

Abstract
A series of 4-aminoquinoline derivatives were synthesized by the reaction of 4-chloro-7-substituted-quinolines with the corresponding mono/dialkyl amines. The structures of the synthesized compounds were confirmed by NMR and FAB-MS spectral and elemental analyses. Subsequently, the compounds were examined for their cytotoxic effects on two different human breast tumor cell lines: MCF7 and MDA-MB468. Although all compounds examined were quite effective on both cell lines, the compound N′-(7-chloro-quinolin-4-yl)-N,N-dimethyl-ethane-1,2-diamine emerged as the most active compound of the series. It was particularly potent against MDA-MB 468 cells when compared to chloroquine and amodiaquine. The compound butyl-(7-fluoro-quinolin-4-yl)-amine showed more potent effects on MCF-7 cells when compared to chloroquine. Therefore, 4-aminoquinoline can serve as the prototype molecule for further development of a new class of anticancer agents.
Keywords: 4-Aminoquinoline, Chloroquine, Cytotoxicity
1. Introduction
The global burden of cancer is huge and growing larger every year. According to the World Health Organization (WHO), more than 11 million people are diagnosed with cancer worldwide every year and this number is expected to increase to 16 million by the year 2020. More than 8 million, or 13% of overall deaths, are directly caused by cancer worldwide every year. The relative rate of cancer-caused mortality is even higher in the developed countries, as it accounts for more than 20% of all deaths [1]. Although cancer therapies have improved significantly in recent years, the development of chemoresistance has severely limited the choice of available anticancer drugs. This clearly highlights the urgent need for novel chemotherapeutic agents for more effective treatment of cancer [2], [3], [4], [5].
The 4-aminoquinoline scaffold is found in the majority of drugs commonly used for the treatment of malaria. The class of compounds containing a 4-aminoquinoline moiety ( Fig. 1) was the first to be used for the successful treatment of malaria, and is still the drug of choice [6], [7], [8]. Chloroquine (CQ) is a well-known anti-malarial drug possessing a 4-aminoquinoline scaffold, and it has been shown to have antiviral effects on the severe acute respiratory syndrome (SARS) causative agents [9], [10], [11]. CQ is currently in clinical trials as an investigational antiretroviral agent in humans with HIV-1/AIDS, who often develop tumors, particularly when survival has been prolonged by antiretroviral treatments [12], [13]. Recently, CQ has also been added to a conventional therapy protocol (i.e., surgery + radiotherapy + chemotherapy) for HIV-1 seronegative glioblastoma patients [14]. We have recently demonstrated that CQ significantly increases the cytotoxic effects on MDA-MB 231 cells when combined with radiation [15]. The present work is an extension of our ongoing efforts towards developing new, effective anti-cancer agents. To this end, we synthesized several 4-aminoquinoline derivatives and examined their cytotoxic effects on MDA-MB 468 and MCF-7 breast cancer cell lines. We found that some of these compounds are very effective.
Fig. 1.
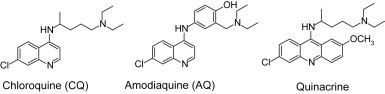
Some of the 4-aminoquinoline drug compounds.
2. Experimental
Melting points (m.p.) were taken in open capillaries on the Complab melting point apparatus. Elemental analysis was performed on a Perkin–Elmer 2400 C, H, N analyzer and values were within the acceptable limits of the calculated values. The 1H spectra were recorded on a DPX-200 MHz Bruker FT-NMR spectrometer using CDCl3 and DMSO-d 6 as solvent. The chemical shifts were reported as parts per million (δ ppm) tetramethylsilane (TMS) as an internal standard. Mass spectra were obtained on a JEOL-SX-102 instrument using fast atom bombardment (FAB positive). The progress of the reaction was monitored on ready-made silica-gel plates (Merck) using chloroform–methanol (9:1) as solvent. Iodine was used as a developing agent or by spraying with the Dragendorff's reagent. Chromatographic purification was performed over a silica gel (100–200 mesh). All chemicals and reagents obtained from Aldrich (USA) were used without further purification.
2.1. General synthetic procedure for butyl-(7-substituted-quinolin-4-yl)-amine (2,3)
A mixture of 7-substituted-4-chloro-quinoline (2.5 mmol) and butyl amine (5 mmol) was heated to 120–130 °C and maintained at this temperature for 6 h, with constantly stirring. The reaction mixture was cooled to room temperature and taken up in dichloromethane. The organic layer was washed with 5% aq. NaHCO3, followed by washing with water and then with brine. The organic layer was dried over anhydrous MgSO4 and solvent was removed under reduced pressure, and the residue was then precipitated by addition of 80:20 hexane:chloroform.
2.2. Synthesis of N1-(7-chloro-quinolin-4-yl)-ethane-1,2-diamine (4)
A mixture of 4,7-dichloroquinoline (2.5 mmol) and ethane-1,2-amine (5 mmol) was heated slowly to 80 °C over 1 h while stirring. The temperature was then increased to 130 °C where it was kept for 7 h while stirring continuously. The reaction mixture was cooled to room temperature and taken up in dichloromethane. The work-up procedure was the same as for compound 2.
2.3. General synthetic procedure for N′-(7-substituted-quinolin-4-yl)-N,N-dimethyl-ethane-1,2-diamine (5–8)
A mixture of 7-substituted-4-chloro-quinoline (2.5 mmol) and N,N-dimethyl-ethane-1,2-diamine (5 mmol) was heated to 120–130 °C, and then maintained at that temperature for 6–8 h while stirring continuously. The reaction mixture was cooled to room temperature and taken up in dichloromethane. The work-up procedure was the same as for compound 2.
2.4. Synthesis of N′-(7-chloro-quinolin-4-yl)-N,N-dimethyl-propane-1,3-diamine (9)
A mixture of 4,7-dichloroquinoline (500 mg, 2.5 mmol) and N,N-dimethyl-propane-1,3-diamine (5 mmol) was heated and maintained at 130 °C for 8 h while stirring continuously. The reaction mixture was cooled to room temperature and taken up in dichloromethane. The organic layer was successively washed with 5% aq. NaHCO3, water, and then brine. The organic layer was dried over anhydrous MgSO4 and solvent was removed under reduced pressure. Finally, the residue was purified by column chromatography over silica gel using chloroform–methanol as eluting agent.
2.5. Synthetic procedure for N,N′-bis-(7-substituted-quinolin-4-yl)-ethane-1,2-diamine (10,11)
A mixture of 7-substituted-4-chloro-quinoline (6.0 mmol) and ethane-1,2-diamine (2.5 mmol) was heated and maintained at 130 °C for 6 h while stirring continuously. The reaction mixture was cooled to room temperature and taken up in dichloromethane. The work-up procedure was the same as for compound 2.
2.6. Biological screening: in vitro cytotoxicity screening
The human cancer cell lines were grown in RPMI 1640 medium containing 10% fetal bovine serum and 5% penicillin:streptomycin. For a typical screening experiment, 5000–10,000 cells were inoculated into 100 μl medium per well of a 96-well microtiter plate. After the inoculation, the microtiter plate was incubated at 37 °C, in 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental drugs. After exposure to drugs for 24 h, cells were fixed in situ with TCA to represent a measurement of the cell population for each cell line at the time of drug addition (T z). Experimental drugs used were solubilized in dimethyl sulfoxide at 400-fold of the desired final maximum test concentration, and were then stored frozen prior to use. At the time of drug addition, an aliquot of the frozen stock was thawed and diluted twice to the desired final maximum test concentration with complete medium. Two-fold log serial dilutions were made to provide a total of seven drug concentrations plus a control. Aliquots of 100 μl of these different drug dilutions were added to the appropriate microtiter wells already containing 100 μl of medium, resulting in the required final drug concentrations. Following drug addition, the plate was incubated for an additional 48 h.
For adherent cells, the assay was terminated by the addition of cold TCA. Cells were fixed in situ by slowly adding 25 μl of cold 50% (w/v) TCA (final concentration, 10% TCA), and were then incubated for 60 min at 4 °C. The supernatant was discarded, and the plate was washed five times with tap water, and then air-dried. Sulforhodamine B (SRB) solution (100 μl) at 0.4% (w/v) in 1% acetic acid was added to each well, and the plate was incubated for 10 min at room temperature. Unbound SRB was removed by washing the plate five times with tap water, followed by air drying. The bound SRB was solubilized with 10 mM trizma base, and the absorbance was read on an automated plate reader at a wavelength of 515 nm. For suspension cells, the methodology was the same except that the assay was terminated by fixing settled cells at the bottom of the wells by gently adding 50 μl of 80% TCA (final concentration, 16% TCA). Using the seven-absorbance measurements (time zero (T z), control growth (C), and test growth in the presence of drug at the five concentration levels (Ti)), the percentage growth was calculated at each of the drug concentrations. Percentage GI was calculated as: [(T i − T z) = (C − T z)] × 100 for concentrations for which Ti > T z; [(Ti − T z)/T z] × 100 for concentrations for which Ti < T z.
Three dose–response parameters were calculated for each experimental agent. GI50 was calculated from [(Ti − T z)/(C − T z)] × 100 = 50, at which concentration a 50% reduction of the net protein increase (as measured by SRB staining) occurs. Values were calculated for each of these parameters if the level of activity was reached. However, if the effect was not reached or was exceeded, the value for that parameter was expressed as greater or less than the maximum or minimum concentration tested.
3. Results and discussion
Compounds described in this report were prepared as outlined in Scheme 1. The synthesis of desired 4-aminoquinoline compounds (2–11) were prepared by aromatic nucleophilic substitution on 7-substituted-4-chloro-quinoline (1) using excess of monoaminoalkane/diaminoalkane in neat conditions with a simple standard work-up procedure as described previously [16]. The alkyl aminoquinoline derivatives (2,3) were prepared by refluxing 7-substituted-4-chloro-quinoline with butyl amine in neat conditions. The aminoquinoline compound (4) was synthesized by refluxing 4,7-dichloroquinoline with an excess of ethane-1,2-diamine in which the temperature was raised slowly and maintained at 80 °C over 1 h while stirring. The temperature was raised further to 130 °C and maintained at that temperature for 7 h with continuous stirring. The dimethyl alkyl aminoquinoline derivatives (5–9) were also synthesized by a single-step reaction, in which appropriate 7-substituted-4-chloro-quinoline reacts with N,N-dimethyl-alkyl-diamine in neat conditions. The bisquinoline compounds (10,11) were obtained by excess of 7-substituted-4-chloro-quinoline reacting with one mole of alkyl diamine during which the temperature was kept at 130 °C for 6 h. The mass spectra of all the synthesized compounds were in conformity with their assigned structures. The mass spectra of these compounds showed molecular ion peaks corresponding to their molecular formulas ( Table 1). Elemental (C, H, N) analysis satisfactorily confirmed elemental compositions and the purity of the synthesized compounds (Table 1). Data from the 1H NMR spectra of these compounds ( Table 2) have been found to be in agreement with their assigned structures.
Scheme 1.
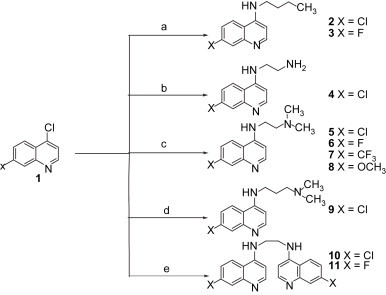
Table 1.
Physiochemical data of the synthesized compounds (2–11)
| Compound no. | Molecular formula | Elemental analysis calculated/founda |
MWb | Yield (%) | M.p. (°C) | ||
|---|---|---|---|---|---|---|---|
| %C | %H | %N | |||||
| 2 | C13H15ClN2 | 66.52/66.58 | 6.44/6.46 | 11.93/11.95 | 236 | 88 | 131–132 |
| 3 | C13H15FN2 | 71.53/71.51 | 6.93/6.91 | 12.83/12.85 | 219 | 82 | 126–127 |
| 4 | C11H12ClN3 | 59.60/59.58 | 5.46/5.44 | 18.95/18.93 | 222 | 85 | 122–123 |
| 5 | C13H16ClN3 | 62.52/62.56 | 6.46/6.44 | 16.83/16.85 | 251 | 76 | 109–110 |
| 6 | C13H16FN3 | 66.93/66.91 | 6.91/6.93 | 18.01/18.03 | 234 | 72 | 106–107 |
| 7 | C14H16F3N3 | 59.36/59.34 | 5.69/5.68 | 14.83/14.82 | 284 | 70 | 112–113 |
| 8 | C14H19N3O | 68.54/68.56 | 7.81/7.79 | 17.13/17.16 | 246 | 74 | 79–80 |
| 9 | C14H18ClN3 | 63.75/65.72 | 6.88/6.90 | 15.93/15.85 | 265 | 71 | 103–105 |
| 10 | C20H16Cl2N4 | 62.67/62.64 | 4.21/4.19 | 14.62/14.66 | 384 | 74 | 122–123 |
| 11 | C20H16F2N4 | 68.56/68.60 | 4.60/4.62 | 15.99/16.02 | 351 | 75 | 109–110 |
Elemental analyses for C, H, N were within 0.4% of the theorectical values.
Molecular weight was determined by mass spectra (FAB-MS m/z [M + H]+).
Table 2.
NMR spectral data of newly synthesized compounds (2–11)
| Compound no. | 1H-NMR (CDCl3) δ ppm |
|---|---|
| 2 | 0.69–0.77 (t, J = 7.2 Hz, 3H, CH3), 1.20–1.26 (m, 2H, CH2), 1.31–1.50 (m, 2H, CH2), 2.98–3.12 (m, 2H, CH2), 4.96 (br s, 1H, NH), 6.13-6.15 (d, J = 5.4 Hz, 1H, Ar-H quinoline), 6.96–7.11 (dd, J = 9.0, 2.2 Hz, 1H, Ar-H quinoline), 7.40–7.44 (d, J = 8.6 Hz, 1H, Ar-H quinoline), 7.68–7.69 (d, J = 1.8 Hz, 1H, 5H quinoline), 8.21–8.24 (d, J = 5.4 Hz, 1H, Ar-H quinoline). |
| 3 | 0.57–0.64 (t, J = 7.2 Hz, 3H, CH3), 1.02–1.32 (m, 2H, CH2), 2.70–7.79 (m, 2H, CH2), 2.92–2.97 (m, 2H, CH2), 5.94–6.00 (d, J = 6.2 Hz, 1H, Ar-H quinoline), 6.35 (br s, 1H, NH), 6.51–6.56 (dd, J = 9.0, 2.2 Hz, 1H, Ar-H quinoline), 6.85–6.87 (d, J = 2.6 Hz, 1H, Ar-H quinoline), 7.62–7.68 (d, J = 9.2 Hz, 1H, 5H quinoline), 7.80–7.73 (d, J = 6.2 Hz, 1H, Ar-H quinoline). |
| 4 | 3.09-3.15 (m, 4H, CH2), 3.27 (br s, 2H, NH2), 5.82 (br s, 1H, NH), 6.38–6.41 (d, J = 5.34 Hz, 1H, Ar-H quinoline), 7.32-7.37 (dd, J = 8.96, 1.72 Hz, 1H, Ar-H quinoline), 7.73–7.77 (d, J = 8.92 Hz, 1H, Ar-H quinoline), 7.94–7.95 (d, J = 1.92 Hz, 1H, Ar-H quinoline), 8.49–8.52 (d, J = 5.34 Hz, 1H, Ar-H quinoline). |
| 5 | 2.32 (s, 6H, N-(CH3)2), 2.67–2.73 (m, 2H, CH2), 3.25–3.31 (m, 2H, CH2), 5.95 (br s, 1H, NH), 6.37–6.40 (d, J = 5.6 Hz, 1H, Ar-H quinoline), 7.35–7.40 (dd, J = 8.8, 2.2 Hz, 1H, Ar-H quinoline), 7.70–7.75 (d, J = 9.2 Hz, 1H, Ar-H quinoline), 7.95–7.96 (d, J = 2.2 Hz, 1H, Ar-H quinoline), 8.53–8.56 (d, J = 5.6 Hz, 1H, Ar-H quinoline). |
| 6 | 2.27 (s, 3H, N-CH3), 2.29 (s, 3H, N-CH3), 2.61–2.69 (m, 2H, CH2), 3.23-3.31 (m, 2H, CH2), 4.81 (br s, 1H, NH), 6.18–6.21 (d, J = 5.2 Hz, 1H, Ar-H quinoline), 6.81–6.87 (dd, J = 9.4, 2.0 Hz, 1H, Ar-H quinoline), 6.96–6.97 (d, J = 2.2 Hz, 1H, Ar-H quinoline), 7.55–7.60 (d, J = 8.8 Hz, 1H, Ar-H quinoline), 8.37–8.40 (d, J = 5.6 Hz, 1H, Ar-H quinoline). |
| 7 | 2.33 (s, 6H, N-(CH3)2), 2.69–2.75 (m, 2H, CH2), 3.27–3.36 (m, 2H, CH2), 6.12 (br s, 1H, NH), 6.45–6.48 (d, J = 5.2 Hz, 1H, Ar-H quinoline), 7.57–7.62 (d, J = 9.2 Hz, 1H, Ar-H quinoline), 7.90–7.95 (d, J = 8.8, 2.2 Hz, 1H, Ar-H quinoline), 8.26 (d, 1H, Ar-H quinoline), 8.61–8.63 (d, J = 5.6 Hz, 1H, Ar-H quinoline). |
| 8 | 2.30 (s, 6H, N-(CH3)2), 2.64–2.70 (m, 2H, CH2), 3.25–3.33 (m, 2H, CH2), 3.93 (s, 3H, OCH3), 5.95 (br s, 1H, NH), 6.29–6.32 (d, J = 5.2 Hz, 1H, Ar-H quinoline), 7.04–7.10 (dd, J = 9.2, 2.2 Hz, 1H, Ar-H quinoline), 7.31–7.33 (d, J = 2.2 Hz, 1H, Ar-H quinoline), 7.66–7.71 (dd, J = 9.2 Hz, 1H, Ar-H quinoline), 8.47–8.49 (d, J = 5.4 Hz, 1H, Ar-H quinoline). |
| 9 | 2.31 (s, 6H, N-(CH3)2), 2.44–2.51 (m, 2H, CH2), 2.69–2.75 (m, 2H, CH2), 3.27–3.33 (m, 2H, CH2), 6.10 (br s, 1H, NH), 6.39–6.42 (d, J = 5.6 Hz, 1H, Ar-H quinoline), 7.37–7.42 (dd, J = 8.8, 2.2 Hz, 1H, Ar-H quinoline), 7.71–7.76 (d, J = 9.2 Hz, 1H, Ar-H quinoline), 7.96–7.97 (d, J = 2.2 Hz, 1H, Ar-H quinoline), 8.55–8.57 (d, J = 5.6 Hz, 1H, Ar-H quinoline). |
| 10 | 2.49–2.51 (m, 4H, CH2), 3.61 (br s, 2H, 2-NH), 6.58–6.61 (d, J = 5.2 Hz, 1H, Ar-H quinoline), 7.45–7.50 (m, 2H, Ar-H quinoline), 7.80 (s, 2H, Ar-H quinoline), 8.20–8.25 (d, J = 9.6 Hz, 1H, Ar-H quinoline), 8.40–8.43 (d, J = 5.6 Hz, 1H, Ar-H quinoline). |
| 11 | 2.47–2.50 (m, 4H, CH2), 3.95 (br s, 2H, 2-NH), 6.56-6.59 (d, J = 5.2 Hz, 1H, Ar-H quinoline), 7.47–7.52 (m, 2H, Ar-H quinoline), 7.82 (s, 2H, Ar-H quinoline), 8.22–8.27 (d, J = 9.6 Hz, 1H, Ar-H quinoline), 8.43–8.45 (d, J = 5.6 Hz, 1H, Ar-H quinoline). |
All the compounds synthesized were evaluated for their cytotoxicity against two cancer cell lines derived from human breast tumors. The drugs at the concentration of 100 μM were diluted to 1.625 μM by two-fold serial dilutions. Following the incubation for 48 h in a drug, the cells were treated with SRB to measure their growth/viability (% of the untreated control) using a spectrophotometer as described previously [17]. The reading of SRB staining is known to accurately reflect the levels of total cellular macromolecules [17]. The GI50 concentration for each derivative was calculated with reference to a standard curve (control cells), which represents the concentration that results in a 50% decrease in cell growth after 3 days of incubation. For each compound, the 50% growth inhibition (GI50) was determined and reported in Table 3. The data for CQ, amodiaquine and mefloquine were included for comparison. The data showed that these derivatives have significant cytotoxicity against the cell lines screened. Among the nine compounds tested, two compounds showed a GI50 range of 7.35–8.73 μM against MDA-MB468 cells, and one compound showed a GI50 at 8.22 μM against MCF-7 cells. The remaining seven compounds showed GI50 values above the 10.85 μM, but not more than 13.72 μM against MDA-MB468 cells. In the case of MCF-7 cells, the six compounds tested showed GI50 in the range of 11.52–14.47 μM, the remaining two compounds showed GI50 values above 36.77 μM, but not more than 51.57 μM. The differences in the GI50 values may be attributable to factors such as the nature of substitution at the seventh positions, the side-chains, and the genetic and biochemical background of the cell lines.
Table 3.
In vitro cytotoxicity of synthesized compounds (2–11)
| Compound no. | GI50 (μM)a |
|
|---|---|---|
| MDA-MB-468 | MCF-7 | |
| 2 | 13.72 ± 0.51 | 11.52 ± 0.25 |
| 3 | 10.85 ± 0.34 | 8.22 ± 0.10 |
| 4 | 11.01 ± 0.29 | 51.57 ± 0.59 |
| 5 | 8.73 ± 0.11 | 36.77 ± 0.45 |
| 6 | 11.47 ± 0.12 | 13.25 ± 0.28 |
| 7 | 12.85 ± 0.15 | 14.47 ± 0.31 |
| 8 | 14.09 ± 0.17 | 12.90 ± 0.27 |
| 9 | 10.86 ± 0.11 | 14.47 ± 0.33 |
| 10 | 7.35 ± 0.10 | 14.80 ± 0.35 |
| 11 | –b | –b |
| Chloroquine | 24.36 ± 0.25 | 20.72 ± 0.23 |
| Amodiaquine | 17.80 ± 0.09 | 13.01 ± 0.27 |
| Mefloquine | 18.25 ± 0.10 | 10.60 ± 0.68 |
GI50, concentration of drug to reduce cell number to 50% of control cultures.
Not determined.
As for compound 2, the replacement of the chloro group with a fluoro group (3) led to an increase in cytotoxicity against MDA-MB-468 cells. It also showed a similar result with MCF-7 cells. In case of the aminoquinoline compound 4 (GI50 = 11.01 μM), cytotoxicity increased by 2-fold in comparison to CQ (GI50 = 24.36 μM) against MDA-MB-468 cells. With MCF-7 cell lines, however, the compound 4 showed lower cytotoxicity (GI50 = 51.57 μM) than CQ (GI50 = 20.72 μM). The dimethyl alkyl aminoquinoline derivatives with the 7-chloro substitution of compound 5 showed drastically increased (5-fold) cytotoxicity (GI50 = 8.73 μM) against MDA-MB-468 cells in comparison to CQ. However, replacement of the chloro group with a fluoro (6, GI50 = 11.47 μM), trifluoromethyl (7, GI50 = 12.85 μM) and a methoxy group (8, GI50 = 14.09 μM) led to a decrease in cytotoxicity against MDA-MB-468 cells. Interestingly, in the MCF-7 cell lines, these results were in reverse order, and the compound substituted with a chloro group (5, GI50 = 36.77 μM) was less active in comparison to that of substituted with a methoxy group (8, GI50 = 12.90 μM). The bisquinoline compound 10 (GI50 = 7.35 μM) showed 3-fold increased activity in MDA-MB-468 cells, compared to CQ. It is interesting to note that the same compound showed lower activity (GI50 = 14.80 M) in the MCF-7 cell line. Compound 11 is insoluble in DMSO; therefore, we were not able to examine cytotoxicity on cultured cells. Further structural modifications are in progress to increase specificity and cytotoxicity on cancer cells by 4-aminoquinoline derivatives.
Acknowledgements
This work was supported by funds from the Canadian Breast Cancer Foundation (CBCF, Ontario Chapter), the Ontario Institute of Cancer Research/Cancer Care Ontario, and the Northern Cancer Research Foundation to HL.
References
- 1.NCI International Portfolio: Addressing the global challenge of cancer 2005. http://www.cancer.gov/nci-international-portfolio/allpages. National Cancer Institue. 2005.
- 2.Broxterman H.J., Georgopapadakou N.H. Anticancer therapeutics: “Addictive” targets, multi-targeted drugs, new drug combinations. Drug Resist Updat. 2005;8:183–197. doi: 10.1016/j.drup.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Broxterman H.J., Georgopapadakou N. Cancer research 2001: drug resistance, new targets and drug combinations. Drug Resist Updat. 2001;4:197–209. doi: 10.1054/drup.2001.0216. [DOI] [PubMed] [Google Scholar]
- 4.Broxterman H.J., Georgopapadakou N.H. New cancer therapeutics: target-specific in, cytotoxics out? Drug Resist Updat. 2004;7:79–87. doi: 10.1016/j.drup.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Broxterman H.J., Lankelma J., Hoekman K. Resistance to cytotoxic and anti-angiogenic anticancer agents: similarities and differences. Drug Resist Updat. 2003;6:111–127. doi: 10.1016/s1368-7646(03)00026-8. [DOI] [PubMed] [Google Scholar]
- 6.O'Neill P.M., Bray P.G., Hawley S.R., Ward S.A., Park B.K. 4-Aminoquinolines–past, present, and future: a chemical perspective. Pharmacol Ther. 1998;77:29–58. doi: 10.1016/s0163-7258(97)00084-3. [DOI] [PubMed] [Google Scholar]
- 7.O'Neill P.M., Ward S.A., Berry N.G., Jeyadevan J.P., Biagini G.A., Asadollaly E. A medicinal chemistry perspective on 4-aminoquinoline antimalarial drugs. Curr Top Med Chem. 2006;6:479–507. doi: 10.2174/156802606776743147. [DOI] [PubMed] [Google Scholar]
- 8.Vangapandu S., Jain M., Kaur K., Patil P., Patel S.R., Jain R. Recent advances in antimalarial drug development. Med Res Rev. 2007;27:65–107. doi: 10.1002/med.20062. [DOI] [PubMed] [Google Scholar]
- 9.Keyaerts E., Vijgen L., Maes P., Neyts J., Van R.M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem Biophys Res Commun. 2004;323:264–268. doi: 10.1016/j.bbrc.2004.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savarino A., Boelaert J.R., Cassone A., Majori G., Cauda R. Effects of chloroquine on viral infections: an old drug against today's diseases? Lancet Infect Dis. 2003;3:722–727. doi: 10.1016/S1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savarino A., Di T.L., Donatelli I., Cauda R., Cassone A. New insights into the antiviral effects of chloroquine. Lancet Infect Dis. 2006;6:67–69. doi: 10.1016/S1473-3099(06)70361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savarino A., Gennero L., Sperber K., Boelaert J.R. The anti-HIV-1 activity of chloroquine. J Clin Virol. 2001;20:131–135. doi: 10.1016/s1386-6532(00)00139-6. [DOI] [PubMed] [Google Scholar]
- 13.Boelaert J.R., Piette J., Sperber K. The potential place of chloroquine in the treatment of HIV-1-infected patients. J Clin Virol. 2001;20:137–140. doi: 10.1016/s1386-6532(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 14.Sotelo J., Briceno E., Lopez-Gonzalez M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144:337–343. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- 15.Zhao H., Cai Y., Santi S., Lafrenie R., Lee H. Chloroquine-mediated radiosensitization is due to the destabilization of the lysosomal membrane and subsequent induction of cell death by necrosis. Radiat Res. 2005;164:250–257. doi: 10.1667/rr3436.1. [DOI] [PubMed] [Google Scholar]
- 16.Solomon V.R., Puri S.K., Srivastava K., Katti S.B. Design and synthesis of new antimalarial agents from 4-aminoquinoline. Bioorg Med Chem. 2005;13:2157–2165. doi: 10.1016/j.bmc.2004.12.051. [DOI] [PubMed] [Google Scholar]
- 17.Skehan P., Storeng R., Scudiero D., Monks A., McMahon J., Vistica D. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]