

Abstract
Previous studies have demonstrated that glycopeptide compounds carrying hydrophobic substituents can have favorable pharmacological (i.e. antibacterial and antiviral) properties. We here report on the in vitro anti-influenza virus activity of aglycoristocetin derivatives containing hydrophobic side chain-substituted cyclobutenedione. The lead compound 8e displayed an antivirally effective concentration of 0.4 μM, which was consistent amongst influenza A/H1N1, A/H3N2 and B viruses, and a selectivity index ≥50. Structural analogues derived from aglycovancomycin were found to be inactive. The hydrophobic side chain was shown to be an important determinant of activity. The narrow structure–activity relationship and broad activity against several human influenza viruses suggest a highly conserved interaction site, which is presumably related to the influenza virus entry process. Compound 8e proved to be inactive against several unrelated RNA and DNA viruses, except for varicella-zoster virus, against which a favorable activity was noted.
Keywords: Glycopeptide antibiotic, Ristocetin, Influenza, Antiviral
Currently available drugs for the treatment of influenza virus infections comprise the M2 ion channel blockers amantadine and rimantadine, and the neuraminidase inhibitors oseltamivir and zanamivir (De Clercq, 2006, Moscona, 2008). Stockpiling of oseltamivir and, to a lesser extent, zanamivir has been advocated in the context of pandemic preparedness (Schünemann et al., 2007), yet the recent isolation of oseltamivir-resistant seasonal influenza virus mutants, even from untreated patients, warrants for continued caution (Lackenby et al., 2008, Van der Vries et al., 2008). Additional anti-influenza virus compounds should be urgently developed, having a novel antiviral target that is highly conserved amongst influenza virus (sub)types and, hence, less prone to genetic variation and resistance selection.
One of the attractive therapeutic strategies would be a blockade of the viral entry into the host cell. The cellular entry process of influenza viruses has been unraveled since many years (reviewed in Skehel and Wiley, 2000). A key role is being played by the viral envelope glycoprotein hemagglutinin (HA), which contains the receptor-binding site for initial attachment to the sialylated cellular receptors, and governs the receptor specificity of human versus avian influenza virus subtypes (Chandrasekaran et al., 2008, Nicholls et al., 2008). In addition, after cellular uptake of the virus by endocytosis, the HA mediates the low pH-induced fusion of the viral envelope with the endosomal membrane, leading to release of the viral ribonucleoprotein in the cytosol. Although the HA has been extensively studied from a biochemical and epidemiological perspective, specific antiviral drugs blocking the HA-receptor interaction remain to be clinically developed. Several reports are available on the in vitro activity of small-molecule inhibitors of influenza virus fusion, which act by preventing the conformational change of the HA at low pH (Russell et al., 2008, Deshpande et al., 2001, Plotch et al., 1999). Unfortunately, their development has been slow due to their inferior activity against some human influenza virus (sub)types, rapid selection for resistance and/or unsatisfactory outcome in animal models (Yagi et al., 1999).
Glycopeptide compounds represent a large series of natural, semisynthetic or fully synthetic compounds, which are widely recognized for their potent activity against Gram-positive bacteria (Nicolaou et al., 1999). Several studies have demonstrated that hydrophobic derivatives of glycopeptide antibiotics (e.g. vancomycin and eremomycin), and/or their aglycones exert antibacterial activity against glycopeptide-resistant Enterococci. (Cooper et al., 1996, Printsevskaya et al., 2002, Pace and Yang, 2006). Remarkably, some of these lipophilic glycopeptide compounds were found to have inhibitory activity against coronaviruses and HIV, the latter being ascribed to inhibition of the HIV entry process (Balzarini et al., 2003, Balzarini et al., 2006, Preobrazhenskaya and Olsufyeva, 2006). We here report on the chemical synthesis, anti-influenza virus activity and structure–activity relationship of novel glycopeptide compounds carrying a hydrophobic side chain on an aglycoristocetin backbone (Fig. 1 ).
Fig. 1.
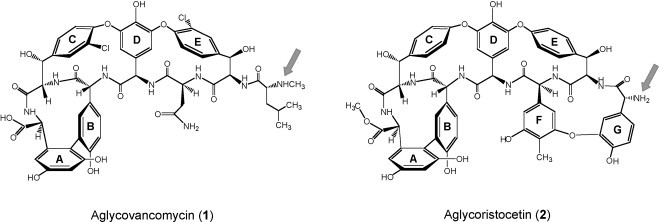
Chemical structures of aglycovancomycin (1) and aglycoristocetin (2). The arrow indicates the secondary amine (aglycovancomycin) and primary amine (aglycoristocetin) functions where the coupling reaction took place.
High-yield synthesis of aglycovancomycin (1) and aglycoristocetin (2) (Fig. 1) was performed as originally described by Wanner et al. (2003) and consisted of deglycosidation of the parent antibiotics with hydrogen fluoride in anisole at neutral pH (Fig. 2 ) (Sztaricskai et al., 2006). The aglycones were converted into the squaric acid amide esters (4 and 5) by coupling with dimethyl squarate (3). This was followed by reaction with primary amines (6a–j) to yield the corresponding asymmetric squaric diamides (7a–g, 8a–j), using a regioselective procedure without the requirement for a protecting group strategy (Tietze et al., 1991, Sztaricskai et al., 2006). The primary amines were: 6-aminohexanol (6a); 6-aminohexanecarboxylic acid (6b); triglycine (6b1); dopamine (6c); N-(4-aminophenyl)piperidine (6d) and 4-phenyl-benzylamine (6e).
Fig. 2.
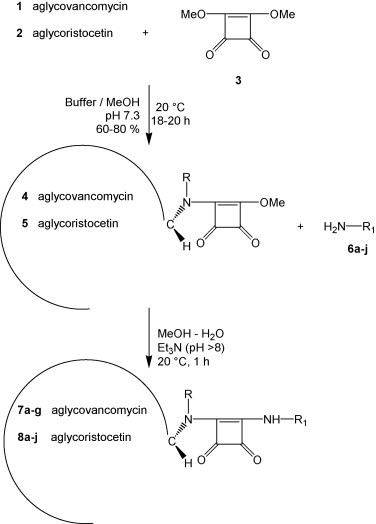
Synthetic pathway of new asymmetric squaric acid diamide compounds derived from aglycovancomycin (R = Me) or aglycoristocetin (R = H). Aglycovancomycin (1) and aglycoristocetin (2) were first converted into their squaric acid amide esters (4 and 5, respectively), followed by conversion to the asymmetric squaric diamides (7a–g and 8a–j, respectively). See Table 1 for the structures of the R1 group, as present in the primary amines 6a–j and the final products 7a–g and 8a–j.
Based on the observation that compound 8e displayed favorable anti-influenza virus activity (see below), subsequent modifications were performed to study the impact of an increasing steric bulk in the rigid aromatic side chain of 7f–g and 8f–h, which were prepared with 1-naphthylamine (6f), 4-aminoterphenyl (6g) or 2-aminoanthracene (6h). It has been shown that introduction of hydrophobic substituents into glycopeptide antibiotics enhances their activity against glycopeptide-resistant bacteria (Cooper et al., 1996, Printsevskaya et al., 2002, Pace and Yang, 2006), but, unfortunately, these products have lower water-solubility. To improve solubility, the squaric acid amide ester 5 was reacted (Sztaricskai et al., 2007b) with d-glucosamine (6i) or d-galactosamine (6j), resulting in the asymmetric squaric diamides 8i and 8j, respectively. In these products, the carbohydrate moiety is linked to the aglycone in an unusual manner, i.e. through a cyclobutandione moiety, and not directly through one of the hydroxyl groups, as in the parent glycopeptide antibiotics.
The structure, reaction yield and physico-chemical data for the aglycones, their squaric acid amide esters and corresponding asymmetric diamides, are summarized in Table 1 . The homogeneity of all compounds was checked by TLC and HPLC, and the structures were confirmed by mass spectrometry.
Table 1.
Chemical structure and physico-chemical properties of the test compounds.a.
| Compound | R | Yield (%) | HPLCbRT | TLCcRf | Formula | Molecular weight |
||
|---|---|---|---|---|---|---|---|---|
| Calculated | Measured MALDI-TOFd (M+Na)+ | |||||||
| Aglycones | ||||||||
| 1 | Me | 90 | 14.68 | (A) 0.62 | C53H52N8O17Cl2 | 1143 | 1165 | |
| 2 | H | 65 | 14.60 | (B) 0.26 | C60H51N7O19 | 1173 | 1196 | |
| Squaric acid amide esters | ||||||||
| 4 | Me |  |
83 | 13.60 | (A) 0.73 | C58H54N8O20Cl2 | 1254 | 1275 |
| 5 | H | 60 | 36.51 | (D) 0.36 | C65H53N7O22 | 1284 | 1306 | |
| Asymmetric squaric diamides | ||||||||
 |
||||||||
| Compound | R | R1 | Yield (%) | HPLCbRT | TLCcRf | Formula | Molecular weight |
|
|---|---|---|---|---|---|---|---|---|
| Calculated | Measured MALDI-TOFd (M+Na)+ | |||||||
| 7a | Me | —(CH2)5–CH2OH | 38 | 12.99 | (B) 0.13 | C63H65N9O20Cl2 | 1139 | 1160 |
| 8a | H | 72 | 13.06 | (B) 0.42 | C70H64N8O22 | 1369 | 1392 | |
| 7b | Me | —(CH2)5–COOH | 34 | 13.02 | (B) 0.12 | C63H63N9O21Cl2 | 1353 | 1374 |
| 8b | H | 38 | 13.03 | (B) 0.24 | C70H62N8O23 | 1383 | 1405 | |
| 8b1 | H | —(Gly)3–COOH | 62 | 10.90 | (A) 0.68 | C70H60N10O25 | 1441 | 1464 |
| 7c | Me | 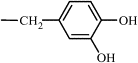 |
68 | 13.28 | (C) 0.29 | C65H61N9O21Cl2 | 1375 (3) | 1396 |
| 8c | H | 87 | ND | (B) 0.43 | C72H60N2O23 | 1405 | 1428 | |
| 8d | H | 73 | 16.68 | (B) 0.59 | C75H65N9O21 | 1428 | 1450 | |
| 7e | Me | 63 | 18.24 | (B) 0.44 | C70H63N9O19Cl2 | 1405 | 1426 | |
| 8e | H | 68 | 18.47 | (B) 0.53 | C77H62N8O21 | 1435 | 1457 | |
| 7f | Me | 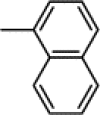 |
58 | 16.70 | (B) 0.28 | C67H59N9O19Cl2 | 1365 | 1386 |
| 8f | H | 54 | 16.75 | (B) 0.73 | C74H58N8O21 | 1395 | 1417 | |
| 7g | Me | 20 | 21.16 | (C) 0.60 | C75H65N9O19Cl2 | 1467 (65) | 1488 | |
| 8g | H | 42 | 21.21 | (C) 0.56 | C82H64N8O21 | 1496 | 1519 | |
| 8h | H | 62 | 19.31 | (B) 0.75 | C78H60N8O21 | 1445 | 1467 | |
| 8i | H | 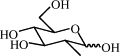 |
75 | 10.07 | (B) 0.34 | C70H62N8O26 | 1431 | 1454 |
| 8j | H | 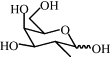 |
69 | 10.05 | (B) 0.35 | C70H62N8O26 | 1431 | 1454 |
After conversion of the aglycons aglycovancomycin (1) and aglycoristocetin (2) into their squaric acid amide esters (4 and 5, respectively), these were converted to the asymmetric squaric diamides (7a–g, derived from aglycovancomycin, and 8a–j, derived from aglycoristocetin).
HPLC conditions: instrument: Waters 600 with UV230nm detection; column: Lichrospher RP-8 (4 mm × 250 mm; 10 μm); injection volume: 20 μl (corresponding to 2 μg compound); solvents: (A) CF3COOH–H2O (pH 2.6) and (B) acetonitrile–H2O; gradient elution from 10 to 90% B; ND: not done.
TLC conditions: silicagel 60F25; solvent systems: (A) nBuOH–Pyr–AcOH–H2O (15:10:3:12); (B) toluene–MeOH–AcOH (1:1:0.01); (C) nBuOH–AcOH–H2O (4:2:2); (D) toluene–MeOH–AcOH (1:1:0.05).
MALDI-TOF MS: instrument: Brucker BIFLEX III. The analytes at a concentration of 5 mg per ml in acetontrile–H2O–0.1% HCOOH (50:50:0.1) were prepared with 2,5-dihydroxybenzoic acid (DHB) matrix (20 mg/ml in DMSO).
The compounds were evaluated for antiviral activity against the following human influenza virus strains: A/Puerto Rico/8/34 (A/H1N1); A/X-31 (A/H3N2) [A/Aichi/2/68 (H3N2) x A/Puerto Rico/8/34 (H1N1)]; A/Hong Kong/7/87 (A/H3N2), and B/Hong Kong/5/72. Virus stocks were inoculated into 10-day-old embryonated hen eggs, followed by harvesting of the allantoic fluid at 48 h post-infection (p.i.) and titration in Madin–Darby canine kidney (MDCK) cells (a kind gift from Dr. M. Matrosovich, Marburg, Germany). For the antiviral assays, MDCK cells were resuspended in infection medium [Ultra-MDCK® medium (Lonza, Basel, Switzerland) supplemented with 2 mM l-glutamine, 0.0225% sodium bicarbonate and 2 μg per mL TPCK (tosylphenylalanylchloromethylketon)-treated trypsin (Sigma, St Louis MO, USA)]. The cells were transferred to 96-well plates at 7500 cells per well and allowed to adhere during 20 h incubation at 35 °C. Then, serial dilutions of the test compounds were added together with virus [multiplicity of infection: 50 CCID50 (cell culture infective dose 50%) per well which corresponds to 0.0003 PFU (plaque-forming units) per cell] and incubated at 35 °C. At 72 h p.i., microscopy was performed to score the virus-induced cytopathic effect (CPE) and compound-induced cytotoxicity. The results were confirmed by a spectrophotometric formazan-based MTS cell viability test (CellTiter 96® AQueous One Solution Cell Proliferation Assay from Promega, Madison, WI, USA). Compound cytostatic activity was determined in uninfected MDCK cells, which were incubated with serial compound dilutions for 72 h, and then subjected to cell counting with a Z1 Coulter Counter® apparatus (Beckman Coulter, Fullerton, CA, USA). The compound concentrations producing 50% antiviral effect (EC50), 50% cytotoxic effect (CC50) or 50% inhibition of cell proliferation (IC50) were calculated by extrapolation, whereas the MCC (minimal cytotoxic concentration) represented the tested compound concentration causing minimal changes in cell morphology.
As shown in Table 2 , several asymmetric squaric diamides derived from aglycoristocetin exerted marked activity against influenza virus, the most potent compounds being the phenylbenzyl derivative 8e [average antiviral EC50: 0.4 μM; selectivity index (SI), defined as the ratio of MCC to EC50: 50]; the hexanol derivative 8a (EC50: 1 μM; SI: 14) and the naphthyl derivative 8f (EC50: 1.4 μM; SI: 10). Their activity was 2- to 5-fold higher than that of the squaric acid amide ester of aglycoristocetin 5 (EC50: 2.4 μM; SI: 42). An intermediate activity (EC50: 5 μM) was observed for the triglycyl derivative 8b1 which, surprisingly, was comparably active as unsubstituted aglycoristocetin 2. The 3,4-dihydroxybenzyl derivative 8c and the d-galactosamine derivative 8j were active against two of the three influenza virus strains tested, and the derivatives containing carboxypentyl (8b) and d-glucosamine (8i) substituents had activity against only one virus strain. The compounds carrying 4-aminophenylpiperidine (8d), terphenyl (8g) and anthracene (8h) substituents were completely inactive. Thus, the intrinsic anti-influenza virus activity of aglycoristocetin is markedly increased by squaric acid amide coupling and addition of a hydrophobic side chain, with the phenylbenzyl group being the optimal substituent. The favorable effect of this side chain appears to depend on different factors, namely: neutral charge (the alcoholic aliphatic derivative 8a is clearly more active than the corresponding carboxypentyl compound 8b) and steric bulkiness (8e and 8f are active while the more bulky compounds 8g and 8h are not).
Table 2.
Antiviral activity in influenza virus-infected MDCKa cells.
| Compound | Antiviral EC50b (μM) |
Cytotoxicityc |
Selectivity indexd | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Influenza A/H3N2 (strain X-31) |
Influenza A/H3N2 (strain A/HK/7/87) |
Influenza B (strain B/HK/5/72) |
MCC (μM) | CC50 (μM) | |||||
| CPE | MTS | CPE | MTS | CPE | MTS | ||||
| 1 | 38 ± 3 | 58 ± 22 | ≥31 | ND | >100 | >100 | >100 | 64 ± 21 | – |
| 2 | 8.4 ± 2.4 | 10.7 ± 2.5 | 4.3 ± 2.9 | 3.2 ± 1.6 | 7.8 ± 2.2 | 6.6 ± 4.4 | 100 | 44 ± 10 | 15 |
| 4 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | ND | – |
| 5 | 3.2 ± 1.2 | nd | 2.0 ± 0.3 | 5.0 ± 3.5 | 1.4 ± 1.3 | 0.5 | 100 | ND | 41 |
| 7a | >100 | >100 | >100 | >100 | >100 | >100 | >100 | ND | – |
| 8a | 1.0 ± 0.9 | 0.6 | 1.1 ± 0.5 | 0.9 | 1.2 | 1.8 | 15 | ND | 14 |
| 7b | >100 | >100 | >100 | >100 | >100 | >100 | >100 | ND | – |
| 8b | >100 | >100 | 5.2 ± 2.0 | 6.4 | >100 | >100 | ≥14 | ND | ≥2 |
| 8b1 | 4.6 ± 3.2 | 10.1 | 2.4 ± 1.4 | 3.4 | 4.2 ± 2.5 | 5.1 ± 4.7 | 20 | ND | 4 |
| 7c | >100 | >100 | >100 | >100 | >100 | >100 | 4 | 11 | – |
| 8c | 2.3 ± 2.4 | 6.6 ± 3.7 | 5.5 ± 5.9 | 2.9 ± 3.3 | >100 | >100 | 20 | 45 ± 33 | 5 |
| 8d | >100 | >100 | >100 | >100 | >100 | >100 | 20 | 80 | – |
| 7e | >100 | >100 | >100 | >100 | >100 | >100 | ≥14 | ND | – |
| 8e | 0.36 ± 0.25 | 0.65 ± 0.58 | 0.32 ± 0.17 | 0.55 ± 0.21 | 0.30 ± 0.16 | 0.31 ± 0.20 | 20 | 47 | 50 |
| 7f | >100 | >100 | >100 | >100 | >100 | >100 | 73 | ND | – |
| 8f | 0.50 ± 0.14 | 0.2 | 1.8 ± 0.9 | 0.9 | 2.9 | 2.4 | 14 | ND | 10 |
| 7g | >100 | >100 | >100 | >100 | >100 | >100 | 0.8 | 0.8 | – |
| 8g | >100 | >100 | >100 | >100 | >100 | >100 | 4 | 10 | – |
| 8h | >100 | >100 | >100 | >100 | >100 | >100 | ≥1 | 16 | – |
| 8i | >100 | >100 | 5.7 ± 2.9 | 6.8 ± 4.6 | >100 | ≥18 | 100 | 48 ± 7 | 16 |
| 8j | >100 | >100 | 4.5 ± 5.2 | 4.6 ± 3.5 | 17 ± 5 | 18 ± 7 | 100 | 51 ± 7 | 9 |
| Ribavirin | 9.0 ± 0.04 | 9.6 ± 2.2 | 8.0 ± 2.2 | 7.6 ± 3.0 | 9.0 ± 0.04 | 7.2 ± 1.9 | ≥100 | >100 | ≥12 |
| Oseltamivir carboxylate | 0.027 ± 0.013 | 0.064 ± 0.078 | 0.26 ± 0.30 | 0.23 ± 0.23 | 11 ± 8 | 5.1 ± 1.8 | >100 | >100 | – |
| Amantadine | 54 ± 33 | 45 ± 21 | 3.9 ± 4.1 | 3.2 ± 4.0 | >100 | >100 | >100 | >100 | – |
Data shown are the mean ± S.D. of 2–7-independent tests; ND: not done.
MDCK: Madin–Darby canine kidney cells.
Antiviral activity was expressed as the EC50 value, defined as the compound concentration producing 50% inhibition of virus replication, as estimated by microscopic scoring of the cytopathic effect (CPE), or by measuring cell viability in the formazan-based MTS assay.
Cytotoxicity was expressed as the minimum cytotoxic concentration (MCC; compound concentration producing minimal changes in cell morphology, as estimated by microscopy), or the 50% cytotoxic concentration (CC50; estimated by the MTS cell viability assay).
Ratio of MCC to average EC50. The ‘–’ symbol means that the compound showed no activity and/or cytotoxicity at the highest concentration tested (100 μM).
The aglycoristocetin backbone structure was shown to be critical for inhibition of influenza virus, since no activity was observed for compound 7e, which represents the aglycovancomycin analogue of 8e. Aglycoristocetin and aglycovancomycin both contain a central heptapeptide core and nonproteinogenic phenolic amino acids, i.e. β-hydroxytyrosine (C and E units), 4-hydroxyphenylglycine (B and D units) and 3,5-dihydroxyphenylglycine (A unit) (Fig. 1). The main structural differences between both glycopeptide aglycones are as follows (Fig. 1): (i) whereas aglycovancomycin has five aromatic rings (A–E) and two known amino acids (l-aspartic acid and N-methyl-d-leucine), aglycoristocetin has seven aromatic moieties (A–G); (ii) the additional F and G rings of aglycoristocetin are interconnected via a diphenylether linkage (constituting the ristomycinic acid moiety); (iii) aglycoristocetin lacks the chloro substituents on the C and E rings, present in aglycovancomycin; (iv) the C-terminal carboxyl function is free in aglycovancomycin, but contains a methyl ester in aglycoristocetin and (v) aglycoristocetin has a primary amine function, whereas aglycovancomycin contains a secondary amine group (Crowley et al., 2004). At present, we cannot speculate on which of these structural components explain the antiviral specificity of the aglycoristocetin derivatives.
Our basic test panel of influenza viruses contained one chimeric virus (A/X-31; H3N2 subtype), one A/H3N2 virus and one B virus strain (Table 2). When the lead compound 8e was further evaluated for activity against A/H1N1 (strain A/Puerto Rico/8/34), its antiviral EC50 value was 0.12 ± 0.04 μM, which is in the same range as that for the A/H3N2 and B strains. Determination of its cytostatic activity in MDCK cells, using a cell counting assay, revealed an IC50 value of 67 ± 19 μM. Thus, 8e emerged as a potent inhibitor of influenza virus replication, with broad activity against different (sub)types and favorable selectivity.
The glycopeptide compounds were evaluated for activity against other viruses besides influenza virus. None of the following RNA viruses was found to be significantly inhibited by any of these glycopeptides, as evaluated by CPE or plaque reduction assay: feline coronavirus [examined in Crandell-Rees feline kidney cells]; vesicular stomatitis virus, Coxsackie B4 virus and respiratory syncytial virus [performed in human epithelial HeLa cells]; parainfluenza-3 virus, reovirus-1, Sindbis virus and Punta Toro virus [tested in African green monkey Vero cells]. In human embryonic lung fibroblast cells infected with various DNA viruses, the only glycopeptide displaying some activity was compound 8e, with antiviral EC50 values of 10 μM (herpes simplex virus type 1; wild-type or thymidine kinase-deficient); 6 μM (herpes simplex virus type 2); >100 μM (cytomegalovirus) and 12 μM (vaccinia virus), and a selectivity index of 10–17. Of note, 8e was found to be highly active against varicella-zoster virus (VZV; wild-type or thymidine kinase-deficient), with an antiviral EC50 value of 0.55 μM and a selectivity index of 180.
The diverse antiviral assays performed in human or animal cell lines permitted to obtain a more detailed insight into the cytotoxicity of the aglycoristocetin derivatives selected from the influenza virus experiments (Table 3 ). All compounds were either not or minimally cytotoxic at a concentration of 70–100 μM. The cytotoxicity values for the lead compound 8e were in the same range as previously obtained in MDCK cells.
Table 3.
Cytotoxicity of selected aglycoristocetin derivatives in human and animal cell lines.a.
| Compound | Cytotoxic concentration |
|||
|---|---|---|---|---|
| MCC (μM)b |
CC50 (μM)b | |||
| HEL | HeLa | Vero | CrFK | |
| 2 | ≥100 | ≥100 | 100 | >100 |
| 5 | 100 | >100 | 100 | ND |
| 8a | >73 | >73 | >73 | >73 |
| 8b | >72 | >72 | >72 | >72 |
| 8b1 | 100 | >100 | >100 | ND |
| 8c | 100 | ≥100 | 100 | >100 |
| 8e | ≥70 | 100 | ≥100 | ≥70 |
| 8f | 72 | >72 | >72 | >72 |
| 8i | ≥100 | >100 | 100 | >100 |
| 8j | ≥100 | >100 | 100 | >100 |
Human embryonic lung (HEL) fibroblasts; human cervix epithelial (HeLa); African green monkey kidney (Vero); and Crandell feline kidney (CrFK) cells; ND: not done.
The cytotoxic concentration was defined as the minimum cytotoxic concentration (MCC) or 50% cytotoxic concentration (CC50); cf. legend to Table 2.
Some glycopeptides in this study were previously evaluated for antibacterial activity (Sztaricskai et al., 2006), with 8e emerging as a highly active compound. Our present anti-influenza virus data thus agree with the view that hydrophobic substitution has a positive impact on the pharmacological (i.e. antibacterial and antiviral) activities of glycopeptide compounds (Printsevskaya et al., 2002, Balzarini et al., 2003, Balzarini et al., 2006).
With regard to the antiviral mode of action, time-of-addition studies suggested that 8e blocks the viral entry process, since optimal anti-influenza virus activity was obtained when the compound was added to MDCK cells 30 min prior to or simultaneously with virus infection. A detailed analysis is currently ongoing to determine the effect of 8e on the virus-receptor interaction (i.e. binding of the viral HA to the sialic acid terminus of cell surface glycans), endocytosis or membrane fusion (i.e. fusion of the viral envelope with the endosomal membrane). Whatever the precise mode of action, the subtype-independent activity of 8e provides strong support that the interaction site of 8e is highly conserved amongst human influenza virus strains. This is consistent with our observation that influenza virus fully retained its sensitivity to 8e after eleven sequential virus passages in MDCK cells in the presence of 8e (at concentrations up to 25 μM). Within human influenza virus HA sequences, only few residues are fully conserved, in particular in the receptor-binding site and fusion peptide (Skehel and Wiley, 2000).
The aglycoristocetin compounds described here and represented by the lead compound 8e are not the first glycopeptides reported to have activity against influenza virus. In 1993, Naruse et al. reported on the isolation, characterization and anti-influenza virus activity of two kistamicin antibiotics (Naruse et al., 1993). Similarly to our compounds, both kistamicins showed strong activity against influenza virus and low activity against herpes simplex virus type 1. The kistamicins were reported to be inactive in HIV syncytium assays (Naruse et al., 1993). The observation that the anti-influenza virus activity of the kistamicins was higher when a lipophilic substituent was present at the terminal amine function, is reminiscent of our findings with the aglycoristocetin derivatives.
In conclusion, the broad and robust anti-influenza virus activity of the aglycoristocetin derivatives described in this study creates a new avenue for the development of anti-influenza virus agents with a novel mode of action. The relatively narrow structure–activity relationship points to a highly specific interaction with the antiviral target, which is probably related to interaction of the influenza virus hemagglutinin with its cellular receptor. Further studies to unravel the structure–activity relationship and precise mode of action are underway in our laboratories.
Acknowledgements
This study was supported by grants from the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO No. 9.0188.07), the International Consortium for Anti-Virals (ICAV) and the Hungarian National Scientific Research Foundation (No. OTKA T 46744). We thank Leentje Persoons, Frieda De Meyer and Vicky Broeckx for dedicated assistance, and Dr. Sándor Kéki (Department of Applied Chemistry, University of Debrecen) for recording the mass spectra. We thank Dr. T. Cihlar (Gilead Sciences, USA) for the generous gift of oseltamivir carboxylate.
Contributor Information
Lieve Naesens, Email: lieve.naesens@rega.kuleuven.be.
Ferenc Sztaricskai, Email: sztarife@delfin.unideb.hu.
References
- Balzarini J., Pannecouque C., De Clercq E., Pavlov A.Y., Printsevskaya S.S., Miroshnikova O.V., Reznikova M.I., Preobrazhenskaya M.N. Antiretroviral activity of semisynthetic derivatives of glycopeptide antibiotics. J. Med. Chem. 2003;46:2755–2764. doi: 10.1021/jm0300882. [DOI] [PubMed] [Google Scholar]
- Balzarini J., Keyaerts E., Vijgen L., Egberink H., De Clercq E., Van Ranst M., Printsevskaya S.S., Olsufyeva E.N., Solovieva S.E., Preobrazhenskaya M.N. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antiviral Res. 2006;72:20–33. doi: 10.1016/j.antiviral.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran A., Srinivasan A., Raman R., Viswanathan K., Raguram S., Tumpey T.M., Sasisekharan V., Sasisekharan R. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat. Biotechnol. 2008;26:107–113. doi: 10.1038/nbt1375. [DOI] [PubMed] [Google Scholar]
- Cooper R.D., Snyder N.J., Zweifel M.J., Staszak M.A., Wilkie S.C., Nicas T.I., Mullen D.L., Butler T.F., Rodriguez M.J., Huff B.E., Thompson R.C. Reductive alkylation of glycopeptide antibiotics: synthesis and antibacterial activity. J. Antibiot. 1996;49:575–581. doi: 10.7164/antibiotics.49.575. [DOI] [PubMed] [Google Scholar]
- Crowley B.M., Mori Y., McComas C.C., Tang D., Boger D.L. Total synthesis of the ristocetin aglycon. J. Am. Chem. Soc. 2004;126:4310–4317. doi: 10.1021/ja039795a. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande M.S., Wei J., Luo G., Cianci C., Danetz S., Torri A., Tiley L., Krystal M., Yu K.L., Huang S., Gao Q., Meanwell N.A. An approach to the identification of potent inhibitors of influenza virus fusion using parallel synthesis methodology. Bioorg. Med. Chem. Lett. 2001;11:2393–2396. doi: 10.1016/s0960-894x(01)00459-0. [DOI] [PubMed] [Google Scholar]
- Lackenby A., Hungnes O., Dudman S.G., Meijer A., Paget W.J., Hay A.J., Zambon M.C. Emergence of resistance to oseltamivir among influenza A (H1N1) viruses in Europe. Eurosurveillance. 2008;13:1–3. doi: 10.2807/ese.13.05.08026-en. [DOI] [PubMed] [Google Scholar]
- Moscona A. Medical management of influenza infection. Annu. Rev. Med. 2008;59:397–413. doi: 10.1146/annurev.med.59.061506.213121. [DOI] [PubMed] [Google Scholar]
- Naruse N., Tenmyo O., Kobaru S., Hatori M., Tomita K., Hamagishi Y., Oki T. New antiviral antibiotics, kistamicins A and B. I. Taxonomy, production, isolation, physico-chemical properties and biological activities. J. Antibiot. 1993;46:1804–1811. doi: 10.7164/antibiotics.46.1804. [DOI] [PubMed] [Google Scholar]
- Nicolaou K.C., Boddy C.N., Bräse S., Winssinger N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. Engl. 1999;38:2096–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Nicholls J.M., Chan R.W., Russell R.J., Air G.M., Peiris J.S. Evolving complexities of influenza virus and its receptors. Trends Microbiol. 2008;16:149–157. doi: 10.1016/j.tim.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Pace J.L., Yang G. Glycopeptides: update on an old successful antibiotic class. Biochem. Pharmacol. 2006;71:968–980. doi: 10.1016/j.bcp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Plotch S.J., O’Hara B., Morin J., Palant O., LaRocque J., Bloom J.D., Lang S.A., DiGrandi M.J., Bradley M., Nilakantan R., Gluzman Y. Inhibition of influenza A virus replication by compounds interfering with the fusogenic function of the viral hemagglutinin. J. Virol. 1999;73:140–151. doi: 10.1128/jvi.73.1.140-151.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preobrazhenskaya M.N., Olsufyeva E.N. Polycyclic peptide and glycopeptide antibiotics and their derivatives as inhibitors of HIV entry. Antiviral Res. 2006;71:227–236. doi: 10.1016/j.antiviral.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printsevskaya S.S., Pavlov A.Y., Olsufyeva E.N., Mirchink E.P., Isakova E.B., Reznikova M.I., Goldman R.C., Branstrom A.A., Baizman E.R., Longley C.B., Sztaricskai F., Batta G., Preobrazhenskaya M.N. Synthesis and mode of action of hydrophobic derivatives of the glycopeptide antibiotic eremomycin and des-(N-methyl-d-leucyl)eremomycin against glycopeptide-sensitive and -resistant bacteria. J. Med. Chem. 2002;45:1340–1347. doi: 10.1021/jm010460i. [DOI] [PubMed] [Google Scholar]
- Russell R.J., Kerry P.S., Stevens D.J., Steinhauer D.A., Martin S.R., Gamblin S.J., Skehel J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 2008;105:17736–17741. doi: 10.1073/pnas.0807142105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schünemann H.J., Hill S.R., Kakad M., Bellamy R., Uyeki T.M., Hayden F.G., Yazdanpanah Y., Beigel J., Chotpitayasunondh T., Del Mar C., Farrar J., Tran T.H., Ozbay B., Sugaya N., Fukuda K., Shindo N., Stockman L., Vist G.E., Croisier A., Nagjdaliyev A., Roth C., Thomson G., Zucker H., Oxman A.D. WHO Rapid Advice Guideline Panel on Avian Influenza WHO rapid advice guidelines for pharmacological management of sporadic human infection with avian influenza A (H5N1) virus. Lancet Infect. Dis. 2007;7:21–31. doi: 10.1016/S1473-3099(06)70684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- Sztaricskai F., Batta G., Herczegh P., Balázs A., Jeko J., Roth E., Szabó P.T., Kardos S., Rozgonyi F., Boda Z. A new series of glycopeptide antibiotics incorporating a squaric acid moiety. Synthesis, structural and antibacterial studies. J. Antibiot. 2006;59:564–582. doi: 10.1038/ja.2006.77. [DOI] [PubMed] [Google Scholar]
- Sztaricskai F., Roth E., Andrei M., Pelyvas I.F., Herczegh P. Application of squaric acid esters in aminodeoxy sugar chemistry. Chem. Lett. 2007;36:1012–1013. [Google Scholar]
- Tietze L.F., Arlt M., Beller M., Glüsenkamp K.-H., Jähde E., Rajewski M.F. Squaric acid diethyl ester: a new coupling reagent for the formation of drug biopolymer conjugates Synthesis of squaric acid ester amides and diamides. Chem. Ber. 1991;124:1215–1221. [Google Scholar]
- Van der Vries E., van den Berg B., Schutten M. Fatal oseltamivir-resistant influenza virus infection. N. Engl. J. Med. 2008;359:1074–1076. doi: 10.1056/NEJMc0803120. [DOI] [PubMed] [Google Scholar]
- Wanner J., Tang D., McComas C.C., Crowley B.M., Jiang W., Moss J., Boger D.L. A new and improved method for deglycosidation of glycopeptide antibiotics exemplified with vancomycin, ristocetin, and ramoplanin. Bioorg. Med. Chem. Lett. 2003;13:1169–1173. doi: 10.1016/s0960-894x(03)00051-9. [DOI] [PubMed] [Google Scholar]
- Yagi S., Ono J., Yoshimoto J., Sugita K., Hattori N., Fujioka T., Fujiwara T., Sugimoto H., Hirano K., Hashimoto N. Development of anti-influenza virus drugs. I. Improvement of oral absorption and in vivo anti-influenza activity of Stachyflin and its derivatives. Pharm. Res. 1999;16:1041–1046. doi: 10.1023/a:1018983715982. [DOI] [PubMed] [Google Scholar]