

Abstract
Aptamers are specific nucleic acid sequences that can bind to a wide range of non-nucleic acid targets with high affinity and specificity. These molecules are identified and selected through an in vitro process called SELEX (systematic evolution of ligands by exponential enrichment). Proteins are the most common targets in aptamer selection. In diagnostic and detection assays, aptamers represent an alternative to antibodies as recognition agents. Cellular detection is a promising area in aptamer research. One of its principal advantages is the ability to target and specifically differentiate microbial strains without having previous knowledge of the membrane molecules or structural changes present in that particular microorganism. The present review focuses on aptamers, SELEX procedures, and aptamer-based biosensors (aptasensors) for the detection of pathogenic microorganisms and viruses. Special emphasis is placed on nanoparticle-based platforms.
Keywords: Aptamers, Aptasensor, Pathogen detection, Complex targets, Nanoparticle-based platforms
1. Introduction
Detection, identification and quantification of microbial pathogens are crucial for public health protection. Areas where detection of microbial pathogens is critical include clinical diagnosis, water and environmental analysis, food safety, and biodefense. Microbial culture-based tests and molecular assays (immunological or nucleic acid technologies) are the most common methodologies currently used (Lazcka et al., 2007). These techniques are either time consuming or require sophisticated equipment and highly trained personnel, hence increasing the analysis cost. Rapid detection techniques should provide reliable, real time, on-field, user-friendly, and inexpensive detection with improved or equivalent sensitivity, specificity and reproducibility of culture-based tests (Alocilja and Radke, 2003). According to Lazcka et al. (2007), biosensor technology is the fastest growing area in rapid pathogen detection. The commonly used biological recognition elements in biosensor platforms are antibodies and nucleic acid probes.
Aptamers are specific nucleic acid sequences that can bind to a wide range of non-nucleic acid targets with high affinity and specificity (Jayasena, 1999). In diagnostic and detection assays, aptamers represent an alternative to antibodies as recognition agents. Aptamers are selected through an in vitro process, which represents lower cost and less batch-to-batch variation than antibody in vivo production. Furthermore, toxins and molecules that do not elicit a good immune response can be used to generate high affinity aptamers (O'Sullivan, 2002, Proske et al., 2005).
Typically, aptamer sequences are identified through the SELEX process (systematic evolution of ligands by exponential enrichment). The methodology consists of screening large random oligonucleotide libraries through iterative cycles of in vitro selection and enzymatic amplification (Ellington and Szostak, 1990, Tuerk and Gold, 1990, Robertson and Joyce, 1990). Briefly, SELEX starts with incubation of the nucleic-acid sequence library with the target (protein, antibody, enzyme, etc.), followed by separation and exponential amplification of the binding oligonucleotides. This process is repeated (typically 8–15 times) using the obtained enriched pool as starting library. Finally, cloning and sequencing of the final specific binding molecules allow identification of the best sequences (Fig. 1 ).
Fig. 1.
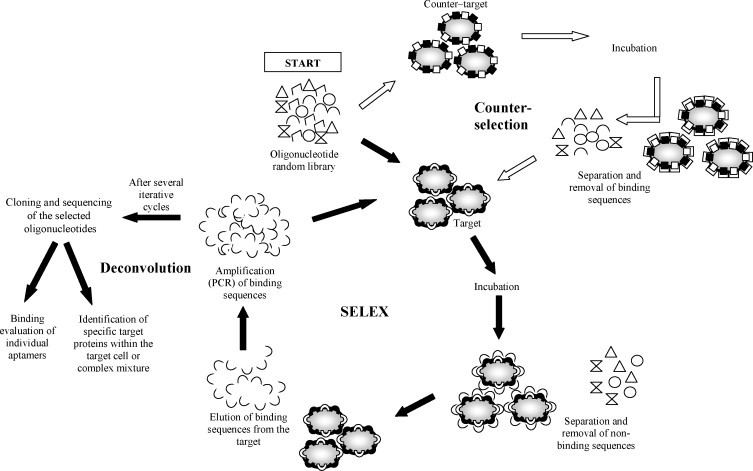
General illustration of complex-target aptamer selection strategies. SELEX procedure: the oligonucleotide pool (random library) is incubated with the target. Binding sequences are partitioned from the non-binding sequences and amplified by PCR. The enriched pool is incubated again with the target. After several iterative cycles, the selected oligonucleotides are cloned and sequenced. Counter-selection step: the random library is incubated with a counter-target (usually closely related to the target) to eliminate the sequences that bind this counter–target and increase the specificity of the random library (remaining sequences) that is incubated with the target. This step is usually conducted over several cycles either before or after the target incubation. Deconvolution step: the cloned and sequenced specific oligonucleotides are analyzed to evaluate binding efficiency and/or determine specific binding sites within the cell wall (usually proteins) or the complex mixture.
Aptamers can be selected for a wide variety of targets, from small molecules to whole cells. SELEX targets include proteins, enzymes, antibodies, antibiotics, and toxins. Complex targets include non-whole-cell (cell surface molecules, membrane fragments, bacterial lysates, and viral particles) and whole-cell targets (Chu et al., 2007, Shamah et al., 2008). Proteins are the most common SELEX targets. Thrombin, an important protease in the coagulation cascade, has been the most studied target; its aptamer sequences have been used as biological recognition agents in the design and development of biosensors (Tombelli et al., 2007). Aptamers are target-conformation specific, which means that if the target protein conformation changes, the binding event will be affected or may not occur. In order to get consistency within the SELEX cycles, it is necessary to use the target in the most stable conformation. Therefore, in some diagnostic and therapeutic applications, target proteins that are expressed on cell membranes cannot be introduced into SELEX as isolated and purified proteins. They need to be presented as cell fragment preparations (non-whole-cell targets) or intact whole cells (Shamah et al., 2008). These targets are commonly called “complex targets.”
It is well documented that due to their high specificity and affinity properties, aptamers can be used as stationary phase or capture molecules in analytical techniques, such as affinity chromatography (Clark and Remcho, 2002, Hamula et al., 2006), capillary electrophoresis, and mass spectrometry (Tombelli et al., 2005b). Clinical applications of aptamers include their use as therapeutic agents (Lee et al., 2006, Ng et al., 2006), anti-infective agents (Ulrich et al., 2002), and as drug delivery molecules (Bagalkot et al., 2007, Xiao et al., 2008). One of the most studied applications is the identification of cell surface markers for cancer detection. Some target examples include lymphoma (Ramos) cells (Tang et al., 2007), leukemia cells (Shangguan et al., 2006), brain tumor microvessel (endothelial) cells (Blank et al., 2001), and brain tumor (glioblastoma) cells (Daniels et al., 2003). Although several aptamer studies have been published about individual protein targets, few aptamers for microorganisms, particularly for whole-cell detection, have been identified. Some examples will be reviewed in the present work. Most of these aptamers have been developed for clinical diagnostics and infection inhibitor agents.
Aptamer detection applications include aptamer-based microarrays (Collett et al., 2005), quantum-dot aptamer complexes (Levy et al., 2005, Hansen et al., 2006), aptamer-functionalized gold nanoparticles (Huang et al., 2005, Kouassi et al., 2007), and aptamer-based biosensors (aptasensors) (Deisingh, 2006, Willner and Zayats, 2007, Fischer et al., 2007, Lee et al., 2008). Electrical and optical aptasensor platforms have been reviewed previously, including aptasensor applications in biosecurity and clinical diagnostics (O'Sullivan, 2002, Li et al., 2008, Song et al., 2008). However, a review on aptamers and aptasensors for detection of microbial pathogens is not yet available. This article provides a review of aptamers, SELEX procedures, and aptasensor designs for microorganism recognition, with special emphasis on whole-cell targets. Nanoparticle-based aptasensors are introduced as a prospective application for microbial and viral pathogen detection.
2. Complex-target SELEX
In aptamer research, cellular detection is a promising area. One of its principal advantages is the ability to target specific pathological cell stages or cell types without having previous knowledge of the membrane molecules or structural changes related to that cell stage or type (Daniels et al., 2003, Hamula et al., 2006). Cellular aptamers can be applied to isolate, concentrate, and identify particular cells from biological matrices (Guo et al., 2006). One of the first models for complex-target SELEX used human red blood cell membranes as target (Morris et al., 1998). In the report, ligands to specific target molecules in the complex mixture were isolated and identified using a deconvolution-SELEX strategy. Deconvolution, a secondary selection step, allows for the partitioning of the aptamer pool generated from the SELEX process that has evolved against multiple targets (Fitter and James, 2005) (Fig. 1). This step involves: (a) evaluation of aptamer's ability to specifically bind, capture, and identify the target (Blank et al., 2001); or (b) incubation of the last selected aptamer pool with the target, in order to identify and isolate potential specific protein targets within the complex mixture, using magnetic capture beads and SDS/PAGE analysis (Morris et al., 1998, Fitter and James, 2005).
Another complex-target SELEX strategy includes the use of genetically modified cells that overexpress a target recombinant protein on the cell surface. The whole cell is introduced into the SELEX process as a target, to isolate aptamers for that particular protein (Cerchia et al., 2005, Ohuchi et al., 2006). The most common application of whole-cell SELEX is the cell-surface biomarker identification for cancer diagnostics (Shangguan et al., 2008). Besides detection, aptamers have other potential applications in cancer therapeutics, such as blocking (neutralizing) agents for whole-cell inhibition differentiation (Chu et al., 2007, Shamah et al., 2008). Aptamers used for cancer cell identification and their potential therapeutic applications have been extensively reviewed (Cerchia et al., 2002, Ireson and Kelland, 2006, Cerchia and Franciscis, 2007).
One of the most critical steps in SELEX is the partitioning of target-binding sequences from non-specific oligonucleotides. In some applications that use complex cellular targets (cell fraction mixtures, cell surface molecules or whole cells), the specific target molecule is unknown before the selection process. Therefore, it is not possible to select aptamers against purified proteins. Besides, there are multiple potential target molecules in cell walls which can be present among species. In order to improve specificity, an additional counter-selection step can be used in the aptamer selection process.
In SELEX, the counter-selection cycle involves incubation of a closely related cell or microorganism with the random nucleotide library, to eliminate the sequences that are non-specific to the target (Fig. 1). This “negative” selection can be conducted after several “positive” cycles with the target of interest (Shangguan et al., 2006), or before the specific selection (Chen et al., 2007). Negative cycles can also include the elimination of nucleotide sequences that bind to solid supports (e.g., membranes, columns, filters or beads), commonly used in partitioning techniques like nitrocellulose membrane filtration, affinity chromatography, magnetic separation, and capillary electrophoresis (Vivekananda and Kiel, 2006).
In complex-target SELEX techniques, the ability to target a specific cell without knowing the precise molecules associated with can be an advantage as well as a disadvantage. A number of additional purification steps of the final aptamer pool may be necessary in order to identify particular family ligands selected against the multiple targets present in complex mixtures. Specific target molecule identification is not always possible. This represents a challenge for further binding optimization, which would be necessary for detection and biosensing aptamer applications.
3. Aptamer development for pathogenic microorganisms and viruses
Aptamers targeting microbial and viral pathogens have been developed for two main purposes: as therapeutics and for pathogen detection. Whole-cell and non-whole-cell targeting approaches have been applied in microbial SELEX. Examples of non-whole-cell approaches include viral and bacterial lysates, and cell membrane preparations. Viral protein aptamers have been used in the molecular analysis of virus replication and in the development of antiviral agents (Zhang et al., 2004, James, 2007, Jang et al., 2008). A summary of the available reports on microbial targets (including whole-cell, non-whole-cell, and toxin targets) is provided in Table 1 . Representative examples of viral aptamers developed for detection or infection inhibition are also included. Whole-cell strategies and targets are discussed in the following section.
Table 1.
Summary of microbial and viral pathogen aptamers.
| Target | Application | Counter-selection step | Aptamer sequence(s) | Identification of specific target molecule(s) | Reference |
|---|---|---|---|---|---|
| Whole-cell targets | |||||
| B. anthracis Sterne strain spores | Detection | – | No sequence reported | Non-identified | Bruno and Kiel (1999) |
| Detection | – | 3 sequences | Non-identified | Kiel et al. (2004b) | |
| Detection | – | 79 sequences from 13 classes | Non-identified | Zhen et al. (2002) | |
| B. thuringiensis spores | Detection | – | 1 sequence | Non-identified | Ikanovic et al. (2007) |
| E. coli DH5α | Detection | – | 1 sequence | Non-identified | So et al. (2008) |
| M. tuberculosis | Anti-mycobacterial agents | M. bovis attenuated strain [Bacillus Calmette-Gue’rin (BCG)] | No sequence reported | Membrane protein | Chen et al. (2007) |
| T. brucei | Anti-parasitic drugs | – | 22 sequences from 3 classes | Parasite flagellar protein | Homann and Goringer (1999) |
| T. cruzi | Invasion inhibitor agents | T. cruzi epimastigotes | 23 sequences from 4 classes | Parasite receptors for the host cell matrix molecules laminin, fibronecitin, thrombospondin and heparin sulfate. | Ulrich et al. (2002) |
| Human Influenza A virus (H3N2) (A/Panama) |
Influenza A virus genotyping and inhibitor agent |
A/Aichi H3N2 virus strain |
2 sequences |
Haemagglutinin (HA1 peptide chain) |
Gopinath et al. (2006) |
| Target | Application | Counter-selection step | Aptamer sequence(s) | Identification of specific target molecule(s) | Reference |
|---|---|---|---|---|---|
| Non-whole-cell targets | |||||
| F. tularensis bacterial protein lysate | Detection | – | No sequence reported | Non-identified | Vivekananda and Kiel (2006) |
| Rous Sarcoma virus (RSV) particles | Virus inhibitor agent | – | 6 sequences | Non-identified | Pan et al. (1995) |
| Vaccinia virus (VACV) particles |
Infection inhibitor agent |
Non-infected Hep2 cells |
1 sequence |
Non-identified |
Nitsche et al. (2007) |
| Target | Application | Counter-selection step | Aptamer sequence(s) | Reference |
|---|---|---|---|---|
| Microbial and viral protein/toxin targets | ||||
| Cholera whole toxin | Detection | – | No sequence reported | Bruno and Kiel (2002) |
| E. coli release factor 1 (RF-1) | Non-sense-suppression-based technology | – | 17 sequences from 3 classes | Sando et al. (2007) |
| Mycobacterium avium subsp. paratuberculosis recombinant MAP0105c gene product | Detection | Maltose-bind protein (MBP) | No sequence reported | Bannantine et al. (2007) |
| S. enterica serovar Typhi IVP pili protein (pre-PilS protein) | Cell invasion inhibitor agent | – | 9 sequences reported | Pan et al. (2005) |
| Shiga toxin | Detection | – | 11 sequences | Kiel et al. (2004a) |
| Staphylococcal enterotoxin B (SEB) | Detection | – | No sequence reported | Bruno and Kiel (2002) |
| Hepatitis C virus (HCV) non-structural protein 3 (NS3) protease | Anti-HCV agents | 3 sequences | Fukuda et al. (2000) | |
| HCV NS3 helicase | Therapeutics and diagnostic | – | 4 sequences | Hwang et al. (2004) |
| HCV NS5B RNA polymerase | Polymerase inhibition | – | 7 sequences from 3 classes | Biroccio et al. (2002) |
| Human immunodeficiency virus 1 (HIV-1) reverse transcriptase (RT) | Reverse transcription inhibition | – | 25 sequences | DeStefano et al. (2006) |
| HIV-1 protein trans-activator of transcription (Tat protein) | Transcription inhibition and detection | – | 4 sequences | Yamamoto et al. (2000) |
| HIV-1 R5 SU glycoptrotein (gp120) | Antiviral | – | 27 sequences | Khati et al. (2003) |
| Influenza A virus (H5N1) HA1 protein | Antiviral | – | 2 sequences | Cheng et al. (2008) |
| SARS coronavirus (SCV) NTPase/Helicase | Anti-SCV agents | – | 6 sequences from 3 classes | Jang et al. (2008) |
3.1. Whole-cell targets
3.1.1. Protozoa
African trypanosomes aptamer selection is one of the first reports for microbial whole-cell targets that included the obtained aptamer sequences, the identification of the particular target molecule location, and the aptamer's secondary structure prediction (Homann and Goringer, 1999). Trypanosoma brucei was used as the model organism for African trypanosomes and extracellular parasites. Three different classes of RNA aptamers were selected. The specific binding site was identified as a protein molecule located in the parasite flagellar pocket. The selected aptamers were not able to specifically identify T. brucei among other trypanosome strains tested (Homann and Goringer, 1999). Further binding analysis and internalization capabilities of the African trypanosomes aptamers have been published (Homann and Ulrich, 2001).
Ulrich and collaborators (2002) reported the use of live Trypanosoma cruzi parasites as SELEX targets for the selection of RNA aptamers with inhibitory activity on T. cruzi cell invasion. T. cruzi is an intracellular parasite for which mediating parasite–host cell molecules that play an important role in the cell adhesion and invasion process have been identified (laminin, thrombospondin, heparin sulfate, and fibronectin) (Ulrich et al., 2002). During the selection cycles, a selective displacement step was applied to the RNA molecules that bound to the T. cruzi surfaces. The four host cell molecules were incubated with the parasite–aptamer complexes and the displaced RNA molecules were used as starting library for the next selection cycle. The obtained RNA ligands specifically bound to the parasite receptors of the host cell matrix molecules. Also, all aptamer classes obtained were able to inhibit T. cruzi invasion in vitro (Ulrich et al., 2002). The development of potential anti-parasitic drugs has been the main application of the trypanosome aptamer research. Post-SELEX optimization has been reported to improve in vivo functionality of the RNA aptamer against African trypanosomes (Adler et al., 2008).
3.1.2. Bacteria
Bacillus anthracis Sterne strain spores have also been used as whole-cell targets in SELEX (Bruno and Kiel, 1999, Zhen et al., 2002; Kiel et al., 2004a, Kiel et al., 2004b). Further post-SELEX optimization (i.e., characterization of aptamer–spore binding reaction, identification of specific spore target molecules, specificity and sensitivity assays) is necessary in order to use the isolated DNA aptamers in the development of detection platforms.
Stratis-Cullum et al. (2005) reported the use of aptamers for the detection of Campylobacter jejuni whole-cells. In the report, aptamer specificity assays showed no cross-reactivity with Salmonella Typhimurium, but limited cross-reactivity with Escherichia coli O157:H7. However, some cross-reactivity was shown with Helicobacter pylori and Listeria sp. at high concentrations (McMasters and Stratis-Cullum, 2006, Stratis-Cullum et al., 2007). In these studies, neither the SELEX process nor the C. jejuni aptamer sequences were detailed in the report.
Aptamers with potential therapeutic application have been identified for Mycobacterium tuberculosis, using a whole-bacterium SELEX strategy (Chen et al., 2007). The selected aptamer sequence, which constituted 30% of the final pool, specifically distinguishes M. tuberculosis cells (H37Rv strain) from M. bovis (the counter-selection cells). The specific target molecules were partially identified as membrane proteins, using a proteinase analysis. M. tuberculosis cells were treated with trypsin and proteinase K before the incubation step with the aptamer sequence. Negative results were obtained for the binding reaction between the treated cells and the aptamer. This suggests that the binding sites were removed by the proteinase treatment and that they were most likely membrane proteins. This proteinase assay can be used for the initial identification of target molecules in whole-cell SELEX approaches.
3.1.3. Viruses
Whole-cell strategies have also been used to obtain aptamers against viruses. The human influenza A virus RNA aptamers obtained by Gopinath et al. (2006) specifically bound and differentiated strains within subtype N3N2. The binding analysis suggested that the aptamers were specific for the haemagglutinin (HA) membrane glycoprotein of the A/Panama virus strain and were able to distinguish this HA from those of other influenza viruses, including strains of the same subtype N3N2 (A/Aichi strain). Post-SELEX optimization assays included determination of binding kinetics, identification of aptamer minimal RNA motif, and determination of inhibitory effect. The obtained aptamers have potential application in influenza A virus genotyping, and inhibition of HA-mediated membrane fusion (Gopinath et al., 2006).
4. Aptasensor platforms
Aptamers represent an alternative to antibodies as recognition elements in biosensors. The aptamer selection can be performed under real matrix conditions, which is particularly useful for environmental and food samples. Aptamers can be modified for immobilization purposes and labeled with reporter molecules, without affecting their affinity. As nucleic acid sequences, aptamers can be subjected to repeated cycles of denaturation and renaturation; this makes it possible to regenerate the immobilized biocomponent function for reuse (Jayasena, 1999, O'Sullivan, 2002).
Besides the advantages of specificity discussed above, aptamer immobilization characteristics are crucial for aptasensor applications. Aptamers can be chemically modified and labeled more easily than antibodies. These modifications facilitate the functionalization of nanoparticles and surfaces. Also, aptamers can undergo conformational changes and become reusable, allowing some of the aptasensor platforms to be recyclable. A major disadvantage is that most of the available aptamers are RNA structures, which are highly sensitive to nuclease degradation. Alternatives to overcome this problem include chemical modifications of the ribose at the 2′ position (Pagratis et al., 1997, Kusser, 2000) and the use of mirror-image analogs that are nuclease resistant (spiegelmers) (Eulberg and Klussmann, 2003).
The introduction of aptamers as potential biorecognition molecules in biosensors was first reviewed by O'Sullivan (2002). Since then, electrical-based (Lee et al., 2008, Willner and Zayats, 2007) and optical-based platforms (Deisingh, 2006, Fischer et al., 2007, Li et al., 2008) have been reviewed, including aptasensors for biosecurity (Fischer et al., 2007) and clinical (Deisingh, 2006) applications. For clinical diagnostics, several aptamer-based biosensors (aptasensors) have been developed. Examples include quartz crystal aptasensors to detect IgE (Liss et al., 2002) and the protein trans-activator of transcription (Tat protein) of human immunodeficiency virus type 1 (HIV-1) (Minunni et al., 2004); a fiber-optic system to detect thrombin (Lee and Walt, 2000); and a multiplex cancer marker detection system (McCauley et al., 2003). In the electrochemical-based detection systems, few aptasensors have been reported (Willner and Zayats, 2007, Lee et al., 2008). Most of these aptasensors use thrombin as a target model for detection.
Electrical aptasensors include: (a) electrochemical platforms, using enzyme labeling detection systems (Ikebukuro et al., 2005, Mir et al., 2006), aptamers functionalized with redox reporters (Bang et al., 2005, Baker et al., 2006), and label-free impedance spectroscopy transduction (Rodriguez et al., 2005, Cai et al., 2006); (b) field-effect transistors (Zayats et al., 2006), using single-walled carbon nanotubes (So et al., 2005); and (c) piezoelectric quartz crystals, using microgravimetric analysis (Liss et al., 2002, Hianik et al., 2005). One of the main advantages of electrical aptasensors is that sensitivity can be enhanced by attaching biocatalytic labels to the aptamer–target complexes, to amplify the detection signal. Furthermore, electrical aptasensors are more convenient for on-field detection applications, since they do not require expensive optical instruments (Willner and Zayats, 2007, Lee et al., 2008). Additionally, it is possible to use label-free and reusable detection systems. In most cases, protein electrical aptasensors can be easily reused after washing off the target protein. This cannot be done with immunological biosensors because both target and sensing element (antibodies) are proteins.
Optical-based aptasensors include aptamers labeled with fluorophores (signaling aptamers) (Jhaveri et al., 2000, Merino and Weeks, 2005), fluorescence resonance energy transfer (FRET) platforms using aptabeacons (Nutiu and Li, 2003, Heyduk and Heyduk, 2005), and optical fibers (Spiridonova and Kopylov, 2002). One of the main disadvantages of using fluorescent labels in optical aptasensors is that their application in complex matrices is limited, due to the interference and quenching of fluorophores by biological components present in the matrix (Li et al., 2008).
4.1. Aptasensors for detection of microorganisms and viruses
Several aptasensors have been developed to detect viral proteins. Minunni et al. (2004) developed an aptasensor platform to detect HIV-1 Tat protein by immobilizing an RNA aptamer on a piezoelectric quartz crystal. Sensitivity, specificity, and reproducibility parameters were quantified. The aptasensor was also compared with the corresponding immunosensor with immobilized anti-Tat antibodies. Both the optimized aptasensor and the immunosensor showed a detection limit of 0.25 ppm (Minunni et al., 2004, Tombelli et al., 2005a). The quartz crystal microbalance (QCM)-based aptasensor has also been compared with the corresponding surface plasmon resonance (SPR)-based aptasensor. The two aptasensors were constructed using biotin–avidin linking onto the gold surface of the transducers (quartz crystals or chips) for the immobilization chemistry. Both platforms showed similar reproducibility, sensitivity and specificity. The linear range of SPR (1–2.5 ppm) was higher that of QCM (0–1.25 ppm) (Tombelli et al., 2005a).
Another viral aptasensor example is the heptatitis C virus (HCV) core antigen detector. Lee et al. (2007) selected and tested the binding affinity of several aptamer sequences. After selection, the core specific aptamer was immobilized in a 96-well plate, using the sol–gel-based immobilization method. Then, the immobilized aptamers on the chip were incubated with recombinant core antigens. After antigen binding, the aptamer–core complexes were incubated with Cy3-labeled secondary antibodies. The platform was able to detect core-specific interaction with the aptamers, using pure recombinant protein as well as human sera matrixes. The results showed that this platform can specifically detect the core antigen from HCV-infected patients’ sera (Lee et al., 2007).
Detection of bacteria using aptasensors is a relatively new area. Recently, two different strategies for whole-cell detection, using quantum dots (QDs) and carbon nanotubes (CNTs), have been reported. Aptamer-functionalized QDs have been used to detect Bacillus thuringiensis spores (Ikanovic et al., 2007). In the study, zinc sulfide-capped cadmium selenide QDs were functionalized with a specific aptamer selected to detect B. thuringiensis. After QD-aptamer incubation with the target, the spores were washed and collected for fluorescence measurement. Several controls with non-functionalized QDs and without spores were tested to measure the non-specific attachment of the QDs to the spores and the fluorescence background noise. The reported sensitivity was 103 CFU/ml. For specificity purposes, spores from B. globigii (B. subtilis var. niger) were also tested. The system could differentiate B. thuringiensis from B. globigii at concentrations above 105 CFU/ml. The second strategy was developed to detect E. coli DH5α, using aptamer-functionalized single-walled carbon nanotube field-effect transistor (SWNT-FET) arrays (So et al., 2008). The binding event between E. coli cells and the aptamer-functionalized FET produced a drop in conductance (>50%) in culture samples with concentrations between 105 and 107 CFU/ml. Specificity assays were conducted with S. Typhimurium.
Aptamer-conjugated nanoparticles have been developed to collect and detect cancer cells from complex matrices (Herr et al., 2006, Smith et al., 2007). Aptamer–magnetic nanoparticles were used for selective cell isolation, and aptamer–fluorescence nanoparticles were used to amplify the detection signal. Fluorescence was detected by confocal microscopy. The system was developed to detect leukemia and lymphoma cells with a detection limit of approximately 250 cells (Smith et al., 2007). These strategies can be potentially applied to the collection, concentration, and detection of microorganisms from complex matrices. Whole-cell aptamer detection platforms represent a promising alternative not only for clinical diagnostics, but also for foodborne and environmental pathogen detection. The principal challenge is the presence of multiple target proteins in the cell wall, which produces difficulty in the post-SELEX evaluation and binding standardization. Future successful application of aptamers in biosensors for whole-cell detection will be dependent upon the SELEX standardization for complex targets and the post-SELEX characterization of the obtained aptamers and their binding reaction with the cells.
4.2. Nanoparticle-based aptasensors
Although nanoparticle-based aptasensors have not yet been used for microbial detection, the following section is presented as an opportunity for biosensor research and development. Recently, several aptasensor platforms have been developed that include the use of nanoparticles, particularly gold nanoparticles (AuNPs) and QDs (Fischer et al., 2007, Li et al., 2008). The optical properties of AuNPs are commonly used in colorimetric detection. The surface resonance frequency of AuNPs can be modified, resulting in different detectable colors (Li et al., 2008). The chromatic changes result from the aptamer-functionalized AuNP aggregation after target recognition. Colorimetric assays do not require sophisticated detection apparatus (Balamurugan et al., 2008), and the detection can be performed in solution, avoiding the disadvantages of platform immobilization.
Liu and Lu (2006) developed two solution-based colorimetric detection systems, using AuNPs functionalized with aptamers to detect adenosine and cocaine. AuNP aggregates linked by oligonucleotide sequences containing the specific aptamer were constructed. In the presence of adenosine or cocaine, the aptamer changed its structure to bind to the target molecule. This resulted in disassembly of the aggregates which produced a change in the color system from purple to red (Fig. 2 ). The color change was instantaneous in the presence of 2 mM adenosine and 1 mM cocaine (Liu and Lu, 2006). In a similar system, successful colorimetric detection was described for platelet-derived growth factor (Huang et al., 2005).
Fig. 2.
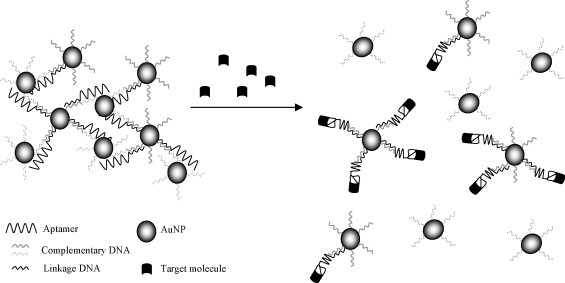
Schematic of gold nanoparticle aptasensor with colorimetric-based detection (adapted from Liu and Lu, 2006). Aptamer-functionalized gold nanoparticles were aggregated using aptamer complementary oligonucleotide sequences. In the presence of the target molecule, the aptamer changed its structure for the binding event. This resulted in disassembly of the aggregates which produced a change in color.
AuNPs can also be used as fluorescence quenchers for optical detection. Using thrombin as a detection model, Wang et al. (2008) investigated three different strategies (adsorption, covalent immobilization, and hybridization) for the AuNP surface modification with aptamers. The thiolated aptamer immobilization provided the best results with the highest constant affinity and the most sensitive detection limit (0.14 nM). For detection, after the thiolated aptamer was immobilized onto gold nanoparticles, dye-labeled complementary DNA was hybridized with the aptamer, which resulted in fluorescence quenching. With the addition of thrombin, the aptamer adopted a different conformation in order to bind with the target. Hence, the dye-labeled DNA was released from the AuNP surfaces, producing a detectable fluorescence signal (Wang et al., 2008).
Nanoparticles functionalized with bio-barcodes (small oligonucleotide sequences) have been used as antibody labels in sandwich detection systems to amplify detection and enhance sensitivity (Nam et al., 2003). The use of AuNP in the amplification of aptamer bio-barcodes has been investigated, using thrombin as target model (He et al., 2007). The proposed platform was based on a sandwich, label-free electrochemical detection. First, thrombin was captured by polyclonal antibodies immobilized in microtiter plates. The Ab–target complex was then recognized by biotin-25polyA-15aptamer-AuNP bio-barcodes. The aptamer was then released and collected from the AuNPs. Finally, the modified aptamers with the polyadenine oligonucleotides (bio-barcodes) were degraded by nuclease or acid, and differential pulse voltammetry was used to detect the well-defined adenine signal (Fig. 3 ). The thrombin detection limit was 0.1 ng/ml (He et al., 2007). Another sandwich-type assay to detect thrombin was used to develop a microgravimetric quartz crystal microbalance (QCM) platform, using AuNP for the detection. The thiolated capture aptamer was immobilized on the QCM electrode. After incubation with the target (thrombin), the aptamer–thrombin complex was recognized by the AuNPs functionalized with the detection aptamer. The AuNP attachment to the QCM surface provided initial thrombin analysis amplification, resulting in a frequency change. Secondary analysis amplification was obtained with the catalytic enlargement of AuNP in the presence of HAuCl4 and NADH, with a sensitivity of 20 nM. The use of aptamer-functionalized AuNPs as catalytic labels for thrombin analysis amplification in solution was also established (Pavlov et al., 2004).
Fig. 3.
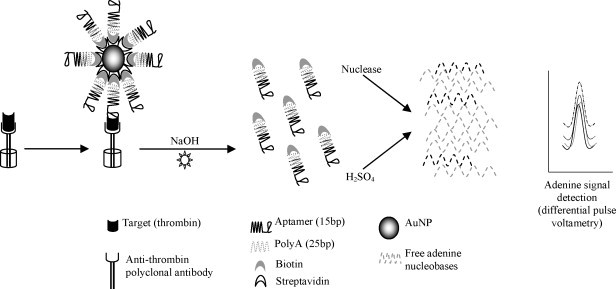
Schematic of gold nanoparticle aptasensor with bio-barcode-based detection (adapted from He et al., 2007). First, the target (thrombin) was captured by the immobilized antibodies. This Ab–target complex was then recognized in a “sandwich” type assay by the aptamer–bio-barcode functionalized gold nanoparticles. The aptamer–bio-barcodes were released and chemically degraded. The free bio-barcode nucleobases produced a specific signal detected by differential pulse voltammetry.
A sandwich-type assay was developed using platinum nanoparticles (PtNP) functionalized with a detection thrombin aptamer. The capture aptamer was immobilized on a gold electrode. After the aptamer–thrombin complex formation on the gold electrode surface, the secondary aptamer attached to the PtNP recognized the complex, and the PtNP catalyzed the H2O2 reduction process used for amperometric detection. This electrocatalytic nanoparticle system showed an improved sensitivity (1 nM) compared with the previous electrochemical aptasensors used for detecting thrombin (Polsky et al., 2006).
Semiconductive nanocrystals (QDs) have been widely used as fluorescent labels in several aptasensor applications. Liu et al. (2007) used two aptamer-functionalized QDs, with different fluorescence emission wavelength for simultaneous detection of adenosine and cocaine, containing AuNPs as quenchers. QDs were assembled with AuNPs by the specific aptamer, quenching the QD fluorescence emission. Addition of the targets (adenosine and cocaine) disassembled the aggregates, resulting in increased fluorescence. The targets were simultaneously detected by fluorescence and colorimetric measurements in solution (Liu et al., 2007). QD–aptamer systems have also been used to detect thrombin (Levy et al., 2005, Choi et al., 2006). Hansen et al. (2006) developed an electrochemical aptasensor using QDs to detect thrombin and lysozyme simultaneously. A gold electrode was functionalized with thiolated aptamers for each protein. QD-tagged proteins (CdS for thrombin and PbS for lysozyme) were bound to the corresponding aptamer into the gold substrate. In the presence of the target proteins, the QD–protein complexes were displaced and the remaining nanocrystals were electrochemically detected. The position and size of the corresponding metal peaks (Cd and Pb) in the voltammograms corresponded to the type and amount of the respective protein target (thrombin and lysozyme) (Hansen et al., 2006).
5. Conclusions
Complex-target SELEX can be used to isolate specific aptamers against microbial pathogens with potential application in molecular diagnostic platforms as well as infection inhibitor agents. One of the main disadvantages of using a whole-cell target approach in SELEX is that the specific target molecule is initially unknown, which can cause specificity problems and can also produce difficulty in the post-SELEX optimization of the binding assay between the obtained aptamer sequence and the target. Some alternatives to overcome these issues are the use of counter-selection steps with closely related species, and the use of deconvolution-SELEX steps to partition the initial aptamer pools against the multiple targets present in cell surfaces.
To date, most of the isolated aptamers against microorganisms have been selected for clinical applications. Aptamer application to detect environmental and foodborne pathogens is a promising area of research. Matrix complexity is one of the greatest challenges in molecular diagnostic application to foodborne pathogen detection. Several components regularly present in food samples can produce interference or cross-reaction in both immunology-based and nucleic acid-based detection systems. Therefore, additional isolation, concentration and/or purification of the microbial target is required before molecular detection, which increases the assay time and cost. However, the ability to select aptamers in real complex matrices represents a way to overcome these issues. Several aptasensor platforms for detecting pathogenic proteins have been developed, especially for viral proteins detection. Aptasensors for microbial whole-cell identification are a promising and challenging application, with potential advantages over existing immunological whole-cell biosensors.
References
- Adler A., Forster N., Homann M., Goringer H.U. Comb. Chem. High Throughput Screen. 2008;11(1):16–23. doi: 10.2174/138620708783398331. [DOI] [PubMed] [Google Scholar]
- Alocilja E.C., Radke S.M. Biosens. Bioelectron. 2003;18(5–6):841–846. doi: 10.1016/s0956-5663(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Bannantine J.P., Radosevich T.J., Stabel J.R., Sreevatsan S., Kapur V., Paustian M.L. Clin. Vaccine Immunol. 2007;14(5) doi: 10.1128/CVI.00022-07. 518–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagalkot V., Zhang L., Levy-Nissenbaum E., Jon S., Kantoff P.W., Langer R., Farokhzad O.C. Nano Lett. 2007;7(10):3065–3070. doi: 10.1021/nl071546n. [DOI] [PubMed] [Google Scholar]
- Baker B.R., Lai R.Y., Wood M.S., Doctor E.H., Heeger A.J., Plaxco K.W. J. Am. Chem. Soc. 2006;128(10):3138–3139. doi: 10.1021/ja056957p. [DOI] [PubMed] [Google Scholar]
- Balamurugan S., Obubuafo A., Soper S.A., Spivak D.A. Anal. Bioanal. Chem. 2008;390(4):1009–1021. doi: 10.1007/s00216-007-1587-2. [DOI] [PubMed] [Google Scholar]
- Bang G.S., Cho S., Kim B.G. Biosens. Bioelectron. 2005;21(6):863–870. doi: 10.1016/j.bios.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Biroccio A., Hamm J., Incitti I., De Francesco R., Tomei L. J. Virol. 2002;76(8):3688–3696. doi: 10.1128/JVI.76.8.3688-3696.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank M., Weinschenk T., Priemer M., Schlusener H.J. J. Biol. Chem. 2001;276(19):16464–16468. doi: 10.1074/jbc.M100347200. [DOI] [PubMed] [Google Scholar]
- Bruno J.G., Kiel J.L. Biosens. Bioelectron. 1999;14(5):457–464. doi: 10.1016/s0956-5663(99)00028-7. [DOI] [PubMed] [Google Scholar]
- Bruno J.G., Kiel J.L. Biotechniques. 2002;32(1):178–183. doi: 10.2144/02321dd04. [DOI] [PubMed] [Google Scholar]
- Cai H., Lee T.M.H., Hsing I.M. Sensor Actuat. B: Chem. 2006;114(1):433–437. [Google Scholar]
- Cerchia L., Hamm J., Libri D., Tavitian B., de Franciscis V. FEBS Lett. 2002;528(1–3):12–16. doi: 10.1016/s0014-5793(02)03275-1. [DOI] [PubMed] [Google Scholar]
- Cerchia L., Ducong F., Pestourie C., Boulay J., Aissouni Y., Gombert K., Tavitian B., de Franciscis V., Libri D. PLoS Biol. 2005;3(4) doi: 10.1371/journal.pbio.0030123. e123- [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cerchia L., Franciscis V. Methods Mol. Biol. 2007;361:187–200. doi: 10.1385/1-59745-208-4:187. [DOI] [PubMed] [Google Scholar]
- Chen F., Zhou J., Luo F.L., Mohammed A.B., Zhang X.L. Biochem. Biophys. Res. Commun. 2007;357(3):743–748. doi: 10.1016/j.bbrc.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Cheng C., Dong J., Yaho L., Chen A., Jia R., Huang L., Guo J., Shu Y., Zhang Z. Biochem. Biophys. Res. Commun. 2008;366(3):670–674. doi: 10.1016/j.bbrc.2007.11.183. [DOI] [PubMed] [Google Scholar]
- Choi J.H., Chen K.H., Strano M.S. J. Am. Chem. Soc. 2006;128(49):15584–15585. doi: 10.1021/ja066506k. [DOI] [PubMed] [Google Scholar]
- Chu T., Ebright J., Ellington A.D. Curr. Opin. Mol. Ther. 2007;9(2):137–144. [PubMed] [Google Scholar]
- Clark S.L., Remcho V.T. Electrophoresis. 2002;23(9):1335–1340. doi: 10.1002/1522-2683(200205)23:9<1335::AID-ELPS1335>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Collett J.R., Cho E.J., Ellington A.D. Methods. 2005;37(1):4–15. doi: 10.1016/j.ymeth.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Daniels D.A., Chen H., Hicke B.J., Swiderek K.M., Gold L. Proc. Natl. Acad. Sci. 2003;100(26):15416–15421. doi: 10.1073/pnas.2136683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisingh A.K. Handb. Exp. Pharmacol. 2006;173:341–357. doi: 10.1007/3-540-27262-3_17. [DOI] [PubMed] [Google Scholar]
- DeStefano J.J., Cristofaro J.V. Nucleic Acids Res. 2006;34(1):130–139. doi: 10.1093/nar/gkj426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington A.D., Szostak J.W. Nature. 1990;346(6287):818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Eulberg D., Klussmann S. Chembiochem. 2003;4(10):979–983. doi: 10.1002/cbic.200300663. [DOI] [PubMed] [Google Scholar]
- Fischer N., Tarasow T.M., Tok J.B.H. Curr. Opin. Chem. Biol. 2007;11(3):316–328. doi: 10.1016/j.cbpa.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Fitter S., James R. J. Biol. Chem. 2005;280(40):34193–34201. doi: 10.1074/jbc.M504772200. [DOI] [PubMed] [Google Scholar]
- Fukuda K., Vishnuvardhan D., Sekiya S., Hwang J., Kakiuchi N., Taira K., Shimotohno K., Kumar P.K.R., Nishikawa S. Eur. J. Biochem. 2000;267(12):3685–3694. doi: 10.1046/j.1432-1327.2000.01400.x. [DOI] [PubMed] [Google Scholar]
- Gopinath S.C.B., Misono T.S., Kawasaki K., Mizuno T., Imai M., Odagiri T., Kumar P.K.R. J. Gen. Virol. 2006;87(3):479–487. doi: 10.1099/vir.0.81508-0. [DOI] [PubMed] [Google Scholar]
- Guo K.T., SchAfer R., Paul A., Gerber A., Ziemer G., Wendel H.P. Stem Cells. 2006;24(10):2220–2231. doi: 10.1634/stemcells.2006-0015. [DOI] [PubMed] [Google Scholar]
- Hamula C.L.A., Guthrie J.W., Zhang H.Q., Li X.F., Le X.C. TRAC-Trend Anal. Chem. 2006;25(7):681–691. [Google Scholar]
- Hansen J.A., Wang J., Kawde A.N., Xiang Y., Gothelf K.V., Collins G. J. Am. Chem. Soc. 2006;128(7):2228–2229. doi: 10.1021/ja060005h. [DOI] [PubMed] [Google Scholar]
- He P.L., Shen L., Cao Y.H., Lia D.F. Anal. Chem. 2007;79(21):8024–8029. doi: 10.1021/ac070772e. [DOI] [PubMed] [Google Scholar]
- Herr J.K., Smith J.E., Medley C.D., Shangguan D.H., Tan W.H. Anal. Chem. 2006;78(9):2918–2924. doi: 10.1021/ac052015r. [DOI] [PubMed] [Google Scholar]
- Heyduk E., Heyduk T. Anal. Chem. 2005;77(4):1147–1156. doi: 10.1021/ac0487449. [DOI] [PubMed] [Google Scholar]
- Hianik T., Ostatna V., Zajacova Z., Stoikova E., Evtugyn G. Bioorg. Med. Chem. Lett. 2005;15(2):291–295. doi: 10.1016/j.bmcl.2004.10.083. [DOI] [PubMed] [Google Scholar]
- Homann M., Goringer H.U. Nucl. Acids Res. 1999;27(9):2006–2014. doi: 10.1093/nar/27.9.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann M., Ulrich H. Bioorg. Med. Chem. 2001;9(10):2571–2580. doi: 10.1016/s0968-0896(01)00032-3. [DOI] [PubMed] [Google Scholar]
- Huang C.C., Huang Y.F., Cao Z.H., Tan W.H., Chang H.T. Anal. Chem. 2005;77(17):5735–5741. doi: 10.1021/ac050957q. [DOI] [PubMed] [Google Scholar]
- Hwang B., Cho J.S., Yeo H.J., Kim J.H., Chung K.M., Hang K., Jang S.K., Lee S.W. RNA. 2004;10(8):1277–1290. doi: 10.1261/rna.7100904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikanovic M., Rudzinski W., Bruno J., Allman A., Carrillo M., Dwarakanath S., Bhahdigadi S., Rao P., Kiel J., Andrews C. J. Fluoresc. 2007;17:193–199. doi: 10.1007/s10895-007-0158-4. [DOI] [PubMed] [Google Scholar]
- Ikebukuro K., Kiyohara C., Sode K. Biosens. Bioelectron. 2005;20(10):2168–2172. doi: 10.1016/j.bios.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Ireson C.R., Kelland L.R. Mol. Cancer Ther. 2006;5(12):2957–2962. doi: 10.1158/1535-7163.MCT-06-0172. [DOI] [PubMed] [Google Scholar]
- James W. J. Gen. Virol. 2007;88(2):351–364. doi: 10.1099/vir.0.82442-0. [DOI] [PubMed] [Google Scholar]
- Jang K.J., Lee N.R., Yeo W.S., Jeong Y.J., Kim D.E. Biochem. Biophys. Res. Commun. 2008;366(3):738–744. doi: 10.1016/j.bbrc.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasena S.D. Clin. Chem. 1999;45(9):1628–1650. [PubMed] [Google Scholar]
- Jhaveri S.D., Kirby R., Conrad R., Maglott E.J., Bowser M., Kennedy R.T., Glick G., Ellington A.D. J. Am. Chem. Soc. 2000;122(11):2469–2473. [Google Scholar]
- Khati M., Schüman M., Ibrahim J., Sattentau Q., Gordon S., James W. J. Virol. 2003;77(23):12692–12698. doi: 10.1128/JVI.77.23.12692-12698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel J.L., Holwitt E.A., Parker J.E., Vivekananda J., Franz V. Proc. SPIE Conf. 2004;5617:382–387. [Google Scholar]
- Kiel J.L., Parker J.E., Holwitt E.A., Vivekananda J. Proc. SPIE Conf. 2004;5416:105–110. [Google Scholar]
- Kouassi G.K., Wang P., Sreevatan S., Irudayaraj J. Biotechnol. Progr. 2007;23(5):1239–1244. doi: 10.1021/bp0602101. [DOI] [PubMed] [Google Scholar]
- Kusser W. Rev. Mol. Biotechnol. 2000;74(1):27–38. doi: 10.1016/s1389-0352(99)00002-1. [DOI] [PubMed] [Google Scholar]
- Lazcka O., Del Campo F.J., Munoz F.X. Biosens. Bioelectron. 2007;22(7):1205–1217. doi: 10.1016/j.bios.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Lee J.F., Stovall G.M., Ellington A.D. Curr. Opin. Chem. Biol. 2006;10(3):282–289. doi: 10.1016/j.cbpa.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Lee J.O., So H.M., Jeon E.K., Chang H., Won K., Kim Y.H. Anal. Bioanal. Chem. 2008;390(4):1023–1032. doi: 10.1007/s00216-007-1643-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M., Walt D.R. Anal. Biochem. 2000;282(1):142–146. doi: 10.1006/abio.2000.4595. [DOI] [PubMed] [Google Scholar]
- Lee S., Kim Y.S., Jo M., Jin M., Lee D., Kim S. Biochem. Biophys. Res. Commun. 2007;358(1):47–52. doi: 10.1016/j.bbrc.2007.04.057. [DOI] [PubMed] [Google Scholar]
- Levy M., Cater S.F., Ellington A.D. Chembiochem. 2005;6(12):2163–2166. doi: 10.1002/cbic.200500218. [DOI] [PubMed] [Google Scholar]
- Li Y.L., Guo L., Zhang Z.Y., Tang J.J., Xie J.W. Sci. Chin. Ser. B-Chem. 2008;51(3):193–204. [Google Scholar]
- Liss M., Petersen B., Wolf H., Prohaska E. Anal. Chem. 2002;74(17):4488–4495. doi: 10.1021/ac011294p. [DOI] [PubMed] [Google Scholar]
- Liu J., Lee J.H., Lu Y. Anal. Chem. 2007;79(11):4120–4125. doi: 10.1021/ac070055k. [DOI] [PubMed] [Google Scholar]
- Liu J.W., Lu Y. Angew. Chem. Int. Ed. 2006;45(1):90–94. doi: 10.1002/anie.200502589. [DOI] [PubMed] [Google Scholar]
- McCauley T.G., Hamaguchi N., Stanton M. Anal. Biochem. 2003;319(2):244–250. doi: 10.1016/s0003-2697(03)00297-5. [DOI] [PubMed] [Google Scholar]
- McMasters S., Stratis-Cullum D.N. Proc. SPIE Conf. 2006;6380 63800B1–63800B8. [Google Scholar]
- Merino E.J., Weeks K.M. J. Am. Chem. Soc. 2005;127(37):12766–12767. doi: 10.1021/ja053189t. [DOI] [PubMed] [Google Scholar]
- Minunni M., Tombelli S., Gullotto A., Luzi E., Mascini M. Biosens. Bioelectron. 2004;20(6):1149–1156. doi: 10.1016/j.bios.2004.03.037. [DOI] [PubMed] [Google Scholar]
- Mir M., Vreeke M., Katakis L. Electrochem. Commun. 2006;8(3):505–511. [Google Scholar]
- Morris K.N., Jensen K.B., Julin C.M., Weil M., Gold L. Prod. Natl. Acad. Sci. 1998;95(6):2902–2907. doi: 10.1073/pnas.95.6.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J.M., Thaxton C.S., Mirkin C.A. Science. 2003;301:1884–1886. doi: 10.1126/science.1088755. [DOI] [PubMed] [Google Scholar]
- Ng E.W.M., Shima D.T., Calias P., Cunningham E.T., Guyer D.R., Adamis A.P. Nat. Rev. Drug Discov. 2006;5(2):123–132. doi: 10.1038/nrd1955. [DOI] [PubMed] [Google Scholar]
- Nitsche A., Kurth A., Dunkhorst A., Panke O., Sielaff H., Junge W., Muth D., Scheller F., Stoclein W., Dahmen C., Pauli G., Kage A. BMC Biotechnol. 2007;7(48) doi: 10.1186/1472-6750-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutiu R., Li Y.F. J. Am. Chem. Soc. 2003;125(16):4771–4778. doi: 10.1021/ja028962o. [DOI] [PubMed] [Google Scholar]
- O'Sullivan C.K. Anal. Bioanal. Chem. 2002;372(1):44–48. doi: 10.1007/s00216-001-1189-3. [DOI] [PubMed] [Google Scholar]
- Ohuchi S.P., Ohtsu T., Nakamura Y. Biochimie. 2006;88(7):897–904. doi: 10.1016/j.biochi.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Pagratis N.C., Bell C., Chang Y.F., Jennings S., Fitzwater T., Jellinek D., Dang C. Nat. Biotechnol. 1997;15(1):68–73. doi: 10.1038/nbt0197-68. [DOI] [PubMed] [Google Scholar]
- Pan Q., Zhang X.L., Wu H.Y., He P.W., Wang F., Zhang M.S., Hu J.M., Xia B., Wu J. Antimicrob. Agents Chemother. 2005;49(10):4052–4060. doi: 10.1128/AAC.49.10.4052-4060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W., Craven R.C., Qiu Q., Wilson C.B., Wills J.W., Golovine S., Wang J. Prod. Natl. Acad. Sci. 1995;92(25):11509–11513. doi: 10.1073/pnas.92.25.11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov V., Xiao Y., Shlyahovsky B., Willner I. J. Am. Chem. Soc. 2004;126(38):11768–11769. doi: 10.1021/ja046970u. [DOI] [PubMed] [Google Scholar]
- Polsky R., Gill R., Kaganovsky L., Willner I. Anal. Chem. 2006;78(7):2268–2271. doi: 10.1021/ac0519864. [DOI] [PubMed] [Google Scholar]
- Proske D., Blank M., Buhmann R., Resch A. App. Microbiol. Biotechnol. 2005;69(4):367–374. doi: 10.1007/s00253-005-0193-5. [DOI] [PubMed] [Google Scholar]
- Robertson D.L., Joyce G.F. Nature. 1990;344(6265):467–468. doi: 10.1038/344467a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez M.C., Kawde A.N., Wang J. Chem. Commun. 2005;(34):4267–4269. doi: 10.1039/b506571b. [DOI] [PubMed] [Google Scholar]
- Sando S., Ogawa A., Nishi T., Hayami M., Aoyama Y. Bioorg. Med. Chem. Lett. 2007;17(5):1216–1220. doi: 10.1016/j.bmcl.2006.12.013. [DOI] [PubMed] [Google Scholar]
- Shamah S.M., Healy J.M., Cload S.T. Acc. Chem. Res. 2008;41(1):130–138. doi: 10.1021/ar700142z. [DOI] [PubMed] [Google Scholar]
- Shangguan D., Li Y., Tang Z., Cao Z.H., Chen H.W., Mallikratchy P., Sefah K., Yang C.J., Tan W. Proc. Natl. Acad. Sci. 2006;103(32):11838–11843. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan D., Meng L., Cao Z.C., Xiao Z., Fang X., Li Y., Cardona D., Witek R.P., Liu C., Tan W. Anal. Chem. 2008;80(3):721–728. doi: 10.1021/ac701962v. [DOI] [PubMed] [Google Scholar]
- Smith J.E., Medley C.D., Tang Z.W., Shangguan D., Lofton C., Tan W.H. Anal. Chem. 2007;79(8):3075–3082. doi: 10.1021/ac062151b. [DOI] [PubMed] [Google Scholar]
- So H.M., Won K., Kim Y.H., Kim B.K., Ryu B.H., Na P.S., Kim H., Lee J.O. J. Am. Chem. Soc. 2005;127(34):11906–11907. doi: 10.1021/ja053094r. [DOI] [PubMed] [Google Scholar]
- So H.M., Park D.M., Jeon E.K., Kim Y.H., Kim B.S., Lee C.K., Choi S.Y., Kim S.C., Chang H., Lee J.O. Small. 2008;4(2):197–201. doi: 10.1002/smll.200700664. [DOI] [PubMed] [Google Scholar]
- Song S.P., Wang L.H., Li J., Zhao J.L., Fan C.H. TRAC-Trend Anal. Chem. 2008;27(2):108–117. [Google Scholar]
- Spiridonova V.A., Kopylov A.M. Biochemistry-Moscow. 2002;67(6):706–709. doi: 10.1023/a:1016110724564. [DOI] [PubMed] [Google Scholar]
- Stratis-Cullum D.N., Wade K.L., Pellegrino P.M. Proc. SPIE Conf. 2005;5994 59940D1–59940D9. [Google Scholar]
- Stratis-Cullum D.N., McMasters S., Sooter L.J., Pellegrino P.M. Proc. SPIE Conf. 2007;6554 65540Z1–65540Z8. [Google Scholar]
- Tang Z., Shangguan D., Wang K., Shi H., Sefah K., Mallikratchy P., Chen H.W., Li Y., Tan W. Anal. Chem. 2007;79(13):4900–4907. doi: 10.1021/ac070189y. [DOI] [PubMed] [Google Scholar]
- Tombelli S., Minunni A., Luzi E., Mascini M. Bioelectrochemistry. 2005;67(2):135–141. doi: 10.1016/j.bioelechem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Tombelli S., Minunni A., Mascini A. Biosens. Bioelectron. 2005;20(12):2424–2434. doi: 10.1016/j.bios.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Tombelli S., Minunni M., Mascini M. Biomol. Eng. 2007;24(2):191–200. doi: 10.1016/j.bioeng.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Tuerk C., Gold L. Science. 1990;249(4968):505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Ulrich H., Magdesian M.H., Alves M.J.M., Colli W. J. Biol. Chem. 2002;277(23):20756–20762. doi: 10.1074/jbc.M111859200. [DOI] [PubMed] [Google Scholar]
- Vivekananda J., Kiel J.L. Lab. Invest. 2006;86(6):610–618. doi: 10.1038/labinvest.3700417. [DOI] [PubMed] [Google Scholar]
- Wang W., Chen C., Qian M., Zhao X.S. Anal. Biochem. 2008;373(2):213–219. doi: 10.1016/j.ab.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Willner I., Zayats M. Angew. Chem. Int. Ed. 2007;46(34):6408–6418. doi: 10.1002/anie.200604524. [DOI] [PubMed] [Google Scholar]
- Xiao Z.Y., Shangguan D.H., Cao Z.H., Fang X.H., Tan W.H. Chem. Eur. J. 2008;14(6):1769–1775. doi: 10.1002/chem.200701330. [DOI] [PubMed] [Google Scholar]
- Yamamoto R., Katahira M., Nishikawa S., Baba T., Taira K., Kumar P.K.R. Genes Cells. 2000;5:371–388. doi: 10.1046/j.1365-2443.2000.00330.x. [DOI] [PubMed] [Google Scholar]
- Zayats M., Huang Y., Gill R., Ma C.A., Willner I. J. Am. Chem. Soc. 2006;128(42):13666–13667. doi: 10.1021/ja0651456. [DOI] [PubMed] [Google Scholar]
- Zhang Z.R., Blank M., Schluesener H.J. Arch. Immunol. Ther. Exp. 2004;52(5):307–315. [PubMed] [Google Scholar]
- Zhen B., Song Y.J., Guo Z.B., Wang J., Zhang M.L., Yu S.Y., Yang R.F. Acta Biochem. Biophys. Sin. 2002;34(5):635–642. [PubMed] [Google Scholar]