

Abstract
Severe acute respiratory syndrome (SARS) has become a global public health emergency. p38 mitogen‐activated protein kinase (MAPK) and its downstream targets are activated in SARS coronavirus (SARS‐CoV)‐infected Vero E6 cells and activation of p38 MAPK enhances the cytopathic effects of SARS‐CoV infection. In addition, weak activation of Akt cannot prevent SARS‐CoV infection‐induced apoptosis in Vero E6 cells. In the present study, we demonstrated that signal transducer and activator of transcription (STAT) 3, which is constitutively phosphorylated at tyrosine (Tyr)‐705 and slightly phosphorylated at serine (Ser)‐727 in Vero E6 cells, was dephosphorylated at Tyr‐705 on SARS‐CoV infection. In addition to phosphorylation of p38 MAPK in virus‐infected cells, other MAPKs, i.e., extracellular signal‐regulated kinase (ERK) 1/2 and c‐Jun N‐terminal kinase (JNK), were phosphorylated. Although inhibitors of ERK1/2 and JNK (PD98059 and SP600125) had no effect on phosphorylation status of STAT3, inhibitors of p38 MAPK (SB203580 and SB202190) partially inhibited dephosphorylation of STAT3 at Tyr‐705. Tyr‐705‐phosphorylated STAT3 was localized mainly in the nucleus in mock infected cells, whereas STAT3 disappeared from the nucleus in virus‐infected cells. As STAT3 acts as an activator of transcription in the nucleus, these results suggest that STAT3 lacks its activity on transcription in SARS‐CoV‐infected Vero E6 cells.
Keywords: Severe acute respiratory syndrome coronavirus, Signal transducer and activator of transcription 3, p38, Extracellular signal-regulated kinase 1/2, c-Jun N-terminal kinase
1. Introduction
Severe acute respiratory syndrome (SARS) is a newly found infectious disease caused by a novel coronavirus, SARS coronavirus (SARS‐CoV) [1, 2]. In late 2002, SARS‐CoV spread from Guangdong Province in China to more than 30 countries. The pathogenesis of SARS in vivo may be mediated by both the effect of viral replication in the target cells and immune responses. Recently, we reported that p38 mitogen‐activated protein kinase (MAPK) plays important roles in the cytopathic effects and apoptosis in SARS‐CoV‐infected cells [3]. Furthermore, weak activation of Akt cannot prevent apoptosis by SARS‐CoV‐infection [4]. Thus, it is necessary to examine the signaling pathways in SARS‐CoV‐infected cells in culture to understand the molecular mechanisms of its pathology in vivo.
MAPKs are signal transducers that respond to extracellular stimulation by cytokines, growth factors, viral infection, and stressors, and in turn regulate cell differentiation, proliferation, survival, and apoptosis [5, 6, 7, 8]. p38 MAPK is strongly activated by stressors and inflammatory cytokines. Our previous study indicated that p38 MAPK phosphorylation in SARS‐CoV‐infected Vero E6 cells reached the maximal level at 18 h post‐infection (h.p.i.) and cytopathic effects (CPEs) were observed from 24 h.p.i. [3]. The CPEs were partially prevented by treatment with the p38 MAPK inhibitor SB203580, strongly suggesting that the p38 MAPK signaling pathway is involved in the control of cell death in SARS‐CoV‐infected Vero E6 cells. On the other hand, signal transducer and activator of transcription (STAT) proteins are transcription factors that mediate cytokines and growth factors. Activation of all STAT proteins is induced by phosphorylation of a single tyrosine residue, leading to dimerization via an intermolecular SH2 phosphotyrosine interaction [9, 10, 11, 12]. STAT3 is a major transcription factor activated in response to cytokines, such as interleukin‐6 (IL‐6) and IL‐10. Inhibition of STAT3 signaling by dominant negative and antisense STAT3 inhibitors resulted in a decrease in cell viability and subsequent apoptosis [13, 14, 15]. Thus, STAT3 is thought to act as an anti‐apoptotic transcription factor. Recent studies indicated that interactions between STATs and some viral proteins cause degradation of STATs in virus‐infected cells. For example, V protein of measles virus forms complexes with STAT1, STAT2, and STAT3, and inhibits extracellular (IL‐6) and intracellular (v‐Src) STAT3‐dependent signaling [16]. Thus, measles virus‐induced degradation of STATs may provide a mechanism for virus‐induced cytokine inhibition that links innate immune evasion to adaptive immune suppression.
In the present study, we found that STAT3, constitutively phosphorylated at tyrosine (Tyr)‐705 and slightly phosphorylated at serine (Ser)‐727 in Vero E6 cells, was dephosphorylated at Tyr‐705 by activation of p38 MAPK on SARS‐CoV infection. Lack of transcriptional activity of STAT3 by viral infection may decrease anti‐apoptotic activity in the cells.
2. Materials and methods
2.1. Cells and virus
Vero E6 cells were routinely subcultured in 75‐cm3 flasks in Dulbecco's modified Eagle's medium (DMEM, Sigma, St. Louis, MO, USA) supplemented with 0.2 mM l‐glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 5% (v/v) fetal bovine serum (FBS), and maintained at 37 °C in an atmosphere of 5% CO2. For use in the experiments, the cells were split once onto 6‐ or 24‐well tissue culture plate inserts and cultured until they reached 100% confluence. The culture medium was changed to 2% FBS containing DMEM prior to virus infection. SARS‐CoV, which was isolated as Frankfurt 1 [17] and kindly provided by Dr. J. Ziebuhr, was used in the present study. Infection was usually performed with a multiplicity of infection (m.o.i.) of 10.
2.2. Treatment with inhibitors
SB203580 and SB202190 as p38 MAPK inhibitors, PD098059 as a MEK inhibitor and SP600125 as a JNK inhibitor were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 or 20 mM. All reagents were purchased from Calbiochem (San Diego, CA, USA). All test wells, including mock treated controls, were treated with 0.25% DMSO (v/v). Vero E6 cells were inoculated with SARS‐CoV at m.o.i. of 10 for 1 h and then cells were treated with inhibitors for 17 h.
2.3. Subcellular fractionation
SARS‐CoV‐infected or mock infected Vero E6 cells at 18 h.p.i. were subjected to subcellular fractionation using a Subcellular Proteome Extraction Kit (Calbiochem) according to the manufacturer's instructions. Each subcellular fraction was then analyzed by Western blotting.
2.4. Western blotting
After virus infection, whole‐cell extracts were electrophoresed on either 12.5% or 10–20% gradient polyacrylamide gels and transferred onto PVDF membranes (Immobilon‐P, Millipore, Bedford, MA, USA). In the present study, we applied two sets of samples to polyacrylamide gels and the membranes were divided into two halves after blotting. The following antibodies, obtained from Cell Signaling Technology Inc. (Beverly, MA, USA), were used in the present study at a dilution of 1:1000: rabbit anti‐phospho STAT3 (Tyr‐705) antibody, rabbit anti‐phospho STAT3 (Ser‐727) antibody, rabbit anti‐p38 MAPK (Thr180/Tyr182) antibody, rabbit anti‐p38 MAPK antibody, rabbit anti‐phospho p44/42 MAPK (Thr202/Tyr204) (=ERK1/2) antibody, rabbit anti‐p44/42 MAPK (=ERK1/2) antibody, rabbit anti‐phospho SAPK/JNK (Thr183/Tyr185) antibody, anti‐SAPK/JNK antibody, anti‐phospho MEK1/2 (Ser217/221) antibody, anti‐MEK1/2 antibody, anti‐phospho SEK1/MKK4 (Thr261) antibody, anti‐SEK1/MKK4 antibody, anti‐phospho MKK7 (Ser271/Thr275) antibody, anti‐MKK7 antibody, anti‐phospho MKK3/MKK6 (Ser189/207) antibody, anti‐MKK3 antibody, anti‐phospho Jak1 (Tyr1022/1023) antibody, anti‐phospho JAK2 (Tyr1007/1008) antibody, and anti‐phospho Tyk2 (Tyr1054/1055) antibody. Mouse anti‐STAT3 antibody (diluted 1:2500), mouse anti‐JAK1 antibody (diluted 1:250), and mouse anti‐Tyk2 antibody (diluted 1:1000), obtained from BD Biosciences, Franklin Lakes, NJ, USA, were also used. Rabbit anti‐JAK2 antibody (C‐20, diluted 1:200) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). After 15‐h incubation with the above antibodies, the membranes were washed with Tris‐borate saline containing 0.1% Tween 20 (0.1% TBS–Tween) and the reactions were detected with a ProtoBlot II AP system (Promega Co., Madison, WI, USA), as described previously [18].
3. Results
3.1. Tyrosine dephosphorylation of STAT3 in SARS‐CoV‐infected cells
As described in our previous report, we compared the cellular protein profiles of SARS‐CoV‐infected (18 h.p.i. when apoptosis was not evident) and mock infected Vero E6 cells by Western blotting using 125 antibodies to investigate cellular responses to SARS‐CoV‐infection [3, 4]. Cellular proteins related to several signaling pathways that responded specifically to SARS‐CoV infection, including p38 MAPK and Akt, were found. In the present study, we examined whether signal transducer and activator of transcription 3 (STAT3) was dysregulated on infection with SARS‐CoV, as reported for measles virus‐infection [16]. Vero E6 cells were infected with SARS‐CoV at m.o.i. of 10, and cellular proteins were harvested at 6, 12, 18, and 24 h.p.i. Western blotting analysis was performed using a series of anti‐STAT3 antibodies that recognized total STAT3 or Tyr‐705‐ or Ser‐727‐phosphorylated forms of STAT3. In Vero E6 cells, STAT3 was phosphorylated constitutively at Tyr‐705, whereas Ser‐727 was only slightly phosphorylated (Fig. 1 ; mock infection). Constitutive activation of STAT3 was observed in breast carcinoma cell lines [19]. Interestingly, Tyr‐705‐phosphorylated STAT3 was not detected after 18 h.p.i. in SARS‐CoV‐infected Vero E6 cells, even though the total amount of STAT3 did not change until 24 h.p.i. (Fig. 1). This result suggested that STAT3 Tyr‐705 was dephosphorylated from 18 to 24 h.p.i. On the other hand, Ser‐727‐phosphorylated STAT3 was slightly increased at the same time points. Based on the hypothesis that tyrosine phosphorylation of STAT is necessary for its activation [9, 10, 11, 12], these results suggest that the level of activation of STAT3 was decreased in SARS‐CoV‐infected Vero E6 cells. Thus, the mechanism of dysregulation of STAT3 by SARS‐CoV is different from that by measles virus in which Measles virus V protein forms complexes with STATs resulting in degradation of STAT3.
Figure 1.
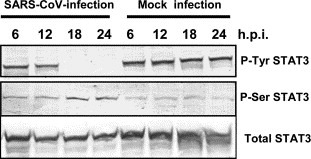
Infection of Vero E6 cells with SARS‐CoV affected the signaling pathway of STAT3. Western blotting analysis of proteins from SARS‐CoV‐infected Vero E6 cells was performed using antibodies that recognized forms of STAT3 phosphorylated at Tyr‐705 or Ser‐727.
3.2. Phosphorylation level of upstream kinases of STAT3
As a general explanation of signal transduction of STAT3, binding of IL‐6 to the receptor induces dimerization of the common gp130 signal transduction subunit of the IL‐6 family of cytokine receptors, and then Janus kinases (JAK1 and 2) and Tyk2 are phosphorylated at tyrosine residues through a conserved membrane‐proximal binding domain [20]. The phosphorylated JAKs and Tyk2 create docking sites for STAT3. Dimeric STAT3 Tyr‐phosphorylated by JAKs and Tyk2 migrates to the nucleus, where STAT3 activates transcription of specific genes. Thus, JAK1, JAK2, Tyk2, and STAT3 are phosphorylated in response to IL‐6. To test the hypothesis that Tyr‐705‐dephosphorylation of STAT3 by SARS‐CoV infection occurred due to a lack of signals from JAK1/2 and Tyk2, the phosphorylation status of these kinases was examined in virus‐infected or mock infected Vero E6 cells. As shown in Fig. 2 , JAK1, JAK2, and Tyk2 were phosphorylated at low levels in mock infected cells, whereas no significant changes in phosphorylation level of these kinases were observed after virus infection. These observations suggest that Tyr‐705 dephosphorylation of STAT3 in virus‐infected cells occurred independent of its upstream kinases. Therefore, total activity of STAT3 may be low in Vero E6 cells. However, it cannot be ruled out that the anti‐phospho JAK1, JAK2 and Tyk2 antibodies used in the present study are difficult to be recognized in the phosphorylated JAK1, JAK2 and Tyk2 in Vero E6 cells as the datasheet included no description of cross‐reactivity with monkey.
Figure 2.

Phosphorylation levels of upstream kinases of STAT3 in virus‐infected cells. Western blotting analyses were performed using anti‐phospho JAK1, JAK2, and Tyk2 antibodies.
3.3. Stimulation of Tyr‐705 phosphorylation of STAT3 by cytokines
Previous studies have indicated that at least six cytokines, IL‐2, IL‐6, IL‐10, IL‐15, IL‐17, and IL‐22, can stimulate activation of STAT3 [21, 22, 23, 24]. Tyr‐705 dephosphorylation of STAT3 in virus‐infected cells may be due to a lack of stimulation by these cytokines after 18 h.p.i. Therefore, it is important to identify the cytokines responsible for stimulating Tyr phosphorylation of STAT3 to understand the mechanism of Tyr‐705‐dephosphorylation of STAT3 in virus‐infected cells. To investigate whether fetal calf serum (FCS) in the medium contains stimulators for STAT3 phosphorylation, Vero E6 cells were cultured in DMEM containing 0%, 2% or 5% FCS for 18 h, subjected to subcellular fractionation, and then the proteins were analyzed by Western blotting using anti‐phospho‐specific antibodies. As shown in Fig. 3A , there were no significant differences in total amount of STAT3 in cultures with various concentrations of FCS in the medium. This result suggests that FCS does not contain components that stimulate Tyr‐phosphorylation of STAT3. We next determined cytokines and their receptors produced in Vero E6 cells using a GEArray Q Series Human Interleukin and Receptor Gene Array (SuperArray Bioscience Corporation, Frederick, MD, USA). Expression level of IL‐22, which has also been reported as a stimulator of STAT3 [23], was very low, and receptors for IL‐22 (IL‐22RA1 and RA2) were obtained as strong and weak signals, respectively (data not shown). Therefore, we examined whether IL‐22 stimulated Tyr‐705 and Ser‐727 phosphorylation of STAT3. As shown in Fig. 4A , the level of Tyr‐705‐phosphorylated STAT3 increased 15 min after treatment with murine IL‐22 (200 ng/ml) (Pepro Tech EC, London, UK), whereas phosphorylation of Ser‐727 was not enhanced, suggesting that it is difficult for Ser‐727 of STAT3 to be phosphorylated in Vero E6 cells stimulated by SARS‐CoV infection and IL‐22. Although the phosphorylation level of p38 MAPK was not changed in SARS‐CoV‐infected Vero E6 cells by treatment with IL‐22 for 20 min at 18 h.p.i., the Tyr‐705‐phosphorylation of STAT3 increased (Fig. 4B). These results suggest that signaling pathway via IL‐22 is not regulated by p38 MAPK in Vero E6 cells.
Figure 3.
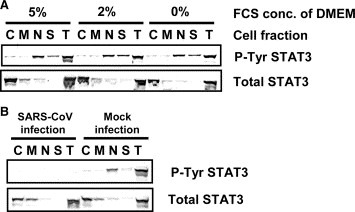
Subcellular localization of Tyr‐phosphorylated STAT3. (A) Vero E6 cells were incubated in DMEM containing 0%, 2% or 5% FCS for 18 h. After subcellular fractionation, Western blotting was performed using anti‐phospho STAT3 (Tyr) antibody. (B) C, M, N, S, and T indicate cytosolic, organelle/membrane, nuclear, cytoskeletal, and total cellular fraction, respectively.
Figure 4.
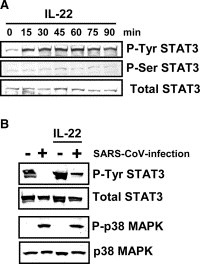
IL‐22 induces Tyr phosphorylation of STAT3. (A) Vero E6 cells were treated with IL‐22 from 0 to 90 min. Cellular proteins were sampled every 15 min. Western blotting was performed using anti‐phospho STAT3 (Tyr and Ser) antibodies. (B) SARS‐CoV‐infected Vero E6 cells were treated with IL‐22 for 20 min at 18 h.p.i., and then, Western blot analysis was performed using anti‐phospho STAT3 (Tyr) and anti‐phospho p38 MAPK.
3.4. Tyr‐705‐phosphorylated STAT3 in the nucleus
To determine the localization of Tyr‐705‐phosphorylated STAT3 in Vero E6 cells, subcellular extraction was performed using a Subcellular Proteome Extraction Kit (Calbiochem) and then Western blotting analyses were performed. As described above, the amounts of total and Tyr‐705‐phosphorylated STAT3 were similar in cells grown in media containing 0%, 2% or 5% FCS. The subcellular localization of STAT3 was not affected by the concentration of FCS (Fig. 3A). Total STAT3 was located mainly in the cytosol, and also in membranes/organelles and the nuclear fractions. On the other hand, Tyr‐705‐phosphorylated STAT3 appeared mainly in the nuclear fraction. We next examined whether Tyr‐705‐phosphorylated STAT3 was not present in the nucleus in SARS‐CoV‐infected Vero E6 cells at 18 h.p.i. As shown in Fig. 3B, Tyr‐705‐phosphorylated STAT3 had clearly disappeared from the nuclear fraction in virus‐infected cells. This result strongly suggested that STAT3 did not act as a transcriptional enhancer in SARS‐CoV‐infected Vero E6 cells after 18 h.p.i.
3.5. Phosphorylation of MAPKs in SARS‐CoV‐infected cells
Our previous report indicated that SARS‐CoV‐infection to Vero E6 induced phosphorylation of p38 MAPK and its downstream targets, HSP‐27, eIF4E and CREB [3]. Phosphorylation of these proteins was prevented by treatment of the cells with the p38 MAPK inhibitor SB20854. MAPKs were reported to induce Ser‐727 phosphorylation of STAT3 [25]. To investigate whether other MAPKs, i.e., ERK1/2 (extracellular signal‐regulated kinase) and JNK, are also phosphorylated in SARS‐CoV‐infected Vero E6 cells, the kinetics of phosphorylation of ERK1/2 and JNK were analyzed by Western blotting. Vero E6 cells were infected with SARS‐CoV at m.o.i. of 10 and the cell extracts were prepared at various time points after infection. Western blotting analysis demonstrated that levels of phosphorylated ERK1/2 and JNK were increased in SARS‐CoV‐infected cells (Fig. 5A ). Two phosphorylated forms of ERK, ERK1 and ERK2, were detected at 12 h.p.i. and accumulated continuously up to 24 h.p.i. The kinetics of accumulation of phosphorylated ERK1/2 and JNK in the infected cells were similar to that of accumulation of phosphorylated p38 MAPK. We next examined whether the upstream MAPK kinases (MAPKKs) were also phosphorylated in SARS‐CoV‐infected Vero E6 cells. The kinases of p38, ERK1/2, and JNK are known as MKK3/6, MEK1/2, and MKK4/7, respectively. As shown in Fig. 5, MKK3/6, MEK1/2, and MKK4/7 were phosphorylated in SARS‐CoV‐infected Vero E6 cells.
Figure 5.
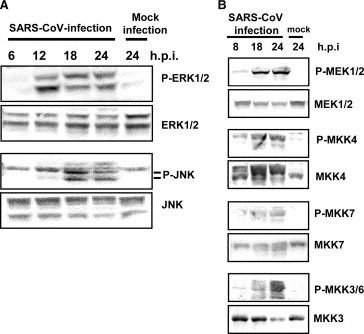
MAPKs phosphorylation in virus‐infected cells. Western blotting analysis of proteins from SARS‐CoV‐infected Vero E6 cells was performed using anti‐phospho ERK1/2, JNK, MEK1/2, MKK4, MKK7, and MKK3/6 antibodies.
3.6. Tyr dephosphorylation of STAT3 by p38 MAPK
To investigate whether Tyr‐705 phosphorylation is regulated by activated MAPKs in SARS‐CoV‐infected Vero E6 cells, the infected cells were treated for 18 h with three MAPK inhibitors: SB203580 (p38 MAPK inhibitor), PD98059 (MEK inhibitor), and SP600125 (JNK inhibitor). Tyr‐705‐ and Ser‐727‐phosphorylated STAT3 were then analyzed by Western blotting. As shown in Fig. 6A , SB203580 did not affect Ser‐727 phosphorylation of STAT3, while SARS‐CoV‐induced dephosphorylation of STAT3 Tyr‐705 was partially inhibited by SB203580. On the other hand, neither PD98059 nor SP600125 affected STAT3 phosphorylation. To confirm whether inhibition of p38 prevents dephosphorylation of STAT3 Tyr‐705, virus‐infected cells were treated with another p38 inhibitor, SB202190. As shown in Fig. 6B, Tyr‐705 dephosphorylation of STAT3 in the infected cells was also partially inhibited by SB202190. These results indicated that activated p38 MAPK in SARS‐CoV‐infected Vero E6 cells regulates Tyr‐705 phosphorylation of STAT3, but not that of Ser‐727.
Figure 6.
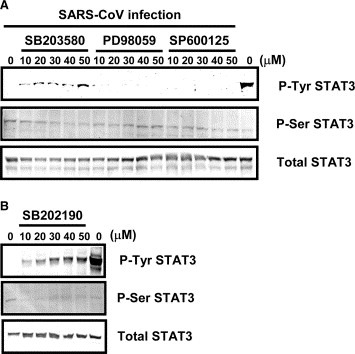
Effects of treatment of SARS‐CoV‐infected Vero E6 cells with p38 MAPK inhibitor. (A) Vero E6 cells were infected with SARS‐CoV at m.o.i. of 10, and then incubated with SB203580, PD98059, and SP600125 at concentrations from 10 to 50 μM for 17 h. Western blotting analyses were performed to detect Tyr‐705‐ and Ser‐727‐phosphorylated forms of STAT3. (B) SB202190 was used as a p38 MAPK inhibitor.
4. Discussion
In the present and previous studies, we reported that the cellular mechanisms by which SARS‐CoV caused the activation of physiological intracellular signaling cascades that lead to the phosphorylation and activation of downstream molecules [3, 4]. We showed here that SARS‐CoV‐infection of permissive Vero E6 cells stimulated p38, ERK1/2, and JNK signaling pathways. The activation of p38 MAPK induces cytopathic effects in Vero E6 cells, whereas ERK1/2 and JNK had no effect (unpublished data). As MAPKKs for p38, ERK1/2, and JNK were all phosphorylated in virus‐infected cells, further studies are needed to identify triggers of the stress‐activated response pathway by viral infection. The present study strongly suggested that the p38 MAPK signaling pathway is upstream of Tyr‐705 dephosphorylation of STAT3. We also demonstrated that infection with SARS‐CoV slightly increased the level of phosphorylation of Ser‐727 STAT3. Although the effect of Ser‐727 phosphorylation on the function of STAT3 in SARS‐CoV‐infected cells remains unresolved, a previous study showed that phosphorylation of Ser‐727 of STAT3 negatively modulates its tyrosine phosphorylation [26]. Thus, the timing of Tyr‐705 dephosphorylation and Ser‐727 phosphorylation may be almost the same in SARS‐CoV‐infected cells. The Ser‐727‐phosphorylated STAT3‐mediated expression of a Bcl‐2 family member, Mcl‐1, is essential for the survival of cells [27], suggesting that Ser‐727‐phosphorylated STAT3 has anti‐apoptotic activity. However, the mechanism through which serine phosphorylation regulates the transcriptional activities of STAT3 is still unclear. On the other hand, one of the important roles of Tyr‐705‐phosphorylated STAT3 is binding to regulatory DNA elements that control the expression of target genes [28, 29]. Suppression of STAT3 expression by siRNA induces apoptosis in several astrocytoma cell lines, and STAT3 is required for the expression of the anti‐apoptotic genes survivin and Bcl‐xL in the A172 glioblastoma cell line [30]. In addition, the role of STAT proteins during viral infection has been the subject of several recent studies. The proteasome‐dependent degradation of STAT1 is induced by V protein of simian virus 5 [31], while type II human parainfluenza virus V protein targets STAT2, and mumps virus V protein targets both STAT1 and STAT3 [32, 33, 34]. In measles virus‐infected cells, the V protein forms complexes with STAT1, STAT2, and STAT3, and inhibits both IL‐6‐ and v‐Src STAT3‐dependent signaling [16]. Thus, a role of V protein as an inhibitor of the STAT3 signaling pathway is advantageous for viral growth. SARS‐CoV may also obtain a growth advantage by Tyr‐705 dephosphorylation of STAT3.
Our recent study indicated that Akt was also activated in response to SARS‐CoV‐replication [4]. Although phosphorylation of serine residue 473 on Akt was detected at least 8 h.p.i., threonine residue 308 was not phosphorylated in virus‐infected Vero E6 cells. A downstream target of Akt, glycogen synthase kinase 3β (GSK‐3β), was slightly phosphorylated, indicating that the level of activation of Akt was very low. The present study showed that IL‐22 can induce Tyr‐705 phosphorylation of STAT3, but not Ser‐727 phosphorylation, similarly to SARS‐CoV infection. It may be difficult for Ser‐727 of STAT3 to be phosphorylated in Vero E6 cells, similarly to Thr‐308 of Akt. Based on these results, we hypothesized that weak activation of Akt cannot prevent apoptosis induced by SARS‐CoV infection in Vero E6 cells. In SARS‐infected Vero E6 cells, both incomplete activation of Akt and STAT3 dephosphorylation via p38 MAPK activation lead to apoptotic cell death. We assume that these are at least part of the mechanisms of the pathogenesis of SARS‐CoV infection.
Acknowledgements
We thank Drs. F. Taguchi (National Institute of Infectious Diseases, Japan) and H. Shima, O. Inanami (Hokkaido University, Japan) for helpful suggestions. We also thank Ms. M. Ogata (National Institute of Infectious Diseases, Japan) for her assistance. This work was supported in part by a grant‐in‐aid from the Ministry of Health, Labor, and Welfare of Japan and the Japan Health Science Foundation, Tokyo, Japan.
Mizutani Tetsuya,Fukushi Shuetsu,Murakami Masaaki,Hirano Toshio,Saijo Masayuki,Kurane Ichiro and Morikawa Shigeru(2004), Tyrosine dephosphorylation of STAT3 in SARS coronavirus-infected Vero E6 cells, FEBS Letters, 577, doi: 10.1016/j.febslet.2004.10.005
References
- 1. Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J., Science, 300, (2003), 1394– 1399. [DOI] [PubMed] [Google Scholar]
- 2. Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L., Science, 300, (2003), 1399– 1404. [DOI] [PubMed] [Google Scholar]
- 3. Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S., Biochem. Biophys. Res. Commun, 319, (2004), 1228– 1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S., Virology, 327, (2004), 169– 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garrington T.P., Johnson G.L., Curr. Opin. Cell. Biol, 11, (1999), 211– 218. [DOI] [PubMed] [Google Scholar]
- 6. Whitmarsh A.J., Davis R.J., Nature, 403, (2003), 255– 256. [DOI] [PubMed] [Google Scholar]
- 7. Chang L., Karin M., Nature, 410, (2001), 37– 40. [DOI] [PubMed] [Google Scholar]
- 8. Kyriakis J.M., Avruch J., Physiol. Rev, 81, (2001), 807– 869. [DOI] [PubMed] [Google Scholar]
- 9. Shuai K., Stark G.R., Kerr I.M., Darnell J.E. Jr., Science, 261, (1993), 1744– 1746. [DOI] [PubMed] [Google Scholar]
- 10. Shuai K., Horvath C.M., Huang L.H., Qureshi S.A., Cowburn D., Darnell J.E. Jr., Cell, 76, (1994), 821– 828. [DOI] [PubMed] [Google Scholar]
- 11. Shuai K., Schindler C., Prezioso V.R., Darnell J.E. Jr., Science, 258, (1992), 1808– 1812. [DOI] [PubMed] [Google Scholar]
- 12. Schindler C., Shuai K., Prezioso V.R., Darnell J.E. Jr., Science, 257, (1992), 809– 813. [DOI] [PubMed] [Google Scholar]
- 13. Rajan P., McKay R.D., J. Neurosci, 18, (1998), 3620– 3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grandis J.R., Drenning S.D., Zeng Q., Watkins S.C., Melhem M.F., Endo S., Johnson D.E., Huang L., He Y., Kim J.D., Proc. Natl. Acad. Sci. USA, 97, (2000), 4227– 4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mora L.B., Buettner R., Seigne J., Diaz J., Ahmad N., Garcia R., Bowman T., Falcone R., Fairclough R., Cantor A., Muro-Cacho C., Livingston S., Karras J., Pow-Sang J., Jove R., Cancer Res, 62, (2002), 6659– 6666. [PubMed] [Google Scholar]
- 16. Palosaari H., Parisien J.-P., Rodriguez J.J., Ulane C.M., Horvath C.M., J. Virol, 77, (2003), 7635– 7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J., J. Gen. Virol, 84, (2003), 2305– 2315. [DOI] [PubMed] [Google Scholar]
- 18. Mizutani T., Kobayashi M., Eshita Y., Shirato K., Kimura T., Ako Y., Miyoshi H., Takasaki T., Kurane T., Kariwa H., Umemura T., Takashima I., Insect. Mol. Biol, 12, (2003), 491– 499. [DOI] [PubMed] [Google Scholar]
- 19. Garcia R., Yu C.-L., Hudnall A., Catlett R., Nelson K.L., Smithgall T., Fujita D.J., Ethier S.P., Jove R., Cell Growth Differ, 8, (1997), 1267– 1276. [PubMed] [Google Scholar]
- 20. Kerr I.M., Costa-Pereira A.P., Lillemeier B.F., Strobl B., FEBS Lett, 546, (2003), 1– 5. [DOI] [PubMed] [Google Scholar]
- 21. Niemand C., Nimmesgern A., Haan S., Fischer P., Schaper F., Rossaint R., Heinrich P.C., Muller-Newen G., J. Immunol, 170, (2003), 3263– 3272. [DOI] [PubMed] [Google Scholar]
- 22. Nielsen M., Nordahl M., Svejgaard A., Odum N., Cytokine, 10, (1998), 735– 738. [DOI] [PubMed] [Google Scholar]
- 23. Subramaniam S.V., Cooper R.S., Adunyah S.E., Biochem. Biophys. Res. Commun, 262, (1999), 14– 19. [DOI] [PubMed] [Google Scholar]
- 24. Lejeune D., Dumoutier L., Constantinescu S., Kruijer W., Schuringa J.J., Renauld J.C., J. Biol. Chem, 277, (2002), 33676– 33682. [DOI] [PubMed] [Google Scholar]
- 25. Haq R., Halupa A., Beattie B.K., Mason J.M., Zanke B.W., Barber D.L., J. Biol. Chem, 277, (2002), 17359– 17366. [DOI] [PubMed] [Google Scholar]
- 26. Chung J., Uchida E., Grammer T.C., Blenis J., Mol. Cell. Biol, 17, (1997), 6508– 6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu H., Ma Y., Cole S.M., Zander C., Chen K.H., Karras J., Pope R.M., Blood, 102, (2003), 344– 352. [DOI] [PubMed] [Google Scholar]
- 28. Wegenka U.M., Buschmann J., Lutticken C., Heinrich P.C., Horn F., Mol. Cell. Biol, 13, (1993), 276– 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yuan J., Wegenka U.M., Lutticken C., Buschmann J., Decker T., Schindler C., Heinrich P.C., Horn F., Mol. Cell. Biol, 14, (1994), 1657– 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Konnikova L., Kotecki M., Kruger M.M., Cochran B.H., BMC Cancer, 3, (2003), 3– 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Didcock L., Young D.F., Goodbourn S., Randall R.E., J. Virol, 73, (1999), 9928– 9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parisien J.-P., Lau J.F., Rodriguez J.J., Sullivan B.M., Moscona A., Parks G.D., Lamb R.A., Horvath C.M, J. Virol, 283, (2001), 230– 239. [DOI] [PubMed] [Google Scholar]
- 33. Nishio M., Garcin D., Simonet V., Kolakofsky D., Virology, 300, (2002), 92– 99. [DOI] [PubMed] [Google Scholar]
- 34. Ulane C.M., Rodriguez J.J., Parisien J.-P., Horvath C.M., J. Virol, 77, (2003), 6385– 6393. [DOI] [PMC free article] [PubMed] [Google Scholar]