

The structural similarities of SARS-CoV main proteinase with HAV 3c protease and HCV Ns3 protease suggest that the available inhibitors in these proteases could be useful for anti-SARS drug design.
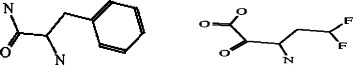
Keywords: SARS-CoV, Main proteinase, Noncanonical interactions, Inhibitor, Binding
Abstract
The main proteinase of SARS-associated coronavirus (SARS-CoV) plays an important role in viral transcription and replication, and is an attractive target for anti-SARS drug development. The important thing is to understand its binding mechanism with possible ligands. In this study, we investigated possible noncanonical interactions, potential inhibitors, and binding pockets in the main proteinase of SARS-CoV based on its recently determined crystal structure. These findings provide a wide clue to searching for anti-SARS drug. Interestingly, we found that similar structure patterns exist in SARS-CoV main proteinase with Poliovirus 3c Proteinase, Rhinovirus 3c Protease, Nsp4 Proteinase From Equine Arteritis Virus, Hepatitis C Virus Ns3 Protease, Hepatitis A Virus 3c Protease, and Dengue Virus Ns3 Protease. It suggests that the available drugs in these viruses could be used to fight SARS disease.
1. Introduction
Since November in 2002, a highly contagious pneumonia, severe acute respiratory syndrome (SARS), has spread rapidly in Asia, North America, and Europe. A new SARS coronavirus (SARS-CoV) has been identified as the etiological agent of the disease. Its seriousness lies in rapid transmission and high fatality (around 15%). However, the origin of SARS-CoV is still unknown, and no effective drug or vaccine is available up to now.
The SARS-CoV replicase encodes two overlapping polyproteins, pp1a and pp1b, which mediate viral replication and transcription. While a special main proteinase, 3C-like proteinase, is responsible for the cleavage of polyproteins, its functional importance make it an attractive target for drug development. Fortunately, the crystal structures of SARS-CoV main proteinase has been determined,[1], [2] which adopt a serine-protease fold and is homologous to the main proteinases from human coronavirus and transmissible gastroenteritis virus.3 The goal of this study is to find potential inhibitors and locate the ligand-binding sites in SARS-CoV main proteinase based on comparison of nonhomologous tertiary structures, hence to provide clues to rational drug design.
2. Materials and methods
The atomic coordinates of SARS-CoV main proteinase were downloaded from Protein Data Bank (ID 1Q2W). NCI program4 was used to identify noncanonical interactions. VAST (http://www.ncbi.nlm.nih.gov/Structure/VAST/vastsearch.html) and DALI (http://www.ebi.ac.uk/dali/) programs were used to find similar structure patterns and in the main proteinase structure of SARS-CoV. The structure alignment was done by CE5 and the structural comparison was performed by LGA.6 The constituents of the binding pocket are determined by those residues that have at least one heavy atom (other than hydrogen) with a distance less than 5 Å from a heavy atom of inhibitor, as did in Chou et al.7 The visualization of 3D structure was generated by PROTEINEXPLORER (http://www.proteinexplorer.org).
3. Results and discussion
The noncanonical interactions in SARS-CoV main proteinase structures are shown in Figure 1 . There are two pairs of main chain-side chain interactions: Glu288 (donor) and Trp 207 (acceptor), Ile152 (donor) and Phe8 (acceptor). There are four pairs of side chain–side chain interactions: Arg40 (donor) and Tyr54 (acceptor), Arg298 (donor) and Phe8 (acceptor), Pro184 (donor) and Phe185 (acceptor), and Tyr126 (donor) and Phe140 (acceptor). These residues are marked as `@' in Figure 2 .
Figure 1.
Noncanonical interactions in the structure of SARS-CoV main proteinase. (A) The residue pairs involved are: Arg298 and Phe 8, Glu288 and Trp207, and Ile152 and Phe8, which are colored blue. (B) The residue pairs involved include: Arg40 and Tyr54, Pro184 and Phe185, and Tyr126 and Phe140, which are colored blue. The Cys-His catalytic dyad (Cys145 and His41) are colored green.
Figure 2.

Structure alignment between SARS-CoV main proteinase (1Q2W) and other proteases: 1L1N (Poliovirus 3c Proteinase), 1CQQ (Rhinovirus 3c Protease), 1MBM (Nsp4 Proteinase From Equine Arteritis Virus), 1DY8 (Hepatitis C Virus Ns3 Protease), 1QA7 (Hepatitis A Virus 3c Protease), and 1DF9 (Dengue Virus Ns3-Protease). The residues marked as `b' indicate similar beta-sheets. The bold residues indicate structurally similar patterns. The residues marked as `@' make noncanonical interactions. The residues marked as `#' make contact with inhibitors.
Among these interactions, Phe8 accepts two N–H⋯π bonds in a sandwich fashion, one donated by a side-side chain Arg298, and one donated by a main-side chian Ile152, as existed in human rac1.8 Taken together with another N–H⋯π interaction between Glu288 and Trp207, these noncanonical bindings connected N-terminus and C-terminus of the enzyme together, then fixed to Ile152 (domain II) and Trp207 (domain III), this makes the domain II and III not flexible due to the loop formed by above interactions in the right and a loop already existed in the left (Fig. 1A), that is stabilizes the structure of the protease. Furthermore, we turn to examine the remaining three pairs of interactions: Arg40 and Tyr54, Pro184 and Phe185, and Tyr126 and Phe140. It can be seen from Figure 1B that these noncanonical bindings will be able to stabilize the protein structure around the active center formed by Cys-His catalytic dyad (Cys145 and His41). On the other hand, after binding, the small helix Arg40 and His41 locate will expect to move up, the loop Phe140 and Cys145 locate and the loop Pro184 and Phe185 locate will expect to move down, hence make the active center more open for substrate binding (Fig. 1B). These results can be used for rational design of mutagenesis experiments and analysis of conservation of interactions at functional sites. In recent years, the noncanonical interactions have been shown to be important for the stability of protein structure[9], [10], [11] and ligand recognition.12
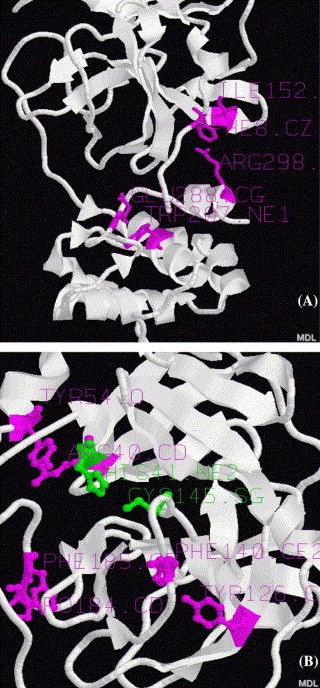
The structurally similar nonhomologous virus proteins identified by VAST and DALI methods focus on 6 proteases: 1CQQ (Rhinovirus 3c Protease With Ag7088 Inhibitor), 1L1N (Poliovirus 3c Proteinase), 1MBM (Nsp4 Proteinase From Equine Arteritis Virus), 1DY8 (Hepatitis C Virus Ns3 Protease with inhibitor FKI), 1QA7 (Hepatitis A Virus 3c Protease with inhibitor NFA) and 1DF9 (Dengue Virus Ns3-Protease Complexed With Mung-Bean Bowman–Birk Inhibitor). The structure-based sequence alignment of the above six proteases with SARS-CoV main protease is shown in Figure 2. The results demonstrate that a number of similar beta sheets (marked by b) exist. In particular, there are three similar structural patterns showing conservative residues (bold representation). The catalytical important residues are included in the first two highly conservative patterns: His-Cys or His-Ser catalytic dyads.[2], [13], [14], [15], [16], [17], [18] In the third pattern, Gly is conservative in 6 proteases except SARS-CoV main proteinase, the significance of mutation from Gly161 to Tyr161 in SARS is unclear. Anyway, the three structural patterns form the common active center of these seven enzymes, as seen in their superpositions (Fig. 3 ).
Figure 3.
The superposition of SARS-CoV main proteinase (1Q2W, white) with other proteases: 1L1N (Poliovirus 3c Proteinase, red), 1CQQ (Rhinovirus 3c Protease, orange), 1MBM (Nsp4 Proteinase From Equine Arteritis Virus, yellow), 1DY8 (Hepatitis C Virus Ns3 Protease, blue), 1QA7 (Hepatitis A Virus 3c Protease, gray), and 1DF9 (Dengue Virus Ns3-Protease, green).
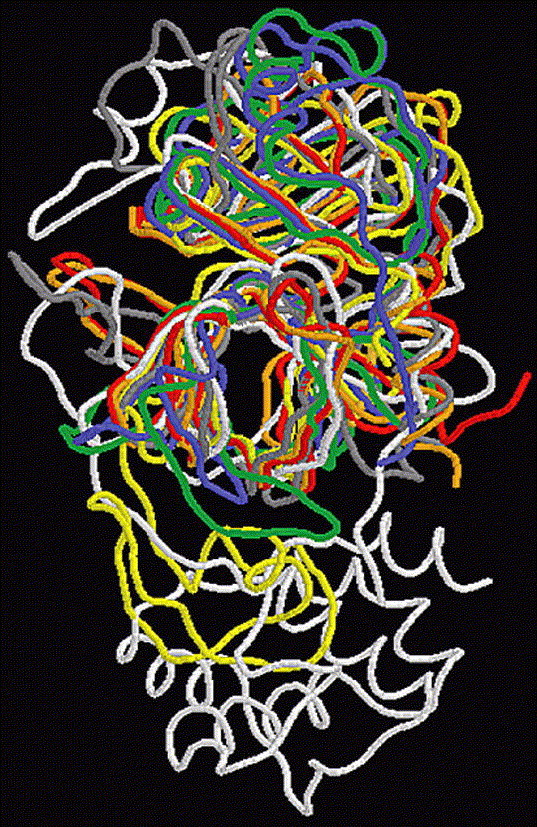
Since these nonhomologous virus proteases share the same active center, we can consider the possibility of applying the inhibitors from these enzymes to SARS-CoV main proteinase. In fact, Anand et al.3 has indicated that the inhibitor AG7088 may be modified to make it useful for SARS therapy. Chou et al.7 conducted the docking studies of KZ7088 (a derivative of AG7088) to the SARS-CoV main proteinase (based on a theoretical model). Here, we studied the possibility of other inhibitors in anti-SARS drug development. Figure 4 shows the structures of docking other inhibitors from HAV, HCV and Dengue virus to SARS-CoV main proteinase, the residues that make contact with these inhibitors are marked as `#' in Figure 2. Furthermore, the corresponding binding pockets are shown in Table 1 . The results reveal that these inhibitors can bind to SARS-CoV main proteinase well.
Figure 4.
The interactions between SARS-CoV main protease (1Q2W, white cartoon) with different inhibitors, binding pockets are represented by blue ball-stick. (A) FKI inhibitor from Hepatitis C Virus Ns3 Protease (yellow spacefill). (B) NFA inhibitor from Hepatitis A Virus 3c Protease (yellow spacefill). (C) Bowman–Birk inhibitor from Dengue Virus Ns3-Protease (yellow stick).
Table 1.
Binding pockets of SARS-CoV main proteinase for different inhibitors
| FKI inhibitor from HCV | NFA inhibitor from HAV | Bowman–Birk inhibitor from Dengue virus | ||
|---|---|---|---|---|
| VAL | 20 MET | 17 GLN | 19 PHE | 140 |
| THR | 25 VAL | 18 VAL | 20 LEU | 141 |
| THR | 26 GLN | 19 THR | 21 ASN | 142 |
| LEU | 27 VAL | 20 CYS | 22 GLY | 143 |
| ASN | 28 THR | 21 GLY | 23 SER | 144 |
| PRO | 39 THR | 24 THR | 24 CYS | 145 |
| ARG | 40 THR | 25 THR | 25 GLY | 146 |
| HIS | 41 THR | 26 HR | 26 SER | 147 |
| VAL | 42 LEU | 27 LEU | 27 TYR | 161 |
| CYS | 44 ASN | 28 ASN | 28 MET | 162 |
| CYS | 117 GLY | 29 PRO | 39 HIS | 163 |
| TYR | 118 HIS | 41 ARG | 40 HIS | 164 |
| ASN | 119 VAL | 42 HIS | 41 MET | 165 |
| SER | 139 GLN | 69 VAL | 42 GLU | 166 |
| PHE | 140 ALA | 116 ILE | 43 LEU | 167 |
| LEU | 141 CYS | 117 CYS | 44 PRO | 168 |
| ASN | 142 TYR | 118 MET | 49 THR | 169 |
| GLY | 143 ASN | 119 LEU | 50 GLY | 170 |
| SER | 144 GLY | 120 ASN | 51 VAL | 171 |
| CYS | 145 SER | 121 PRO | 52 HIS | 172 |
| GLY | 146 PRO | 122 ASN | 53 ALA | 173 |
| SER | 147 LEU | 141 TYR | 54 GLY | 174 |
| TYR | 161 ASN | 142 LEU | 57 PHE | 181 |
| HIS | 163 GLY | 143 VAL | 114 PRO | 184 |
| HIS | 164 SER | 144 CYS | 117 PHE | 185 |
| MET | 165 CYS | 145 TYR | 118 VAL | 186 |
| GLU | 166 GLY | 146 ASN | 119 ASP | 187 |
| LEU | 167 SER | 147 TYR | 126 ARG | 188 |
| VAL | 171 | THR | 135 GLN | 189 |
| HIS | 172 | ILE | 136 THR | 190 |
| ALA | 173 | LYS | 137 ALA | 191 |
| PHE | 181 | GLY | 138 GLN | 192 |
| ASP | 187 | SER | 139 ALA | 193 |
| ALA | 194 | |||
Thus, the results above suggest that there is a link of SARS-CoV to human rhinovirus, Poliovirus, Arteritis virus, HAV, HCV, and Dengue virus. The inhibitors from these viruses could be modified to make them useful for SARS therapy. In fact, Cinatl et al.19 reported that human interferons, a medicine widely used in HCV patients, was useful for the treatment of SARS. Anyway, whether relationships between SARS-CoV and these pathogens exist or not, the present results provide a wide clue to search for anti-SARS inhibitors.
References
- 1.Bonanno, J. B., Fowler, R., Gupta, S., hendle, J., Lorimer, D., Romero, R., Sauder, M., Wei, C. L., Liu, E. T., Burley, S. K., Harris, T. X-ray crystal structure of the SARS coronavirus main protease. (in press) (http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1Q2W)
- 2.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., ye S., Pang H., Gao G., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 4.Babu M.M. NCI: a server to identify non-canonical interactions in protein structures. Nucleic Acids Res. 2003;31:3345–3348. doi: 10.1093/nar/gkg528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shindyalov I.N., Bourne P.E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 6.Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou K., Wei D., Zhong W. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirshberg M., Stockley R.W., Dodson G., Webb M.R. The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nat. Struct. Biol. 1997;4:147–152. doi: 10.1038/nsb0297-147. [DOI] [PubMed] [Google Scholar]
- 9.Fabiola G.F., Krishnaswamy S., Nagarajan V., Pattabhi V. C–H⋯O hydrogen bonds in beta sheets. Acta Crystallogr., Sect. D. 1997;53:316–320. doi: 10.1107/S0907444997000383. [DOI] [PubMed] [Google Scholar]
- 10.Senes A., Ubarretxena-Belandia I., Engelman D.M. The C-H⋯O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babu M.M., Singh S., Balaram P. A C–H⋯O hydrogen bond stabilized polypeptide chain reversal motif at the C terminus of helices in proteins. J. Mol. Biol. 2002;322:871–880. doi: 10.1016/s0022-2836(02)00715-5. [DOI] [PubMed] [Google Scholar]
- 12.Kryger G., Silman I., Sussman J.L. Structure of acetylcholinesterase complexed with E2020: implications for the design of new anti-alzheimer drugs. Structure. 1999;7:297–307. doi: 10.1016/s0969-2126(99)80040-9. [DOI] [PubMed] [Google Scholar]
- 13.Barrette-Ng I.H., Ng K.K., Mark B.L., Van Aken D., Cherney M.M., Garen C., Kolodenko Y., Gorbalenya A.E., Snijder E.J., James M.N. Structure of arterivirus nsp4. The smallest chymotrypsin-like proteinase with an alpha/beta C-terminal extension and alternate conformations of the oxyanion hole. J. Biol. Chem. 2002;277(42):39960–39966. doi: 10.1074/jbc.M206978200. [DOI] [PubMed] [Google Scholar]
- 14.Bergmann E.M., Cherney M.M., Mckendrick J., Frormann S., Luo C., Malcolm B.A., Vederas J.C., James M.N. Crystal structure of an inhibitor complex of the 3C proteinase from hepatitis A virus (HAV) and implications for the polyprotein processing in HAV. Virology. 1999;265(1):153–163. doi: 10.1006/viro.1999.9968. [DOI] [PubMed] [Google Scholar]
- 15.Di Marco S., Rizzi M., Volpari C., Walsh M.A., Narjes F., Colarusso S., De Francesco R., Matassa V.G., Sollazzo M. Inhibition of the hepatitis C virus NS3/4A protease. The crystal structures of two protease-inhibitor complexes. J. Biol. Chem. 2000;275(10):7152–7157. doi: 10.1074/jbc.275.10.7152. [DOI] [PubMed] [Google Scholar]
- 16.Mosimann S.C., Cherney M.M., Sia S., Plotch S., James M.N. Refined X-ray crystallographic structure of the poliovirus 3C gene product. J. Mol. Biol. 1997;273(5):1032–1047. doi: 10.1006/jmbi.1997.1306. [DOI] [PubMed] [Google Scholar]
- 17.Murthy H.M., Judge K., DeLucas L., Padmanabhan R. Crystal structure of Dengue virus NS3 protease in complex with a Bowman–Birk inhibitor: implications for flaviviral polyprotein processing and drug design. J. Mol. Biol. 2000;301(4):759–767. doi: 10.1006/jmbi.2000.3924. [DOI] [PubMed] [Google Scholar]
- 18.Matthews D.A., Dragovich P.S., Webber S.E., Fuhrman S.A., Patick A.K., Zalman L.S., Hendrickson T.F., Love R.A., Prins T.J., Marakovits J.T., Zhou R., Tikhe J., Ford C.E., Meador J.W., Ferre R.A., Brown E.L., Binford S.L., Brothers M.A., DeLisle D.M., Worland S.T. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U.S.A. 1999;96(20):11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cinatl J., Morgenstern B., Chandra P., Rabenau H., Doerr H.W. Treatment of SARS with human interferons. The Lancet. 2003;362:293–294. doi: 10.1016/S0140-6736(03)13973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]