with < or equal to 0.3 were classified as Tolerated; RMSF measures flexibility of polypeptide chain by calculating the fluctuation of C-alpha atoms coordinating from their average position [44]. RMSF values illustrate the difference in residue flexibility of protein segments between wildtype and mutant that correlates with the different intermolecular hydrogen bonds and hydrophobic contacts observed during analysis. All variants with RMSF value >0.25 were classified as Deleterious and <0.25 as Tolerated; Rg shows the shape of a molecule at specific instance through comparing the hydrodynamic radius of the native protein structure with that of substituted variants [45]. Rg measures the distance of the atoms of the structure from either its center of gravity or an axis for the compactness of each protein structure. If a protein is stably folded, it will likely maintain a relatively steady value of Rg but the value will change if a protein unfolds. Variants with Rg score >1.7 were classified as Deleterious and <1.7 as Tolerated; NH-bond provides information of hydrogen bonds, either internally between protein–protein or externally between peptide and surrounding solvent [46]. The presence of a hydrogen bond is inferred from the distance between a donor-H - acceptor pair and the donor – H – acceptor angle. As hydrogen bonds are important in maintaining steady configuration of protein, NH bond analysis of native and mutant form of the protein helps to determine the liaison between flexibility and NH bond formation. Variants with the number of NH bond <300 were classified as Deleterious and >300 as Tolerated; SASA defines the surface accessibility of protein for solvent binding [47]. SASA value indicates the relative expansion of mutant structures and increased intrinsic flexibility that reduces the likelihood of stable binding. Variants with solvent surface area >100 nm2 were classified as Deleterious and <100 as Tolerated. All simulations were performed through GROMACS version 5.0. The protein complex was at center of 100*100*100 Å triclinic grid, which was solvated with SPC water model and neutralized with 5 Na+ ions. Equilibration of protein complex along with energy minimization utilizing the OPLS-AA/L force field was carried out at constant pressure of 1 atm and temperature (NPT) of 298 K with time interval of 2 fs using leap-frog integrator. Verlet cut‐off scheme was used to relax the unfavorable contacts between molecules. Particle Mesh Ewald method was used to treat the long-range electrostatic interactions. Energy-minimized systems were equilibrated till 100 ps at constant volume and temperature (NVT) with pressure 1 atm. A modified Berendsen thermostat v‐rescale [48] was applied for temperature coupling in combination with the Parrinello–Rahman dynamics for pressure coupling [49]. LINCS algorithm was applied to constrain the bond lengths [50]. Trajectory frames of MDS were saved every 15 ps. Analysis of the trajectory files was performed on different statistical parameters using various inbuilt scripts of GROMACS. XMGRACE program was utilized to generate the corresponding plots.
2.3. Protein docking
Variants identified as Deleterious through MDS were analyzed in terms of the change in binding affinity and mode of interaction with BACH1 phospho-protein involving in DNA repair through protein–protein docking with ClusPro server [51]. The docked molecules (BRCT and BACH1) were ranked according to the RMSD value of the lowest clusters.
2.4. Covariance analysis
Characterization and comparative analysis of the overall protein motion were performed using the Essential Dynamics method to give an improved outlook of large‐scale collective motions and confined fluctuations of protein structure [52]. It characterizes the phase space behavior of protein on the basis of eigenvectors, the principal components that sort out the essential motions of a macromolecule in possible subspace. Eigenvectors calculate a converged trajectory of protein complex simulation, which gives insight of the movement of C‐alpha atoms representing the amino acid residues. Covariance matrix for the atomic coordinates of 213 C‐alpha atoms was calculated by the following equations:
where σ is a symmetric 3 N × 3 N matrix and r is a diagonal matrix, which contains the masses of atoms in the instances of weighted analysis, representing the unit matrix with regards to non‐mass weighted matrix.
Where R represents the transformation into a new coordinate system and columns in R depicts the eigenvectors.
The contribution of atom j toward fluctuation of ith mode is defined by the following equation:
where, represents component vectors of jth atom for ith mode.
Eigenvalues corresponding to each eigenvector explain the energetic contribution of that principal component to protein motion. For a long‐term MDS, only the first few modes are able to delineate the global collective fluctuations.
2.5. Statistical analysis
Statistical analysis was performed using the Principal Component Analysis, Wilcox test function [53] in R. The datasets were classified into Deleterious and Tolerated for VUS and Pathogenic and Benign for control group. Plots of the receiver operating characteristic (ROC) curve of the classifier and the calculation of the area under the curve (AUC) were fulfilled using the verification package. The ROC curve demonstrates the sensitivity (Se, true positive rate) for any possible change in the number of variants (n) as function of (1 − Sp), Sp is defined as specificity or false negative rate.
3. Results
3.1. Protein modelling with VUS integration and classification based on structural and conformational changes in BRCT
We selected the crystal structure of the BRCA1 BRCT repeat region (PDB ID: 1JNX) to classify the 131 variants within the phospho-peptide motif of the BRCT domain. The comparative modelling through Modeller program included alignment of the query sequence to the known 3D template (PDB ID:1JNX), along with the shifting of spatial features such as Cα-Cα distances and main-chain / side-chain dihedral angles from the template to target, on the basis of the number of spatial restraints. The output was a 3D model for the targeted sequence comprising all main-chain and side-chain heavy atoms. The 131 variants within the BRCT repeats were located at the 42 amino acid residues: N1647, S1651, M1652, S1655, G1656, E1661, F1662, M1663, V1665, A1669, H1686, M1689, K1690, T1691, D1692, C1697, R1699, L1701, K1702, F1704, G1706, A1708, S1715, Y1716, W1718, T1720, I1723, K1724, L1729, G1738, D1739, G1748, P1749, R1751, A1752, G1763, I1766, M1775, M1783, V1809, L1839, Y1853 (Table 1). Phospho-dependent interacting proteins such as BACH1 directly interact with tandem BRCT repeats upon its phosphorylation at Ser990. Studies have established a “two-knob” model to illustrate the binding of the phospho-dependent proteins with the conserved hydrophobic cleft of BRCT repeats [12]. Two amino acids represented as knobs, i.e. BACH1 phosphorylated at pSer990 and Phe993, anchor through the N and C-terminal of the BRCT repeats. pSer990 interacts forming hydrogen bonds at N-terminal whereas Phe993 interacts through van der Waals at C-terminal.
Table 1.
The 42 residues positions in BRCT and the corresponding VUS.
| Amino acid position | Amino acid | Codon | VUS altering phosphopeptide binding with BACH1 |
|---|---|---|---|
| 1647 | N | AAT | 1 (K) |
| 1651 | S | TCA | 1 (P) |
| 1652 | M | ATG | 1 (T) |
| 1655 | S | TCA | 6 (T,P,A,Y,C,F) |
| 1656 | G | GGA | 6 (R,C,S,V,A,D) |
| 1661 | E | GAA | 1 (K) |
| 1662 | F | TTC | 1 (S) |
| 1663 | M | ATG | 1 (K) |
| 1665 | V | GTG | 5 (M,L,G,A,E) |
| 1669 | A | GCT | 5 (T,S,P,G,D) |
| 1686 | H | CAC | 1 (R) |
| 1689 | M | ATG | 2 (R, T) |
| 1690 | K | AAG | 1 (Q) |
| 1691 | T | ACG | 1 (A, I) |
| 1692 | D | GAC | 7 (N,Y,H,G,A,V,E) |
| 1697 | C | TGC | 1 (Y) |
| 1699 | R | AGA | 3 (G,P,L) |
| 1701 | L | CTG | 1 (M) |
| 1702 | K | AAG | 6 (E,Q,I,T,R,N) |
| 1704 | F | TTC | 6 (V,L,I,S,C,Y) |
| 1706 | G | GGA | 1 (E) |
| 1708 | A | GCT | 1 (V) |
| 1715 | S | TCA | 7 (C,R,G,N,I,T,R) |
| 1716 | Y | TAC | 1 (C) |
| 1718 | W | TGG | 5 (G,R,L,S,C) |
| 1720 | T | ACG | 2 (S,A) |
| 1723 | I | ATT | 1 (N) |
| 1724 | K | AAG | 1 (N) |
| 1729 | L | CTG | 1 (Q) |
| 1738 | G | GGA | 6 (R,G,R,E,A,V) |
| 1739 | D | GAC | 7 (H,N,Y,A,V,G,E) |
| 1748 | G | GGA | 1 (C) |
| 1749 | P | CCA | 6 (T,S,A,L,Q,R) |
| 1751 | R | AGA | 4 (G,P,Q,L) |
| 1752 | A | GCT | 6 (S,P,T,V,E,G) |
| 1763 | G | GGA | 1 (V) |
| 1766 | I | ATT | 1 (V) |
| 1775 | M | ATG | 6 (L,V,R,T,W,I) |
| 1783 | M | ATG | 6 (L,V,K,R,T,I) |
| 1809 | V | GTG | 3 (I,D,A) |
| 1839 | L | CTG | 2 (S,F) |
| 1853 | Y | TAC | 4 (D,N,H,F) |
| Total | 131 |
We first carried out simulations for the 10 known Pathogenic (D1692H, D1692Y, D1692N, A1708E, S1715R, S1715N, W1718S, W1718C, M1775K, M1775R) and 10 known benign (P1637L, M1652I, M1652T, F1662S, L1664P, E1682K, G1706A, T1720A, R1751Q, V1804A) variants from ClinVar. We observed full agreement with their existing classification. The rationale to select the control variants (Pathogenic and Benign) in our study was to utilize these to define the conditions to discriminate Deleterious and Tolerated variants. The cut-off scores determined from the 10 known pathogenic and benign variants for each MDS parameter were set to differentiate the variants into Deleterious and Tolerated classes (Supplementary Table 1). We then used the conditions to test all 131 variants. The followings are the detailed description for the results from each MDS program:
RMSD: It calculates the trajectories of the native and variant structures. Native BRCT structure stabilized around 0.3–0.4 nm with a deviation around 15 ns, as compared to the variants G1656R, K1702T with an unstable trajectory starting right from the equilibrium phase up to the productive phase, i.e. up to 40 ns as compared to the variants A1708S, G1738E, P1749Q with low RMSD values of less unstable as compared to G1656R and K1702T (Fig. 2A, B). RMSD predicted 78 variants as Deleterious and 53 variants as Tolerated;
Fig. 2.

Examples of VUS classification by individual MDS programs. The x-axis represents the time period in each program. A, B. RMSD. The results show the RMSD of native and mutant protein complexes for a time period of 40 ns. The y-axis represents the RMSD for the native and variants structures. A. Native, G1738E, A1708S, G1656R, K1702T; B. Native, P1749Q, M1775T, L1839S, Y1853D. C, D. RMSF. The results show the RMSF profile of native and mutant protein complexes for a time period of 40 ns. The y-axis represents the RMSF score for native and variant structures. C. Native, G1738E, A1708S, G1656R, K1702T; D. Native, P1749Q, M1775T, L1839S, Y1853D. E, F. Rg. The results show the Rg profile of native and mutant protein complexes for a time period of 40 ns. The y-axis represents the Rg score for native and variant structures. E: Native, A1708S, G1738E, G1656R, K1702T; F: Native, P1749Q, M1775T, Y1853D, L1839S. G, H. NH-bond. The results show the number of intermolecular hydrogen bonds in native and mutant protein structures for a time period of 40 ns. The y-axis represents the no. of H bond for the native and variant structures. G: Native, A1708S, G1738E, G1656R, K1702T; H: Native, P1749Q, M1775T, Y1853D, L1839S. I, J. SASA. The results show the SASA profile of the native and mutant protein complexes for a time period of 40 ns. The y-axis represents the area in nm2. I. Native, G1738E, K1702T, G1656R, A1708S: J. Native, Y1853D, P1749Q, M1775T, L1839S.
RMSF: It calculates values for the native and variant protein structures to determine the effect of substitutions in BRCT structure towards the dynamic behavior of residues. The main chain RMSF calculated over trajectories and averaged over the native and variant scores shows that the amino acid fluctuations were mostly in the region 1650–1730 and 1740 to 1780. G1656R and K1702T demonstrated high flexibility at their respective amino acid substitution positions with 0.55 and 0.48 RMSF scores respectively, whereas P1749Q having RMSF score of 0.40 demonstrated less degree of resilience as compared to the native protein (Fig. 2C, D). RMSF predicted 77 variants as Deleterious and 51 variants as Tolerated;
Rg: Rg is defined as the mass-weighted root mean square distance of collected atoms from their common center of mass. We utilized the Rg to test the compactness of the native and variant protein structure. All VUS had higher Rg values compared to the native BRCT structure with Rg score of 1.78, with P1749Q as the highest, signifying that the amino acid substitution resulted in larger hydrodynamic radius. i.e. less compact protein structure (Fig. 2E, F). Rg predicted 76 variants as Deleterious and 53 variants as Tolerated;
NH bond: Hydrogen bonds are considered to play vital roles in molecular recognition and overall stability of protein structure. NH-bond analyses the inter-molecular H bond in native and variant protein structure. The number of hydrogen bond decreased in all VUS as compared to the native BRCT repeats: the number of h-bond was 300–340 in the native BRCT, whereas the numbers fallen below 100 in P1749Q, M1775T and K1702T, and between 150 and 220 in G1738E, G1656R, A1708S, L1839S and Y1853D (Fig. 2G, H). NH bond predicted 77 variants as Deleterious and 52 variants as Tolerated;
SASA: It measures the relative expansion of protein structures. The value was the lowest of around 100 nm2 for the native BRCT but increased in multiple variants, such as the highest ones of around 145 nm2 for P1749Q, Y1853D, and L1839S, and higher ones between 110 and 120 nm2 for M1775T, G1738E, A1708S and K1702T (Fig. 2I, J). SASA predicted 74 variants as Deleterious and 53 variants as Tolerated;
Covariance analysis: It measures the overall strenuous motion within both native and variant protein structure. A total of 800 eigenvectors for 190 amino acid residues of BRCT repeats were generated for PCA analysis. Flexibility of all protein structures including the wild type and the mutants was measured by calculating the trace values for diagonalized covariance matrix. The sum of eigen values (trace of diagonalized matrix) was found to have increased strenuous motion for G1656R, K1702T, G1738E, M1775T and Y1853D compared to A1708S, P1749Q and L1839S. Covariance analysis elaborates the positive and negative-correlated motions in the protein (Fig. 3) and in the agreement with MD analysis. Covariance predicted 78 variants as deleterious and 53 variants as Tolerated;
Fig. 3.

Covariance correlated motion changes between BRCT mutant structures measured by Principal Component Analysis. It shows the diagonalized covariance matrix of mutants for the eight variants that G1656R, K1702T, G1738E, M1775T and Y1853D have increased strenuous motion comparing to A1708S, P1749Q and L1839S.
Each of the MDS programs above classified the 131 variants into Deleterious and Tolerated groups following its own parameters. In order to provide high reliability of the variant classification, we set a cut-off condition that each classification must be supported by the same results from at least 5 of the 6 individual MDS programs. Under this condition, the 131 variants were classified into 78 (59.5%) Deleterious variants and 53 (40.4%) Tolerated variants (Supplementary Table 1).
Substantial changes of structural conformation can be visualized for most of the VUS classified as Deleterious by each MDS program. For example, variant M1663K, R1699G, A1708S and M1775L mapped to the hydrophobic core of the protein and disrupted the folding as well as structure of BRCT repeats; A1708E completely buried within the hydrophobic core by changing α-amino acid alanine to a negatively charged glutamic acid, thereby, destabilized the structure of BRCT repeats; P1749A and M1775R disrupted the interaction resulting in loss of function; S1655T, S1655Y, S1655F, G1656R, G1656S, G1656A, K1702T, K1702N, K1702Q caused translational shift, moved BRCT1 away from BRCT2, resulting in the loss of hydrogen bonding and disrupting the hydrophobic cleft; F1704L, F1704I, F1704C, M1775T, M1775V, M1775L, L1839S resulted in vertical conformation, thereby, altered interaction between phospho-proteins as well as loss of three hydrogen bonds at position R1699 of BRCT repeats; M1775T and L1839S changed polar and hydrophilicity affecting the packing or folding in local vicinity, causing distortions in the conserved surface cleft pocket (Fig. 4). Many variants affected residues in the hydrophobic core of the BRCT repeats and disrupted its structural integrity. Covariance analysis clearly showed that mutants occupied more area in conformational space with high trace value than the native protein. The trace values of the native and deleterious mutant changes in cα atoms showed the flexible nature of the structure, and expansion of motion in deleterious mutants was observed from the range of eigen vectors in conformational space.
Fig. 4.

Amino acid substitutions at specific phosphor-peptide binding position causing structural change. A. Native structure with M1775; B. Changed structure in M1775T; C. Native structure with L1839; D. Changed structure in L1839S.
Eight variants (G1656R, K1702T, A1708S, G1738E, P1749Q, M1775T, L1839S, Y1853D) were identified as the top Deleterious ones based on individual scores of MDS parameters through comparing with native BRCT structure as well as the known pathogenic and benign variants (control group). Further analyzing their changes in binding pattern for phospho-peptide interaction with BACH1 phospho-protein (PDBID: 2IHC) showed that the Deleterious variants demonstrated a loss of binding affinity to BACH1 when comparing to the native BRCT (Supplementary Table 2).
In summary, by using MDS, we were able to classify 69 VUS into 40 Deleterious and 29 Tolerated variants, 20 VUS with conflicting interpretations into 14 Deleterious and 6 Tolerated, and 42 unclassified variants into 24 Deleterious and 18 Tolerated variants (Fig. 5, Supplementary Table 3). The top eight Deleterious variants (c.4966G>C, G1656R) (c.5105A>C, K1702T) (c.5122G>T, A1708S) (c.5217T>G, G1738E) (c.5246C>A, P1749Q) (c.5324T>C, M1775T) (c.5516T>C, L1839S) c.5557T>G, Y1853D) are strong candidates as Pathogenic variants (Fig. 2), of which seven were VUS and only one (c.5105A>C, K1702T) was unclassified variant.
Fig. 5.

Histogram fitting distribution curve showing variant distribution frequency for 131 variants through respective MD simulation program (A) RMSD (B) RMSF (C) Rg (D) SASA (E) NH-bonds (F) Covariance.
3.2. Statistical analysis
Statistical analysis was performed for the classified 131 variants, along with the 10 known Pathogenic and 10 known Benign controls. All the datasets were observed being well discriminated in the scores plot between the two groups, i.e., Deleterious with the Pathogenic, and Tolerated with the Benign, respectively (Fig. 6). PC1 represented 98.5% of the explained variance compared to PC2, which represents 1.5% of the variance. Thus, the datasets were well defined and discriminated across the score’s plots, indicating that the PCs were the clear representation of inter-group variance.
Fig. 6.

Principal component analysis (PCA) of the BRCT variants for four groups. Red and purple circles represent the Tolerated (Benign) variants whereas green and cyan circles represent Deleterious (Pathogenic) variants clustered together respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Sensitivity (Se) and Specificity (Sp) for each MDS program was calculated along with the area under the curve AUC using the pROC function (Table 2). The prediction accuracy for respective MD program to classify variants as Deleterious or Tolerated was further validated through receiver-operating characteristic (ROC) curve. Area under the curve (AUC) was calculated for each MDS program (Fig. 7) representing its ability to correctly classify the selected variants as Deleterious or Tolerated. Area under the ROC curve is statistically significant (area >0.5) for all MDS programs, thus, we conclude that the variants were not randomly classified.
Table 2.
Sensitivity, specificity and AUC values for respective MDS program.
| No. | pROC | RMSD | RMSF | Rg | SASA | Nh-bond | Covariance |
|---|---|---|---|---|---|---|---|
| 1 | Sensitivity (Se) | 0.9358 | 0.9102 | 0.8974 | 0.7948 | 0.8076 | 0.8126 |
| 2 | Specificity (Sp) | 0.8679 | 0.8867 | 0.83018 | 0.7735 | 0.7735 | 0.8084 |
| 3 | Area under curve (AUC) | 0.9019 | 0.8985 | 0.8638 | 0.7842 | 0.7906 | 0.8289 |
Fig. 7.

Receiver Operating Characteristic (ROC) curve demonstrating AUC for respective MD simulation program (A) RMSD (B) RMSF (C) Rg (D) SASA (E) NH-bond (F) Covariance.
We further calculated the p-value and the 95% confidence interval (CI) for scores of Deleterious and Tolerated variants, in order to verify the statistical significance of different parameters used in simulation studies, utilizing the Wilcox test function. The results showed that the p-values for all parameters were statistically significant with p < 0.05 (Table 3) and the estimated values for each MDS program were within the 95% CI interval.
Table 3.
p-value and 95% confidence interval (CI) scores for respective MDS program.
| No. | Methods | p-value | Estimate | Lower 95% | Upper 95% |
|---|---|---|---|---|---|
| 1 | RMSD | 5.10E-22 | 0.299923373 | 0.200040408 | 0.299959413 |
| 2 | RMSF | 1.51E-17 | 0.100004389 | 0.099970979 | 0.149994106 |
| 3 | Rg | 3.57E-22 | 0.199945107 | 0.100078943 | 0.199916133 |
| 4 | SASA | 8.04E-23 | 10.00001104 | 10.00006462 | 19.99996245 |
| 5 | NH-bond | 3.53E-21 | −110.0000377 | −120.0000138 | −100.0000281 |
| 6 | Co-variance | 2.82E-23 | 4.210045407 | 3.400055817 | 4.310000299 |
3.3. Comparison of VUS classified by different assays
We compared our classification for all 131 variants with these classified by different methodologies, including the saturation genome editing that predicted the hypothetical variations in BRCA1 including BRCT repeats [34], experimental assays [36], [54], [55], and the classifications by major BRCA1 variant databases (ENIGMA, ClinVar, LOVD, BIC, and UMD). The results showed that our classification is highly concordance with these classifications: 90 of 131 (68.70%) variant classification were consistent with the saturation genome editing data, 32 (82%) of 39 variant classification were consistent with different functional assays [54]. For the 89 VUS unclassified in BRCA1 databases, we were able to classify 75 (84.2%) as Deleterious or Tolerated, with 14 (15.7%) consistent between our analysis and BRCA1 databases (Table 4). We also compared between our data and the data reported on the classification of 102 VUS in BRCA1 BRCT study [55]. Of the 17 shared VUS between the two studies, 6 variants were classified as Deleterious/Pathogenic and 5 variants classified as Tolerated/Benign by both studies. Further, we also compared our data with a recent study comprising of 248 BRCA1 variants annotated through functionally validated sequence-based computational prediction models [36]. Of the 38 variants shared in both studies, 20 (52.6%) were classified as Deleterious/Damaging and 5 (16.6%) as Tolerated/Neutral by both studies (Supplementary Table 3).
Table 4.
Comparison of BRCT variant classification by different assays.
| Result | Classification |
||
|---|---|---|---|
| SGE (33) | Structure (13) | BRCA1 databases* | |
| A. Summary of the comparison result | |||
| Total | 131 | 39 | 89 |
| Same | 90 (68.70%) | 32 (82%) | 14 (15.7%) |
| Benign | 33 | 10 | 2 |
| Deleterious | 57 | 22 | 12 |
| Difference | 41 (31.29%) | 7 (17.9%) | 75 (84.2%) |
| Benign | 21 | 3 | 33 |
| Deleterious | 20 | 4 | 42 |
| Not available | 0 | 92 | 42 |
| Variants reported |
Classified by BRCA1 databases* | By current study | |
| cDNA | Amino acid | ||
| B. Examples of variants classified by BRCA1 databases and current study | |||
| c.4951T>C | p.S1651P | Uncertain Significance (2) | Deleterious |
| c.4963T>A | p.S1655T | Uncertain Significance (5) | Deleterious |
| c.4963T>G | p.S1655A | Uncertain Significance (5) | Deleterious |
| c.4964C>A | p.S1655Y | Uncertain Significance (2) | Deleterious |
| c.4966G>C | p.G1656R | Uncertain Significance (2) | Deleterious |
| c.4967G>C | p.G1656A | Uncertain Significance (2) | Deleterious |
| c.4981G>A | p.E1661K | Uncertain Significance (2) | Deleterious |
| c.4988T>A | p.M1663K | Uncertain Significance (1) | Deleterious |
| c.4993G>T | p.V1665L | Uncertain Significance (2,5) | Deleterious |
| c.5005G>C | p.A1669P | Uncertain Significance (1) | Deleterious |
| c.5066T>C | p.M1689T | Uncertain Significance (2) | Deleterious |
| c.5068A>C | p.K1690Q | Uncertain Significance (2,5) | Deleterious |
| c.5071A>G | p.T1691A | Uncertain Significance (2,5) | Deleterious |
| c.5072C>T | p.T1691I | Unceratin Significance (1,5) | Deleterious |
| c.5075A>C | p.D1692A | Uncertain Significance (2) | Deleterious |
| c.5090G>A | p.C1697Y | Uncertain Significance (2) | Deleterious |
| c.5096G>T | p.R1699L | Uncertain Significance (2) | Deleterious |
| c.5101C>A | p.L1701M | Uncertain Significance (2) | Deleterious |
| c.5110T>C | p.F1704L | Uncertain Significance (2) | Deleterious |
| c.5112T>G | p.F1704L | Uncertain Significance (2) | Deleterious |
| c.4955T>C | p.M1652T | Uncertain Significance (1) | Tolerated |
| c.4967G>A | p.G1656D | Uncertain Significance (2) | Tolerated |
| c.4993G>A | p.V1665M | Uncertain Significance (2,5) | Tolerated |
| c.5005G>T | p.A1669S | Uncertain Significance (2,5) | Tolerated |
| c.5075A>T | p.D1692V | Uncertain Significance (2,5) | Tolerated |
| c.5076T>A | p.D1692E | Uncertain Significance (2) | Tolerated |
| c.5096G>C | p.R1699P | Uncertain Significance (2) | Tolerated |
| c.5111T>A | p.F1704Y | Uncertain Significance (2) | Tolerated |
| c.5122G>A | p.A1708T | Uncertain Significance (2) | Tolerated |
| c.5147A>G | p.Y1716C | Uncertain Significance (1, 2) | Tolerated |
| c.5152T>G | p.W1718G | Uncertain Significance (2) | Tolerated |
| c.5154G>A | p.W1718S | Uncertain Significance (2) | Tolerated |
| c.5158A>T | p.T1720S | Uncertain Significance (1) | Tolerated |
| c.5168T>A | p.I1723N | Uncertain Significance (1, 3) | Tolerated |
| c.5172A>T | p.K1724N | Uncertain Significance (2) | Tolerated |
| c.5186T>A | p.L1729Q | Uncertain Significance (2) | Tolerated |
| c.5215G>A | p.D1739N | Uncertain Significance (2) | Tolerated |
| c.5242G>T | p.G1748C | Uncertain Significance (1, 2) | Tolerated |
| c.5243G>T | p.G1748V | Uncertain Significance (1) | Tolerated |
| c.5245C>A | p.P1749T | Uncertain Significance (2) | Tolerated |
| c.5251C>G | p.R1751G | Uncertain Significance (2) | Tolerated |
*BRCA databases: 1: ENIGMA; 2: ClinVar; 3: LOVD; 4: BIC; 5: UMD.
4. Discussion
The presence of large quantity of VUS is an obstacle in applying BRCA1 information for clinical cancer applications. Classification of VUS remains a difficult task although multiple approaches have been developed to address the issue [35]. While experiment-based assays, e.g., through measuring homology damage repair and partner protein–protein interaction, can provide functional evidence for VUS classification, they are labor-intensive, time consuming, costly and difficult to scale-up to test large number of variants; computational approaches based on evolution conservation and/or molecular features have also been applied to classify VUS. However, the results are largely predictive and different programs often generate contradictive results. Recently, CRISPR-Cas9 technique was applied for functional classification of BRCA1 variants including VUS. By using saturated genome editing in BRCA1 coding exons and testing their functional impact on the viability of haploid cells, this approach provides a truly high-throughput manner for comprehensive classification of BRCA1 VUS [34]. However, viability of haploid cells affected by the edited BRCA1 may not reflect the actual physical pathogenicity of VUS, as haploid cells contain only one-copy BRCA1 whereas the natural BRCA1 variant carriers are nearly exclusively heterozygotic carrying a varied-copy and an intact-copy of BRCA1. Therefore, lethal effects caused by a variant in haploid cells may not actually happen in heterozygotic diploid cells. Further, measuring the viability of haploid cells doesn’t be necessary to reflect the pathogenic effects of variants, which promotes long-term oncogenic transformation rather than immediate lethal effects on the variant-carrying cells. New approaches are in demand in order to provide evidence for VUS classification in BRCA1 and other cancer predisposition genes. The use of structure-based computational simulation approaches can be a very promising means for the purpose, as it uses the well-established protein structure as the reference to determine the influences of a variant on the structure and is computational-based to allow high-throughput analysis [55], [56], [57], [58], [59]. Its power is well reflected by our current study in using MDS to characterize the variant-induced structural changes in BRCA1 BRCT repeats.
Based on the trajectory analysis using RMSD, RMSF, Rg, inter molecular NH bond analysis and SASA programs, the conformational changes from the native BRCT structure allow classification of VUS and unclassified variants into Deleterious or Tolerated variants. The eight well-known Deleterious variants (G1656R, K1702T, A1708S, G1738E, P1749Q, M1775T, L1839S, Y1853D) provide examples for the conformational change detected by each program, as reflected by high average deviation in RMSD, fluctuations in RMSF, high Rg results in flexibility, loss of hydrogen bonds and overall motion by the covariance vectors. 3D conformational dynamics in G1738E, M1775T, V1809S sites showed their +3-specificity pocket, and the interface between the N- and C- terminal BRCT repeats. In G1738E and M1775T, their positively charged side chains adopted a vertical orientation to block the binding of the Phe side chain at the +3-pocket, thereby, inhibited phospho-protein binding to the motif. Although V1809S occurred far from the +3-pocket, it caused the shift of M1775 position to block the phospho-protein binding to the motif, resulting in conformational change.
Further, graphical representation between the Deleterious and Tolerated variants classified through respective MDS program demonstrated fitted distribution histogram curve (Fig. 5). Fitted distribution curve further demonstrated the probability distribution for selected variants and its maximum likelihood through each MDS program.
MDS for the 10 Pathogenic and 10 Benign controls in our study demonstrated a clear distinction in scores for each MDS program (RMSD, RMSF, Rg, SASA, NH-bond, Covariance) analyzed for a time period of 40 ns (Fig. 8). These scores were used as benchmark scoring towards classification of the 131 variants as Deleterious or Tolerated respectively. Variants classified by respective MDS program were also evaluated through AUC of the ROC curve (Fig. 7). The AUC value for each MDS program was found to be >0.5, implying that the variants are not randomly classified. Also, the p-value was <0.05 along with the estimate scores for each program within the 95% confidence interval.
Fig. 8.

MD score of 10 known Pathogenic and 10 Benign variants by each MDS assay. It shows clear distinction of scores between the Pathogenic and Benign variants.
5. Conclusion
Our study demonstrates that protein structure-based approach can be a powerful means to classify VUS and unclassified variants into Deleterious or Tolerated variants in cancer predisposition genes. Data from such studies should provide solid evidence to further classify pathogenicity of the variants to promote their clinical applications in cancer prevention and treatment.
CRediT authorship contribution statement
Siddharth Sinha: Data curation, Resources, Software, Visualization, Methodology, Formal analysis. San Ming Wang: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Validation, Writing - review & editing.
Acknowledgments
Acknowledgements
We are thankful for the critical comments on the manuscript by Prof. Shirley Siu and English editing by Shanmuga Priya Bhaskaran. We are also thankful to Information and Communication Technology Office (ICTO), University of Macau for providing the High-Performance Computing Cluster resource and facilities for the study.
Competing interests
The authors declare no competing financial interests.
Funding
This work was supported by a grant from Macau Science and Technology Development Fund (085/2017/A2), a grant from The University of Macau (SRG2017‐00097‐FHS), an Innovation grant (FHSIG/SW/0007/2020P) and a startup fund and from Faculty of Health Sciences University of Macau (SMW).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.03.013.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Huen M.S.Y., Sy S.M.H., Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010;11:138–148. doi: 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson B.O., Yip C.H., Smith R.A., Shyyan R., Sener S.F. Guideline implementation for breast healthcare in low-income and middle-income countries: overview of the Breast Health Global Initiative Global Summit 2007. Cancer. 2008;113:2221–2243. doi: 10.1002/cncr.23844. [DOI] [PubMed] [Google Scholar]
- 3.Bhaskaran S.P., Chandratre K., Gupta H., Zhang L., Wang X. Germline variation in BRCA1/2 is highly ethnic-specific: evidence from over 30,000 Chinese hereditary breast and ovarian cancer patients. Int J Cancer. 2019;145(4):962–973. doi: 10.1002/ijc.32176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frank T.S., Deffenbaugh A.M., Reid J.E., Hulick M., Ward B.E. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol. 2002;20:1480–1490. doi: 10.1200/JCO.2002.20.6.1480. [DOI] [PubMed] [Google Scholar]
- 5.Lon S.E., Eccles D.M., Easton D., Foulkes W.D., Genuardi M. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291**. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richards S., Aziz N., Bale S., Bick D., Das S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Studer R.A., Dessailly B.H., Orengo C.A. Residue mutations and their impact on protein structure and function: detecting beneficial and pathogenic changes. Biochem J. 2013;449:581–594. doi: 10.1042/BJ20121221. [DOI] [PubMed] [Google Scholar]
- 8.Sheng Z.-Z., Zhao Y.-Q., Huang J.-F. Functional evolution of BRCT domains from binding DNA to protein. Evol Bioinfo Online. 2011;7:87–97. doi: 10.4137/EBO.S7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang B., Matsuoka S., Ballif B.A., Zhang D., Smogorzewska A. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou B.B., Elledge S.J. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 11.Khanna K.K., Jackson S.P. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 12.Yu X., Chini C.C., He M., Mer G., Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 13.Wu Q., Paul A., Su D., Mehmood S., Foo T.K. Structure of BRCA1-BRCT/abraxas complex reveals phosphorylation-dependent BRCT dimerization at DNA damage sites. Mol Cell. 2016;61:434–448. doi: 10.1016/j.molcel.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.di Masi A., Gullotta F., Cappadonna V., Leboffe L., Ascenzi P. Cancer predisposing mutations in BRCT domains. IUBMB Life. 2011;63:503–512. doi: 10.1002/iub.472. [DOI] [PubMed] [Google Scholar]
- 15.Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spurdle A.B., Healey S., Devereau A., Hogervorst F.B.L., Monteiro A.N.A. ENIGMA–evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33:2–7. doi: 10.1002/humu.21628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fokkema I.F., den Dunnen J.T., Taschner P.E. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum Mutat. 2005;26:63–68. doi: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- 18.Cline M.S., Liao R.G., Parsons M.T., Paten B., Alquaddoomi F. BRCA challenge: BRCA exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018;14:e1007752. doi: 10.1371/journal.pgen.1007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M.X., Kwan J.S., Bao S.Y., Yang W., Ho S.L. Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasidharan Nair P., Vihinen M. VariBench: a benchmark database for variations. Hum Mutat. 2013;34:42–49. doi: 10.1002/humu.22204. [DOI] [PubMed] [Google Scholar]
- 22.Bendl J., Stourac J., Salanda O., Pavelka A., Wieben E.D. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014;10 doi: 10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams R.S., Green R., Glover J.N. Crystal structure of the BRCT repeat region from the breast cancer-associated protein BRCA1. Nat Struct Biol. 2001;8:838–842. doi: 10.1038/nsb1001-838. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X., Morera S., Bates P.A., Whitehead P.C., Coffer A.I. Structure of an XRCC1 BRCT domain: a new protein-protein interaction module. EMBO J. 1998;17:6404–6411. doi: 10.1093/emboj/17.21.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manke I.A., Lowery D.M., Nguyen A., Yaffe M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 26.Clapperton J.A., Manke I.A., Lowery D.M., Ho T., Haire L.F. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat Struct Mol Biol. 2004;11:512–518. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- 27.Dore A.S., Furnham N., Davies O.R., Sibanda B.L., Chirgadze D.Y. Structure of an Xrcc4-DNA ligase IV yeast ortholog complex reveals a novel BRCT interaction mode. DNA Repair (Amst) 2006;5:362–368. doi: 10.1016/j.dnarep.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Wu P.Y., Frit P., Meesala S., Dauvillier S., Modesti M. Structural and functional interaction between the human DNA repair proteins DNA ligase IV and XRCC4. Mol Cell Biol. 2009;29:3163–3172. doi: 10.1128/MCB.01895-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.UniProt: a hub for protein information. Nucleic Acids Res 43;2015:D204–212. [DOI] [PMC free article] [PubMed]
- 30.Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D. Comparative protein structure modeling using Modeller. Curr Protocols Bioinformatics. 2006;5 doi: 10.1002/0471250953.bi0506s15. Chapter 5: Unit-5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams R.S., Lee M.S., Hau D.D., Glover J.N. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat Struct Mol Biol. 2004;11:519–525. doi: 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- 32.Mirkovic N., Marti-Renom M.A., Weber B.L., Sali A., Monteiro A.N. Structure-based assessment of missense mutations in human BRCA1: implications for breast and ovarian cancer predisposition. Cancer Res. 2004;64:3790–3797. doi: 10.1158/0008-5472.CAN-03-3009. [DOI] [PubMed] [Google Scholar]
- 33.Lee M.S., Green R., Marsillac S.M., Coquelle N., Williams R.S. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010;70:4880–4890. doi: 10.1158/0008-5472.CAN-09-4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Findlay G.M., Daza R.M., Martin B., Zhang M.D., Leith A.P. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–222. doi: 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woods N.T., Baskin R., Golubeva V., Jhuraney A., De-Gregoriis G. Functional assays provide a robust tool for the clinical annotation of genetic variants of uncertain significance. NPJ Genom Med. 2016;1:Pii:16001. doi: 10.1038/npjgenmed.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hart S.N., Hoskin T., Shimelis H., Moore R.M., Feng B. Comprehensive annotation of BRCA1 and BRCA2 missense variants by functionally validated sequence-based computational prediction models. Genet Med. 2019;21:71–80. doi: 10.1038/s41436-018-0018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beroud C., Letovsky S.I., Braastad C.D., Caputo S.M., Beaudoux O. BRCA share: a collection of clinical BRCA gene variants. Hum Mutat. 2016;37:1318–1328. doi: 10.1002/humu.23113. [DOI] [PubMed] [Google Scholar]
- 38.Varma A.K., Brown R.S., Birrane G., Ladias J.A. Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry. 2005;44:10941–10946. doi: 10.1021/bi0509651. [DOI] [PubMed] [Google Scholar]
- 39.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 40.Sippl M.J. Recognition of errors in three-dimensional structures of proteins. Proteins: Struct Funct Bioinformatics. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 41.Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karplus M. Molecular dynamics simulations of biomolecules. Acc Chem Res. 2002;35:321–323. doi: 10.1021/ar020082r. [DOI] [PubMed] [Google Scholar]
- 43.Dong Y-w, Liao M-l, Meng X-l, Somero G.N. Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc Natl Acad Sci U S A. 2018;115:1274. doi: 10.1073/pnas.1718910115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benson N.C., Daggett V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J Phys Chem B. 2012;116:8722–8731. doi: 10.1021/jp302103t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daidone I., Amadei A., Roccatano D., Nola A.D. Molecular dynamics simulation of protein folding by essential dynamics sampling: folding landscape of horse heart cytochrome c. Biophys J. 2003;85:2865–2871. doi: 10.1016/S0006-3495(03)74709-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheu S.-Y., Yang D.-Y., Selzle H.L., Schlag E.W. Energetics of hydrogen bonds in peptides. Proc Natl Acad Sci U S A. 2003;100:12683. doi: 10.1073/pnas.2133366100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang D., Lazim R. Application of conventional molecular dynamics simulation in evaluating the stability of apomyoglobin in urea solution. Sci Rep. 2017;7:44651. doi: 10.1038/srep44651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berendsen H.J.C., Postma J.P.M., Gunsteren W.F.V., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 49.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 50.Hess B., Kutzner C., van der Spoel D., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 51.Kozakov D., Hall D.R., Xia B., Porter K.A., Padhorny D. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12:255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amadei A., Linssen A.B., Berendsen H.J. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 53.Yao F., Coquery J., Lê Cao K.-A. Independent Principal Component Analysis for biologically meaningful dimension reduction of large biological data sets. BMC Bioinf. 2012;13 doi: 10.1186/1471-2105-13-24. 24–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee M.S., Edwards R.A., Thede G.L., Glover J.N. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem. 2005;280:32053–32056. doi: 10.1074/jbc.C500273200. [DOI] [PubMed] [Google Scholar]
- 55.Fernandes V.C., Golubeva V.A., Di Pietro G., Shields C., Amankwah K. Impact of amino acid substitutions at secondary structures in the BRCT domains of the tumor suppressor BRCA1: implications for clinical annotation. J Biol Chem. 2019;294:5980–5992. doi: 10.1074/jbc.RA118.005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Porta-Pardo E., Hrabe T., Godzik A. Cancer3D: understanding cancer mutations through protein structures. Nucleic Acids Res. 2015;43(Database issue):D968–D973. doi: 10.1093/nar/gku1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCoy M.D., Shivakumar V., Nimmagadda S., Jafri M.S., Madhavan S. SNP2SIM: a modular workflow for standardizing molecular simulation and functional analysis of protein variants. BMC Bioinf. 2019;20(1):171–178. doi: 10.1186/s12859-019-2774-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCoy M.D., Madhavan S., Nimmagadda S., Klimov D., Jafri M.S. Translational applications of protein structure simulation: predicting phenotype of missense variants. Biophys J. 2019;116(3):13a. [Google Scholar]
- 59.Jafri MS, Mccoy M. Mining All Atom Simulations for Diagnosing and Treating Disease. US Patent App. 16/225,789.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.