

Abstract
Computational analysis of RNA secondary structure is a classical field of biosequence analysis, which has recently gained momentum due to the manyfold regulatory functions of RNA that have become apparent. We present five recent computational approaches that address the problems of synoptic folding space analysis, pseudoknot prediction, structure alignment, comparative structure prediction, and miRNA target prediction. All these programs are in current use and are available via the Bielefeld Bioinformatics Server at http://bibiserv.techfak.uni-bielefeld.de.
Keywords: RNA secondary structure, RNA interference, miRNA target prediction, RNA structure comparison, Consensus structure prediction, RNA pseudoknots
1. Introduction
1.1. Minimum-free-energy RNA folding and its shortcomings
RNA secondary structure analysis has been an important task in molecular biology throughout the recent years. Computational structure prediction based on a thermodynamic model has been around since the first version of the Mfold program (Zuker and Stiegler, 1981) became available. Both the program and the model have been refined several times. In the Vienna RNA package (Hofacker et al., 1994, Hofacker, 2003) RNAfold provides an alternative implementations of the same model, with a wide spectrum of additional features. Based on dynamic programming algorithms, these programs perform a complete evaluation of all feasible structures for a given RNA sequence, and determine the one with minimal free energy. Therefore, this approach is called MFE folding, for short. The widespread use of these programs demonstrates that the research community has learned to live with the shortcomings of MFE folding, which are well known:
-
(1)
The MFE structure is not guaranteed to be the native one. For sequences longer than (say) 400 bases, it will almost surely be wrong. On the other hand, it will also contain some correct structural elements – given just the MFE structure and no additional clues, we do not know which parts of the prediction are good. And also, the native structure is typically a member of the near-optimal folding space, but again, we do not know which one.
-
(2)
For reasons of computational complexity, standard folding programs disregard pseudoknots, which are an important structural feature. Since the pseudoknots are not contained in the folding space evaluated by the programs at all, they cannot be found among the near-optimals either.
-
(3)
Better results can be obtained with a comparative approach, using the fact that structure is conserved more than sequence with homologous RNA molecules that share the same function. When a large number of well-aligned sequences is available, statistical methods yield good results (Doshi et al., 2004). When only a few sequences are given, and when their alignment is difficult, much more sophisticated methods are required.
-
(4)
RNA molecules do not fold in isolation, and contacts with proteins or other RNA molecules may play an important role. Furthermore, RNA–RNA interaction is guided by the same thermodynamics as is the self-interaction that creates the structure of a single sequence, but its correct modeling again requires specialized approaches.
1.2. Computational approaches that go beyond MFE folding
A large number of contributions has been made addressing the above problems. We only list a few for each category, adding in the ones to be presented below in the appropriate place:
-
(1)
The fact that the truth may lie buried in the near-optimal folding space has prompted researchers to augment their folding programs to also compute near-optimal structures. There are basically three approaches: Mfold provides a heuristic sample of near-optimal structures, taking care that the structures produced are not too similar. RNAsubopt (Wuchty et al., 1998) produces a complete account of the near-optimal folding space up to a certain energy threshold. In the first case, we do not know if an “interesting” structure is overlooked in the heuristic example, in the second case, we are confronted with a large number of structures, as the near-optimal folding space grows exponentially with sequence length as well with the allowed energy interval. Sfold (Ding and Lawrence, 2003) samples structures according to their probabilities derived from Boltzmann statistics. A sample size of 1000 structures is usually sufficient for good results. With subsequent clustering of the sampled structures according to structural similarity, a small number of centroids can be returned that can be taken as a representative ensemble of potentially relevant structures (Ding et al., 2005). The MFE structure is not guaranteed to be contained in the sample, but can be easily added by traditional methods. This approach comes close to a complete, non-heuristic and yet useful (because of the compact result) analysis of the folding space. A more direct way to achieve the same goal is chosen by the program RNAshapes (Giegerich et al., 2004b), to be presented below. Here the folding space is subdivided into families of structures that share the same abstract shape, and for each family, a representative structure is determined.
-
(2)
The problem of pseudoknots has been widely studied, as it is also of great theoretical interest. The general case has been proved to be an NP-hard problem (Lyngsø and Pedersen, 2001, Akutsu, 2000), and hence computationally intractable. Some researchers have abandoned the thermodynamic model and resorted to graph algorithms or genetic algorithms (Tabaska et al., 1998, Shapiro and Navetta, 1994). The program pknots by Rivas and Eddy (1999) implements thermodynamic folding of a rather large subclass of pseudoknots in O(n 6) time and O(n 4) space, which implies that it can be used only for very short sequences. The program pknotsRG, to be presented below, restricts the class of pseudoknots further, but still recognizes many pseudoknots arising in practice, and runs in O(n 4) time and O(n 2) space. A filtering approach has recently be presented (Huang et al., 2005) that combines a pre-filter with the folding programs, which sometimes substantially gains in efficiency.
-
(3)
Comparative structure prediction from unaligned sequences must solve the task of simultaneous alignment and thermodynamic folding with a combined objective function to be optimized. As multiple sequence alignment under the common sum-of-pairs score by itself is NP-complete (Wang and Jiang, 1994), one cannot hope to obtain an efficient non-heuristic algorithm. The Sankoff algorithm (Sankoff, 1985), which gives the exact solution to this problem, has hence served as a source of derived approaches, for example, DNYALIGN (Mathews and Turner, 2002) and FOLDALIGN (Gorodkin et al., 1997). These programs must make severe restrictions to the underlying model to achieve practical runtime. Even so, the results are often quite moderate; see (Gardner and Giegerich, 2004) for a recent survey. In this situation, the recent tool RNAforester (Höchsmann et al., 2003, Höchsmann et al., 2004), which allows for a meaningful way of aligning structures directly (without use of a sequence alignment), has opened a new road that circumvents the dilemma of the Sankoff approach. The program RNAcast (Reeder and Giegerich, 2005) efficiently determines the consensus shapes of a set of sequences, and from their representative structures, a multiple alignment can be obtained with RNAforester. Both RNAforester and RNAcast are described below.
-
(4)
When the sites are known where RNA molecules interact with other molecules, this can be incorporated into the folding algorithms. The aforementioned folding programs provide options to specify that a certain base must pair or should not pair. Although such information can be obtained by experimental or comparative methods, most often it is not available. General RNA–RNA interaction has been recently studied by Andronescu et al. (2005). A special case of great current interest is the interaction between a miRNA or a siRNA with its target RNA. As this process is guided by thermodynamics, early approaches have modeled this situation by linking the small RNA to the end of its target, and then folding them jointly with a standard folding program (Stark et al., 2003) As this approach tends to produce artefacts, a proper model of hybridizing a small RNA to a larger target has been developed and implemented in the program RNAhybrid. This program, already widely used for miRNA and siRNA target prediction, is presented below.
2. RNAshapes: comprehensive analysis of representative structures
2.1. Motivation and background
A common problem in RNA secondary structure analysis is the huge number of suboptimal structures. Many of these are similar, whereas only few show differences that are biologically relevant.
The idea of abstract shape analysis (Giegerich et al., 2004b) is to classify structures upon their composition from structural elements, such as helices, multiloops, internal loops and bulge loops. For each shape class one representative structure is reported. This provides a synoptic and non-heuristic overview of the structure space. Furthermore, this partitioning allows to deduce properties of each class, such as its probability or number of members.
2.2. Model
The secondary structure of an RNA is defined by its base pairs. A widely used representation of a secondary structure is the Dot-Bracket-Notation, also known as “Vienna”-string, where a base pair is represented by a pair of parentheses and unpaired bases by dots, see Fig. 1 (b). We use a similar representation for shapes. A paired region is represented by a pair of square brackets and unpaired regions by the underscore character, see Fig. 1(c). The member of the shape class having lowest free energy is called shape representative structure or shrep for short.
Fig. 1.
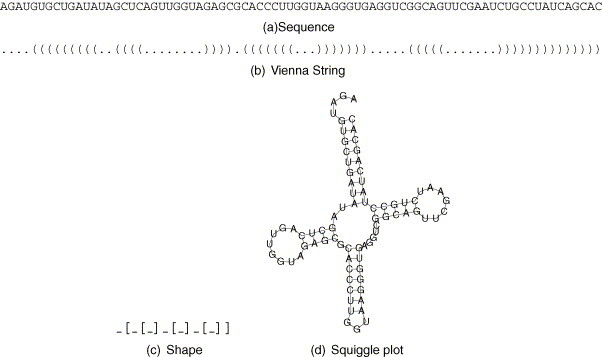
Sequence (a), secondary structure represented as a Vienna string (b), the abstract shape (c) and the secondary structure as a so called squiggle plot (d) of a tRNA.
We can also compute the probability of a shape as the sum of the probabilities of its members, according to Boltzmann statistics. The calculation of the probability of an individual structure follows McCaskill (1990) and makes use of the partition function known from thermodynamics. A manuscript describing the complete probabilistic shape analysis is currently in preparation.
2.3. Implementation/tool
The method of shape analysis is implemented in the tool RNAshapes. This tool computes shapes together with shreps, calculates probabilities of shapes, performs stochastic sampling of structures, and includes complete suboptimal folding and prediction of MFE structure.
2.4. Example
Transfer RNAs (tRNAs) are one of the best analyzed RNA families. Various experiments have revealed the biological active structure of tRNAs which is known as the cloverleaf structure. In contrast to this we found that out of 99 tRNA sequences from the Rfam database (Griffiths-Jones et al., 2003), only 30 have a cloverleaf as their predicted MFE structure (data not shown). The biological explanation for this is that tRNAs possess modified bases which may on the one hand be no longer capable of forming base pairs, or on the other hand are able to interact in a different way. This alters the free energy of the predicted conformation such that it rises above the free energy of the cloverleaf (or vice versa), letting the latter achieve the energetical optimum. For structure prediction, when the modifications are unknown, current practice is to calculate suboptimal structures for a certain energy range and to subsequently search (by eye or by a simple pattern matching algorithm) for the cloverleaf structure in the list of suboptimals. For tRNAs this means that about 50–300 structures have to be checked. To give an example we chose the Natronobacterium pharaonis tRNA for alanine (gb: AB003409.1/96-167). The predicted MFE structure is one hairpin with three internal loops, as depicted in Fig. 2 (a). The cloverleaf structure, shown in Fig. 2(c) appears at position 104 in the energy sorted list of 199 suboptimals, produced by RNAsubopt. Using RNAshapes, we obtain three shapes in an energy range of 5 kcal/mol above the MFE, of which the rank 3 shrep is the cloverleaf structure. Furthermore, the probabilities indicate that without the aforementioned modifications it is highly unlikely that this tRNA attains the cloverleaf shape. The output of RNAshapes and the squiggle plots for the shreps are shown in Fig. 2.
Fig. 2.

Predicted shreps for Natronobacterium pharaonis tRNA-ala in an energy range of 5 kcal/mol above the mfe. This energy range holds 199 structures.
3. pknotsRG: prediction of structures possibly containing pseudoknots
3.1. Motivation and background
Pseudoknots play an important role in many biological processes. They build the catalytic core of some ribozymes and are an important building block of many structural RNAs. Pseudoknots are involved in telomerase activity and they can stimulate efficient ribosomal frameshifting, a mechanism used by a wide range of RNA viruses.
Standard RNA folding programs neglect pseudoknots for reasons of efficiency. While the standard methods need time proportional to the cube of the input sequence length, pseudoknot prediction is much more demanding. The algorithm presented in this section needs O(n 4) time and O(n 2) space, where n is the sequence length. The algorithm by Rivas and Eddy (1999), which is able to predict a larger class of pseudoknots, needs O(n 6) time and O(n 4) space.
3.2. Model
Most of the currently known pseudoknots are rather simple ones. They consist of only two helices, interacting in a crosswise fashion, as shown in Fig. 3 . If we allow the unpaired strands ( in Fig. 3) to build secondary structures internally in an arbitrary way, including multiloops and pseudoknots, we call this class simple recursive pseudoknots.
Fig. 3.
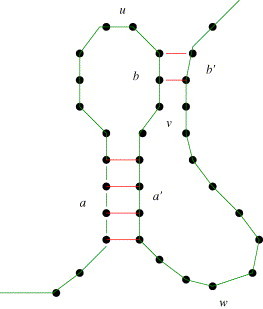
A simple pseudoknot formed by helices a–a′ and b–b′. If the inner components contain additional secondary structures, it is a simple recursive pseudoknot.
Our model further restricts this class by three canonization rules:
-
•
Rule 1: the 5′ and 3′ part of each pseudoknot helix must have the same length and must not have an interruption. This disallows bulges and internal loops inside pseudoknot stems.
-
•
Rule 2: the pseudoknot helices must have maximal length, i.e. if there is a possible base pair at either end, it must be closed.
-
•
Rule 3: if due to Rule 2 the helices would overlap, the first helix (a–a′) is prioritized and the second one is shortened.
We call the resulting class canonical simple recursive pseudoknots (csr-pk).
The canonization rules are motivated by the following observations: if a pseudoknot contains a bulge in one of the stems, it is not canonical. However, there must be a pseudoknot with a smaller stem without the bulge that serves as a canonical representative. This representative may have a higher energy, but usually the differences are small.
Rule 2 is justified by the fact that the energy model strongly favors helix extension. The only exception is, when bases of the pseudoknot stem compete with internal structures of the inner parts .
Finally, the decision of Rule 3 will have only a minor effect, since the same bases are either stacked on helix a–a′ or b–b′.
In an exhaustive analysis of the pseudoknot database PseudoBase,1 it was shown, that out of 212 known pseudoknots, 172 are simple recursive (Reeder and Giegerich, 2004). Almost 80% of these are even canonical simple recursive pseudoknots. This shows the abundance of the class csr-pk within all validated structures.
3.3. Implementation/tool
The program pknotsRG extends the usual RNA folding programs by the class of simple recursive pseudoknots. It uses the current thermodynamic energy model by the Turner group (Mathews et al., 1999). pknotsRG provides three basic modes:
-
(1)
Standard MFE folding with pseudoknots.
-
(2)
Enforced folding: the best structure in the folding space that contains at least one pseudoknot is reported.
-
(3)
Local folding: the pseudoknot with the best energy to length ratio is predicted.
All modes can also be run in a suboptimal fashion, where all suboptimal solution up to a user-defined threshold are computed.
The program is available as download, as interactive tool and WebService, which allows for an easy integration into other software.
3.4. Example
Recently, an unusual three-stemmed pseudoknot has been identified that promotes ribosomal frameshift in the SARS Coronavirus (Plant et al., 2005). The pseudoknot is thought to pause the ribosome during translation, which then shifts back by one nucleotide on the “slippery site”. This special pseudoknot seems to be conserved in all Coronaviruses, and thus could be a target for anti-viral therapeutics.
The three-stem topology is predicted by pknotsRG as the optimal structure with an energy of −31.8 kcal/mol (Fig. 4 ).
Fig. 4.
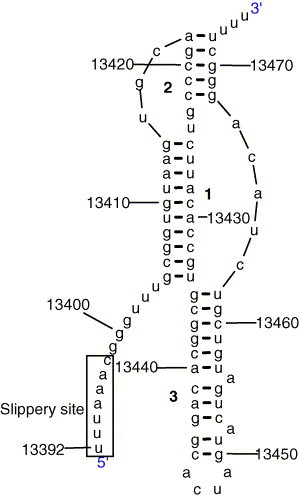
A unusual three-stem pseudoknot in the ribosomal frameshift signal of SARS Coronavirus, predicted by pknotsRG.
4. RNAforester: structure comparison, local similarity, and multiple alignment
4.1. Motivation and background
Comparative analysis of coding regions, i.e. regions where the order of nucleotides codes for proteins, has been studied extensively. Today, there are numerous examples of RNA genes and motifs where the structure instead of the sequence determines the function (and for sure, there are a lot of unknown ones today). Where the selective pressure acts on the function, often the structure instead of the sequence is conserved. In spite of all its success, purely sequence based comparative analysis gets to its limit when structural conservation is of interest. In contrast, RNAforester is a tool that aligns the structures of RNA molecules.
4.2. Model
From the viewpoint of computer modeling, RNA secondary structures are naturally represented as trees or forests. The parent and sibling relationship of nodes is determined by the nesting of base-pair bonds. The 5′–3′ orientation of an RNA molecule imposes the order among sibling nodes. This produces a forest structure in general (where a forest is simply a sequence of trees) but a virtual root node can always turn a forest into a tree. Different tree representations of RNA structures that vary in their resolution have been proposed. See Fig. 5 for an example. Given the tree-like nature of RNA secondary structures, every distance measure on trees can be applied to compare RNA secondary structures.
Fig. 5.

(a) Shows a secondary structure with colored components that indicate the relation between the representations. (b) Shows the natural tree representation where internal nodes correspond to base pairs and leaves correspond to unpaired bases.
4.2.1. Alignment of trees and global similarity of structures
The tree alignment distance was introduced by Jiang et al. (1995). Let Σ be an alphabet and let denote the set of Σ-labeled trees. We define the alignment alphabet as the tuple alphabet where for each of its elements at least one component is different from λ. Formally, . A tree alignment A is an element of . Its component-wise projections A|1 and A|2 are elements of . For some is the forest that results from the deletion of all nodes with label() = λ. (Deleting node in T means that the children of node become the children of the parent node of . Moreover, if has any siblings, the deletion preserves the preorder relation of these nodes.) A tree alignment A is an alignment of trees F and G, iff π(A|1) = F and π(A|2) = G. See Fig. 6 for an illustration of the tree alignment model.
Fig. 6.
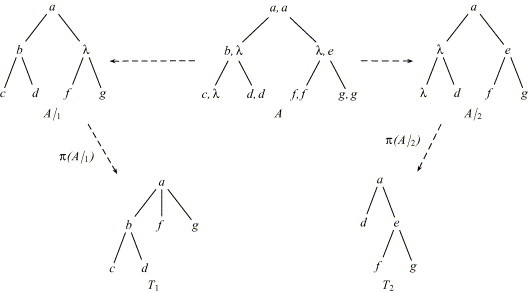
A is an alignment of T1 and T2.
Given this notation of tree alignment, similarity of trees can be defined in a way analogous to sequence similarity: the similarity score σ of an alignment A is the sum of scoring contributions for its node labels. The alignment similarity σ TA of T 1 and T 2 is the maximum score that an alignment of T 1 and T 2 can achieve. An alignment of T 1 and T 2 is optimal if it achieves this score. These definitions generalize to forests in a straightforward way.
4.2.2. Local similarity in RNA secondary structures
Calculating global similarity of RNA secondary structures is not sufficient when the focus is to find similar substructures. This holds particularly if the substructures are far apart in sequence due to their 5′ positions. Local similarity means finding the maximal similarity between two substructures. If these substructures are extended, the score decreases. This requires a scoring scheme that balances positive and negative scoring contributions. Otherwise, the similarity of the whole structure always achieves the maximum score. With suitable generalized definitions we can perform a BLAST- or Smith-Waterman-like local similarity search on RNA structures. A dynamic programming algorithm that solves this problem was proposed in (Höchsmann et al., 2003) and is implemented in RNAforester.
4.2.3. Multiple alignment of RNA secondary structures
An alignment of forests is a forest and, hence, can again be aligned in the forest alignment model. This makes virtually every progressive strategy that was reported for multiple sequence alignment applicable to multiple forest alignments. The idea of the algorithms persists even if the type of the alignment is not a sequence. The concept of sequence profiles can be naturally extended to forests, which results in a profile representation of RNA secondary structures. This data structure and a progressive profile alignment algorithm for RNA secondary structures were proposed in (Höchsmann et al., 2004) and are implemented in RNAforester.
4.3. Implementation/tool
RNAforester is a command line based tool for comparing RNA secondary structures. It supports the computation of (local) pairwise and multiple alignment of structures based on the tree alignment model (Jiang et al., 1995) and the extensions and algorithms presented in (Höchsmann et al., 2003, Höchsmann et al., 2004). The user interface follows the philosophy of the Vienna RNA Package (Hofacker et al., 1994) and the program is part of the forthcoming Vienna RNA Package Version 1.6. An online version of RNAforester and the source code distribution is available at the Bielefeld Bioinformatics Server (BiBiServ).
4.4. Examples
In Fig. 7 (a) the local alignment of the 5′UTRs of the human ferritin heavy chain mRNA (D28463) and the mouse ferritin heavy chain mRNA (M60170) is displayed. Here, the IRE is not only conserved in structure but also in sequence. In contrast, the local structure alignment of the 5′UTR of the human ferritin heavy chain mRNA and the 3′UTR of the human transferrin receptor mRNA (X01060) (see Fig. 7(b)) shows numerous compensatory mutations in the IRE. This shows that our approach can discover arbitrary conserved structural motifs in a larger structure, independent of their position and primary sequence. Fig. 8 shows an example of a multiple structure alignment and its progressive calculation.
Fig. 7.
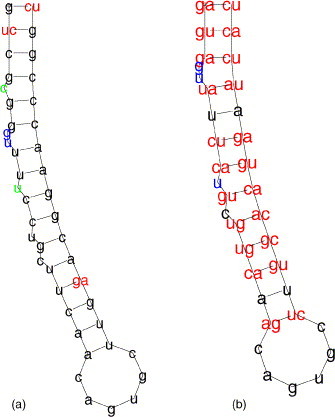
(a) Local structure alignment of the human and mouse ferritin heavy chain 5′UTRs. (b) Local structure alignment of the human ferritin heavy chain 5′UTR and the human transferrin receptor 3′UTR.
Fig. 8.
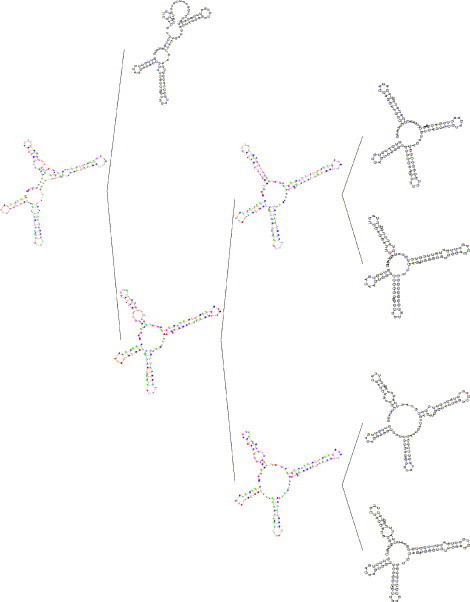
A progressive profile alignment of RNA secondary structures.
5. RNAcast: consensus structure prediction from multiple sequences
5.1. Motivation and background
Most RNAs exert their function by structure, not by sequence. Thus a family of RNAs with the same function has most likely a conserved tertiary and secondary structure. Determining the secondary structure by comparative methods usually gives better results than methods based on only one sequence. The best results were achieved by statistical analysis of covariation and sequence conservation within a RNA family (Gutell et al., 1992). However, these methods need a large family and a very good, hand crafted alignment. For the comparative analysis of only a few related RNAs, the algorithm formulated by Sankoff (1985) is thought to be the ideal approach. It simultaneously aligns and folds a set of RNA sequences. Unfortunately, this method is exponential in the number of sequences and practical only for two sequences of moderate length. An intrinsic reason why the Sankoff algorithm is so expensive is the implicit multiple sequence alignment. The key to a practical, yet non-heuristic algorithm for consensus prediction is to find the consensus without constructing a sequence alignment. If desired, an alignment can be derived from the consensus afterwards with a structural alignment program, such as RNAforester.
5.2. Model
We redefine the problem addressed by the Sankoff algorithm: a consensus structure for k sequences s 1, …, s k is a set of secondary structures x 1, …, x k, such that x 1, …, x k have the same abstract shape.
Since the shape abstracts from sequence content and explicit loop and helix lengths, it lends itself perfectly to a comparative approach: shapes of different structures and different sequences can be meaningfully compared for identity, using fast exact matching rather than alignment methods.
This outlines our method termed RNAcast:
-
•
Step 1: for RNA sequences s 1, …, s k, enumerate the near-optimal shape spaces . For each shape , also compute the shape representative structure , which is the best way sequence s i can fold into this particular shape.
-
•
Step 2: identify the shapes that occur in all shape spaces, i.e. that all sequences can fold into. This yields a list of all common shapes, together with their respective shreps: .
-
•
Step 3: finally, rank each common shape p i by adding the individual free energies of . The best scoring common shape is the consensus shape, and its shreps constitute our consensus structure prediction.
5.3. Implementation/tool
The idea of the RNA consensus abstract shapes technique is implemented in the tool RNAcast. For a set of (related) RNA sequences, it computes a consensus shape, and for each sequence, the shape representative structure (shrep). RNAcast predictions are unaligned structures. If an alignment is of interest, it can be computed afterwards with a multiple structure alignment program, such as RNAforester.
RNAcast consensus structure predictions are most reliable for sets of 5–10 sequences, not longer than 200 nucleotides. If fewer sequences are used, individual sequences may have a too strong overall influence. The strength of the comparative approach does not show to advantage. On the other hand, with more than 10 sequences in the input set, the user should check for potential outliers, since these may prohibit the finding of the native folding.
5.4. Example
RNAcast has been evaluated on several RNA families, including micro RNAs, tRNAs, the RNA of the signal recognition particle, several spliceosomal RNAs and other. It shows a significant increase in accuracy over single sequence RNA folding and performs as good as the heuristic implementation of the Sankoff approach by Mathews and Turner (2002), while being a lot faster.
As an example, we apply RNAcast to five signal recognition particle RNAs of Homo sapiens, Rattus norvegicus, Gallus gallus, Canis lupus and Anopheles gambiae (accession numbers: X04248, AC091616, AB073218, J01853, BX044091), each of length approximately 300 bases, taken from the SRP Database.2 The predicted shreps are all very similar to the published reference structure (not shown). The consensus structure, computed by RNAforester afterwards from RNAcast's prediction clearly shows all the common features of the SRP (see Fig. 9 ). This particular computation took less than 20 s on current hardware, which demonstrates the efficiency of RNAcast.
Fig. 9.

RNA of the signal recognition particle: (a) structure of the dog SRP as found in the SRP database, (b) RNAforester consensus produced from an RNAcast prediction of five SRP sequences: human, chicken, rat, dog and mosquito. The predicted consensus structure clearly shows the conserved features of the SRP.
6. RNAhybrid: prediction of miRNA targets
6.1. Motivation and background
MicroRNAs (miRNAs) are short RNAs of around 22 nt that post-transcriptionally regulate the expression of target genes by binding to the target mRNAs. In plants, this binding usually leads to cleavage and subsequent degradation of the target, in animals the target is usually not cleaved, but blocked from translation into protein. Although several thousand miRNAs have been defined, only a small, albeit increasing, number of targets is known. The increase in known miRNA/target relationships is also due to the use of computational prediction methods, one of which is RNAhybrid (Rehmsmeier et al., 2004). A related situation is the design of small interfering RNAs (siRNAs), in which RNAhybrid could be used to reduce off-target effects by checking for possible hybridizations of designed siRNAs and genes other than the ones to be targeted.
6.2. Model
RNAhybrid extends the classical RNA secondary structure prediction to two sequences. A small query RNA, such as a microRNA, is hybridized to a potential target RNA in a way that optimizes the free energy of the hybridization site. Secondary structure outside a hybridization site is not considered, and intra-molecular base pairings are not allowed. The sizes of bulge loops and internal loops can be restricted if from the nature of the RNA/RNA interaction this appears reasonable. This can be the case, for example, in the prediction of microRNA targets in plants, where microRNAs usually bind nearly perfectly to the target, the hybridizations showing only small loops of 1 nucleotide, if any. As a further refinement, hybridizations can be forced to have uninterrupted base pairing for specified regions, for example from nucleotide 2 to nucleotide 7 in the microRNA. Such “seeds” have been suggested as necessary for functioning microRNA/target interactions.
Important in any prediction method is an assessment of statistical significance. In the RNAhybrid rationale, this is accounted for in a stepwise manner in which p-values are calculated for, first, individual binding sites, second, multiple binding sites of one microRNA in a target, and third, binding sites of one microRNA in orthologous targets across multiple species. p-Values for individual binding sites are based on extreme value distributions that are estimated in a microRNA-specific way; p-values of multiple binding sites are modeled with a Poisson approximation that considers individual good binding sites as rare events; multi-species p-values are based on the effective number of orthologous sequences, which describes the statistical dependence of these and which avoids biased overestimates of statistical significance.
6.3. Implementation/tool
RNAhybrid is written in C and is available as source distribution for download as a program bundle. For small query RNAs and constant maximal loop sizes, the time and memory efficiency of the algorithm is linear in the length of the target. There will be a new version available soon that integrates the complete prediction rationale in one program and also contains a Graphical User Interface to facilitate the use of the program. A version that allows for the easy prediction of energetically favorable binding sites is available as an online tool on the BiBiServ.
6.4. Example
Fig. 10 shows a prediction of Drosophila melanogaster miR-2b binding sites in the sickle 3′UTR sickle, with grim and reaper, constitutes a group of pro-apoptotic genes which have been demonstrated to be targets of miRNA-2 (Stark et al., 2003). In this reference, however, only grim and reaper were among the predictions. In (Rehmsmeier et al., 2004), sickle was predicted as possible target of miR-2b.
Fig. 10.

Two potential target sites of Drosophila melanogaster miR-2b in the sickle 3′UTR. The upper part shows an ASCII output of RNAhybrid, the lower part illustrates the predicted hybridization sites graphically, with the microRNAs shown in green and the target sites shown in red with their 5′-ends additionally marked.
7. Conclusion
The tools presented here have already acquired a widespread usership. All programs are available for download at the BiBiServ, http://bibiserv.techfak.uni-bielefeld.de. In addition, RNAforester is distributed as part of the Vienna RNA Package (Hofacker et al., 1994), starting with version 1.6. All programs are also available as online tools at BiBiServ, and Table 1 shows their usage counts for January 2004 through July 2005. For online use, we normally restrict the size of jobs to be submitted, but exceptions may be negotiated.
Table 1.
Tool access statistics on the BiBiServ until July 2005
| RNAshapes | RNAhybrid | pknotsRG | RNAforester | RNAcasta | |
|---|---|---|---|---|---|
| 2004 | 914 | 3906 | 836 | 721 | – |
| 2005 | 1477 | 7710 | 530 | 1367 | 31 |
BiBiServ statistics only account for uses from outside of Bielefeld University.
Published July 2005.
Note that often the tools can be used in context. For example, a predicted miRNA target site should be checked for accessibility – it should not be hidden in helical regions of the target molecule structure. This requires a run of RNAshapes on the targets predicted by RNAhybrid. An important aspect of our future work will be to support the use of these tools in an integrated fashion.
Acknowledgements
Four of the five tools presented here have been developed with the method of algebraic dynamic programming (Giegerich et al., 2004a). The authors gratefully acknowledge the contributions of Peter Steffen to the efficient implementation of this programming technique.
Footnotes
References
- Akutsu T. Dynamic programming algorithms for RNA secondary structure prediction with pseudoknots. Discrete Appl. Math. 2000;104:45–62. [Google Scholar]
- Andronescu M., Zhang Z.C., Condon A. Secondary structure prediction of interacting RNA molecules. J. Mol. Biol. 2005;345(5):987–1001. doi: 10.1016/j.jmb.2004.10.082. [DOI] [PubMed] [Google Scholar]
- Ding Y., Chan C.Y., Lawrence C.E. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA. 2005;11(8):1157–1166. doi: 10.1261/rna.2500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Lawrence C.E. A statistical sampling algorithm for RNA secondary structure prediction. Nucl. Acids Res. 2003;31(24):7280–7301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi K., Cannone J., Cobaugh C., Gutell R. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinformatics. 2004;5(105) doi: 10.1186/1471-2105-5-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner P.P., Giegerich R. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinformatics. 2004;5(140) doi: 10.1186/1471-2105-5-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegerich R., Meyer C., Steffen P. A discipline of dynamic programming over sequence data. Sci. Comput. Program. 2004;51(3):215–263. [Google Scholar]
- Giegerich R., Voss B., Rehmsmeier M. Abstract shapes of RNA. Nucl. Acids Res. 2004;32(16):4843–4851. doi: 10.1093/nar/gkh779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodkin J., Heyer L.J., Stormo G.D. Finding the most significant common sequence and structure motifs in a set of RNA sequences. Nucl. Acids Res. 1997;25(18):3724–3732. doi: 10.1093/nar/25.18.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R. Rfam: an RNA family database. Nucl. Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell R.R., Power A., Hertz G.Z., Putz E.J., Stormo G.D. Identifying constraints on the higher-order structure of RNA: continued development and application of comparative sequence analysis methods. Nucl. Acids Res. 1992;20(21):5785–5795. doi: 10.1093/nar/20.21.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höchsmann M., Töller T., Giegerich R., Kurtz S. Proc. of CSB ’03. 2003. Local similarity in RNA secondary structures; pp. 159–168. [PubMed] [Google Scholar]
- Höchsmann M., Voss B., Giegerich R. Pure multiple RNA secondary structure alignments: a progressive profile approach. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2004;1(1):53–62. doi: 10.1109/TCBB.2004.11. [DOI] [PubMed] [Google Scholar]
- Hofacker I.L. Vienna RNA secondary structure server. Nucl. Acids Res. 2003;31(13):3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer S., Tacker M., Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- Huang C.-H., Lu C.L., Chiu H.-T. A heuristic approach for detecting RNA H-type pseudoknots. Bioinformatics. 2005;21(17):3501–3508. doi: 10.1093/bioinformatics/bti568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T., Wang J.T.L., Zhang K. Alignment of trees an alternative to tree edit. Theor. Comput. Sci. 1995;143(1):137–148. [Google Scholar]
- Lyngsø R.B., Pedersen C.N.S. RNA pseudoknot prediction in energy based models. J. Comp. Biol. 2001;7:409–428. doi: 10.1089/106652700750050862. [DOI] [PubMed] [Google Scholar]
- Mathews D.H., Sabina J., Zuker M., Turner D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Mathews D.H., Turner D.H. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J. Mol. Biol. 2002;317(2):191–203. doi: 10.1006/jmbi.2001.5351. [DOI] [PubMed] [Google Scholar]
- McCaskill J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- Plant E.P., Prez-Alvarado G.C., Jacobs J.L., Mukhopadhyay B., Hennig M., Dinman J.D. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 2005;3(6):e172. doi: 10.1371/journal.pbio.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder J., Giegerich R. Design, implementation and evaluation of a practical pseudoknot folding algorithm based on thermodynamics. BMC Bioinformatics. 2004;5:104. doi: 10.1186/1471-2105-5-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder J., Giegerich R. Consensus shapes: an alternative to the Sankoff algorithm for RNA consensus structure prediction. Bioinformatics. 2005;21(17):3516–3523. doi: 10.1093/bioinformatics/bti577. [DOI] [PubMed] [Google Scholar]
- Rehmsmeier M., Steffen P., Höchsmann M., Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E., Eddy S.R. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol. 1999;285:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- Sankoff D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J. Appl. Math. 1985;45(5):810–825. [Google Scholar]
- Shapiro B.A., Navetta J. A massively parallel genetic algorithm for RNA secondary structure prediction. J. Supercomput. 1994;8:195–207. [Google Scholar]
- Stark A., Brennecke J., Russell R.B., Cohen S.M. Identification of Drosophila microRNA targets. PLoS Biol. 2003;1(3):e60. doi: 10.1371/journal.pbio.0000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabaska J.E., Cary R.B., Gabow H.N., Stormo G.D. An RNA folding method capable of identifying pseudoknots and base triples. Bioinformatics. 1998;14(8):691–699. doi: 10.1093/bioinformatics/14.8.691. [DOI] [PubMed] [Google Scholar]
- Wang L., Jiang T. On the complexity of multiple sequence alignment. J. Comp. Biol. 1994;1(4):337–348. doi: 10.1089/cmb.1994.1.337. [DOI] [PubMed] [Google Scholar]
- Wuchty S., Fontana W., Hofacker I.L., Schuster P.F. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1998;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Zuker M., Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary informations. Nucl. Acids Res. 1981;9(1):133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]