

Abstract
Crystallographic studies have shown that the coiled-coil motif occurs in several viral membrane-fusion proteins, including HIV-1 gp41 and influenza virus hemagglutinin. Here, the LearnCoil-VMF program was designed as a specialized program for identifying coiled-coil-like regions in viral membrane-fusion proteins. Based upon the use of LearnCoil-VMF, as well as other computational tools, we report detailed sequence analyses of coiled-coil-like regions in retrovirus, paramyxovirus and filovirus membrane-fusion proteins. Additionally, sequence analyses of these proteins outside their putative coiled-coil domains illustrate some structural differences between them. Complementing previous crystallographic studies, the coiled-coil-like regions detected by LearnCoil-VMF provide further evidence that the three-stranded coiled coil is a common motif found in many diverse viral membrane-fusion proteins. The abundance and structural conservation of this motif, even in the absence of sequence homology, suggests that it is critical for viral-cellular membrane fusion. The LearnCoil-VMF program is available at http://web.wi.mit.edu/kim
Keywords: coiled coil, computational methods, viral membrane fusion, retrovirus, paramyxovirus, filovirus
Keywords: HIV-1, human immunodeficiency virus type 1; SIV, simian immunodeficiency virus; MoMLV, Moloney murine leukemia virus; CAEV, caprine arthritis encephalitis virus; FIV, feline immunodeficiency virus; BIV, bovine immunodeficiency virus; EIAV, equine infectious anemia virus; PDB, Protein Data Bank
Introduction
The surface glycoproteins of enveloped viruses are essential to viral cell entry and replication. These envelope proteins mediate both the initial virion attachment to the cell, as well as the subsequent fusion of viral and cellular membranes; these processes result in the release of viral contents into the host cell. Insight into the membrane fusion process has been advanced by structural studies Wilson et al 1981, Bullough et al 1994, Rey et al 1995, Fass et al 1996, Chan et al 1997, Weissenhorn et al 1997, Weissehorn et al 1998b, Malashkevich et al 1998, Malashkevich et al 1999, Caffrey et al 1998. Remarkably, these studies suggest that rather diverse enveloped viruses share similar mechanisms for membrane fusion Chan and Kim 1998, Hughson 1997.
The structures of the membrane-fusion proteins of influenza virus (hemagglutinin HA2), Moloney murine leukemia virus (MoMLV TM), human and simian immunodeficiency viruses (HIV-1 and SIV gp41), and Ebola virus (Ebola GP2) reveal significant similarity: in their putative fusogenic (i.e., fusion-active) conformations, all five proteins contain a parallel homotrimeric coiled coil adjacent to the fusion-peptide regions (Figure 1). At the carboxyl end of this coiled coil, the polypeptide chains reverse direction, and the base of the coiled coil is supported by additional structures. In HIV-1 and SIV gp41, influenza HA2 and Ebola GP2, this support comes from three extended helices packed on the exterior of the coiled coil. In MoMLV TM, a short helix and a more extended region in each monomer serve to stabilize the coiled coil. In all five structures, the C-terminal residues extend back towards the N-terminal end of the coiled coil to form a hairpin-like structure.
Figure 1.
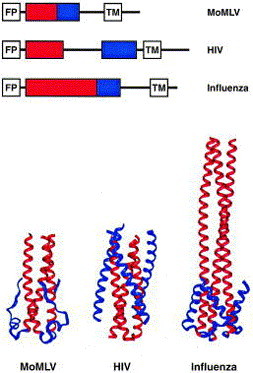
Common structural elements between MoMLV TM, HIV gp41 and influenza HA2. The top panel shows schematic maps of the MoMLV, HIV and influenza sequences. For each, the position of the coiled coil is shown in red, and the position of the supporting structures is shown in blue. The bottom panel shows the structures of MoMLV TM residues 45–98 (Fass et al., 1996), HIV gp41 residues 546–581 (Chan et al., 1997), and influenza HA2 residues 40–129 (Bullough et al., 1994). The interior coiled coils are shown in red, and the exterior supporting structures are shown in blue.
Do other enveloped viruses share similar structures for membrane fusion? Computational methods provide a quick way to begin addressing this question for a large, diverse group of viruses. The lack of sequence similarity among viral membrane-fusion proteins requires computational tools more sensitive than alignment-based methods. Fortunately, the coiled-coil structure common to HIV-1 gp41, SIV gp41, influenza HA2, MoMLV TM, and Ebola GP2 is a motif for which several prediction schemes have already been developed Parry 1982, Lupas et al 1991, Berger et al 1995, Wolf et al 1997. Nevertheless, these methods cannot be used alone to recognize the coiled coils in membrane-fusion proteins. For example, the coiled-coil region in HIV-1 (Chan et al., 1997) is scored with low likelihood (⩽10 %) using either PairCoil (Berger et al., 1995), MultiCoil (Wolf et al., 1997) or COILS (Lupas et al., 1991). Several authors have previously noted heptad repeat patterns visually in viral membrane-fusion proteins Groot et al 1987, Gallaher et al 1989, Delwart et al 1990, Chambers et al 1990. However, the probability of a heptad repeat pattern occurring by chance in a protein sequence is significant (Brendel & Karlin, 1989), and predicting coiled coils “by eye” may therefore lead to significant over-prediction. For this reason, reliable computational methods are necessary in evaluating whether these patterns actually correspond to coiled-coil-like structures.
Here, an iterative algorithm LearnCoil Berger and Singh 1997, Singh et al 1998 is used as the primary method to detect potential coiled-coil-like regions in viral membrane-fusion proteins (see Methods). In the algorithm, an initial evaluation of viral membrane-fusion protein sequences was made using the PairCoil algorithm and a probability table estimated from a two-stranded and three-stranded coiled-coil database Berger et al 1995, Wolf et al 1997. Then, those sequences with likelihoods above zero were selected by a randomized procedure to update the table. This process was repeated until convergence. After convergence, the final tables built from the viral membrane-fusion proteins were incorporated into a program, LearnCoil-VMF.
The LearnCoil-VMF program detects coiled-coil-like regions in many viral membrane-fusion proteins, including most retrovirus and paramyxovirus membrane-fusion proteins examined. These proteins are quite diverse, with no apparent sequence similarity between most pairs of viruses in different families, or between some viruses within the same family. The LearnCoil-VMF program also helps further characterize the overall core structure of these viral membrane-fusion proteins. The exterior of the HIV-1 gp41 core is made up of three extended amphipathic helices that contain a heptad repeat and wrap around the interior coiled coil in a left-handed direction (Figure 1); the LearnCoil-VMF program identifies these coiled-coil-like helices in HIV-1 as well as in other viruses in the lentivirus genus of retroviruses. Intriguingly, LearnCoil-VMF also identifies two coiled-coil-like regions in many paramyxovirus membrane-fusion proteins. A recent result shows that peptides containing the two predicted coiled-coil-like regions in the paramyxovirus simian parainfluenza virus 5 (SV5) interact with each other to form an α-helical trimeric complex (Joshi et al., 1998).
The inability of previous coiled-coil recognition methods to identify the coiled-coil-like structures found in many viral membrane-fusion proteins indicates that, although these proteins contain an apparent heptad repeat, their coiled-coil-like regions have some subtle statistical differences from other known coiled coils. This work thus helps characterize the coiled-coil-like regions found in viral membrane-fusion proteins. Additionally, because coiled-coil recognition methods are primarily limited by the lack of a sufficiently large and diverse database of coiled-coil sequences (Wolf et al., 1997), this work suggests improvements for existing methods for coiled-coil recognition.
Results
Coiled-coil-like regions are detected using LearnCoil-VMF in many viral membrane-fusion proteins, including many retrovirus envelope proteins, paramyxovirus fusion proteins, orthomyxovirus hemagglutinins, coronavirus spike proteins, arenavirus glycoproteins, and baculovirus envelope glycoproteins. (See Methods for a description of the LearnCoil-VMF algorithm.) Here, we focus on the membrane-fusion proteins of retroviruses, paramyxoviruses and filoviruses. Protein sequences from any virus family may be submitted at the LearnCoil-VMF webpage.
Retroviruses
LearnCoil-VMF finds coiled-coil-like regions in membrane-fusion proteins from all retrovirus genera. Detailed analysis is given for the lentivirus, mammalian type C, avian type C, type D and BLV-HTLV type retrovirus genera (Murphy et al., 1995).
LearnCoil-VMF detects two coiled-coil-like regions in most of the envelope proteins from the lentivirus genus of retroviruses Table 1, Table 2.† Note the overall sequence diversity in Table 1, Table 2; even though pairs of sequences are quite similar, there is not a single conserved residue in either Table. The subsequences shown are aligned (using the output of LearnCoil-VMF) to the N36/C34 fragment crystallized for HIV-1 gp41 (Chan et al., 1997) and SIV gp41 (Malashkevich et al., 1998). For the envelope protein sequences shown in Table 1, Table 2, two coiled-coil-like regions are detected by LearnCoil-VMF in HIV-1, SIV, feline immunodeficiency virus (FIV), visna virus, and caprine arthritis encephalitis virus (CAEV), but only one coiled-coil-like region is found in equine infectious anemia virus (EIAV) and bovine immunodeficiency virus (BIV). However, the sequence features shown in Table 2 shared between EIAV and BIV and the other lentiviruses suggests that a second helix exists in EIAV and BIV, although the LearnCoil-VMF method is not able to detect it. In particular, the heptad repeat is maintained in the N-terminal portion of these subsequences, as it is in the other lentivirus sequences given. Furthermore, in both the HIV-1 and SIV structures, there are two tryptophan residues in the C-terminal helix that fit into a conserved pocket found in the interior three-stranded coiled coil Chan et al 1997, Chan et al 1998, Malashkevich et al 1998; these residues (corresponding to columns 1 and 4 in Table 2) are conserved in EIAV, while the first tryptophan residue is conserved in BIV and the second is replaced by a leucine residue. Finally, for an alignment of the lentivirus envelope sequences, the PHD secondary structure program predicts the region shown in Table 2 as helical, with 85% of the predictions with reliability index at least 8 (the three-state accuracy for predictions with reliability index 8 or higher is 91.1%) Rost and Sander 1993, Rost and Sander 1994, Rost et al 1994. ‡ Thus, computational methods argue that for the lentivirus membrane-fusion proteins, there are two coiled-coil-like regions that form a six-helical bundle structure similar to that found in HIV-1.
Table 1.
Lentivirus N-terminal coiled coils
| Virus | Add dist | Sequence | Likelihoods | |||
|---|---|---|---|---|---|---|
| L | P | C | M | |||
| bcdefgabcdefgabcdefgabcdefgabcdefgab | ||||||
| HIV-1 | 36 | SGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL | 9 | 0 | 0 | 1 |
| SIV | 34 | AGIVQQQQQLLDVVKRQQELLRLTVWGTKNLQTRVS | 9 | 0 | 0 | 0 |
| Visna | 37 | QSLANATAAQQNVLEATYAMVQHVAKGVRILEARVA | 9 | 0 | 0 | 0 |
| CAEV | 37 | QTLANATAAQQDALEATYAMVQHVAKGVRILEARVA | 9 | 0 | 0 | 0 |
| FIV | 36 | ATHQETIEKVTEALKINNLRLVTLEHQVLVIGLKVE | 9 | 3 | 1 | 3 |
| EIAV | 39 | NHTFEVENSTLNGMDLIERQIKILYAMILQTHADVQ | 9 | 0 | 1 | 2 |
| BIV | 39 | ERVVQNVSYIAQTQDQFTHLFRNINNRLNVLHRRVS | 5 | 0 | 0 | 0 |
For the lentiviruses, two helical regions are found; the lentivirus N-terminal helix is shown here. In all Tables, L, P, C and M indicate the highest likelihood for the shown sequence fragment for (respectively) LearnCoil-VMF, PairCoil, COILS and Multicoil. (0 represents likelihoods less than 0.1; 1 represents likelihoods at least 0.1 and less than 0.2, etc.) For Multicoil, the likelihoods given are the total coiled-coil likelihoods (i.e. the sum of the dimer and trimer likelihoods). In all Tables lower-case letters represent positions in the heptad repeat. Add dist is the number of amino acid residues from the proteolytic maturation cleavage site to the beginning of the shown subsequence. The N36 fragment of HIV-1 gp41 (Chan et al., 1997) is shown, and the other lentivirus sequences are aligned to it. Abbreviations and GenBank accession numbers (Benson et al., 1998) for both lentivirus Tables are: HIV-1, human immunodeficiency virus 1 (119452); SIV, simian immunodeficiency virus (119496); Visna, visna virus (543528); CAEV, caprine arthritis encephalitis virus (399432); FIV, feline immunodeficiency virus (544245); EIAV, equine infectious anemia virus (119407); BIV, bovine immunodeficiency virus (119399).
Table 2.
The lentivirus C-terminal helices
| Virus | Add dist2 | Sequence | Likelihoods | |||
|---|---|---|---|---|---|---|
| L | P | C | M | |||
| abcdefgabcdefgabcdefgabcdefgabcdef | ||||||
| HIV-1 | 47 | WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL | 9 | 3 | 8 | 1 |
| SIV | 43 | WQEWERKVDFLEANITALLEEAQIQQEKNMYELQ | 9 | 1 | 9 | 5 |
| Visna | 47 | WQQWEEEIEQHEANLSQLLREAALQVHIAQRDAQ | 9 | 0 | 8 | 5 |
| CAEV | 47 | WQQWERELQGYDGNLTMLLRESARQTQLAEEQVR | 9 | 0 | 7 | 0 |
| FIV | 50 | LGEWYNQTKELQQKFYEIIMNIEQNNVQVKKGLQ | 9 | 0 | 4 | 0 |
| EIAV | 44 | WDDWVSKMEDLNQEILTTLHGARNNLAQSMITFN | 0 | 0 | 0 | 0 |
| BIV | 44 | WSDLQDEYDKIEEKILKIRVDWLNSSLSDTQDTF | 0 | 0 | 0 | 0 |
Add dist2 column is the distance from the end of the region in Table 1 to the start of this region. The C34 fragment of HIV-1 gp41 (Chan et al., 1997) is shown, and the other lentivirus sequences are aligned to it.
Recently, to test these predictions for visna virus, peptides were constructed corresponding to the sequences shown in Table 1, Table 2. These peptides were crystallized and in fact form a six-helical bundle structure that is very similar to HIV-1 and SIV gp41 (V.N. Malashkevich & P.S.K., unpublished results).
For envelope proteins from the retrovirus genera mammalian type C, avian type C, type D and BLV-HTLV type, there is a single predicted coiled-coil region using the LearnCoil-VMF, PairCoil, MultiCoil, and COILS programs (Table 3). A fragment of the envelope protein of MoMLV, a mammalian type C retrovirus, has been studied using X-ray crystallography (Fass et al., 1996); the subsequences shown in Table 3 are aligned to the coiled-coil region found in this crystal structure. All four programs identify the coiled-coil regions in the mammalian type C retroviruses and the type D viruses; however, only LearnCoil-VMF identifies coiled coils in the avian C and BLV-HTLV retroviruses.
Table 3.
Representative envelope proteins for retrovirus genera mammalian C, avian C, D and BLV-HTLV
| Virus | Add dist | Sequence | Likelihoods | |||
|---|---|---|---|---|---|---|
| L | P | C | M | |||
| Mam C | gabcdefgabcdefgabcdefgabcdefgabcd | |||||
| MoMLV | 46 | DLREVEKSISNLEKSLTSLSEVVLQNRRGLDLL | 9 | 7 | 9 | 9 |
| FeLV | 46 | DIQALEESISALEKSLTSLSEVVLQNRRGLDIL | 9 | 6 | 5 | 9 |
| GALV | 47 | DLRALQDSVSKLEDSLTSLSEVVLQNRRGLDLL | 9 | 6 | 3 | 9 |
| ARV | 41 | DVQALSGTINDLQDQIDSLAEVVLQNRRGLDLL | 9 | 6 | 9 | 8 |
| Avian C | ||||||
| RSV | 50 | QANLTTSLLGDLLDDVTSIRHAVLQNRAAIDFL | 9 | 0 | 0 | 0 |
| D | ||||||
| MPMV | 41 | DVQAISSTIQDLQDQVDSLAEVVLQNRRGLDLL | 9 | 6 | 6 | 9 |
| BLV-HTLV | ||||||
| BLV | 41 | DQQRLITAINQTHYNLLNVASVVAQNRRGLDWL | 9 | 0 | 0 | 0 |
| HTLV-1 | 41 | DISQLTQAIVKNHKNLLKIAQYAAQNRRGLDLL | 9 | 0 | 0 | 0 |
| HTLV-2 | 41 | DISHLTQAIVKNHQNILRVAQYAAQNRRGLDLL | 9 | 0 | 0 | 0 |
| PTLV | 41 | DIDHLTRAIVKNHDNILRVAQYAAQNRRGLDLL | 9 | 0 | 0 | 0 |
For these sequences, there is only one predicted coiled coil. Add dist is the distance from the cleavage site to start of the subsequence shown. The subsequences shown correspond to the coiled coil in the Moloney murine leukemia virus crystal structure (Fass et al., 1996) and are aligned using subsequence QNRRGLDLL (Delwart et al., 1990). Abbreviations and GenBank accession numbers (Benson et al., 1998) are: MoMLV, Moloney murine leukemia virus (119478); FeLV, feline leukemia virus (119418); GALV, gibbon ape leukemia virus (119426); ARV, avian reticuloendotheliosis virus (119396); RSV, Rous sarcoma virus (119487); MPMV, Mason-Pfizer monkey virus (119482); BLV, bovine leukemia virus (119401); HTLV, human T-lymphotropic virus (119464 and 119467); PTLV, primate T-lymphtropic virus (632274).
Interestingly, there is some computational evidence that the mammalian type C, avian type C, and type D retroviruses also contain a second helix. In particular, for an alignment of these sequences, the PHD secondary structure program finds a second helix approximately 25 residues C-terminal to the region shown in Table 3, with length 18. (For MoMLV, this places the start of the helix five residues C-terminal to the fragment solved by X-ray crystallography (Fass et al., 1996).) Additionally, in all of these sequences except Rous sarcoma virus, the same region scores as coiled-coil-like when using the MultiCoil program with windows of length 14 (individual likelihoods varying from 0.31 to 0.71); this suggests a short amphipathic helix. This computational evidence is consistent with circular dichroism (CD) experiments on MoMLV that show additional helical content C-terminal of the coiled-coil structure (Fass & Kim, 1995). Additionally, these retrovirus envelope sequences share sequence similarity with Ebola GP2, and recent X-ray crystallography studies reveal a helix in this region Weissehorn et al 1998b, Malashkevich et al 1999.
Paramyxoviruses
LearnCoil-VMF detects two coiled-coil-like regions (likelihoods ⩾0.5) in 15 out of the 20 sequences listed in Table 4, Table 5. An additional four viruses have two scoring regions, although the second regions are given likelihoods less than 0.5 by LearnCoil-VMF. Human parainfluenza virus 1 is the only sequence for which only one coiled-coil-like region is found; however, this sequence aligns well with the other paramyxovirus sequences in its genus, and thus it is expected to contain a second coiled-coil-like region as well. As in the lentivirus membrane-fusion proteins, LearnCoil-VMF finds one coiled-coil-like region soon after the cleavage site and fusion peptide, and the other directly preceding the final transmembrane domain. The number of residues between the two coiled-coil-like regions in all the paramyxoviruses is substantial (more than 265 residues in the paramyxoviruses, compared to 50 or fewer residues in the lentiviruses); in fact, there is no apparent sequence similarity between the paramyxovirus and lentivirus sequences. An additional third coiled-coil region is found using all four programs in bovine respiratory syncytial virus and human respiratory syncytial virus; however, this region is before the cleavage site, in the F2 glycoprotein (data not shown). The significance of this region is unknown.
Table 4.
Paramyxovirus N-terminal helix
| Virus | Add dist | Sequence | Likelihoods | |||
|---|---|---|---|---|---|---|
| L | P | C | M | |||
| Paramyx | gabcdefgabcdefgabcdefgabcdefgabcdefgabcdefgabc | |||||
| BPIV-3 | 27 | EAKQAKSDIEKLKEAIRDTNKAVQSIQSSVGNLIVAVKSVQDYVNN | 9 | 8 | 9 | 8 |
| HPIV-3 | 27 | EAKQARSDIEKLKEAIRDTNKAVQSVQSSIGNLIVAIKSVQDYVNK | 9 | 8 | 9 | 8 |
| HPIV-1 | 30 | EAREARKDIALIKDSIIKTHNSVELIQRGIGEQIIALKTLQDFVNN | 7 | 0 | 1 | 0 |
| Sendai | 30 | EAREAKRDIALIKESMTKTHKSIELLQNAVGEQILALKTLQDFVND | 9 | 4 | 9 | 6 |
| Morbilli | ||||||
| CDV | 27 | QSNLNAQAIQSLRTSLEQSNKAIEEIREATQETVIAVQGVQDYVNN | 9 | 6 | 7 | 7 |
| Measles | 27 | QSMLNSQAIDNLRASLETTNQAIEAIRQAGQEMILAVQGVQDYINN | 9 | 0 | 4 | 3 |
| PPRV | 27 | QSLMNSQAIESLKTSLEKSNQAIEEIRLANKETILAVQGVQDYINN | 9 | 6 | 8 | 5 |
| PDV | 27 | QSNLNAQAIQSLRASLEQSNKAIDEVRQASQNIIIAVQGVQDYVNN | 9 | 5 | 7 | 8 |
| RPV | 27 | QSMMNSQAIESLKASLETTNQAIEEIRQAGQEMVLAVQGVQDYINN | 9 | 0 | 5 | 7 |
| Rubula | ||||||
| HPIV-2 | 27 | KANANAAAINNLASSIQSTNKAVSDVIDASRTIATAVQAIQDHING | 9 | 0 | 1 | 0 |
| HPIV-4a | 27 | KAQENAKLILTLKKAATETNEAVRDLANSNKIVVKMISAIQNQINT | 9 | 2 | 6 | 2 |
| HPIV-4b | 27 | KAQENAQLILTLKKAAKETNDAVRDLTKSNKIVARMISAIQNQINT | 9 | 5 | 3 | 2 |
| SV5 | 27 | KANENAAAILNLKNAIQKTNAAVADVVQATQSLGTAVQAVQDHINS | 9 | 4 | 2 | 1 |
| PRV | 27 | RANKNAEKVEQLSQALGETNAAISDLIDATKNLGFAVQAIQNQINT | 9 | 5 | 4 | 5 |
| Mumps | 27 | QAQTNARAIAAMKNSIQATNRAVFEVKEGTQQLAIAVQAIQDHINT | 9 | 0 | 0 | 0 |
| NDV | 27 | QANQNAANILRLKESITATIEAVHEVTDGLSQLAVAVGKMQQFVND | 9 | 2 | 0 | 4 |
| Pneumo | ||||||
| BRSV | 20 | KVLHLEGEVNKIKNALLSTNKAVVSLSNGVSVLTSKVLDLKNYIDK | 9 | 2 | 1 | 1 |
| HRSV | 20 | KVLHLEGEVNKIKNALLSTNKAVVSLSNGVSVLTSKVLDLKNYINN | 9 | 2 | 1 | 1 |
| PVM | 20 | KTVQLESEIALIRDAVRNTNEAVVSLTNGMSVLAKVVDDLKNFISK | 9 | 0 | 4 | 0 |
| TRTV | 24 | KTIRLEGEVKAIKNALRNTNEAVSTLGNGVRVLATAVNDLKEFISK | 9 | 4 | 5 | 2 |
Add dist is the distance between the cleavage site and beginning of the region shown. Abbreviations and GenBank accession numbers (Benson et al., 1998) for both Table 4, Table 5: paramyx, genus paramyxovirus; morbilli, genus morbillivirus; rubula, genus rubulavirus; pneumo, genus pneumovirus; BPIV, bovine parainfluenza (1353202); HPIV, human parainfluenza (138273, 138268, 138269, 1255649, and 1255651); Sendai (138276); CDV, canine distemper (138249); measles (138254); PPRV, peste-des-petits-ruminants (1085797); PDV, phocine distemper (138267); RPV, rinderpest (138275); SV5, simian parainfluenza 5 (335117); PRV, porcine rubulavirus (1808667); mumps (138259); NDV, Newcastle disease (465403); BRSV, bovine respiratory syncytial (138248); HRSV, human respiratory syncytial (138250); PVM, pneumonia virus of mice (549309); TRTV, turkey rhinotracheitis (138283).
Table 5.
Paramyxovirus C-terminal helix
| Virus | Add dist2 | Sequence | Likelihoods | |||
|---|---|---|---|---|---|---|
| L | P | C | M | |||
| Paramyx | defgabcdefgabcdefgabcdefgabcd | |||||
| BPIV-3 | 275 | ISMELNKAKLELEESKEWIKKSNQKLDSV | 9 | 3 | 0 | 1 |
| HPIV-3 | 275 | ISIELNKAKSDLEESKEWIRRSNQKLDSI | 9 | 3 | 0 | 2 |
| HPIV-1 | 275 | ISLNLASATNFLEESKIELMKAKAIISAV | 0 | 0 | 0 | 0 |
| Sendai | 275 | ISLNLADATNFLQDSKAELEKARKILSEV | 3 | 5 | 7 | 4 |
| Morbilli | ||||||
| CDV | 268 | ISLDRLDVGTNLGNALKKLDDAKVLIDSS | 9 | 2 | 7 | 0 |
| Measles | 268 | ISLERLDVGTNLGNAIAKLEDAKELLESS | 9 | 3 | 6 | 3 |
| PPRV | 268 | ISLEKLDVGTNLGNAVTRLENAKELLDAS | 9 | 0 | 2 | 0 |
| PDV | 268 | ISLERLDVGTNLGSALKKLDDAKVLIESS | 9 | 1 | 6 | 1 |
| RPV | 268 | ISLEKLDVGTNLWNAVTKLEKAKDLLDSS | 5 | 0 | 0 | 0 |
| Rubula | ||||||
| HPIV-2 | 275 | LSNQINSINKSLKSAEDWIADSNFFANQA | 9 | 0 | 0 | 0 |
| HPIV-4a | 275 | LSTDLNQYNQLLKSAEDHIQRSTDYLNSI | 9 | 0 | 6 | 5 |
| HPIV-4b | 275 | LSTDLNQYNQLLKSAENHIQRSNDYLNSI | 9 | 0 | 8 | 3 |
| SV5 | 275 | ISQNLAAVNKSLSDALQHLAQSDTYLSAI | 9 | 0 | 0 | 0 |
| PRV | 275 | ISGNLIAVNNSLSSALNHLATSEILRNEQ | 2 | 0 | 0 | 0 |
| Mumps | 275 | ISTELSKVNASLQNAVKYIKESNHQLQSV | 9 | 6 | 3 | 0 |
| NDV | 275 | ISTELGNVNNSISNALDKLEESNSKLDKV | 9 | 6 | 9 | 4 |
| Pneumo | ||||||
| BRSV | 287 | FDASIAQVNAKINQSLAFIRRSDELLHSV | 9 | 0 | 0 | 0 |
| HRSV | 287 | FDASISQVNEKINQSLAFIRRSDELLHNV | 9 | 0 | 0 | 0 |
| PVM | 285 | FDVAIRDVEHSINQTRTFFKASDQLLDLS | 4 | 0 | 0 | 0 |
| TRTV | 285 | FNVALDQVFESIDRSQDLIDKSNDLLGAD | 3 | 0 | 0 | 0 |
Add dist2 is the distance from the end of the region shown in Table 4 to the beginning of this region.
Recently it has been shown that two peptides, each containing one of the heptad repeat regions of simian parainfluenza virus 5 (SV5), interact and form a helical complex that consists of a trimer of heterodimers (Joshi et al., 1998). The first peptide (denoted N1) contains the region shown in Table 4, along with seven N-terminal residues and ten C-terminal residues, and the second peptide (denoted C1) contains the region shown in Table 5, along with 14 N-terminal residues.
Interestingly, for both the lentiviruses and the paramyxoviruses, the percentage of β-branched residues in a and d positions found in the first coiled-coil-like region is approximately twice the percentage found in the second coiled-coil-like region, † with a high percentage of β-branched residues in both the a and d positions in the first coiled-coil-like region. This is consistent with the observation that β-branched residues in both buried positions favor formation of the trimer oligomeric state of coiled coils (Harbury et al., 1993).
Filoviruses
As anticipated earlier (Fass et al., 1996), recent structural studies have suggested that the Ebola GP2 glycoprotein is structurally analogous to MoMLV TM Weissenhorn et al 1998a, Weissehorn et al 1998b, Malashkevich et al 1999. LearnCoil-VMF finds a coiled-coil-like region in the Ebola GP2 viral membrane-fusion protein, but not in the closely related Marburg virus GP (Table 6). The Ebola and Marburg virus glycoproteins are very similar to the non-filovirus Rous sarcoma virus membrane-fusion protein; remarkably, in the putative coiled-coil region, these filovirus proteins are more similar at a sequence level to the Rous sarcoma virus envelope protein than are any of the retrovirus sequences in Table 1, Table 3. In fact, the sequence similarity between the Rous sarcoma virus envelope protein and Ebola GP2 extends throughout the membrane-fusion domain of these sequences (Gallaher, 1996).‡ Thus, sequence analysis suggests that the Ebola GP2 contains a coiled coil, flanked by a short amphipathic helix on its outer core, and this is in agreement with recent structural studies Weissehorn et al 1998b, Malashkevich et al 1999. Although LearnCoil-VMF does not find a coiled-coil-like region in the Marburg virus GP, its sequence similarity to Ebola within this region (Table 6) suggests a similar structure.
Table 6.
The filoviruses and Rous sarcoma virus
| Virus | Sequence | Likelihoods | |||
|---|---|---|---|---|---|
| L | P | C | M | ||
| gabcdefgabcdefgabcdefgabcdefgabcd | |||||
| Ebola | LANETTQALQLFLRATTELRTFSILNRKAIDFL | 8 | 0 | 0 | 0 |
| Marburg | LANQTAKSLELLLRVTTEERTFSLINRHAIDFL | 0 | 0 | 0 | 0 |
| Rous | QANLTTSLLGDLLDDVTSIRHAVLQNRAAIDFL | 9 | 0 | 0 | 0 |
| ∗∗ ∗ ∗ ∗ ∗ ∗ ∗∗ ∗∗∗∗∗ | |||||
Shown is an alignment between the putative coiled-coil region of the viral membrane-fusion proteins of Ebola, Marburg and the non-filovirus Rous sarcoma virus. Residues that are identical in all three viruses are indicated with asterisks. The regions shown correspond to the coiled-coil in MoMLV TM (Table 3). GenBank accession numbers (Benson et al., 1998) are: Ebola virus, 290392; Marburg virus, 2459880; and Rous sarcoma virus, 119487.
Other virus families
Virus families in the iteration test set for which no coiled-coil regions are found in their putative membrane-fusion proteins by LearnCoil-VMF include flaviviridae (e.g., yellow fever virus and tick-borne encephalitis virus), rhabdoviridae (e.g., vesicular stomatitis virus), and togaviridae (e.g., eastern equine encephalitis virus). In the case of tick-borne encephalitis virus, the membrane-fusion protein indeed does not contain a coiled coil, at least in the native (i.e., non-fusogenic) state (Rey et al., 1995).
PDB sequences
In order to test the discriminative power of LearnCoil-VMF, sequences in the Protein Data Bank (PDB) were evaluated with the final learned tables. The homodimeric GCN4 coiled coil and its mutants, the Fos-Jun heterodimeric coiled coil, the Max homodimeric coiled coil, the trimeric chicken cartilage matrix coiled coil, the pentameric COMP coiled coil, and the antiparallel seryl-tRNA synthetase coiled coil all have likelihoods greater than 0.5 using both LearnCoil-VMF and PairCoil. None of these proteins are included in the databases used by LearnCoil-VMF; their identification suggests similarities between the identified coiled-coil-like regions in viral membrane-fusion proteins and traditional coiled coils.
The following lists all non-coiled-coil structures in the PDB (June 1998 release) that are given greater than 0.5 likelihood by LearnCoil-VMF but not PairCoil (PDB accession ID, description, Learn-Coil-VMF likelihood, PairCoil likelihood): 4hb1, designed four helical bundle, 0.51, 0.33; 1rmv, ribgrass mosaic virus coat protein, >0.9, 0.16; 1wer, ras-gtpase-activating domain of human P120gap, 0.78, 0.40; 1zak, adenylate kinase from maize, 0.61, 0.21. The scoring regions for all these sequences correspond to primarily helical regions in the X-ray crystal structures. Thus, the LearnCoil-VMF program is not overly permissive in identifying coiled-coil-like regions.
Discussion
The LearnCoil-VMF program has detected coiled-coil-like regions in many viral membrane-fusion proteins. These detected regions are likely to correspond to coiled-coil-like structures for several reasons. For membrane-fusion proteins with solved X-ray crystal structures, LearnCoil-VMF detects only those regions that have been established as coiled-coil-like. Additionally, other protein sequences in the PDB with known coiled-coil structures, such as GCN4 (PDB accession ID 2zta), still score as coiled-coil-like using the LearnCoil-VMF program. On the other hand, although some non-coiled-coil sequences from the PDB are detected by the LearnCoil-VMF algorithm, the vast majority are not, indicating specificity in the learned tables. Finally, despite the significant sequence diversity in many viral membrane-fusion proteins within a particular genus, LearnCoil-VMF is usually consistent in recognizing similar regions within them. For example, although the lentivirus sequences in Table 1 vary considerably, the coiled-coil-like regions that the LearnCoil-VMF program identifies are all similarly located within the protein sequences (i.e., following the protein maturation cleavage site).
Complementing previous crystallographic studies, the coiled-coil-like regions detected by LearnCoil-VMF give further evidence that the three-stranded coiled coil is a common motif found in many diverse viral membrane-fusion proteins, including those of many retroviruses, paramyxoviruses and filoviruses. Additionally, for many of these proteins, there is computational evidence for the existence of helices that pack against the outside of the coiled coil. For many of the retrovirus membrane-fusion proteins, such as those in the mammalian type C, avian type C, and type D genera, secondary structure prediction indicates a short, amphipathic helix following the N-terminal coiled-coil-like region. For the lentivirus membrane-fusion proteins, the two coiled-coil-like regions found by LearnCoil-VMF, in conjunction with the HIV-1 and SIV gp41 crystal structures, suggest amphipathic helices packed against the length of the three-stranded coiled-coil core. Two similarly placed coiled-coil-like regions are also found in the paramyxovirus membrane-fusion proteins, and studies on SV5 indicate that the two coiled-coil-like regions interact to form a trimer of heterodimers (Joshi et al., 1998) (see also note added in proof).
Other experimental evidence hints at a mechanistic similarity between the paramyxovirus membrane-fusion proteins and the HIV-1 and SIV gp41 structures. In particular, synthetic C-peptides (peptides that overlap the second heptad repeat region of HIV-1 gp41) have been shown to be potent inhibitors of HIV-1 infection Jiang et al 1993, Wild et al 1994, Lu et al 1995, Kilby et al 1998, Chan et al 1998. It is thought that the C-peptide acts in a dominant-negative manner, with the synthetic peptides binding to the central coiled coil Lu et al 1995, Chen et al 1995, Chan and Kim 1998. Like HIV-1, many paramyxoviruses are inhibited by peptides that overlap the second heptad repeat region Rapaport et al 1995, Lambert et al 1996, Yao and Compans 1996, Joshi et al 1998 (i.e., peptides overlapping those shown in Table 5).
By analogy to influenza HA2, some of the coiled-coil-like regions found by LearnCoil-VMF may only be helical in the fusogenic state of the protein. In influenza HA2, a region that corresponds to a loop in the X-ray structure of native HA2 (Wilson et al., 1981) “springs” into a helical conformation in its fusogenic state Carr and Kim 1993, Bullough et al 1994. This loop-to-helix region in influenza HA2 is predicted as coiled-coil-like using all four prediction programs. Furthermore, in the case of HIV-1, although the gp41 helical core is extremely stable, synthetic C peptides inhibit HIV-1 infection and syncytia formation at very low concentration, thus giving evidence that the gp41 core structure is not present in the native state of this protein (for a review, see Chan & Kim, 1998).
Coiled coils have also been predicted Carr and Kim 1993, Spring et al 1993, Inoue et al 1992 and experimentally found to be a dominant feature Sutton et al 1998, Hayashi et al 1994, Chapman et al 1994, Fasshauer et al 1997, Lin and Scheller 1997, Hanson et al 1997, Weber et al 1998, Nicholson et al 1998 of the SNARE proteins that mediate membrane-fusion events in neurotransmission and protein trafficking. The general purpose coiled-coil prediction programs PairCoil, MultiCoil and COILS identify some but not all of the regions that make up the four-stranded parallel coiled coil in the crystal structure of the core of the synaptic-fusion complex (Sutton et al., 1998). Although LearnCoil-VMF is not as effective in identifying these coiled-coil regions, an iterative approach such as LearnCoil may be useful in designing a specialized program for identifying these coiled-coil regions in as yet unidentified proteins involved in other, non-viral membrane-fusion events.
Methods
A collection of 588 putative membrane-fusion proteins for enveloped viruses was gathered using the Entrez protein browser (Benson et al., 1998). For each virus family (Murphy et al., 1995), the following protein sequences were gathered (Fields et al., 1996): arenavirus, GP-C glycoprotein; baculovirus, glycoproteins gp64 and gp67; bunyavirus, G2 glycoprotein; coronavirus, S spike protein; filovirus, GP peplomar glycoprotein; flavivirus, E protein; herpesvirus, gH glycoprotein; orthomyxovirus, hemagglutinin; paramyxovirus, F protein; retrovirus, envelope protein; rhabdovirus, G protein; and togavirus, E1 glycoprotein. Obvious closely related sequences were eliminated by scoring each sequence with the PairCoil program and allowing only one representative sequence into the iteration test set from those that have the same sequence score. This left a total of 266 sequences in the iteration test set.
The primary method used for coiled-coil detection is the LearnCoil program. The LearnCoil program is a general iterative method that extends the two-stranded coiled-coil prediction program PairCoil to improve identification of other types of coiled coils (Berger & Singh, 1997). Previously, the LearnCoil program has been used to identify coiled-coil-like regions in histidine kinase proteins (Singh et al., 1998). Iterative approaches similar to LearnCoil have been applied to sequence alignment and motif recognition Tatusov et al 1994, Lawrence et al 1993, Attwood and Findlay 1993, Gribskov 1992, Dodd and Egan 1990. Most recently, the position-specific iterated BLAST (PSI-Blast) procedure has been developed to detect weak but biologically relevant sequence similarities during database searches (Altschul et al., 1997).
The basic method and its application to viral membrane-fusion proteins are first briefly outlined, and then described below in more detail. Further description of the method and its cross-validation testing can be found elsewhere (Berger & Singh, 1997). The LearnCoil program iteratively scans the 266 test sequences of putative viral membrane-fusion proteins. In each iteration, the algorithm scores all the test sequences (if the sequence was identified in the previous iteration, its effects are removed before scoring) and converts each score into a likelihood as in the pairwise correlation method PairCoil (Berger et al., 1995). Using these likelihoods, a subset of the sequences are chosen to build a database of potential coiled-coil-like regions. At the end of the iteration, these selected sequences are used to update the parameters to the scoring procedure. This iterative procedure repeats until the number of residues in each subsequent database changes by less than 3 %. In the final iteration, regions that have likelihood of at least 0.5 are selected for the final database. Since the procedure is randomized, the algorithm was run five times on the iteration test sequences. This gave five learned probability tables, which were averaged and then used along with the PairCoil scoring method to build the LearnCoil-VMF program. LearnCoil-VMF was thus designed as a specialized program for identifying coiled-coil-like regions in viral membrane-fusion proteins. As in each iteration of the LearnCoil algorithm, for each viral membrane-fusion protein, its reported LearnCoil-VMF likelihood is computed after removing the effects of the sequence from the averaged probability table.
Scoring
The LearnCoil program uses the scoring method of PairCoil (Berger et al., 1995) as a subroutine. It uses probability estimates (see below) of each residue pair existing in each pair of heptad repeat positions. To obtain normalized probabilities, each probability for a given pair of heptad repeat positions distance i apart is divided by the corresponding distance-i probability for sequences in Genpept. Normalized singles probabilities are computed similarly. A residue propensity in a given heptad repeat position incorporates correlations between that residue and the residues that follow at distances i = 1, i = 2 and i = 4 (chosen empirically). For normalized probabilities P, the propensity of kth residue is given by:13lnP(k,k+1)P(k,k+2)P(k,k+4)P(k+1)P(k+2)P(k+4) Windows of length 30 are considered, and a window score for a particular heptad-repeat position is the sum of the residue propensities in the window. Finally, the residue score for the kth residue is the maximum window score over all windows containing it in all seven possible heptad repeat positions, and each sequence score is the maximum score over all its residues. Each score is converted into a likelihood in the interval [0,1] using the methods described by Berger et al 1995, Berger and Singh 1997. Mathematical justification of the scoring subroutine can be found in Berger (1995).
Selection
During each iteration, the LearnCoil algorithm builds a new database of potential coiled-coil regions. At the start of each iteration, this new database contains no residues. Once each sequence in the iteration test set is scored, it is selected for the new database with probability proportional to its likelihood. That is, a number is drawn uniformly at random from the interval [0,1], and if the number drawn is less than or equal to the likelihood of the sequence, then the sequence is selected for the new database. If a sequence is selected, regions in the sequence where the residues have a likelihood of greater than or equal to either the sequence likelihood or 0.5 are included in the new database. At the ends of scoring regions, window effects are handled by requiring that consecutive residue likelihoods do not drop off by more than 0.1.
Estimating probabilities
In the first iteration, probabilities are estimated from a database consisting of dimeric (Berger et al., 1995) and trimeric (Wolf et al., 1997) coiled coils†. At the end of each iteration, estimates of the singles and pairwise probabilities are updated using the new database. The new probabilities are a weighted average of the probabilities computed from the original database and the probabilities computed from the database selected in this iteration. The original database is weighted 0.05, and the selected database is weighted 0.95. These updated probabilities affect the scoring of sequences in the next iteration. In each iteration after the first, if a sequence is included in the database built in the previous iteration, it is removed from the database and the probabilities are adjusted before that sequence is scored.
Other computational methods used for coiled-coil detection are PairCoil (Berger et al., 1995), MultiCoil (Wolf et al., 1997) and COILS (Lupas et al., 1991).‡ Additionally, the PHD program Rost and Sander 1993, Rost and Sander 1994, Rost et al 1994 is used for secondary structure predictions. For all coiled-coil prediction methods, a likelihood of greater than or equal to 0.5 is taken as a prediction of a coiled-coil-like structure.
Acknowledgements
M.S. thanks the Charles A. King Trust and the Medical Foundation, and the Program in Mathematics and Molecular Biology at the Florida State University with funding from the Burroughs Wellcome Fund Interfaces Program. B.A.B. thanks NSF Career Award CCR-9501997. This research was supported by the NIH (GM44162 to P.S.K.). Thanks to Jodi Harris for assistance with the Figure.
Footnotes
Edited by F. E. Cohen
For all Tables, the virus species in each Table are organized using standard virus taxonomy (Murphy et al., 1995). In each Table, representative sequences from the iteration test set are shown. Typically, the regions shown are the only scoring regions found by Learn-Coil-VMF in the sequences, although the actual boundaries of the scoring regions vary. For most retrovirus and paramyxovirus sequences shown, the precursor glycoprotein gene product is presumed to be cleaved at a RX(K/R)R site. (Each retrovirus and paramyxovirus protein is initially synthesized as a precursor that is subsequently cleaved into two subunits. In each case, the subunit following the cleavage site is responsible for membrane fusion.)
Predictions by PHD on the BIV and EIAV subsequences individually also indicate helices, although for BIV, a helix is predicted only in the N-terminal portion of the subsequence. It is possible that shorter helices would not be detected by the LearnCoil-VMF program.
Lentiviruses N-terminal helix: 54 % of a and d positions in Table 1 are β-branched; lentivirus C-terminal helix: 21 % (Table 2); paramyxovirus N-terminal helix: 60 % (Table 4); paramyxovirus C-terminal helix: 33 % (Table 5)
However, the Marburg GP differs substantially from both the Ebola GP and the Rous sarcoma virus envelope protein in the region preceding the fusion peptide. An alignment of the three proteins fails to identify a similarly placed cleavage site in the Marburg virus GP, although RX(K/R)R motifs occur 85 residues N-terminal to the fusion peptide, as well as immediately N-terminal to the putative coiled-coil domain shown in Table 6
Since they are included in the iteration test set, viral membrane-fusion proteins were removed from the initial trimeric coiled-coil database.
The COILS program implemented is as described by Lupas et al. (1991), with 28-long windows. The COILS program with other window sizes or tables may give somewhat different results than those reported here.
References
- Altschul S., Madden T., Schaffer A., Zhang J., Zhang Z., Miller W., Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood T.K., Findlay J.B.C. Design of a discriminating fingerprint for G-protein-coupled receptors. Protein Eng. 1993;6:167–176. doi: 10.1093/protein/6.2.167. [DOI] [PubMed] [Google Scholar]
- Benson D., Boguski B., Lipman D., Ostell J., Ouellette B. GenBank. Nucl. Acids Res. 1998;26:1–7. doi: 10.1093/nar/26.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B. Algorithms for protein structural motif recognition. J. Comput. Biol. 1995;2:125–138. doi: 10.1089/cmb.1995.2.125. [DOI] [PubMed] [Google Scholar]
- Berger B., Singh M. An iterative method for improved protein structural motif recognition. J. Comput. Biol. 1997;4:261–273. doi: 10.1089/cmb.1997.4.261. [DOI] [PubMed] [Google Scholar]
- Berger B., Wilson D.B., Wolf E., Tonchev T., Milla M., Kim P.S. Predicting coiled coils using pairwise residue correlations. Proc. Natl Acad. Sci. USA. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel V., Karlin S. Too many leucine zippers? Nature. 1989;341:574–575. doi: 10.1038/341574a0. [DOI] [PubMed] [Google Scholar]
- Bullough P.A., Hughson F.M., Skehel J.J., Wiley D.C. Structure of influenza hemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Caffrey M., Cai M., Kaufman J., Stahl S., Wingfield P., Covell D., Gronenborn A., Clore G. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr C., Kim P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- Chambers P., Pringle C., Easton A. Heptad repeat regions are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J. Gen. Virol. 1990;71:3075–3080. doi: 10.1099/0022-1317-71-12-3075. [DOI] [PubMed] [Google Scholar]
- Chan D., Kim P.S. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- Chan D., Fass D., Berger J.M., Kim P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- Chan D., Chutkowski C., Kim P.S. Evidence that a prominent cavity in the coiled coil of HIV-1 gp41 is an attractive drug target. Proc. Natl Acad. Sci. USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E., An S., Barton N., Jahn R. SNAP-25, a t-SNARE which binds to both syntaxin and synaptobrevin via domains that may form coiled coils. J. Biol. Chem. 1994;269:27427–27432. [PubMed] [Google Scholar]
- Chen C., Matthews T., McDanal C., Bolognesi D., Greenberg M. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: implication for viral fusion. J. Virol. 1995;69:3771–3777. doi: 10.1128/jvi.69.6.3771-3777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwart E., Moialos G., Gilmore T. Retroviral envelope glycoproteins contain a leucine zipper-like repeat. AIDS Res. Hum. Retroviruses. 1990;6:703–706. doi: 10.1089/aid.1990.6.703. [DOI] [PubMed] [Google Scholar]
- Dodd I., Egan J.B. Improved detection of helix-turn-helix DNA-binding motifs in protein sequences. Nucl. Acids Res. 1990;18:5019–5026. doi: 10.1093/nar/18.17.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass D., Kim P.S. Dissection of a retrovirus envelope protein reveals structural similarity to influenza hemagglutinin. Curr. Biol. 1995;5:1377–1383. doi: 10.1016/s0960-9822(95)00275-2. [DOI] [PubMed] [Google Scholar]
- Fass D., Harrison S., Kim P.S. Retrovirus envelope domain at 1.7 Angstrom resolution. Nature Struct. Biol. 1996;3:465–469. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- Fasshauer D., Bruns D., Shen B., Jahn R., Brünger A. A structural change occurs upon binding of syntaxin to SNAP-25. J. Biol. Chem. 1997;272:4582–4590. doi: 10.1074/jbc.272.7.4582. [DOI] [PubMed] [Google Scholar]
- Fields B., Knipe D., Howley P. Fields Virology. 3rd edit. Lippincott-Raven; Philadelphia: 1996. Editors of. [Google Scholar]
- Gallaher W. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell. 1996;85:477–478. doi: 10.1016/s0092-8674(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Gallaher W., Ball J., Garray R., Griffen M., Montelaro R. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res. Hum. Retroviruses. 1989;5:431–440. doi: 10.1089/aid.1989.5.431. [DOI] [PubMed] [Google Scholar]
- Gribskov M. Translational initiation factors IF-1 and eIF-2α share an RNA-binding motif with prokaryotic ribosomal protein S1 and polynucleotide phosphorylase. Gene. 1992;119:107–111. doi: 10.1016/0378-1119(92)90073-x. [DOI] [PubMed] [Google Scholar]
- Groot R., Luytjes W., Horzinke M., van der Zeijst B., Spaan W., Lenstra J. Evidence for a coiled-coil structure in the spike proteins of coronaviruses. J. Mol. Biol. 1987;196:963–966. doi: 10.1016/0022-2836(87)90422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson P., Roth R., Morisaki H., Jahn R., Heuser J. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- Harbury P.B., Zhang T., Kim P.S., Alber T. A switch between two-, three- and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Hayashi T., McMahon H., Yamasaki S., Binz T., Hata Y., Sudhof T., Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughson F. Enveloped viruses: a common mode of membrane fusion? Curr. Biol. 1997;7:R565–R569. doi: 10.1016/s0960-9822(06)00283-1. [DOI] [PubMed] [Google Scholar]
- Inoue A., Obata K., Akagawa K. Cloning and sequence analysis of cDNA for a neuronal cell membrane antigen, HPC-1. J. Biol. Chem. 1992;267:10613–10619. [PubMed] [Google Scholar]
- Jiang S., Lin K., Neurath A. HIV-1 inhibition by a peptide. Nature. 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- Joshi S., Dutch R., Lamb R. A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology. 1998;248:20–34. doi: 10.1006/viro.1998.9242. [DOI] [PubMed] [Google Scholar]
- Kilby J., Hopkins S., Venetta T., DiMassimo B., Cloud G., Lee J., Alldredge L., Hunter E., Lambert D., Bolognesi D., Matthews T., Johnson M., Nowak M., Shaw G., Saag M. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nature Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- Lambert D., Barney S., Lambert A., Guthrie K., Medinas R., Davis D., Bucy T., Erickson J. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl Acad. Sci. USA. 1996;93:2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C., Altschul S., Boguski M., Liu J., Neuwald A., Wootton J. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Lin R., Scheller R. Structural organization of the synaptic exocytosis core complex. Neuron. 1997;19:1087–1094. doi: 10.1016/s0896-6273(00)80399-2. [DOI] [PubMed] [Google Scholar]
- Lu M., Blacklow S., Kim P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nature Struct. Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- Lupas A., van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Malashkevich V.N., Chan D., Chutkowski C., Kim P.S. Crystal structure of the simian immunodeficiency virus (SIV) gp41 core: conserved helical interactions underlie the broad inhibitory activity of gp41 peptides. Proc. Natl Acad. Sci. USA. 1998;95:9134–9139. doi: 10.1073/pnas.95.16.9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malashkevich V.N., Schneider B.J., McNally M., Millhollen M., Pang J., Kim P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9 Angstrom resolution. Proc. Natl Acad. Sci. USA. 1999;96:2662–2667. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy F., Fauquet C., Bishop D., Ghabrial S., Jarvis A., Martelli G., Mayo M., Summers M. Virus Taxonomy: Classification and Nomenclature of Viruses. Sixth Report of the International Committee on Taxonomy of Viruses. Springer Verlag; Wien and New York: 1995. Editors of. [Google Scholar]
- Nicholson K., Munson M., Miller R., Filip T., Fairman R., Hughson F. Regulation of SNARE complex assembly by an N-terminal domain of the t-SNARE Ssolp. Nature Struct. Biol. 1998;5:793–802. doi: 10.1038/1834. [DOI] [PubMed] [Google Scholar]
- Parry D.A.D. Coiled coils in alpha-helix-containing proteins: analysis of residue types within the heptad repeat and the use of these data in the prediction of coiled-coils in other proteins. Biosci. Rep. 1982;2:1017–1024. doi: 10.1007/BF01122170. [DOI] [PubMed] [Google Scholar]
- Rapaport D., Ovadia M., Shai Y. A synthetic peptide corresponding to a conserved heptad repeat domain is a potent inhibitor of Sendai virus-cell fusion: an emerging similarity with functional domains of other viruses. EMBO J. 1995;14:5524–5531. doi: 10.1002/j.1460-2075.1995.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey F., Heinz F., Mandl C., Kunz C., Harrison S. The envelope glycoprotein from tick-borne encephalitis virus at 2 Angstrom resolution. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- Rost B., Sander C. Prediction of protein structure at better than 70 % accuracy. J. Mol. Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- Rost B., Sander C. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins: Struct. Funct. Genet. 1994;19:55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- Rost B., Sander C., Schneider R. PHD-an automatic mail server for protein secondary structure. CABIOS. 1994;10:53–60. doi: 10.1093/bioinformatics/10.1.53. [DOI] [PubMed] [Google Scholar]
- Singh M., Berger B., Kim P.S., Berger J., Cochran A. Computational learning reveals coiled coil-like motifs in histidine kinase linker domains. Proc. Natl Acad. Sci. USA. 1998;95:2738–2743. doi: 10.1073/pnas.95.6.2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring J., Kato M., Bernfield M. Epimorphin is related to a new class of neuronal and yeast vesicle targetting proteins. Trends Biochem. Sci. 1993;18:124–125. doi: 10.1016/0968-0004(93)90018-i. [DOI] [PubMed] [Google Scholar]
- Sutton R., Fasshauer D., Jahn R., Brünger A. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Angstrom resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Tatusov R., Altschul S., Koonin E. Detection of conserved segments in proteins: Iterative scanning of sequence databases with alignment blocks. Proc. Natl Acad. Sci. USA. 1994;91:12091–12095. doi: 10.1073/pnas.91.25.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T., Zemelman B., McNew J., Westermann B., Gmachl M., Parlati F., Sollner T., Rothman J. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Dessen A., Harrison S., Skehel J., Wiley D. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Calder L., Wharton S., Skehel J., Wiley D. The central structural feature of the membrane fusion protein subunit from the Ebola virus glycoprotein is a long triple-stranded coiled coil. Proc. Natl Acad. Sci. USA. 1998;95:6032–6036. doi: 10.1073/pnas.95.11.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissehorn W., Carfi A., Lee K., Skehel J., Wiley D. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- Wild C., Shugars D., Greenwell T., McDanal C., Matthews T. Peptides corresponding to a predictive α-helical domain of human immunodeficiency virus type 1 are potent inhibitors of virus infection. Proc. Natl Acad. Sci. USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson I., Skehel J., Wiley D. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Ångstrom resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Wolf E., Kim P.S., Berger B. Multicoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 1997;6:1179–1189. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q., Compans R. Peptides corresponding to the heptad repeat sequence of human parainfluenza virus fusion protein are potent inhibitors of virus infection. Virology. 1996;223:103–112. doi: 10.1006/viro.1996.0459. [DOI] [PubMed] [Google Scholar]
Further reading
Note added in proof
Since submission of this manuscript, crystal structures for the cores of the HTLV-1 TM subunit (Kobe et al., 1999) and the SV5 F protein (Baker et al., 1999) have been obtained. These structures are in agreement with the LearnCoil-VMF results described here.
- Baker K., Dutch R., Lamb R., Jardetzky T. Mol. Cell. 1999;3:309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- Kobe B., Center R., Kemp B., Poumbouris P. Proc. Natl Acad. Sci. USA. 1999;96:4319–4324. doi: 10.1073/pnas.96.8.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]