

Graphical abstract
A new class of compounds that have been designed and synthesized based on a natural product, quercetin-3-β-galactoside, has been discovered to be effective inhibitors of SARS coronavirus 3CL protease (SARS-CoV 3CLpro), which could be developed as drug leads for SARS.
Keywords: SARS-CoV 3CLpro, Virtual screening, Quercetin-3-β-galactoside, Molecular modeling, Structure–activity relationships
Abstract
The 3C-like protease (3CLpro) of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) is one of the most promising targets for discovery of drugs against SARS, because of its critical role in the viral life cycle. In this study, a natural compound called quercetin-3-β-galactoside was identified as an inhibitor of the protease by molecular docking, SPR/FRET-based bioassays, and mutagenesis studies. Both molecular modeling and Q189A mutation revealed that Gln189 plays a key role in the binding. Furthermore, experimental evidence showed that the secondary structure and enzymatic activity of SARS-CoV 3CLpro were not affected by the Q189A mutation. With the help of molecular modeling, eight new derivatives of the natural product were designed and synthesized. Bioassay results reveal salient features of the structure–activity relationship of the new compounds: (1) removal of the 7-hydroxy group of the quercetin moiety decreases the bioactivity of the derivatives; (2) acetoxylation of the sugar moiety abolishes inhibitor action; (3) introduction of a large sugar substituent on 7-hydroxy of quercetin can be tolerated; (4) replacement of the galactose moiety with other sugars does not affect inhibitor potency. This study not only reveals a new class of compounds as potential drug leads against the SARS virus, but also provides a solid understanding of the mechanism of inhibition against the target enzyme.
1. Introduction
The emergence of severe acute respiratory syndrome (SARS) in November 2002 followed by its rapid transmission posed serious challenges to public health globally.1, 2, 3, 4, 5 By July 31, 2003, a total of 8096 SARS cases and 774 SARS-related fatalities were reported worldwide.6 Although there have been no further reports of new SARS cases since April 2004, the disease should still be regarded as a potential serious health threat. Therefore, there is still need to study the biology, epidemiology and pathogenesis of the SARS-CoV and develop effective therapeutics for its treatment as part of strategic measures to manage the impact and severity of future outbreaks.7
SARS-CoV, which bears similarities to other coronaviruses, is an enveloped, positive-strand RNA virus that contains a single-stranded RNA genome of ∼29,700 nucleotides.8, 9 This large viral genome is predominated by two overlapping open reading frames that are connected by a ribosomal frame shift site to encode two replicase polyproteins, pp1a and pp1ab, that mediate viral replication. The 3C-like protease (3CLpro)10 plays a key role in the proteolytic release of replicative proteins from their replicase precursor polyproteins. In view of its essentiality in viral replication, the 3CLpro represents an attractive target for the development of therapeutics against SARS-CoV.
Since the emergence of SARS, much concerted efforts had been made in solving the crystal structure of SARS-CoV 3CLpro,11, 12, 13 characterizing the structural determinants of enzyme catalysis or maturation mechanism,14, 15, 16, 17, 18 performing structure-based inhibitor design, virtual screening,19, 20, 21, 22 and biological activity assays against SARS-CoV 3CLpro or viral entry.7, 13, 23, 24, 25, 26, 27, 28, 29, 30 To date, a number of small molecules with inhibitory activity toward SARS-CoV 3CLpro or viral replication have been identified. However, no effective small molecule agent has thus far been validated as a drug to treat SARS. Hence, the identification of new chemical classes of inhibitors and characterization of their interaction mode with SARS-CoV 3CLpro remains a relevant pursuit in the search for chemotherapeutic agents for the disease.
Quercetin, first reported in 1955,31 is a water soluble flavanoid that serves as a building block for other members of the flavonoid family. This natural compound has been reported to have ameliorative effects on a host of disorders including cancer, renal, and cardiovascular disease,32, 33, 34, 35 and have inhibitory activity toward SARS-CoV 3CLpro or viral replication.36, 37
In this work, quercetin-3-β-galactoside (1) was identified as a potent inhibitor of SARS-CoV 3CLpro by molecular docking studies and enzymatic inhibition assays. This compound showed inhibitory activity against SARS-CoV 3CLpro with IC50 of 42.79 ± 4.97 μM in a competitive mode. Molecular modeling strongly suggested that the residue Q189 plays a key role in the binding between quercetin-3-β-galactoside and SARS-CoV 3CLpro. To confirm this binding model, site-directed mutagenesis of the 3CLpro protease was performed. Comparative studies between SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A using CD spectroscopy, fluorescence spectra, and enzyme activity assays were conducted to determine the mechanistic effects of the Q189A mutation on enzyme–inhibitor interaction. The results indicated that while SARS-CoV 3CLpro Q189A retained the same level of biological activity as its wild-type counterpart, the inhibitory potency of quercetin-3-β-galactoside on SARS-CoV 3CLpro Q189A was decreased due to a reduction in binding affinity. To further explore the mode of binding interaction between the natural product and SARS-CoV 3CLpro, and discover more potent inhibitors, we systematically designed, with the aid of structural information from molecular modeling, and synthesized eight quercetin-3-β-galactoside derivatives (2a–h). We then tested their inhibitory activities against SARS-CoV 3CLpro. The results showed that removing the hydroxy groups on the quercetin moiety (2f–h) substantially decreased inhibitory activity; acetoxylation of the sugar moiety (2a) was unfavorable for the inhibitory activity of the compound; and introduction of a large sugar substituent on 7-hydroxy of quercetin (2e) did not adversely affect the ability of the inhibitor to functionally disrupt the SARS-CoV 3CLpro protease activity. All the experimental results were in good agreement with the molecular binding model. Thus, this work not only demonstrates quercetin-3-β-galactoside and its derivatives as a new class of SARS-CoV 3CLpro inhibitors, but also clarifies the mode of binding interaction between the flavonoids and SARS-CoV 3CLpro. The elucidation of a legible pharmacophore of SARS-CoV 3CLpro inhibitors lays the foundation for future rational drug design.
2. Results and discussion
2.1. Molecular docking result and possible inhibition mechanism of quercetin-3-β-galactoside against SARS-CoV 3CLpro
A bioflavanoid, quercetin-3-β-galactoside (1), was discovered by virtual screening as a potent binder to the catalytic pocket of SARS-CoV 3CLpro. The predicted binding free energy, inhibitory constant, and the predicted Log P are −9.24 kcal/mol, 1.70 × 10−7, and 0.43, respectively. Figure 1 shows the details of the specific interactions between quercetin-3-β-galactoside and the enzyme active site. Clearly, the molecule forms hydrophobic interactions with residues Leu141, Asn142, Met165, and Glu166 of SARS-CoV 3CLpro (Fig. 1A). Meanwhile, the inhibitor forms six hydrogen bonds with residues in the vicinity of the catalytic binding pocket (Fig. 1). The N atom of main chain of Glu166 forms two hydrogen bonds with quercetin-3-β-galactoside with distances of 2.88 and 3.28 Å, respectively, while the Oε1 and Nε2 atoms of the Gln189 side chain form four hydrogen bonds with distances of 2.62, 2.69, 3.44, and 3.68 Å, respectively. This binding model points to Gln189 as a crucial residue providing the key interaction elements for the binding of quercetin-3-β-galactoside with SARS-CoV 3CLpro. Logically then, substitution of the polar Gln189 residue to an aliphatic alanine residue would reduce the number of hydrogen bond interaction sites and thus significantly weaken the binding affinity. From the crystal structure of SARS-CoV 3CLpro,12 it is shown that Gln189 lies in the long loop region (residues 185–200), separated from the active sites of SARS-CoV 3CLpro. Therefore, the Q189A mutation was not expected to adversely affect the catalytic capability of the enzyme but rather lead to a significant drop in its binding affinity to quercetin-3-β-galactoside. Thus, this mutational model provides a good index for assessing the correctness of our proposed docking released binding mechanism. In addition, it can be observed from Figure 1B that there is ample spatial capacity within the binding pocket of the enzyme around the 7-hydroxy group of quercetin, suggesting that this site can accommodate significant structural modification with larger chemical moieties around 7-hydroxy of quercetin (2e).
Figure 1.
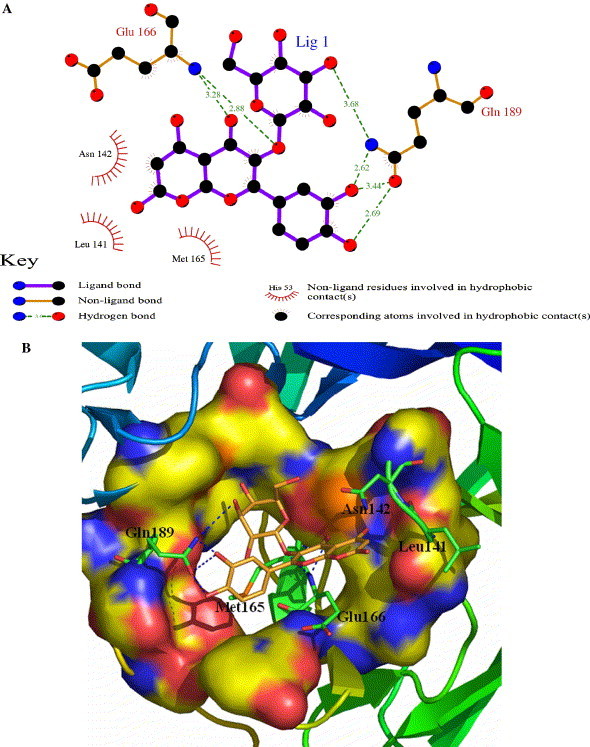
The main interactions between quercetin-3-β-galactoside and SARS-CoV 3CLpro. (A) The binding conformations of quercetin-3-β-galactoside and SARS-CoV 3CLpro. The N atom of main chain of Glu166 forms two hydrogen bonds with quercetin-3-β-galactoside. The side chain of Gln189 accepts four strong hydrogen bonds with quercetin-3-β-galactoside, respectively. (B) Three dimensional representation of the binding between quercetin-3-β-galactoside and SARS-CoV 3CLpro. The blue dash denotes hydrogen bonds.
2.2. Characterization of SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A
The His-tag recombinant SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A were successfully expressed in Escherichia coli strain M15 and purified by affinity chromatography to yield 12 and 16 mg of the purified proteins per liter of culture, respectively. The purity and molecular weight of the two proteases were assessed by SDS–PAGE. CD spectra of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A were recorded on a JASCO-810 spectropolarimeter collected with a thermal controller. Far-UV spectra of the two proteins give a positive peak at 196 nm that is characteristic of a β-sheet structure. Negative peaks at 209 and 222 nm obtained for SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A, respectively, are typical of an α-helical structure (Fig. 2 A). Thus, the CD spectra demonstrated that the secondary structure of SARS-CoV 3CLpro Q189A is similar to that of SARS-CoV 3CLpro.14 The evidence from CD analysis excludes the possibility of structural misfolding resulting from the mutagenesis of Gln189. Figure 2B shows the fluorescence spectra of the two proteases to be identical with a major peak at the wavelength of 330 nm.
Figure 2.
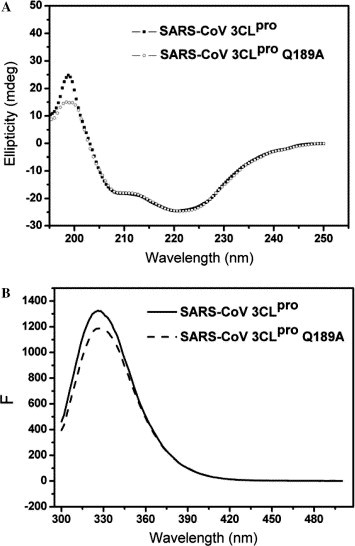
Characterization studies of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A proteases. (A) CD spectra of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A Far-UV CD spectra of SARS-CoV 3CLpro (–■–) and SARS-CoV 3CLpro Q189A (–○–) at 25 °C. Protein concentration used in CD experiments is 10 μM, and the two protein samples were prepared in 20 mM Tris–HCl, pH 7.4, 120 mM NaCl. (B) Fluorescence spectra of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A fluorescence spectra of SARS-CoV 3CLpro (—) and SARS-CoV 3CLpro Q189A (…) at 25 °C. Protein concentration used in the experiment is 10 μM, and two protein samples were prepared in 20 mM Tris–HCl, pH 7.4, 120 mM NaCl.
The enzymatic activity of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A was determined by cleavage of the fluorogenic substrate Dabcyl-KNSTLQSGLRKE-Edans according to the published method.28 Based on the double reciprocal plots of the initial velocity (v 0) and substrate concentration of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A (Figs. 3 A and B), the kinetic parameters (K m and k cat) were obtained. The determined K m values of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A are 49.38 ± 5.80 and 38.17 ± 2.60 μM, respectively. The k cat values of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A are 0.16 ± 0.01 and 0.14 ± 0.01 min−1. Therefore, the k cat/K m values of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A (3.2 ± 0.2 and 3.7 ± 0.3 mM min−1) are very close to each other, indicating that the activities of the two proteinases are similar in vitro. Hence, experimental data from CD spectroscopy, fluorescence spectra, and enzyme activity assay indicated that SARS-CoV 3CLpro mutant was structurally and functionally similar to the wild-type protease.
Figure 3.
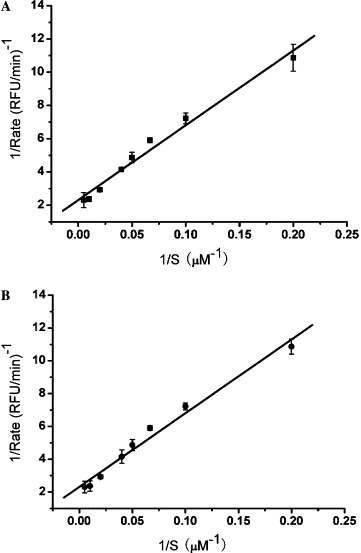
Kinetic parameters determination of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A. The enzymatic activity of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A was measured by the fluorescence resonance energy transfer (FRET) method. SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A (1 μM) and the fluorogenic substrate with different concentrations from 5 to 200 μM were incubated at 25 °C. The fluorescence intensity was monitored continuously for 1 h. The Km and kcat values were calculated by Lineweaver–Burk plot. (A) The double reciprocal plot of SARS-CoV 3CLpro, (B) the double reciprocal plot of SARS-CoV 3CLpro Q189A.
2.3. Binding affinity of quercetin-3-β-galactoside to SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A
The binding affinity of quercetin-3-β-galactoside to SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A in vitro was determined using surface plasmon resonance (SPR) biosensor technology. For kinetic analysis on the Biacore3000 instrument, various concentrations of quercetin-3-β-galactoside were injected for 120 min at a flow rate of 30 μL/min to allow for interaction with SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A immobilized on the sensor chip surface. The binding curves of quercetin-3-β-galactoside with SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A are shown in Figure 4 . For the SARS-CoV 3CLpro (Fig. 4A), quercetin-3-β-galactoside results in a significant and dose-dependent increase in SPR response units (RU) and presents characteristic fast-binding and slow-dissociation curves. The concentration series of quercetin-3-β-galactoside are fitted by the 1:1 Langmuir binding model in the Biacore3000 evaluation software for binding affinity determination. The dissociation constant (K D) between quercetin-3-β-galactoside and SARS-CoV 3CLpro was determined as 38.41 ± 2.06 μM. This result is consistent with our molecular modeling prediction that Q189A mutation would lead to a significant drop in binding affinity. While the residue is essential for the binding of the natural product, it is not a key residue for the function of the 3CL protease. Therefore, the modeled interaction mode between the natural product and the protease is accurate.
Figure 4.
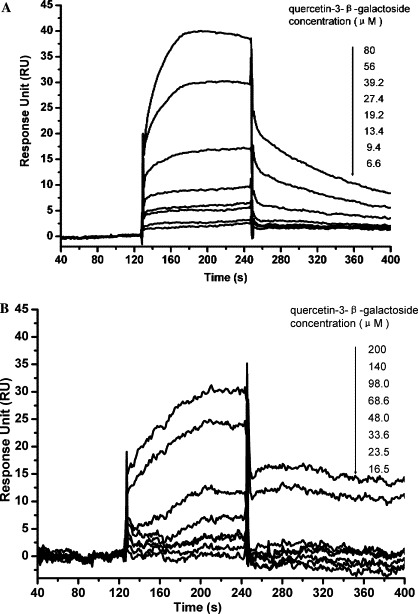
Binding affinity of quercetin-3-β-galactoside to SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A determined by SPR. Specificity and concentration-dependent binding and real-time binding affinity measurement of quercetin-3-β-galactoside to immobilized SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A (A and B) using Biacore 3000. For SARS-CoV 3CLpro, the concentrations of quercetin-3-β-galactoside were 80, 56, 39.2, 27.4, 19.2, 13.4, and 9.4 μM, while for SARS-CoV 3CLpro Q189A, the concentrations of quercetin-3-β-galactoside were 200, 140, 98, 68.6, 48.0, 33.6, 23.5, and 16.5 μM from top to bottom. Quercetin-3-β-galactoside was injected for 60 s, and dissociation was monitored for more than 120 s, respectively.
2.4. Design and synthesis of quercetin-3-β-galactoside derivatives (2a–h)
With the validation of our binding model between quercetin-3-β-galactoside and SARS-CoV 3CLpro, and taking into account the structural and functional similarity but contrasting binding interaction mode of the protease and its mutant (Fig. 1), we designed and synthesized compound 2a (Scheme 1 ) to determine if unprotected hydroxy sites on the sugar moiety are necessary for ligand–enzyme interaction, compounds 2f–h (Scheme 2, Scheme 3 ) for demonstrating the significance of the four hydroxy groups of the quercetin compound 1 (Scheme 1) as key interactive elements, and compound 2e for proving that significant spatial volume within the binding pocket of the enzyme around the 7-hydroxy group of quercetin available to accommodate large chemical moieties. Meanwhile, compounds 2b–d were prepared to determine if the type of sugar moiety would affect inhibitory activity. The details of the synthesis procedures and structural characterization of the compounds 1 and 2a–h are depicted in Scheme 1, Scheme 2, Scheme 3, and described in Section 4.
Scheme 1.
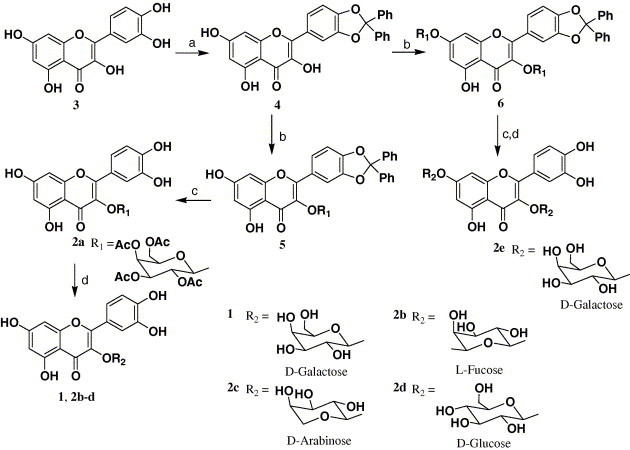
Synthesis of compounds 2a–2e. Reagents and conditions: (a) Ph2CCl2, 180 °C, 5 min; (b) O-acetylglycosyl bromide, K2CO3, DMF; (c) 10% Pd/C, H2; (d) i—MeONa, MeOH, ii—H+ resin.
Scheme 2.
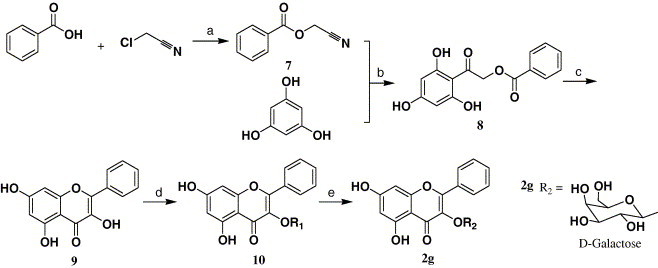
Synthesis of compound 2g. Reagents and conditions: (a) Et3N, CH2Cl2; (b) HCl (g), Et2O; (c) i—Bz2O, PhCO2Na, ii—KOH, EtOH, CO2 (g); (d) acetobromo-α-d-galactose, K2CO3, DMF; (e) i—MeONa, MeOH, ii—H+ resin.
Scheme 3.
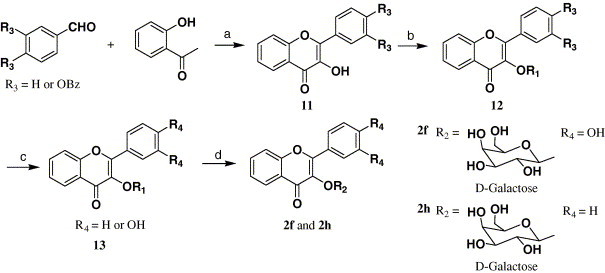
Synthesis of compounds 2f and 2h. Reagents and conditions: (a) i—KOH, EtOH, dioxane, ii—NaOH, 30% H2O2, EtOH; (b) acetobromo-α-d-galactose, K2CO3, DMF; (c) 10% Pd/C, H2; (d) i—MeONa, MeOH, ii—H+ resin.
2.5. Inhibitory activity of quercetin-3-β-galactoside (1) and its derivatives 2a–h
The inhibitory activities of quercetin-3-β-galactoside (1) toward the proteolytic activity of SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A were measured by FRET assay using a peptide substrate labeled with a pair of fluorogenic dyes. The inhibition of SARS-CoV 3CLpro slowly increased in the concentration range from 0 to 500 μM of quercetin-3-β-galactoside (Fig. 5 A). The curve finally reached steady state, which was dependent on the concentration of inhibitors. The IC50 value of the natural product in inhibiting the catalytic activity of SARS-CoV 3CLpro was calculated to be 42.79 ± 4.97 μM by fitting the dose–response curve using a logistic derivative equation in Origin 7.0.28 However, the IC50 value of quercetin-3-β-galactoside against SARS-CoV 3CLpro Q189A was estimated to be 127.89 ± 10.06 μM (Fig. 5B), which is 2-fold larger than that of SARS-CoV 3CLpro. Meanwhile, the double reciprocal plot of velocity as a function of substrate at varying inhibitor concentrations shown in Figure 6 suggests that quercetin-3-β-galactoside inhibits SARS-CoV 3CLpro in a competitive mode as well.
Figure 5.
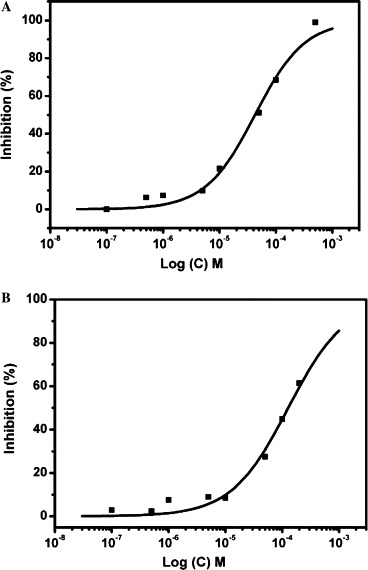
Dose–response plot for quercetin-3-β-galactoside on the proteolytic activity inhibition of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A. Inhibition of cleavage was measured by FRET using a peptide substrate labeled with a pair of fluorogenic dyes. IC50 values were calculated from the curves using nonlinear regression analysis. IC50 value for quercetin-3-β-galactoside on SARS-CoV 3CLpro is 42.79 ± 4.97 μM (A). IC50 value for quercetin-3-β-galactoside on SARS-CoV 3CLpro Q189A is 127.89 ± 10.06 μM (B).
Figure 6.
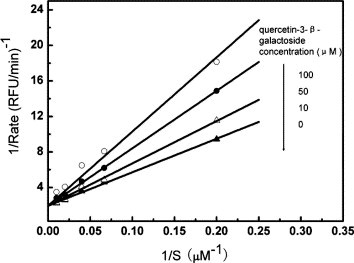
Enzyme kinetic analysis of SARS-CoV 3CLpro by quercetin-3-β-galactoside. The reaction was performed in the presence of different concentrations of quercetin-3-β-galactoside (▴, 0 μM; ▵, 10 μM; ○, 50 μM; •, 100 μM) at varying concentrations of the fluorogenic substrate (from 5 to 100 μM). Lineweaver–Burk plots of kinetic data were fitted to obtain the Ki value of 35.66 ± 4.57 μM.
The inhibition assays of compounds 2a–h against SARS-CoV 3CLpro were performed in the same manner as described for compound 1. The results was summarized in Table 1 . For the primary assay, the percent inhibitions of the compounds at 50 μM were measured. Of the synthetic derivatives tested, four of these—namely 2b, 2c, 2d, and 2e, displayed remarkable inhibitory action on the catalytic activity of SARS-CoV 3CLpro (expressed as percent inhibition at 50 μM > 50%), indicating that these are good candidate inhibitors of SARS-CoV 3CLpro. The IC50 values determined for these compounds were 24.14 ± 4.32, 31.62 ± 2.43, 48.85 ± 8.15, and 61.46 ± 9.13 μM, respectively.
Table 1.
The inhibitory activities and IC50 values of compounds 1 and 2a–h against SARS-CoV 3CLpro
| Compound | Inhibitory rate at 50 μM (%) | IC50 (μM) |
|---|---|---|
| 2a | 12.4 | — |
| 2b | 57.4 | 24.14 ± 4.32 |
| 2c | 49.4 | 31.62 ± 2.43 |
| 2d | 57.5 | 48.85 ± 8.15 |
| 2e | 53.0 | 61.46 ± 9.13 |
| 2f | 30.1 | — |
| 2g | 18.7 | — |
| 2h | NA | — |
| 1 | 41.8 | 42.79 ± 4.97 |
2.6. Structure and activity relationship of quercetin-3-β-galactoside derivatives (2a–h)
From the foregoing, the binding model of quercetin-3-β-galactoside with SARS-CoV 3CLpro provided valuable insights into the structural determinants involved in compound–target interaction, leading to rational design and synthesis of eight quercetin-3-β-galactoside derivatives containing systematic chemical variations. Biological evaluation of inhibitory activities of these analogues (Table 1) provides the basis to formulate the structure–activity relationship (SAR) The key elements observed are: (1) removing the hydroxy groups of the quercetin moiety (2f–h) substantially decreases the inhibitory activity of the derivatives, (2) acetoxylation of sugar moiety (2a) abolishes inhibitory functions, (3) introduction of a large sugar substituent on 7-hydroxy of quercetin (2e) can be tolerated, and (4) replacement of the galactose moiety with other sugars, such as fucose (2b), arabinose (2c), and glucose (2d), had no evident effect on inhibitor potency. These observations were in good agreement with the molecular binding model.
3. Conclusion
A natural compound, quercetin-3-β-galactoside that has been shown to be effective for treating allergies and preventing heart disease and cancer, was identified as a new class of inhibitors against SARS-CoV 3CLpro through molecular docking, SPR and FRET bioassays, and mutagenesis studies. Both molecular modeling and G189A mutation revealed that Gln189 plays a key role in the binding of quercetin-3-β-galactoside to SARS-CoV 3CLpro. Furthermore, comparative studies of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A employing CD spectroscopy, fluorescence spectra, and enzyme activity assay revealed no evidence that secondary structure and biological activity of the protease are affected by the mutation. Eight new derivatives of the quercetin-3-β-galactoside containing systematic chemical variations to binding elements were rationally designed under the guidance of molecular modeling and successfully synthesized. The bioassay data obtained from these new compounds demonstrate that the four hydroxy groups of the quercetin moiety are key determinants of the bioactivity of this type of inhibitors, that the spatial dimensions of the binding pocket in the vicinity of the sugar moiety and 7-hydroxy site of quercetin can accommodate large structural modifications, and that the hydroxyl groups of the sugar moiety should be unprotected to allow compound–target interaction by hydrogen bonding. Our data provide new insights into the essential interactions of quercetin-3-β-galactoside with the binding pocket of SARS-CoV 3CLpro, thus leading to a legible pharmacophore of SARS-CoV 3CLpro inhibitors that will aid future inhibitor design.
4. Experimental procedures
4.1. Materials
All solvents and reagents with reagent grade or ultra-pure quality were purchased commercially and were used without further purifications. The vector pQE30 and the bacterial strain E. coli strain M15 were purchased from Qiagen. The chelating column and protein molecular weight marker for SDS–PAGE were from Amersham Pharmacia Biotech. Amicon Ultra-4 centrifugal filter devices were purchased from Millipore. The QuickChange site-directed mutagenesis kit was purchased from Stratagene (La Jolla, Calif). Analytical thin-layer chromatography (TLC) was performed on HSGF 254 (150–200 μm thickness, Yantai Huiyou Company, China). Yields were not optimized. Melting points of the compounds were measured in a capillary tube on a SGW X-4 melting point apparatus without correction. Nuclear magnetic resonance (NMR) spectra were obtained from a Brucker AMX-400 NMR (IS as TMS). Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low- and high-resolution mass spectra (LRMS and HRMS) were obtained using electrospray (ESI) produced by LCQ-DECA spectrometer.
4.2. Virtual screening
The crystal structure of SARS-CoV 3CLpro (PDB entry: 1UK4) was used as the target for virtual screening on our in-house drug-like filtered ACD database (17,000 molecules) by using DOCK4.0.38 Residues around the binding site were isolated for constructing the grids for docking screening, and the pocket composed by these residues is large enough to include residues of the binding pocket. During the docking calculations, Kollman-all-atom charges39 were assigned to the protein, and Geisterger–Hückel charges40 were assigned to the small molecules in the database. The conformational flexibility of the compounds from the database was considered during the docking. In the DOCK simulation, the ligand–receptor binding energy was taken to be approximately the sum of the van der Waals and electrostatic interaction energies. After the initial orientation and scoring evaluation, grid-based rigid body minimization was carried out for the ligand to locate the nearest local energy minimum within the SARS-CoV 3CLpro binding site. The top-3000 molecules obtained from the DOCK search were selected for further analyses. These molecules were re-scored by Sybyl/Cscore and the scoring function of AutoDock3.0.41 Finally, a natural product, quercetin-3-β-galactoside, was selected for bioassay, based on the scoring results.
4.3. Synthesis of quercetin-3-β-galactoside and its derivatives
4.3.1. 2-(2,2-Diphenylbenzo[d][1,3]dioxol-5-yl)-3,5,7-trihydroxy-4H-chromen-4-one (4)
A mixture of quercetin 3 (1 g, 3 mmol) and Ph2CCl2 (1.7 mL, 8.9 mmol) was stirred at 180 °C for 10 min. The reaction mixture was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:4, v/v), to afford 4 (0.5 g, 37%) as a yellow solid. Mp 239–240 °C, 1H NMR (DMSO) δ: 6.22 (1H, d), 6.50 (1H, d), 7.26 (1H, d), 7.44–7.60 (10H, m), 7.80–7.83 (2H, m).
4.3.2. 2-(2,2-Diphenylbenzo[d][1,3]dioxol-5-yl)-5,7-dihydroxy-3-tetraacetylgalactosyl-4H-chromen-4-one (5) and 2-(2,2-diphenylbenzo[d][1,3]dioxol-5-yl)-5-hydroxy-3,7-di(tetraacetylgalactosyl)-4H-chromen-4-one (6)
A mixture of 4 (0.3 g, 0.64 mmol), acetobromo-α-d-galactose (0.4 g, 0.97 mmol), and K2CO3 (0.12 g, 0.9 mmol) in DMF (5 mL) was stirred at rt for 10 h under argon. The reaction mixture was poured into H2O and extracted with EtOAc. The combined organic layer was washed, dried, filtered, and condensed. The crude product obtained was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:4, 1:2 v/v), to afford 5 (0.16 g, 31%) and 6 (0.18 g, 25%), respectively. MS-ESI 797 [M+H]+ and MS-ESI 1127 [M+H]+.
4.3.3. 2-(3′,4′-Dihydroxyphenyl)-5,7-dihydroxy-3-β-d-galactosyl-4H-chromen-4-one (1)
A mixture of 5 (160 mg, 0.2 mmol), 10% palladium on charcoal (20 mg), and EtOH (20 mL) was stirred at 25 °C for 24 h in an atmosphere of hydrogen. The catalyst was filtered and the filtrate was concentrated to dryness. The crude product obtained was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:1 v/v), to afford 2a (0.1 g, 79%). Mp 108–110 °C, 1H NMR(CDCl3) δ: 1.89 (3H, s), 2.01(3H, s), 2.10 (3H, s), 2.26 (3H, s), 3.89–4.02 (3H, m), 5.15 (1H, dd), 5.42 (2H, m), 5.82 (1H, d), 6.26 (1H, s), 6.37 (1H, s), 6.98 (1H, d), 7.51 (1H, d), 7.79 (1H, s), MS-ESI 633 [M+H]+.
To a solution of the 2a (50 mg, 0.079 mmol) in methanol (10 mL) was added NaOMe (5 mg, 0.09 mmol) and the resultant solution was stirred at rt for 1 h. The solution was neutralized by passage down a Dowex 50 (H+) ion-exchange resin. The resin was filtered and the filtrate was concentrated to afford 1 (30 mg, 81%). Mp 212–215 °C, 1H NMR(DMSO) δ: 3.20–3.62 (7H, m), 5.37 (1H, d), 6.19 (1H, d), 6.40 (1H, d), 6.82 (1H, d), 7.51 (1H, d), 7.66 (1H, d). MS-ESI 465 [M+H]+, HRMS (ESI) m/z: Calcd C21H21O12 [M+H]+ 465.1053. Found: 465.1067.
4.3.4. 2-(3′,4′-Dihydroxyphenyl)-5,7-dihydroxy-3-β-l-fucosyl-4H-chromen-4-one (2b)
In the same manner as described for 1, 2b was prepared from 4 and acetobromo-α-l-fucose, yield 80% (last step). Mp 172–174 °C, 1H NMR (DMSO) δ: 1.02 (3H, d), 3.30–3.60 (5H, m), 5.36 (1H, d), 6.23 (1H, d), 6.44 (1H, d), 6.86 (1H, d), 7.56 (1H, d), 7.71 (1H, d). MS-ESI 449 [M+H]+, HRMS (ESI) m/z: Calcd C21H21O11 [M+H]+ 449.1084. Found: 449.1087.
4.3.5. 2-(3′,4′-Dihydroxyphenyl)-5,7-dihydroxy-3-β-d-arobinosyl-4H-chromen-4-one (2c)
In the same manner as described for 1, 2c was prepared from 4 and acetobromo-α-d-arabinose, yield 60% (last step). Mp 42–44 °C, 1H NMR(DMSO) δ: 3.20–3.80 (6H, m), 5.30 (1H, d), 6.23 (1H, d), 6.44 (1H, d), 6.87 (1H, d), 7.66 (1H, d), 7.69 (1H, d). MS-ESI 435 [M+H]+, HRMS (ESI) m/z: Calcd C20H19O11 [M+H]+ 435.0927. Found: 435.0909.
4.3.6. 2-(3′,4′-Dihydroxyphenyl)-5,7-dihydroxy-3-β-d-glucosyl-4H-chromen-4-one (2d)
In the same manner as described for 1, 2d was prepared from 4 and acetobromo-α-d-glucose, yield 48% (last step). Mp 172–174 °C, 1H NMR (DMSO) δ: 3.1–3.8 (7H, m), 5.49 (1H, d), 6.22 (1H, d), 6.43 (1H, d), 6.87 (1H, d), 7.61 (2H, m). MS-ESI 465 [M+H]+, HRMS (ESI) m/z: Calcd C21H21O12 [M+H]+ 465.1033. Found: 465.1067.
4.3.7. 2-(3′,4′-Dihydroxyphenyl)-5-hydroxy-3,7-di(β-d-galactosyl)-4H-chromen-4-one (2e)
In the same manner as described for 1, 2e was prepared from 6, yield 56% (two steps). Mp 165–168 °C, 1H NMR (DMSO): δ 3.30–3.70 (12H, m), 5.06 (1H, d), 5.43 (1H, d), 6.46 (1H, s), 6.76 (1H, s), 6.86 (1H, d), 7.57 (1H, s), 7.70 (1H, d); ESI-MS m/z: 627 [M+H]+. HRMS (ESI) m/z: Calcd C27H31O17 [M+H]+ 627.1561. Found: 627.1560.
4.3.8. Cyanomethyl benzoate (7)
A mixture of benzoic acid (4.9 g, 40 mmol), 2-chloroacetonitrile (4.5 g, 60 mmol), and Et3N (8 g, 80 mmol) in CH2Cl2 (25 mL) was stirred at rt for 48 h. The reaction mixture was poured into H2O, and extracted with EtOAc. The combined organic layer was washed, dried, filtered, and condensed, to afford 7 (5.2 g, 81%).
4.3.9. 3,5,7-Trihydroxy-2-phenyl-4H-chromen-4-one (9)
Compound 8 was synthesized from compound 7 and phloroglucinol using Chen’s method.42 A mixture of 8 (2 g, 6.9 mmol), benzoic anhydride (8.1 g, 35.8 mmol), and sodium benzoate (1.6 g, 11.2 mmol) was stirred at 170 °C for 3 h. To the reaction mixture were added KOH (5 g) and EtOH (51 mL). After refluxing for 0.5 h, the greater part of the alcohol was evaporated, the residue was dissolved in water and subsequently precipitated as a brown solid by saturating the liquid with carbon dioxide, collected, washed, and dried, to afford 9 (1.1 g, 59%). Mp 175–176 °C, 1H NMR (DMSO) δ: 6.25 (1H, d), 6.50 (1H, d), 7.61 (3H, m), 8.20 (2H, m).
4.3.10. 2-Phenyl-5,7-dihydroxy-3-β-d-galactosyl-4H-chromen-4-one (2g)
In the same manner as described for 1, 2g was prepared from 9, yield 66% (last step). Mp 162–164 °C, 1H NMR (DMSO): δ 3.30–3.58 (5H, m), 3.66 (1H, d), 5.46 (1H, d), 6.26 (1H, d), 6.48 (1H, d), 7.54 (3H, m), 8.17 (2H, m); ESI-MS m/z: 433 [M+H]+. HRMS (ESI) m/z: Calcd C21H21O10 [M+H]+ 433.1135. Found: 433.1138.
4.3.11. 2-(3′,4′-bis(Benzyloxy)phenyl)-3-hydroxy-4H-chromen-4-one (11)
Compound 11 was synthesized using the Frederique’s method43 from 2-hydroxyacetophenone and 3,4-dibenzyloxybenzaldehyde.
4.3.12. 2-(3′,4′-Dihydroxyphenyl)-3-β-d-galactosyl-4H-chromen-4-one (2f)
In the same manner as described for 1, 2f was prepared from 11 (R3 = OBz), yield 61% (last step). Mp 156–158 °C, 1H NMR (DMSO): δ 3.30–3.7 (6H, m), 5.45 (1H, d), 6.86 (1H, d), 7.52 (1H, t), 7.63 (1H, d), 7.74 (2H, d), 7.83 (1H, t), 8.10 (1H, d); ESI-MS m/z: 433 [M+H]+. HRMS (ESI) m/z: Calcd C21H21O10 [M+H]+ 433.1135. Found: 433.1175.
4.3.13. 2-Phenyl-3-β-d-galactosyl-4H-chromen-4-one (2h)
In the same manner as described for 1, 2h was prepared from 11 (R3 = H), yield 64% (last step). Mp 175–177 °C, 1H NMR (DMSO): δ 3.30–3.8 (6H, m), 5.53 (1H, d), 7.57 (4H, m), 7.78 (1H, d), 7.86 (1H, t), 8.12 (1H, d), 8.23 (2H, d); ESI-MS m/z: 401 [M+H]+. HRMS (ESI) m/z: Calcd C21H21O8 [M+H]+ 401.1236. Found: 401.1211.
4.4. Site-directed mutagenesis of SARS-CoV 3CLpro
Since Q189 was predicted from molecular modeling to be a key residue for inhibitory activity, site-directed mutagenesis was performed by a recombination PCR method previously described to test out this hypothesis. The plasmid, pQE30-SARS_3CLpro, was constructed according to Sun et al.44 The plasmid carrying the gene encoding the SARS-CoV 3CLpro Q189A mutant was prepared with the mutagenesis kit using pQE30-SARS_3CLpro as a template. A forward primer with a sequence of 5′-GGT CCA TTT GTT GAC AGA GCA ACT GCA CAG GCT GCA GG-3′ and a reverse primer 5′-CC TGC AGC CTG TGC AGT TGC TCT GTC AAC AAA TGG ACC-3′ were designed to amplify the DNA fragment encoding the entire mutant protease. Subsequently, QuickChange site-directed mutagenesis was performed in accordance with manufacturer’s instructions. The mutation was verified by DNA sequencing. After sequence analysis, the recombinant plasmid, designated pQE30-SARS_3CLpro Q189A, was transformed into competent E. coli strain M15 cells.
4.5. Protein preparation
Escherichia coli strain M15 cells containing pQE30-SARS_3CLpro were grown in 20 mL LB medium containing ampicillin (100 μg mL−1) and kanamycin (25 μg mL−1) at 37 °C overnight and then inoculated into 1 L LB supplemented with both antibiotics. The expression of SARS-CoV 3CLpro was induced by the addition of 0.5 mM of isopropyl β-d-thiogalactoside (IPTG). The cells were harvested by centrifugation at 8000g, 4 °C for 10 min after induction for 5 h at 22 °C. The pellet was washed, frozen, and then disrupted by sonication against the binding buffer (20 mM Tris–HCl, 0.5 M NaCl, and 10 mM imidazole, pH 8.0). The lysate was centrifuged at 18,000g for 30 min at 4 °C to pellet the cellular debris. The supernatant was then added to a HiTrap Ni2+ chelating column (1 mL) pre-equilibrated with the binding buffer, followed by washing with 20 mL of washing buffer (20 mM Tris–HCl, 0.5 M NaCl, and 60 mM imidazole, pH 8.0). The protease of interest was eluted with 10 mL elution buffer (20 mM Tris–HCl, 0.5 M NaCl, and 300 mM imidazole, pH 8.0). The freshly prepared protease was dialyzed against the appropriate buffer as required. The purified SARS-CoV 3CLpro was concentrated by Amicon Ultra-4 centrifugal filter devices (Millipore) with molecular cutoff at 10 kDa. The expression and purification of SARS-CoV 3CLpro Q189A were performed using the same procedure as for SARS-CoV 3CLpro. The purity and molecular weights of SARS-CoV 3CLpro and SARS-CoV 3CLpro Q189A were verified by SDS–PAGE.
4.6. Circular dichroism (CD) and fluorescence spectral analyses
CD spectra of the wild and mutant type protease were determined on a JASCO-810 spectropolarimeter collected with thermal controller according to Chen et al.16 The protein sample was prepared in 20 mM sodium phosphate, pH 7.4, 100 mM NaCl at 25 °C at a concentration of 10 μM. Far-UV CD spectra from 195 to 250 nm were collected with 1 nm bandwidth using a 0.1-cm path length cuvette and normalized by subtracting the baseline recorded for the buffer under identical conditions. The fluorescent measurement was performed on a HITACHI F-2500 fluorescence spectrophotometer connected with a thermostat. Two proteases were prepared in the same buffer and were diluted to the final concentration of 10 μM. The emission fluorescence intensity was monitored separately from 300 to 500 nm wavelengths upon excitation at 280 nm (slit width 2.5 nm) at 25 °C. The scan speed was 300 nm/min and the PMT voltage was 400 V. Each measurement was repeated three times, and the final result was the average of the three independent scans.
4.7. Enzyme activity assay
The activity of SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A was measured by cleavage of the fluorogenic substrate Dabcyl-KNSTLQSGLRKE-Edans based on the reported protocols.28 Briefly, the fluorescence intensity was monitored on a GENios microplate reader (TECAN) and the enzyme activity was determined by the increase in fluorescence upon continuous monitoring of the reactions in 96-well black microplates (BMG LABTECH) with the wavelengths of 340 and 488 nm for excitation and emission, respectively. Kinetic parameters (K m and k cat) of SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A were determined by incubation of the fluorogenic substrate with varied concentrations ranging from 5 to 200 μM, and 1 μM SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A at 25 °C. The reaction velocity (v 0) for each substrate concentration was averaged from three assay results. K m and k cat values were calculated by using a Lineweaver–Burk plot.
4.8. Inhibition activity assay
The inhibition activity of quercetin-3-β-galactoside or its derivatives against SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A was measured by a quenched fluoresence resonance energy transfer (FRET) assay with the fluorogenic substrate Dabcyl-KNSTLQSGLRKE-Edans. A stock solution of the substrate was dissolved in sterile H2O and stored at −20 °C. The SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A (1 μM) and the test compounds (from 0.1 to 500 μM) were incubated for 2 h at 4 °C and the reaction was initiated by adding the substrate to the desired final concentration (10 μM). The enzyme activity was determined by the increase in fluorescence upon continuous monitoring of the reactions in 96-well black microplates (BMG) using wavelengths of 340 and 488 nm for excitation and emission, respectively. Reactions were run with continuous monitoring of fluorescence for 60 min at 25 °C on a GENios (TECAN). The initial velocities of the inhibited reactions were plotted against the different inhibitor concentrations to obtain the IC50 values by fitting according to the analysis method as previously reported.28
4.9. Surface plasmon resonance (SPR) assay
Interaction studies between quercetin-3-β-galactoside and SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A were performed with the surface plasmon resonance based biosensor instrument Biacore 3000 (Biacore AB, Uppsala, Sweden). Quercetin-3-β-galactoside was dissolved in DMSO as a 20 mM stock solution for the Biacore experiments. SARS-CoV 3CLpro or SARS-CoV 3CLpro Q189A was immobilized on the sensor surface by the standard primary amine coupling reaction to the carboxymethylated matrix dextran of a sensor chip CM5 (Biacore AB, Uppsala, Sweden). Equilibration of the baseline was completed by continuous flow of HBS-EP running buffer (10 mM Hepes, 150 mM NaCl, 3 mM EDTA, and 0.005% (v/v) surfactant P20, pH 7.4) through the chip overnight. One of the four serial flow cells was activated for 7 min by injecting a 1:1 fresh mixture of 0.2 M N-ethyl-N′-dimethylaminopropyl carbodiimide (EDC) and 50 mM N-hydroxysuccinimide (NHS) at 25 °C. SARS-CoV 3CLpro was diluted with 10 mM sodium acetate buffer at pH 4.3 to a concentration of 25 μg mL−1; SARS-CoV 3CLpro Q189A was diluted with 10 mM sodium acetate buffer at pH 5.65 to a final concentration of 16 μg mL−1 and immobilized to the surface of sensor chip CM5, respectively. Finally, unreacted protease was blocked by injecting 1 M ethanolamine–HCl at pH 8.5 for 7 min, resulting in immobilized densities of 4000 RU. After stabilizing the baseline, Biacore data were collected at 25 °C with HBS–EP (containing 0.4% DMSO) as the running buffer at a constant flow of 30 μL/min. All the sensorgrams were processed by using automatic correction for non-specific bulk refractive index effects. The equilibrium constants (K Ds) evaluating the protein–inhibitor binding affinity were determined by the 1:1 Langmuir binding fitting model.
Acknowledgments
We gratefully acknowledge financial support from Shanghai Key R&D Program (Grants 036505003 and 05JC14092), National Key R&D Program (Grant 2005BA711A04), and 973 Program (Grant 2004CB518901).
References and notes
- 1.Drosten C., Preiser W., Gunther S., Schmitz H., Doerr H.W. Trends Mol. Med. 2003;9:325–327. doi: 10.1016/S1471-4914(03)00133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fouchier R.A., Kuiken T., Schutten M., van Amerongen G., van Doornum G.J., van den Hoogen B.G., Peiris M., Lim W., Stohr K., Osterhaus A.D. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt G.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., Lui S.F., Szeto C.C., Chung S., Sung J.J. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 4.Poutanen S.M., Low D.E., Henry B., Finkelstein S., Rose D., Green K., Tellier R., Draker R., Adachi D., Ayers M., Chan A.K., Skowronski D.M., Salit I., Simor A.E., Slutsky A.S., Doyle P.W., Krajden M., Petric M., Brunham R.C., McGeer A.J. N. Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 5.Tsang K.W., Ho P.L., Ooi G.C., Yee W.K., Wang T., Chan-Yeung M., Lam W.K., Seto W.H., Yam L.Y., Cheung T.M., Wong P.C., Lam B., Ip M.S., Chan J., Yuen K.Y., Lai K.N. N. Engl. J. Med. 2003;348:1977–1985. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- 6.Anonymous, World Health Organization, 2004, <http://www.who.int/csr/sars/country/table2003_2009_2023/en//>.
- 7.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 9.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 10.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 12.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuo C.J., Chi Y.H., Hsu J.T., Liang P.H. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C., Wei P., Fan K., Liu Y., Lai L. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- 16.Chen S., Chen L., Tan J., Chen J., Du L., Sun T., Shen J., Chen K., Jiang H., Shen X. J. Biol. Chem. 2005;280:164–173. doi: 10.1074/jbc.M408211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi J., Wei Z., Song J. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu M.F., Kuo C.J., Chang K.T., Chang H.C., Chou C.C., Ko T.P., Shr H.L., Chang G.G., Wang A.H., Liang P.H. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou K.C., Wei D.Q., Zhong W.Z. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajnarayanan R.V., Dakshanamurthy S., Pattabiraman N. Biochem. Biophys. Res. Commun. 2004;321:370–378. doi: 10.1016/j.bbrc.2004.06.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong B., Gui C.S., Xu X.Y., Luo C., Chen J., Luo H.B., Chen L.L., Li G.W., Sun T., Yu C.Y., Yue L.D., Duan W.H., Shen J.K., Qin L., Shi T.L., Li Y.X., Chen K.X., Luo X.M., Shen X., Shen J.H., Jiang H.L. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 22.Liu Z., Huang C., Fan K., Wei P., Chen H., Liu S., Pei J., Shi L., Li B., Yang K., Liu Y., Lai L. J. Chem. Inf. Model. 2005;45:10–17. doi: 10.1021/ci049809b. [DOI] [PubMed] [Google Scholar]
- 23.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 24.Hsu J.T., Kuo C.J., Hsieh H.P., Wang Y.C., Huang K.K., Lin C.P., Huang P.F., Chen X., Liang P.H. FEBS Lett. 2004;574:116–120. doi: 10.1016/j.febslet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu C.Y., Jan J.T., Ma S.H., Kuo C.J., Juan H.F., Cheng Y.S., Hsu H.H., Huang H.C., Wu D., Brik A., Liang F.S., Liu R.S., Fang J.M., Chen S.T., Liang P.H., Wong C.H. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kao R.Y., To A.P., Ng L.W., Tsui W.H., Lee T.S., Tsoi H.W., Yuen K.Y. FEBS Lett. 2004;576:325–330. doi: 10.1016/j.febslet.2004.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C.N., Lin C.P., Huang K.K., Chen W.C., Hsieh H.P., Liang P.H., Hsu J.T. Evid. Based Complement. Altern. Med. 2005;2:209–215. doi: 10.1093/ecam/neh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L., Gui C., Luo X., Yang Q., Gunther S., Scandella E., Drosten C., Bai D., He X., Ludewig B., Chen J., Luo H., Yang Y., Zou J., Thiel V., Chen K., Shen J., Shen X., Jiang H. J. Virol. 2005;79:7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y.C., Huang V., Chao T.C., Hsiao C.D., Lin A., Chang M.F., Chow L.P. Biochem. Biophys. Res. Commun. 2005;333:194–199. doi: 10.1016/j.bbrc.2005.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shie J.J., Fang J.M., Kuo C.J., Kuo T.H., Liang P.H., Huang H.J., Yang W.B., Lin C.H., Chen J.L., Wu Y.T., Wong C.H. J. Med. Chem. 2005;48:4469–4473. doi: 10.1021/jm050184y. [DOI] [PubMed] [Google Scholar]
- 31.Horhammer L., Volz E. Arch. Pharm. Ber. Dtsch. Pharm. Ges. 1955;288:58–60. doi: 10.1002/ardp.19552880203. [DOI] [PubMed] [Google Scholar]
- 32.Mouria M., Gukovskaya A.S., Jung Y., Buechler P., Hines O.J., Reber H.A., Pandol S.J. Int. J. Cancer. 2002;98:761–769. doi: 10.1002/ijc.10202. [DOI] [PubMed] [Google Scholar]
- 33.Singh D., Chander V., Chopra K. Drug Chem. Toxicol. 2004;27:145–156. doi: 10.1081/dct-120030729. [DOI] [PubMed] [Google Scholar]
- 34.Hayek T., Fuhrman B., Vaya J., Rosenblat M., Belinky P., Coleman R., Elis A., Aviram M. Arterioscler. Thromb. Vasc. Biol. 1997;17:2744–2752. doi: 10.1161/01.atv.17.11.2744. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Saura M.F., Galisteo M., Villar I.C., Bermejo A., Zarzuelo A., Vargas F., Duarte J. Mol. Cell. Biochem. 2005;270:147–155. doi: 10.1007/s11010-005-4503-0. [DOI] [PubMed] [Google Scholar]
- 36.Yi L., Li Z., Yuan K., Qu X., Chen J., Wang G., Zhang H., Luo H., Zhu L., Jiang P., Chen L., Shen Y., Luo M., Zuo G., Hu J., Duan D., Nie Y., Shi X., Wang W., Han Y., Li T., Liu Y., Ding M., Deng H., Xu X. J. Virol. 2004;78:11334–11339. doi: 10.1128/JVI.78.20.11334-11339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin C.W., Tsai F.J., Tsai C.H., Lai C.C., Wan L., Ho T.Y., Hsieh C.C., Chao P.D. Antiviral Res. 2005;68:36–42. doi: 10.1016/j.antiviral.2005.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ewing T.J., Makino S., Skillman A.G., Kuntz I.D. J. Comput. Aided Mol. Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 39.Cornell W.D., Cieplak D., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 40.Gasteiger J., Marsili M. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 41.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. J. Comput. Chem. Biol. 1998;19:1639–1662. [Google Scholar]
- 42.Chen J.J., Yang W.W., Pan X.F., Li Y.L., Tan Z. Acta Chim. Sin. 1987;45:503–505. [Google Scholar]
- 43.van Acker F.A., Hageman J.A., Haenen G.R., van Der Vijgh W.J., Bast A., Menge W.M. J. Med. Chem. 2000;43:3752–3760. doi: 10.1021/jm000951n. [DOI] [PubMed] [Google Scholar]
- 44.Sun H., Luo H., Yu C., Sun T., Chen J., Peng S., Qin J., Shen J., Yang Y., Xie Y., Chen K., Wang Y., Shen X., Jiang H. Protein Expr. Purif. 2003;32:302–308. doi: 10.1016/j.pep.2003.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]