

Abstract
Thiazolides are polypharmacology agents with at least three mechanisms of action against a broad spectrum of parasites, bacteria and viruses. In respiratory viruses they inhibit the replication of orthomyxoviridae and paramyxoviridae at a post-translational level. Nitazoxanide 1a, the prototype thiazolide, was originally developed as an antiparasitic agent and later repurposed for the treatment of viral respiratory infections. The second generation thiazolides following nitazoxanide, such as the 5-chloro analogue RM-5038 2a, are also broad-spectrum antiviral agents as we have reported. Both 1a and its effective circulating metabolite, tizoxanide 1b, are 5-nitrothiazole derivatives, while RM-5038 2a and its de-acetyl derivative RM-4848 2b are the corresponding 5-chloro derivatives. Recently 1a has completed phase II-III clinical trials in the United States, Canada, Australia and New Zealand in a total of 2865 adults and adolescents of at least 12 months of age with viral acute respiratory illness. Since its biodisposition is primarily seen in the gastro-intestinal tract, its efficacy in systemic viral diseases requires relatively high oral doses. The chemical synthesis of new derivatives with a better systemic absorption was therefore urgently needed. In order to improve their systemic absorption, new amino-ester prodrug derivatives of 1b and RM4848 2b were prepared and tested for their animal pharmacology, pharmacokinetics and toxicology. RM-5061 8a in rats showed 7-fold higher blood concentration compared to 1a: absolute bioavailability increased from 3 to 20%, with a good safety profile in animal safety pharmacology and toxicology.
Keywords: Antiviral agents, Prodrugs, Chemical synthesis, Stability, Biodisposition, Oral absorption, Pharmacology, Toxicology
Graphical abstract

Highlights
-
•
An effective phenolic prodrug for the antiviral agent tizoxanide and a 5-Cl analogue is described.
-
•
These derivatives employ the amino-acid L-tertiary-leucine.
-
•
The stability of this prodrug significantly exceeds that of the Val or Ile analogues.
-
•
Good blood levels are obtainable by oral or IV administration.
-
•
The compounds show a good safety profile.
1. Introduction
Nitazoxanide 1a, the first of thiazolides discovered in 1975 by Rossignol and Cavier is a broad-spectrum antiparasitic agent effective against protozoa, nematodes, cestodes and trematodes [1]. It was registered throughout Latin America, Egypt, India and Bangladesh for the treatment of intestinal protozoa and helminths while in the United States the drug was approved for the treatment of two intestinal protozoa, Cryptosporidium parvum and Giardia intestinalis [2], [3], [4]. The antiprotozoal activity of 1a against anaerobic organisms such as some protozoa and bacteria is due to its interference with the pyruvate ferredoxin oxidoreductase (PFOR) enzyme-dependent electron transfer reaction, which is essential to anaerobic metabolism. Against helminths and Mycobacterium tuberculosis it disrupts membrane potential and homeostasis of intramicro-organisms [5], [6], [7], [8]. Thiazolides also inhibit protein disulfide isomerase (PDI) and possess a broad spectrum of activity against parasites and viruses. In Neospora caninum, an apicomplexan emerging protozoa related to Cryptosporidium parvum, 1a binds to the NcPDI blocking the protozoan replication [9]. In respiratory viruses 1a inhibits the replication of respiratory viruses belonging to the classes of orthomyxoviridae and paramyxoviridae at a post-translational level.[10], [11]. 1a and a number of structurally related thiazolides, e. g. RM5038 2a are also active against both hepatitis B and C viruses at low micromolar concentrations [12], [13], [14]. For example, against a range of respiratory viruses including H1N1 influenza A strains, 1a exhibits IC50 values of 0.3–1.0 μg/mL; against a range of flaviviridae including hepatitis C, from 0.05 to 0.5 μg/mL; against hepatitis B (hepadnaviridae), 0.06 μg/mL.
Moving to in vivo antiviral activity, the antiviral activity of 1a and tizoxanide 1b, its active circulating metabolite, was confirmed in well-controlled clinical trials carried out in more than 3000 patients in the treatment of gastroenteritis caused by rotavirus and norovirus, uncomplicated viral respiratory infections caused by influenza A and B and, alone or combined with pegylated-interferon with and without ribavirin, in the treatment of chronic hepatitis C [15], [16], [17], [18].
1a is only partially absorbed from the gastro-intestinal tract: 14C-1a given to human volunteers showed that 33% of the oral dose was excreted via urine and 64% was excreted in faeces. This is a perfect bio-disposition profile for a drug intended to treat intestinal pathogens, but much less desirable for a systemic antiviral agent. Upon oral absorption it is immediately metabolized into deacetyl derivative 1b, which is subsequently metabolized in the liver as tizoxanide-glucuronide and rapidly eliminated via urine [19], [20]. Ideally, the treatment of viral systemic infections as opposed to parasites or viruses infecting the intestinal tract calls for a compound with a better oral biodisposition and metabolism than 1a but ideally liberating in the blood stream the same active circulating metabolite, 1b and its inactive glucuronide. A considerable amount of safety and efficacy data have been accumulated for these derivatives in the United States and abroad during the last 20 years.
In summary, the thiazolides are typified by nitazoxanide 1a, Scheme 1 , and its active circulating metabolite tizoxanide 1b as shown. Among a large number of analogues synthesised, the chloro analogue RM5038 2a and, similarly, its circulating metabolite RM4848 2b also have very good broad-spectrum antiviral activity [12], [13], [14].
Scheme 1.

Thiazolide structures.
Although the O-acetates such as 1a are satisfactorily taken up by passive absorption, behaving effectively as ester prodrugs, the oral bioavailability of 1a in the absence of food is typically ≤30% [20]. We therefore set out to design a robust prodrug form of 1a/1b, so as to improve both the oral absorption and the solubility properties of the parent drug.
We were impressed by amino-acid based prodrug esters such as the antiviral agent valacyclovir 3, Scheme 2 [21], [22] which improves the oral bioavailability of acyclovir 4 from <20% to 54% and greatly improves its aqueous solubility; valacyclovir enters cells via the h-PEPT 1 transporter [23]. Initially, therefore, we prepared the direct analogue of tizoxanide, namely valyl ester 5 (Scheme 3 ). This derivative was readily prepared as the HCl salt shown [cf. Scheme 4 ], but unfortunately its stability proved inadequate. After 3 weeks' storage at room temperature, hydrolysis of 5 was significant, with about 20% release of the parent drug 1b by NMR evidence.
Scheme 2.

Valacyclovir, as its HCl salt, and acyclovir.
Scheme 3.
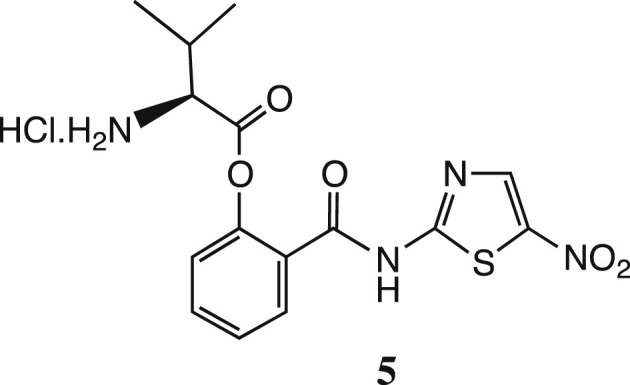
The L-valyl ester prodrug of tizoxanide.
Scheme 4.

Synthesis of L-tert-leucyl thiazolide prodrugs. Reagents: i) Boc-Tle-OH 6, EDC, DMAP, THF or DMF, 65%; ii) HCl-dioxane, 0–20 °C, 95%. Abbreviation: Tle = L-tert-leucine, (2S)- 2-amino-3,3-dimethylbutanoic acid [24].
1.1. Chemistry
We considered that a more stable ester should result on increasing the steric bulk of the amino-acid side chain, and therefore turned next to the corresponding derivative of L-tert-leucine [24], Scheme 4. Reaction of 1b with Boc-Tle-OH [25] 6 using EDC catalysed by DMAP in THF afforded protected ester 7a in satisfactory yield after chromatography. We later found that DMF was a superior solvent, especially for 1b, and performed well on scale-up, delivering a 90% yield of 7a. Deprotection was very conveniently achieved by treatment of 7a with HCl-dioxane, as the HCl salt RM5061 8a of the product could be crystallised directly from the reaction medium and was obtained in excellent yield. Importantly, RM5061 8a showed no appreciable hydrolysis after standing at 20 °C for three months (<1% of 1b seen by NMR analysis and by HPLC); its aqueous solubility was approximately 5 mg/mL [16]. It was of paramount importance to assay the chiral purity of RM5061 8a; for comparison, valacyclovir is typically obtained from acyclovir in about 92% e.e. [26]. From Boc-Tle-OH (S-enantiomer), using DMF as solvent, RM5061 8a was obtained in 99% purity by HPLC and the e.e. was 99.8%. Starting from the corresponding derivative of D-tert-leucine, by chiral HPLC analysis the e. e. of ent- 8a was determined to be 99.5%.
As noted above, the 5′-chlorothiazolide RM4848 2b is another important compound: its corresponding pro-drug was similarly made, Scheme 1. The intermediate 7b was isolated as a crystalline solid and deprotection again proceeded smoothly to afford HCl salt RM5064 8b as a white solid in excellent yield. This product showed very similar stability behaviour to RM5061 8a (<1% hydrolysis after 3 months at 20 °C) and its aqueous solubility was greater, approximately 20 mg/mL.
The stability of valacyclovir has been extensively studied [27]. In order to probe further the issues of stability and chiral purity of products, we also prepared the isoleucine and allo-isoleucine derivatives 9 and 10, Scheme 5 (see supporting information). No scrambling of NMR peaks could be seen in either product at the NMR detection level; here, epimerisation would have generated diastereoisomeric mixtures, possibly to different extents for the two products. As might have been intuitively expected, the stabilities of these compounds by NMR, ca.5% hydrolysis after 3 months at 20 °C, were greater than 5 but less than RM5061 8a/RM5064 8b.
Scheme 5.

Isoleucyl and allo-isoleucyl prodrugs.
1.2. Comparative pharmacokinetics of nitazoxanide 1a, RM-5061 8a, RM-5038 2a and RM-5064 8b in rats
Thiazolides are poorly soluble compounds. In rodents they are inefficiently absorbed from the gastrointestinal tract: most of the compounds given orally remain in the gut. In order to assess the absolute bioavailability of our new amino-acid ester derivatives, RM-5061 8a and RM-5064 8b, in comparison with 1a and RM5038 2a we carried out four parallel studies, each one including six Sprague-Dawley rats weighing about 300 g divided into two groups of three animals. In each of the four studies a group of three rats was treated with a single oral dose of each of the four compounds while the second group of three rats received a single intravenous injection of each of the same four test compounds.
For each of the four compounds the oral dose was calculated to be 30 mg/kg while the intravenous injection was 6 mg/kg, viz. 5 times less. Serial blood samples were obtained from each animal at 5, 10, 15, 30 min and 1, 2, 4, 8, 23, and 24 h post-dose. As noted above [19], tizoxanide glucuronide 11a, Scheme 6 , is the major in vivo metabolite of 1b and significant concentrations of 11a were noted after 5 min upon oral administration of either 1a or RM-5061 8a; this was also the time of maximum plasma concentration.
Scheme 6.
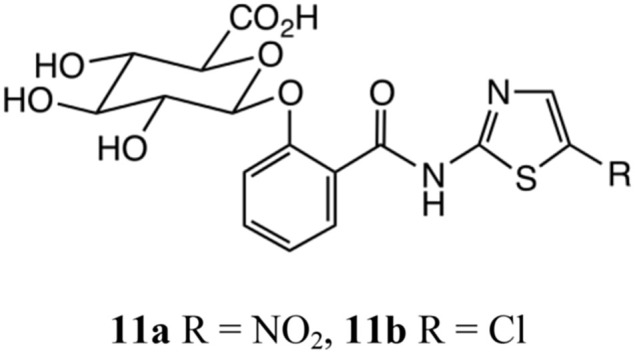
Thiazolide glucuronides.
The comparison of the AUC of 1b calculated from the pharmacokinetics parameters obtained after oral and intravenous administrations of 1a and RM-5061 8a showed a 2.8% absolute bioavailability for 1a, 2.04 versus 70.5 after correction for the dose given to 30 mg/kg, and 20% for RM-5061 8a, 3.12 versus 26.0 after correction for the dose to 30 mg/kg. Interestingly, the chloro-derivative are much less bioavailable: the comparison of the AUCs of RM4848 2b shows essentially no oral absorption for RM5038 2a while the amino-ester derivative, RM5064 8b, shows a 22% absolute bioavailability (0.23 versus 17.4 and 0.71 versus 3.15 for corrected values of the intravenous dose). This again demonstrates a better pharmacokinetic profile for the amino-acid ester derivatives than the corresponding O-acetyl compounds. Here again, the glucuronide metabolite 11b [12] was quickly observed on administration of RM5038 2a or RM 5064 8b and is the main in vivo metabolite of RM4848 2b. The bioavailabilities of the four compounds are summarised in Table 1 . For detailed post oral and intravenous blood levels, see supporting information.
Table 1.
Absolute bioavailabilities F for compounds 1a, 8a, 2a and 8b.
| Compound | Bioavailability F% |
|---|---|
| Nitazoxanide 1a | 2.8 |
| RM5038 2a | ∼0 |
| RM5061 8a | 20 |
| RM5064 8b | 22 |
We therefore decided to proceed with the complete pre-clinical development of RM 5061 8a in order to perform phase 1 human trials. Full pharmacokinetic data for all four compounds 1a, RM 5038 2a, RM 5061 8a and RM 5064 8b are given in supporting information. Additionally, the new derivatives are active in vitro antivirals in their own right, equivalent to the parent thiazolides. Full details will be published separately.
1.3. Safety pharmacology
Two safety pharmacology studies evaluated the effect of RM-5061 8a on the central nervous system and on the respiratory function respectively in the rats.
Single oral doses of 100, 300 and 1000 mg/kg of RM5061 8a were administered to three groups of 10 rats, 5 males and 5 females, by oral gavage: one group of untreated animals was kept as a control. There were no abnormal signs recorded at 4 and 24 h after treatment at the 100 and 300 mg/kg doses but at the 1000 mg/kg dose level there was decreased activity, decreased abdominal tone, labored breathing, tremors and no pain responses observed in all animals in the group suggesting that this dose level was producing CNS toxicity but without mortality of the animals treated at this high dose.
In a second study, single oral doses of 100, 300 and 1000 mg/kg of RM5061 8a were given by oral gavage to three groups of six conscious rats, 3 males and 3 females to study the effects of the test drugs on respiratory function. One group of untreated animals was kept as controls. Some minor changes on the respiratory rate expressed as breaths per minute and the tidal volume were observed for the low 100 mg/kg and the 300 mg/kg oral doses without affecting the Minute Volume dose, but more pronounced effects on the three parameters recorded and described above were observed at the 1000 mg/kg dose suggesting that this level of RM5061 8a has some effects on the respiratory function of the rats. However, no mortality was recorded at the three dose levels tested. The dosing schedules are summarised in Table 2 .
Table 2.
Dosing schedules for safety pharmacology study of RM5061 8a in rats. One untreated group was kept as a control; for full details, see supporting information.
| Indication | Grouping | Dose employed mg/kg | ||
|---|---|---|---|---|
| CNS | 3 groups, 10 rats each (5M/5F) | 100 | 300 | 1000 |
| Respiratory | 3 groups, 6 rats each (3M/3F) | 100 | 300 | 1000 |
1.4. Animal toxicology of RM5061 8a
Two sub-acute toxicity studies of RM 5061 8a were carried out in Sprague-Dawley rats and Beagle dogs. The first study involved dosage of rats at three separate dose levels of 10, 30 and 75 mg/kg of RM 5061 8a once a day for 28 consecutive days, to evaluate systemic exposure to the drug. No terminal adverse effects were noted with the rats. Bright yellow urine was commonly observed, linked to the formulations of RM 5061 8a, which were yellow suspensions. Weight losses were observed, especially in the male group, but these were within acceptable limits. It was concluded that the no observed adverse effect level (NOAEL) was achieved with a dose of 10 mg/kg. Full toxicological details are given in supporting information; the dosing and group numbers are summarised in Table 3 .
Table 3.
Dosing schedules for sub-acute toxicology study of RM5061 8a in rats. The doses shown were administered for 28 consecutive days; for full details, see supporting information.
| Indication | Grouping | Dose employed mg/kg | ||
|---|---|---|---|---|
| Systemic exposure to drug | 4 groups, ≤ 30 rats each (15M/15F) | 10 | 30 | 75 |
The second study evaluated the systemic exposure of RM-5061 8a when administered orally via gelatin capsule at three separate dose levels of 5, 15 and 25 mg/kg once a day for 28 consecutive days to Beagle dogs. Again, there were no early deaths with the dogs during the study period. Bright yellow urine was commonly observed and there were cases of emesis, with yellow particulate material in a few cases. However, emesis was not observed in any control animal. In both animal studies, exposure to 1b and 11a was apparent. Based on the overall study data, the high dose level of 25 mg/kg/day of RM 5061 8a administered in a single gelatin capsule for 28 consecutive days to Beagle dogs was considered a no observed adverse effect level (NOAEL). Full details are given in supporting information: the dosing and group numbers are summarised in Table 4 .
Table 4.
Dosing schedules for sub-acute toxicology study of RM5061 8a in beagle dogs. The doses shown were administered for 28 consecutive days; for full details, see supporting information.
| Indication | Grouping | Dose employed mg/kg | |||
|---|---|---|---|---|---|
| Systemic exposure to drug | 4 groups, ≤10 dogs each (5M/5F) | 0 | 10 | 30 | 75 |
2. Conclusions
Nitazoxanide 1a was originally designed as a broad-spectrum antiparasitic drug for the treatment of intestinal protozoan and helminthic infections, for which the drug has been marketed around the world for more than 15 years. It was recently re-purposed as a broad-spectrum antiviral agent in the treatment of viral acute respiratory infections. Phase III clinical trials carried out in 2865 adults and adolescents with uncomplicated influenza A and B showed that the drug reduced the duration of the influenza illness when compared to placebo with a p value < 0.05. Additionally the studies showed that nitazoxanide compared to placebo was effective in the treatment of the common cold caused by rhinovirus and coronavirus. Further studies in patients with viral respiratory infections at risk of developing complications, children, and adults and children with severe acute respiratory infections (SARI) are currently underway. It was important to identify new derivatives with better systemic absorption, and we have now shown that RM-5061 8a is a second prodrug for tizoxanide 1b. RM5061 8a is more soluble and better absorbed in laboratory animals than 1a and is now undergoing Phase I clinical trials. It may provide an oral dose effective at a lower dosage, and more importantly an injectable form of tizoxanide that nitazoxanide was unable to do.
3. Experimental
3.1. Chemistry
Organic extracts were washed finally with satd. aq. NaCl and dried over anhydrous Na2SO4 prior to rotary evaporation at <30 °C. Analytical thin-layer chromatography was performed using Merck Kieselgel 60 F 254 silica plates. Preparative column chromatography was performed on Merck 938S silica gel. Unless otherwise stated, 1H and 13C NMR spectra were recorded on CDCl3 solutions using either Bruker 250 or 400 MHz (100 MHz for 13C) instruments with tetramethylsilane as internal standard. Both low- and high-resolution mass spectra were obtained by direct injection of sample solutions into a Micromass LCT mass spectrometer operated in the electrospray mode, +ve or -ve ion as indicated. CI mass spectra (NH3) were obtained on a Fisons Instruments Trio 1000. Analytical HPLC was performed using an Ascentia Express C-18 column, eluting with a gradient of 10–100% MeCN aq.+ 0.1% v/v CF3CO2H and monitored at 345 nm. Chiral HPLC was performed using a Chiralpak AD-H column, eluting with n-C7H16: PriOH, 4:1. Microanalytical data were obtained using an Elementar Vario micro cube instrument.
3.1.1. (2S)-[2-[(5-nitro-1,3-thiazol-2-yl)carbamoyl]phenyl]-2-(t-butoxycarbonyl)amino-3,3-dimethylbutanoate (7a)
A mixture of Boc-Tle-OH 2 (0.21 g, 0.97 mmol) and tizoxanide 1b (0.25 g, 0.94 mmol) was stirred at 20 °C in anhydrous THF (7.5 mL). N-ethyl-N′-3-(dimethylamino)propyl carbodiimide. HCl (EDC; 0.19 g, 1 mmol) was added in one portion, followed immediately by 4-dimethylaminopyridine (DMAP; 0.12 g, 1 mmol). After 20 h, the mixture was filtered through Celite and the precipitate washed with further THF, then diluted with ethyl acetate (25 mL). The combined filtrate and washings were washed with 7% aq. citric acid, saturated aq. NaHCO3, water and brine, then dried over anhydrous Na2SO4. Evaporation afforded a yellow foam which was chromatographed on silica gel, being applied in CH2Cl2 and eluted with 1:1 ethyl acetate: hexane. Appropriate fractions were combined and evaporated to afford the title compound 7a as an off-white solid (280 mg, 64%); 1H NMR [400 MHz, (CD3)2SO] δ H1.02 (9 H, s, Me3C), 1.40 (9 H, s, Me3CO), 4.05 (1 H, d, J = 7.6 Hz, CHNH), 7.25 (1H, d, J = 8.0 Hz, ArH), 7.31 (1 H, d, J = 7.6 Hz, CHNH), 7.47 (1 H, t, J = 8.0 Hz, ArH), 7.70 (1 H, t, J = 8.0 Hz, ArH), 7.78 (1 H, d, J = 8.0 Hz, ArH), 8.70 (1 H, s, thiazole 4-H) and 13.67 (1 H, br s, NH); 13C NMR [100 MHz, (CD3)2SO] δ C 26.9, 28.6, 34.0, 63.5, 79.0, 123.4, 126.6, 127.2, 129.9, 133.6, 142.6, 143.0, 148.4, 156.3, 162.4, 165.8 and 170.5; m/z (ES + ve mode) 501 (MNa+, base peak). Found: m/z, 501.1417. C21H26N4O7SNa requires m/z, 501.1420.
We later found that, by using DMF as solvent and 1.5 eq. of both EDC and DMAP, all reagents could be fully dissolved and a conversion of 88% of 7a was obtained after 6 h at 0 °C; the final isolated yield was very similar.
3.1.2. (2S)-[2-[(5-chloro-1,3-thiazol-2-yl)carbamoyl]phenyl]-2-(t-butoxycarbonyl)amino-3,3-dimethylbutanoate (7b)
This compound was prepared similarly to 7a; from 2b (0.51 g, 2 mmol) was obtained 7b (0.62 g, 67%) as a solid which could be crystallised from EtOAc-hexane. Found: C, 53.8; H, 5.5; N, 9.1: S, 6.5. C21H26ClN3O3S requires C, 53.9; H, 5.6; N, 9.0: S, 6.85%; 1H NMR (400 MHz, CDCl3) δ H1.10 (9 H, s, Me3CC), 1.43 (9 H, s, Me3CO), 4.30 (1 H, d, J = 7.6 Hz, CHNH), 5.28 (1 H, br d, J = 7.6 Hz, CHNH), 6.82 (1 H, s, thiazole 4-H), 7.35–7.45 (2 H, m, 2xArH), 7.62 (1 H, t, J = 8.0 Hz, ArH), 7.89 (1 H, d, J = 8.0 Hz, ArH) and 11.66 (1 H, br s, NH); 13C NMR (100 MHz, CDCl3) δ C 26.6, 28.2, 34.4, 62.7, 80.3, 120.8, 123.2, 125.9, 126.4, 130.1, 133.1, 134.5, 148.3, 155.7, 156.6, 163.3 and 170.0; m/z (ES + ve mode) 490, 492 (MNa+, base peaks for 35Cl/37Cl). Found: m/z, 490.1166. C21H26 35ClN3O3SNa requires m/z, 490.1179.
3.1.3. (S)-[2-[(5-nitro-1,3-thiazol-2-yl)carbamoyl]phenyl]-2-amino-3,3-dimethylbutanoate, hydrochloride RM-5061 (8a)
The preceding Boc derivative 7a (0.254 g, 0.53 mmol) was suspended in CH2Cl2 (5 mL) and 4 M HCl in dioxane (2 mL) was added with stirring at 20 °C. A solution resulted after a few minutes, but solid soon began to precipitate. After 16 h, the reaction was diluted with ether, briefly stirred, then cooled to 0 °C to complete precipitation; filtration afforded the title compound RM5061 8a (0.205 g, 93%); 1H NMR [400 MHz, (CD3)SO] δ H 1.10 (9 H, s, Me3C), 4.00 (1 H, br s, CHNH3 +), 7.54 (1 H, d, J = 8.0 Hz, ArH), 7.62 (1 H, t, J = 8.0 Hz, ArH), 7.75 (1 H, t, J = 8.0 Hz, ArH), 7.85 (1 H, d, J = 8.0 Hz, ArH), 8.73 (1 H, s, thiazole 4-H), 8.86 (3 H, br s, NH3 +) and 13.85 (1 H, br s, NH); 13C NMR [100 MHz, (CD3)SO] δ C 26.6, 33.9, 61.5, 124.0, 126.6, 127.1, 130.0, 133.7, 142.6, 143.0, 147.8, 162.2, 165.8 and 167.5; m/z (ES + ve mode) 379 (base peak, ammonium ion). Found: C, 46.1; H, 4.6; N, 13.6. C16H19N4O5SCl requires C, 46.3; H, 4.6; N, 13.5%; Found: m/z, 379.1060. C16H19N4O5S requires m/z, 379.1076.
The highest ee's were observed when DMF was used in the coupling step. Following deprotection, RM5061 8a was obtained with an HPLC area purity of 99.0% and a chiral purity of 99.8%. The corresponding (R) enantiomer, viz. derived from D-tert-leucine, was similarly made; this material had an HPLC purity of 99.5% and a chiral purity of 99.5%.
3.1.4. (S)-[2-[(5-chloro-1,3-thiazol-2-yl)carbamoyl]phenyl]-2-amino-3,3-dimethylbutanoate, hydrochloride RM-5064 (8b)
This compound was prepared similarly to 8a. From 7b (600 mg, 1.28 mmol) there was obtained HCl salt RM 5064 8b (490 mg, 94%); 1H NMR [400 MHz, (CD3)SO] δ H 1.07 (9 H, s, Me3C), 3.96 (1 H, d, J = 7.6 Hz, CHNH), 7.47 (1 H, t, J = 8.0 Hz, ArH), 7.56 (1 H, d, J = 8.0 Hz, ArH), 7.60 (1 H, s, 4′-H), 7.68 (1 H, t, J = 8.0 Hz, ArH), 7.75 (1 H, d, J = 8.0 Hz, ArH) and 8.82 (3 H, br s, NH3 +); 13C NMR [100 MHz, (CD3)SO] δ C 26.6, 33.8, 61.6, 119.1, 123.8, 127.0, 127.5, 129.7, 133.0, 136.2, 147.7, 156.3, 164.8 and 167.4; m/z (ES + ve mode) 368 (base peak, ammonium ion). Found: m/z, 368.0833. C16H19N3O3S35Cl requires m/z, 370.0806.
For the synthesis and characterization of compounds (5a), (9) and (10) and their Boc precursors, together with details of the pharmacokinetic and toxicological methods employed, see Supporting Information.
Acknowledgements
We are grateful to Romark Laboratories LC for funding this work at the University of Liverpool (AVS) and at Calvert Laboratories (DN, SDS).
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2016.09.080.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Rossignol J.-F., Cavier R. 2-Benzamido-5-Nitrothiazoles. Chem. Abstr. 1975;83:28216n. [Google Scholar]
- 2.Rossignol J.-F., Maisonneuve H. Nitazoxanide in the treatment of Taenia saginata and Hymenolepis nana. Am. J. Trop. Med. Hyg. 1984;33:511–512. doi: 10.4269/ajtmh.1984.33.511. [DOI] [PubMed] [Google Scholar]
- 3.Doumbo O., Rossignol J.-F., Pichard E., Traore H., Dembele M., Diakite M., Traore F., Diallo D. Nitazoxanide in the treatment of cryptosporidial diahorrea and other intestinal parasitic infections associated with acquired immunodeficiency syndrome in tropical Africa. Am. J. Trop. Med. Hyg. 1997;56:637–639. doi: 10.4269/ajtmh.1997.56.637. [DOI] [PubMed] [Google Scholar]
- 4.Rossignol J.-F. Nitazoxanide in the treatment of acquired immune deficiency syndrome-related cryptosporidiosis: results of the United States compassionate programme in 365 patients. Aliment. Pharmacol. Ther. 2006;24:887–894. doi: 10.1111/j.1365-2036.2006.03033.x. [DOI] [PubMed] [Google Scholar]
- 5.Fox L.M., Saravolatz M.D. Nitazoxanide: a new thiazolide antiparasitic agent. Clin. Infect. Dis. 2005;40:1173–1180. doi: 10.1086/428839. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman P.S., Sisson G., Croxen M.A., Welch K., Harman W.D., Cremades N., Morash M.G. Antiparasitic drug nitazoxanide inhibits the pyruvate oxidoreductases of Helicobacter pylori, selected anaerobic bacteria and parasites, and Campylobacter jejuni. Antimicrob. Agents Chemother. 2007;51:868–876. doi: 10.1128/AAC.01159-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atherton R.R.D. 2003. Mechanism of Action of Nitazoxanide and Related Drugs against Helminths. Thesis submitted for the degree of Doctor of Philosophy of the University of London. [Google Scholar]
- 8.De Carvalho L.P.S., Dardy C.M., Rhee K.Y., Nathan C. Nitazoxanide disrupts membrane potential and Intrabacterial homeostasis of Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2011;2:849–854. doi: 10.1021/ml200157f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Müller J., Arunasalam N., Müller N., Hemphill A. Neospora caninum: functional inhibition of protein disulfide isomerase by the broad-spectrum anti-parasitic drug nitazoxanide and other thiazolides. Exp. Parasitol. 2008;118:80–88. doi: 10.1016/j.exppara.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Rossignol J.F., La Frazia S., Chiappa L., Ciucci A., Santoro M.G. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at post-translational level. J. Biol. Chem. 2009;284:29798–29808. doi: 10.1074/jbc.M109.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piacentini S., La Frazia S., Rossignol J.F., Santoro G. Oral Presentation at the XVIII International Symposium on Respiratory Viral Infections. The Macrae Foundation; Lisbon, Portugal: 2016. Antiviral activity of nitazoxanide against paramyxoviridae infection: effect on the fusion (F) protein. March 31- April 2. [Google Scholar]
- 12.Stachulski A.V., Pidathala C., Row E.C., Sharma R., Berry N.G., Iqbal M., Bentley J., Allman S.A., Edwards G., Helm A., Hellier J., Korba B.E., Semple J.E., Rossignol J.-F. Thiazolides as novel antiviral agents. 1. Inhibition of hepatitis B virus replication. J. Med. Chem. 2011;54:4119–4132. doi: 10.1021/jm200153p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stachulski A.V., Pidathala C., Row E.C., Sharma R., Berry N.G., Lawrenson A.S., Moores S.L., Iqbal M., Bentley J., Allman S.A., Edwards G., Helm A., Hellier J., Korba B.E., Semple J.E., Rossignol J.-F. Thiazolides as novel antiviral agents. 2. Inhibition of hepatitis C virus replication. J. Med. Chem. 2011;54:8670–8680. doi: 10.1021/jm201264t. [DOI] [PubMed] [Google Scholar]
- 14.Korba B.E., Montero A.B., Farrar K., Gaye K., Mukerjee S., Ayers M.S., Rossignol J.F. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antivir. Res. 2008;77:56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Rossignol J.F. Nitazoxanide, a first-in-class broad-spectrum antiviral agent. Antivir. Res. 2014;110:94–103. doi: 10.1016/j.antiviral.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossignol J.-F., Abu-Zekry M., Hussein A., Santoro M.G. Effect of nitazoxanide for treatment of severe rotavirus diarrhoea: randomized double-blind placebo-controlled trial. Lancet. 2006;368:124–129. doi: 10.1016/S0140-6736(06)68852-1. [DOI] [PubMed] [Google Scholar]
- 17.Haffizula J., Hartman A., Hoppers M., Resnick H., Samudrala S., Ginocchio C., Bardin M., Rossignol J.-F. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: a double-blind, randomized, placebo-controlled, phase 2b/3 trial. Lancet Infect. Dis. 2014;14:609–618. doi: 10.1016/S1473-3099(14)70717-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rossignol J.F., Elfert A., El-Gohary Y., Keefe E.B. Improved virologic response in chronic hepatitis C genotype 4. Patients given nitazoxanide, peginterferon and ribavirin. Gastroenterology. 2009;136:856–862. doi: 10.1053/j.gastro.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 19.Rossignol J.-F., Stachulski A.V. Syntheses and antibacterial activites of tizoxanide, an N-(Nitrothiazolyl)salicylamide, and its O-aryl glucuronide. J. Chem. Res. (S) 1999:44–45. [Google Scholar]
- 20.Broekhuysen J., Stockis A., Lins R.L., De Graeve J., Rossignol J.F. Nitazoxanide: pharmacokinetics and metabolism in man. Int. J. Clin. Pharmacol. Ther. 2000;38:387–394. doi: 10.5414/cpp38387. [DOI] [PubMed] [Google Scholar]
- 21.Rauutio J., Kumpulainen H., Heimbach T., Oliyai R., Oh D., Järvinen T., Savolainen J. Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 22.Beauchamp L.M., Orr G.F., Demiranda P., Burnette T., Krenitsky T., A. Amino-acid ester prodrugs of acyclovir. Antivir. Chem. Chemother. 1992;3:157–164. [Google Scholar]
- 23.Guo A., Hu P., Balimane P.V., Leibach F.H., Sinko P.J. Interactions of a nonpeptidic drug, valacyclovir, with the human intestinal peptide transporter (hPEPT1) expressed in a mammalian cell line. J. Pharmacol. Exp. Ther. 1999;289:448–454. [PubMed] [Google Scholar]
- 24.Galande A.K., Bramlett K.S., Trent J.O., Burris T.P., Wittliff J.L., Spatola A.F. Potent inhibitors of LXXLL-based protein-protein interactions. Chem. Bio. Chem. 2005;6:991–1998. doi: 10.1002/cbic.200500083. Tertiary leucine has not infrequently been incorporated into peptide sequences. [DOI] [PubMed] [Google Scholar]
- 25.Yu S.-W., Lee H.-Y., Cho B.-H., An K.-M., Ryu J.-S., Lee Y.-H., Kang J.-H. Synthesis and biological evaluation of N-formyl hydroxylamine derivatives as potent peptide deformylase inhibitors. Bull. Korean Chem. Soc. 2006;27:1075–1078. [Google Scholar]
- 26.Raju V.V.N.K.V.P., Vedantham R., Khunt M.D., Mathad V.T., Dubey P.K., Chakravarthy A.K. An efficient and large scale process for synthesis of valacyclovir. Asian J. Chem. 2010;22:4092–4098. [Google Scholar]
- 27.Granero G.E., Amidon G.L. Stability of valacyclovir: implications for its oral bioavailability. Int. J. Pharm. 2006;317:14–18. doi: 10.1016/j.ijpharm.2006.01.050. The stability of valacyclovir has been studied over a wide pH range. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.