

Graphical abstract
Keywords: Nonnucleoside reverse transcriptase inhibitors, NNRTIs, HIV, Uracil, Benzophenone, Cinnamyl
Abstract
A series of phenyloxyethyl and cinnamyl derivatives of substituted uracils were synthesized and found to exhibit potent activity against HIV-RT and HIV replication in cell culture. In general, the cinnamyl derivatives proved superior to the phenyloxyethyl derivatives, however 1-[2-(4-methylphenoxy)ethyl]-3-(3,5-dimethylbenzyl)uracil (19) exhibited the highest activity (EC50 = 0.27 μM) thus confirming that the 3-benzyluracil fragment in the NNRTI structure can be regarded as a functional analogue of the benzophenone pharmacophore typically found in NNRTIs.
1. Introduction
Recent reports from the UNAIDS reveal that there are 34 million HIV-infected patients worldwide.1 The number of annual AIDS-related deaths is estimated to be 2.5–3.3 million, or 25 million deaths overall.1 Despite the development of resistance to many currently used therapeutics, HIV reverse transcriptase (HIV-RT) remains one of the major biological targets in the search of new drugs against HIV. HIV-RT plays a key role at early stages of viral replication by catalyzing the RNA-dependent synthesis, ribonuclease H and DNA-dependent DNA synthesis of proviral double-stranded DNA.2 Compounds inhibiting HIV-RT are categorized as nucleoside/nucleotide (NRTI) and nonnucleoside (NNRTI) inhibitors. Nucleoside/nucleotide inhibitors bind in the active site of HIV-RT, while the NNRTIs comprise a structurally diverse spectrum of compounds capable of initiating conformational changes in HIV-RT by binding allosterically, which then results in the loss of HIV-RT polymerase activity.3
The first generation of NNRTIs (nevirapine,4 delavirdine,5 efavirenz6) displayed potent activity and low toxicity. However due to the high genetic variability of the virus, resistant strains with one or more point mutations in the RT binding pocket rapidly developed, and these first generation drugs proved to be problematic.7 In contrast, second generation FDA-approved drugs including etravirine (TMC125)8 and rilpivirine (TMC278)9 proved to be highly active against both wild-type and the most frequent mutant HIV strains with enhanced safety profiles. Etravirine and rilpivirine are classified as diarylpyrimidines. More recently biaryl esters such as MK-4965 have further increased the structural diversity of the NNRTI inhibitors (Fig. 1 ).10 These biaryl ethers are also active against both wild-type and mutant RTs at nanomolar concentrations. Notably, the common feature of these two NNRTI types, as well as many others, particularly, the benzophenone derivatives 1 11 is the presence of three aromatic rings connected by short linkers (Fig. 2 ).
Figure 1.
Figure 2.
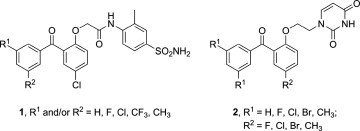
Related to this, the uracil ring system has found many applications in the construction of therapeutically useful molecules. It is more attractive than a benzene ring because of increased potential to form polar intermolecular interactions and hydrogen bonding interactions, both of which can improve enzyme affinity and the pharmacokinetic properties of target compounds. Drawing upon the structural features of other highly active NNRTIs,12, 13 we previously reported14 a series of benzophenone derivatives of pyrimidines 2 with potent antiretroviral activity. Like the original benzophenones 1 reported by GlaxoSmithKline (Fig. 2)11, 12 our compounds featured replacement of the distal benzene ring by uracil. As a logical extension of our previous investigations, herein we have prepared a new series of compounds with the central aromatic system replaced by uracil (Fig. 3 ).
Figure 3.
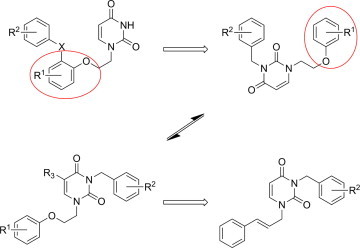
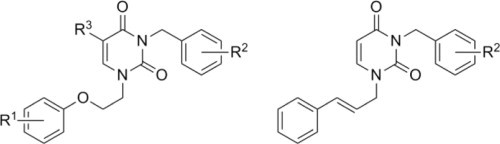
Two groups of N1,N3-disubstituted uracil derivatives were designed as potential HIV-RT inhibitors.
As shown in Figure 3, the N1-atom of uracil is connected to the aryl fragment by a three-atom linker containing either an ether (oxygen) or a double bonded (alkenyl) moiety.
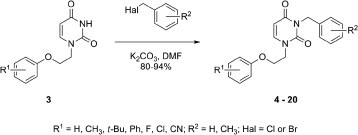
The generalized synthesis of the first group of target compounds is shown in Scheme 1 . The various 1-[2-(phenoxy)ethyl]uracils 3 were obtained according to literature procedures.15 Each uracil was then treated with the appropriate benzyl halide in DMF in the presence of K2CO3 to give the target compounds 4–20 (80–94%). In addition, 3-benzyl-1-[2-(4-methylphenoxy)ethyl]thymine (21) was obtained in a yield of 87% from 1-[2-(4-methylphenoxy)ethyl]thymine and benzyl chloride.
Scheme 1.
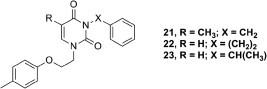
Next, with a goal of studying the bridge linking the phenyl residue and the pyrimidine N3 we synthesized the 3-(2-phenylethyl)- (22) and 3-(1-phenylethyl)-derivatives of 1-[2-(4-methylphenoxy)ethyl] uracil (23). In an analogous manner as before, starting with 3 (R1 = 4-Me), treatment with 2- or 1-chloroethylbenzene gave derivatives 22 and 23 (Fig. 4 ), respectively. Interestingly, the yield of compound 22 did not exceed 35% due to the highly favorable elimination of hydrogen chloride which was not observed in the case of 1-chloroethylbenzene.
Figure 4.
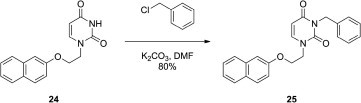
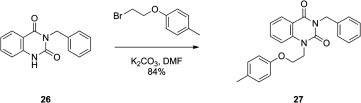
The corresponding naphthyl and quinazolyl derivatives 25 and 27 were also sought. Compound 25 was prepared using the addition of a benzyl substituent to the N-3 of uracil of 1-[2-(2-naphthoxy)ethyl]uracil (24) synthesized previously in our laboratories15 (Scheme 2 ). The synthesis of analogue 27 is shown in Scheme 3 . Employing 3-benzylquinazolin-2,4(1Н,3Н)-dione (26), 27 was synthesized as described by Kirincich16 using amination of benzoxazin-2,4(1Н,3Н)-dione with benzylamine in ethylene glycol. The resulting 3-benzyl derivative was then treated with 1-bromo-2-(4-methylphenoxy)ethane in DMF in the presence of K2CO3 to give the target 3-benzyl-1-[2-(4-methylphenoxy)ethyl]quinazolin-2,4(1Н,3Н)-dione (27) in 84% yield.
Scheme 2.
Scheme 3.
Synthesis of the unsaturated targets was then undertaken. In that regard, 3-benzyl-1-cinnamyluracils 29–34 and their 3-(1-naphthylmethyl) derivative 35 were synthesized in two steps as shown in Scheme 4 . Next, 1-cinnamyluracil (28) was prepared by condensation of 2,4-bis(trimethylsilyloxy)pyrimidine with cinnamyl bromide. The reaction mixture was refluxed in 1,2-dichloroethane for 20 h to give 28 in a 90% yield. As noted by TLC, 1-cinnamyluracil (28) was the only product. Although different synthetic routes were employed, the physicochemical and spectral data matched those published previously.17, 18, 19 It should be noted that Malik et al.20, 21 initially incorrectly assigned the structure of 1-cinnamyluracil (28) to its isomerization product22 when 28 and some of its analogues were synthesized using I2 as a catalyst. The product of the isomerization is apparently the oxazolo[3,2-a]pyrimidin-7-one heterocyclic system as previously reported by Skaric.22
Scheme 4.
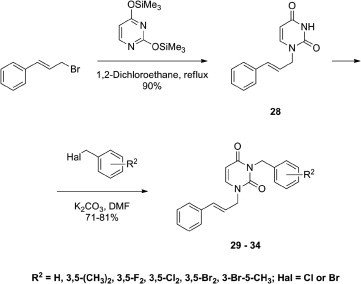
Next, 1-cinnamyluracil (28) was treated with the corresponding benzyl halides in DMF solution in the presence of K2CO3 (1.5 molar excess) at room temperature.23 This reaction afforded the corresponding 3-benzyl-1-cinnamyl derivatives of uracil 29–34 with yields ranging between 71% and 81%.
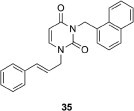
1-Cinnamyl-3-(1-naphthylmethyl)uracil (35, shown in Figure 5 ) was obtained in a similar manner with a yield of 72%.
Figure 5.
The structures of the synthesized compounds and their purity were confirmed by NMR (1Н and 13С) spectroscopy, TLC and mass-spectrometry.
The results of the biological studies revealed that several oxyethyl (4–23, 25 and 27), and cinnamyl derivatives (29–35) demonstrated anti-HIV-1 activity in MT-4 cell culture and proved to be inhibitors of the recombinant RT in vitro. However, their activity was lower than structurally related benzophenone analogues 1 and 2 11, 12, 14, 24 or several known NNRTIs.10, 16, 25 Analysis of the structure–activity relationship study revealed that the highest activity was displayed for the uracils bearing a 3,5-disubstituted benzyl group.26, 27 In that regard, 1-[2-(4-methylphenoxy)ethyl]-3-(3,5-dimethylbenzyl)uracil (19) exhibited the highest activity (EC50: 0.27 μM) and the selectivity index was greater than 1270 (see Table 1 ). This compound also showed the most pronounced inhibitory activity toward HIV-1.
Table 1.
Antiviral activity of the studied compounds against HIV-1
| Compd | R1 = X | R2 | R3 | CC50 (μM)a | HIV-1 (IIIB) wild type |
RT |
|
|---|---|---|---|---|---|---|---|
| EC50 (μM)b | SIc | Ki (μM)d | |||||
| 4 | H | H | H | 193 | 9.5 | 20 | 68 |
| 5 | 2-Me | H | H | 117 | >117 | — | >100 |
| 6 | 3-Me | H | H | 207 | 31 | 6.7 | >100 |
| 7 | 4-Me | H | H | 187 | 11 | 17 | 52 |
| 8 | 4-t-Bu | H | H | 63 | >63 | — | 15 |
| 9 | 4-Ph | H | H | 31 | 8.3 | 3.8 | 8.1 |
| 10 | 4-Cl | H | H | 36 | >36 | — | 22 |
| 11 | 4-F | H | H | 187 | 27 | 6.9 | 71 |
| 12 | 4-CN | H | H | 166 | 43 | 3.9 | 51 |
| 13 | 3,4-Me2 | H | H | 60 | >60 | — | >100 |
| 14 | 3,5-Me2 | H | H | 46 | >46 | — | >100 |
| 15 | 4-Me | 2-Me | H | 46 | >46 | — | >100 |
| 16 | 4-Me | 3-Me | H | ⩾38 | 3.4 | ⩾11 | 6.6 |
| 17 | 4-Me | 4-Me | H | 250 | >250 | — | 47 |
| 18 | 4-Me | 2,5-Me2 | H | >343 | 5.2 | >66 | 10 |
| 19 | 4-Me | 3,5-Me2 | H | >343 | 0.27 | >1270 | 0.26 |
| 20 | 4-Me | 2,4,6-Me3 | H | 133 | >133 | — | >100 |
| 21 | — | — | Me | > 356 | >356 | — | 1 |
| 22 | — | — | H | 129 | >129 | — | 6.5 |
| 23 | — | — | H | 132 | 6.5 | 20 | 12 |
| 25 | — | — | H | 37 | >37 | — | 1 |
| 27 | — | — | — | 25 | >25 | — | 1 |
| 29 | H | H | H | 160 | 7.85 | 20 | 29 |
| 30 | H | 3,5-Me2 | H | 257 | 1.27 | 202 | 0.55 |
| 31 | H | 3,5-F2 | H | 31 | 8.4 | 4 | 12 |
| 32 | H | 3,5-Cl2 | H | 49 | 1.99 | 24 | 0.67 |
| 33 | H | 3,5-Br2 | H | 231 | 1.72 | 134 | 0.31 |
| 34 | H | 3-Br-5-Me | H | 100 | 1.36 | 73 | 0.31 |
| 35 | H | — | H | 293 | 13 | 22 | 6.6 |
| Nevirapine | >4 | 0.048 | >400 | 7.2 | |||
CC50, cytotoxic concentration; the concentration affording 50% death of noninfected MT-4 cells.
EC50, effective concentration; the concentration affording 50% inhibition of virus replication in MT-4 cells.
SI, selectivity index, ratio CC50/IC50.
Ki, inhibition constant; the concentration of a non-competitive RT inhibitor ensuring 50% inhibition of the enzymatic activity.
RT, thus confirming the mechanism of anti-HIV-1 activity for the described compounds. This implies that the 3-benzyluracil fragment in the NNRTI structure can be regarded as a functional analogue of the benzophenone pharmacophore, although the latter ensures higher antiviral activity. In general, the cinnamyl derivatives showed a stronger inhibitory activity against HIV and HIV-RT than the phenyloxyethyl derivatives, although the highest activity was demonstrated by the phenoxyethyl-containing compound 19 (Table 1).
The most active compounds were then tested against a double mutant of HIV-1 RT, K103N/Y181C, in MT-4 cell culture. However, neither of them showed any activity in the range of the concentrations studied. This may be due to the replacement of the anilide fragment in the linker region, as compared to the benzophenone NNRTIs, which apparently results in a loss of stacking interactions with the Tyr181 residue. Analysis of the inhibitory activity of several compounds against a panel of single- and double-mutated RTs revealed that N1,N3-disubstituted uracils retained pronounced activity against the L100I and the G190A mutants (Table 2 ). They also inhibit RTs with a V106A or Y181C substitution, although at much higher concentrations.
Table 2.
Inhibitory activity of the selected compounds against mutant RTs
| HIV-1 RT |
Ki (μМ) |
||||||
|---|---|---|---|---|---|---|---|
| 19 | 30 | 32 | 33 | 34 | NVP | EFV | |
| WT | 0.26 | 0.55 | 0.67 | 0.31 | 0.31 | 7.2 | 0.01 |
| L100I | 0.13 | 0.76 | 0.92 | 0.36 | 0.24 | 273 | 0.08 |
| K103N | >26 | 12 | 15.5 | 9.4 | 6.9 | >2000 | 0.58 |
| V106A | 7.5 | 2.9 | 11 | 4.2 | 2.3 | >2000 | 0.05 |
| Y181C | 169 | 75 | 43 | 25 | 20 | >2000 | 0.03 |
| G190A | 0.36 | 0.4 | 0.55 | 0.3 | 0.18 | >2000 | 0.06 |
| K103N/Y181C | >26 | >100 | >100 | >100 | 85 | >2000 | 0.14 |
The compounds were also tested as potential inhibitors of a large panel of viruses including Feline Corona Virus, Herpes simplex virus-1, Herpes simplex virus-2, Vaccinia virus, Vesicular stomatitis virus, Coxsackie virus B4, Respiratory syncytial virus, Influenza A H1N1 subtype, Influenza A H3N2 subtype, Influenza B, Para-influenza-3 virus, Reovirus-1, Sindbis virus, Punta Toro virus, varicella-zoster virus and human cytomegalovirus, but found inactive, except for the four cinnamyl derivatives that exhibited notable antiviral activity towards human cytomegalovirus (HCMV) (Table 3 ).
Table 3.
Anti-HCMV activity of the synthesized compounds
| Compd | IC50 (μM) (SI) |
CC50 (μM) | |
|---|---|---|---|
| AD169 | Davis | ||
| 31 | 37 (1.38) | 45 (1.13) | 51 |
| 32 | 9.4 (11) | 12 (9) | ⩾100 |
| 34 | 16 (6) | ⩾20 (5) | ⩾100 |
| 35 | 14 (7) | 6.4 (16) | ⩾100 |
| Ganciclovir | 6.3 (21) | 5.7 (23) | 132 |
| Cidofovir | 0.98 (64) | 1.0 (63) | 63 |
In order to rationalize the anti-HIV activity observed, molecular modeling studies were employed. Cross-docking was performed with AutoDock Vina 1.1.2 and MGLTools 1.5.4 software.28 Preliminary studies show the ability to reproduce experimentally observed binding modes in various HIV-1 RT/NNRTI complexes (PDB entries 2BE2, 3DLE, 3DLG, 3DM2, 3DOK, 3DRP, 3DYA) with an average RMSD (root mean square deviation) value as low as 0.67 Å. The accuracy of predicted modes was also ascertained by visual observations according to the Interactions-Based Accuracy Classification protocol.29
The most active compound in the series (19) was docked into HIV-1 WT RT (3DYA13) and K103N RT (3DOK 11) mutants and the results are shown in Figure 6 . An excellent correlation between the benzyl of 19 and the corresponding moiety of the original 3DYA ligand was revealed. The uracil ring also forms a close hydrophobic contact with the Leu100 residue (omitted for clarity), while the phenoxy core is sandwiched between the Val106 and Pro236 residues. However, a complete lack of interactions with the phenoxyethyl side chain was also noted.
Figure 6.
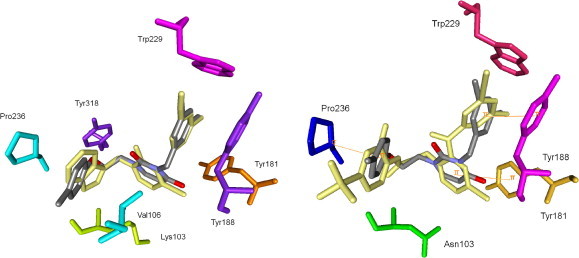
Predicted binding modes for 19 overlaid with original ligands (pale-yellow colored) in 3DYA (left) and 3DOK (right) crystal structures. Only key residues are shown. Orange lines indicate stacking interactions.
In comparison to the benzophenone derivative GW78248, significant differences in the benzyl and benzoyl cores’ orientations were observed, although the overall binding mode was retained. Strong correlation between the benzophenone’s and uracil’s carbonyl groups is also noteworthy and hypothesized to be a consequence of steric preferences. As depicted in Figure 6, unfavorable π-stacking interactions associated with mutations at Tyr181 and Tyr188 are also predicted, along with the lack of H-bonding at Asn103. These findings may explain the dramatic loss in activity against K103N and some other mutant HIV strains. As a result, further optimization of this region should be pursued. Cinnamyl derivatives 28–34 were modeled in analogous manner and follow similar binding patterns (data not shown).
As seen from Table 1, Table 2, the presence of the methyl groups in positions (3 and 5) of the proximal phenyl ring and a polar group in position 4 of the distal phenyl ring (R1) lead to the best binding for ligands 7–12 and 17–19. This can be explained by the interactions of the groups of ring R2 with Trp229 (Fig. 3), while proton substitution in position 4 disrupts the favorable CH–π stacking (compounds 17 and 20). In contrast, the extended groups in position 4 of ring R1 are not favorable as they reach solvent accessible area at the entrance of the pocket. The absence of binding for compound 20 is due to steric restrictions between the methyl group in the ortho position of the R2 ring and the carbonyl oxygens in the central uracil ring R3 since they are densely packed in the pocket. However it appears there are still enough degrees of freedom to properly orientate the R2 ring when only one methyl group in the ortho position is present (compound 18).
2. Conclusion
A series of phenyloxyethyl and cinnamyl derivatives of substituted uracils were synthesized and found to be inhibitory against HIV-RT and HIV replication in cell culture. In general, the cinnamyl derivatives proved somewhat superior to the phenyloxyethyl derivatives, however inhibition of mutant HIV-1 RT was less pronounced when additional NNRTI-characteristic mutations were present in the enzyme. Moreover, the double mutant K103N/Y181C was detrimental for anti-HIV-1 activity. Some cinnamyl derivatives showed moderate anti-HCMV activity, however this was not the case for the phenyloxyethyl derivatives, which were essentially inactive. Efforts are currently underway to improve the promising activity noted by several of the compounds. New structural modifications guided by additional docking efforts are currently being explored. For example, it may prove that the N1-side chain can be optimized for maximizing interactions with the amino acid landscape by use of H-bond donors and/or introducing conformational restrictions. These results will be reported elsewhere as they are obtained.
3. Experimental
3.1. General
Activated DNA was purchased from GE Healthcare (Little Chalfont, UK) [α-32P]dATP (5000 Ci/mmol) was from Izotop (Moscow, Russia). Ni-NTA-agarose resin and Rosetta (DE3) Escherichia coli strain were from Novagen (Madison, WI). All other reagents of highest grade were obtained from Sigma (St. Louis, MI). All chemicals were obtained from commercial sources and used without further purification unless otherwise noted. Anhydrous DMF, isopropanol, and ethylene glycol were purchased from Sigma–Aldrich Co. Anhydrous acetone, CH2Cl2, 1,2-dichloroethane, and ethyl acetate were obtained by distillation over P2O5. NMR spectra were registered on a Bruker Avance 400 spectrometer (400 MHz for 1H and 100 MHz for 13C) in CDCl3 or DMSO-d 6 with tetramethylsilane as an internal standard. Mass spectra were registered on Hewlett Packard 5973 MSD and FINNIGAN POLARIS Q mass-spectrometers (70 eV, the ion chamber temperature 250 °С). TLC was performed on Merck TLC Silica gel 60 F254 plates eluted with ethyl acetate and developed with iodine. Melting points were determined in glass capillaries on a Mel-Temp 3.0 (Laboratory Devices Inc., США). Yields refer to spectroscopically (1H and 13C NMR) homogeneous materials.
3.1.1. 3-Benzyl-1-[2-(phenoxy)ethyl]uracil (4)
A suspension of 1-[2-(phenoxy)ethyl]uracil15 (3) (0.85 g, 3.66 mmol) of and К2СО3 (0.6 g, 4.34 mmol) in DMF (8 mL) was stirred at 80 °С for 1 h. Benzyl chloride (0.44 mL, 3.82 mmol) was added and the mixture stirred at the same temperature for 24 h. The reaction mixture was cooled to room temperature, poured into cold H2O (250 mL) and kept at 4 °С for 10 h. The resulting precipitate was filtered and recrystallized from mixture of acetone (30 mL) and H2O (10 mL) to give colorless crystals which were filtered and air-dried to give 4 (1.05 g, 89%), mp: 101–103 °C, R f 0.50 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 4.12 (2H, t, J = 4.5 Hz, CH2), 4.20 (2H, t, J = 4.5 Hz, CH2), 5.15 (2H, s, CH2Ph), 5.77 (1H, d, J = 8 Hz, H-5), 6.88 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.01 (1H, t, J = 7.3 Hz, H-4′), 7.27–7.35 (7H, m, H-3′, H-5′, C6H5), 7.50 (1H, d, J = 7 Hz, H-6). 13C NMR (CDCl3): δ 43.9, 48.8, 65.1, 100.9, 114.0, 121.2, 127.2, 128.0, 128.5, 129.3, 136.4, 143.1, 151.1, 157.5, 162.6. MS (ES+): m/z (%) 229.1 (65), 91.3 (100).
3.1.2. 3-Benzyl-1-[2-(2-methylphenoxy)ethyl]uracil (5)
Was synthesized in a similar manner as 4 to give 5 (1.2 g, 3.57 mmol, 88%) as colorless crystals, mp: 108–109 °C, R f 0.52 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.21 (3H, s, CH3), 4.16 (2H, t, J = 5.3 Hz, CH2), 4.22 (2H, t, J = 5.0 Hz, CH2), 5.17 (2H, s, CH2Ph), 5.79 (1H, d, J = 7.8 Hz, H-5), 6.78 (1H, d, J = 8.2 Hz, H-6′), 6.94 (1H, dt, J = 7.4 and 0.8 Hz, H-4′), 7.17–7.20 (2H, m, H-5′, H-6′), 7.29–7.36 (5H, m, C6H5), 7.51 (1H, dd, J = 8 and 1.2 Hz, H-6). 13C NMR (CDCl3): δ 16.0, 43.9, 49.0, 64.9, 100.8, 110.3, 120.8, 126.0, 126.6, 127.2, 128.0, 128.6, 130.6, 136.4, 143.3, 151.1, 155.6, 162.6. MS (ES+): m/z (%) 229.1 (75), 91.1 (100).
3.1.3. 3-Benzyl-1-[2-(3-methylphenoxy)ethyl]uracil (6)
Was synthesized in a similar manner as 4 to give 6 (1.13 g, 3.36 mmol, 83%) as colorless crystals, mp: 102–103 °C, R f 0.53 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.36 (3H, s, CH3), 4.11 (2H, t, J = 4.8 Hz, CH2), 4.20 (2H, t, J = 4.8 Hz, CH2), 5.16 (2H, s, CH2Ph), 5.78 (1H, d, J = 8 Hz, H-5), 6.69 (1H, d, J = 8.2 Hz, H-4′), 6.72 (1H, s, H-2′), 6.84 (1H, d, J = 7.6 Hz, H-6′), 7.19 (1H, t, J = 7.8 Hz, H-5′), 7.29–7.35 (5H, m, C6H5), 7.51 (1H, d, J = 6.9 Hz, H-6). 13C NMR (CDCl3): δ 21.1, 43.9, 48.8, 65.1, 100.9, 110.8, 115.0, 122.0, 127.2, 128.0, 128.5, 129.0, 136.4, 139.4, 143.2, 151.1, 157.6, 162.6. MS (ES+): m/z (%) 229.1 (77), 91.1 (100).
3.1.4. 3-Benzyl-1-[2-(4-methylphenoxy)ethyl]uracil (7)
Was synthesized in a similar manner as 4 to give 7 (1.1 g, 3.27 mmol, 81%) as white lamellar crystals, mp: 99–101 °C, R f 0.52 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.31 (3H, s, CH3), 4.11 (2H, t, J = 4.6 Hz, CH2), 4.20 (2H, t, J = 4.5 Hz, CH2), 5.15 (2H, s, CH2Ph), 5.77 (1H, d, J = 7.8 Hz, H-5), 6.77 (2H, d, J = 8.5 Hz, H-3′, H-5′), 7.10 (2H, d, J = 8.3 Hz, H-2′, H-6′), 7.26–7.34 (5H, m, C6H5), 7.49 (1H, d, J = 7.1 Hz, H-6). 13C NMR (CDCl3): δ 20.1, 43.9, 48.8, 65.3, 100.9, 113.9, 127.2, 128.0, 128.5, 129.7, 130.4, 136.4, 143.1, 151.1, 155.4, 162.6. MS (ES+): m/z (%) 229.1 (73), 91.1 (100).
3.1.5. 3-Benzyl-1-[2-(4-tert-butylphenoxy)ethyl]uracil (8)
Was synthesized in a similar manner as 4 to give 8 (0.75 g, 1.98 mmol, 82%) as white crystals, mp: 121–122 °C, R f 0.58 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 1.34 (9H, s, CH3), 4.12 (2H, t, J = 4.2 Hz, CH2), 4.22 (2H, t, J = 4.3 Hz, CH2), 5.16 (2H, s, CH2Ph), 5.77 (1H, d, J = 7.8 Hz, H-5), 6.83 (2H, d, J = 6.7 Hz, H-3′, H-5′), 7.29–7.36 (7H, m, C6H5, H-2′, H-6′), 7.51 (1H, d, J = 8.1 Hz, H-6). 13C NMR (CDCl3): δ 31.1, 43.9, 48.9, 65.2, 100.9, 113.5, 126.0, 127.2, 128.0, 128.6, 136.4, 143.2, 143.9, 151.1, 155.3, 162.6. MS (ES+): m/z (%) 377.8 (1) [M+], 229.1 (88), 91.1 (100).
3.1.6. 3-Benzyl-1-[2-(4-phenylphenoxy)ethyl]uracil (9)
Was synthesized in a similar manner as 4 to give 9 (1.1 g, 2.76 mmol, 85%) as white crystals, mp: 125–126 °C, R f 0.53 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 4.14 (2H, t, J = 4.5 Hz, CH2), 4.27 (2H, t, J = 4.5 Hz, CH2), 5.18 (2H, s, CH2Ph), 5.80 (1H, d, J = 8 Hz, H-5), 6.95 (2H, d, J = 8.8 Hz, H-3′, H-5′), 7.28–7.38 (7H, m, C6H5, H-2′, H-6′), 7.45–7.48 (2H, aromatic H), 7.52–7.59 (4H, m, aromatic H, H-6). 13C NMR (CDCl3): δ 43.9, 48.8, 65.3, 101.0, 114.4, 126.4, 126.5, 127.3, 127.6, 127.9, 128.1, 128.4, 128.6, 134.3, 136.4, 140.1, 143.1, 143.9, 151.1, 157.1, 162.6. MS (ES+): m/z (%) 397.9 (1) [M+], 229.1 (100), 91.2 (97).
3.1.7. 3-Benzyl-1-[2-(4-chlorophenoxy)ethyl]uracil (10)
Was synthesized in a similar manner as 4 to give 10 (1.63 g, 4.57 mmol, 94%) as white crystals, mp: 102–103 °C, R f 0.49 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 4.10 (2H, d, J = 4.4 Hz, CH2), 4.17 (2H, d, J = 4.4 Hz, CH2), 5.13 (2H, s, CH2Ph), 5.76 (1H, d, J = 7.8 Hz, H-5), 6.78 (2H, d, J = 9 Hz, H-3′, H-5′), 7.22 (2H, d, J = 9 Hz, H-2′, H-6′), 7.26–7.34 (7H, m, C6H5), 7.48 (1H, d, J = 8.2 Hz, H-6). 13C NMR (CDCl3): δ 43.9, 48.8, 65.4, 101.0, 115.3, 126.0, 127.2, 128.0, 128.5, 129.1, 136.3, 143.0, 151.1, 156.1, 162.5. MS (ES+): m/z (%) 229.1 (69), 91.1 (100).
3.1.8. 3-Benzyl-1-[2-(4-fluorophenoxy)ethyl]uracil (11)
Was synthesized in a similar manner as 4 to give 11 (1.7 g, 4.99 mmol, 89%) as long needle crystals, mp: 95–96 °C, R f 0.56 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 4.10 (2H, d, J = 4.2 Hz, CH2), 4.16 (2H, d, J = 4.4 Hz, CH2), 5.14 (2H, s, CH2Ph), 5.76 (1H, d, J = 8 Hz, H-5), 6.76–6.80 (2H, aromatic H), 6.94–6.99 (2H, aromatic H), 7.25–7.34 (4H, m, aromatic H), 7.47–7.49 (2H, m, aromatic H, H-6). 13C NMR (CDCl3): δ 43.9, 48.8, 65.8, 100.9, 115.1, 115.5, 115.7, 127.2, 128.0, 128.5, 136.4, 143.1, 151.1, 153.7, 156.0, 158.4, 162.5. MS (ES+): m/z (%) 229.1 (81), 91.1 (100).
3.1.9. 3-Benzyl-1-[2-(4-cyanophenoxy)ethyl]uracil (12)
Was synthesized in a similar manner as 4 to give 12 (0.95 g, 2.73 mmol, 88%) as needle crystals, mp: 126–127.5 °C, R f 0.36 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 4.13 (2H, d, J = 4.6 Hz, CH2), 4.25 (2H, d, J = 4.6 Hz, CH2), 5.11 (2H, s, CH2Ph), 5.76 (1H, d, J = 7.8 Hz, H-5), 6.90 (2H, d, J = 8.8 Hz, H-3′, H-5′), 7.23–7.31 (4H, m, aromatic H), 7.44–7.46 (2H, m, aromatic H, H-6), 7.53–7.56 (2H, m, aromatic H). 13C NMR (CDCl3): δ 43.9, 48.7, 65.4, 101.2, 104.4, 114.8, 118.5, 127.3, 128.0, 128.5, 133.7, 136.3, 142.8, 151.1, 160.7, 162.4. MS (ES+): m/z (%) 229.1 (100), 91.1 (94).
3.1.10. 3-Benzyl-1-[2-(3,4-dimethylphenoxy)ethyl]uracil (13)
Was synthesized in a similar manner as 4 to give 13 (1.25 g, 3.57 mmol, 93%) as white prismatic crystals, mp: 111–112.5 °C, R f 0.64 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.23 (3H, s, CH3), 2.26 (3H, s, CH3), 4.11 (2H, d, J = 4.3 Hz, CH2), 4.19 (2H, d, J = 4.4 Hz, CH2), 5.16 (2H, s, CH2Ph), 5.78 (1H, d, J = 7.8 Hz, H-5), 6.63 (1H, dd, J = 8.2 and 2.6 Hz, H-5′), 6.71 (1H, d, J = 2.4 Hz, H-2′), 7.05 (1H, d, J = 8.3 Hz, H-6′), 7.27–7.36 (5H, m, C6H5), 7.51 (1H, d, J = 6.7 Hz, H-6). 13C NMR (CDCl3): δ 18.4, 19.6, 43.9, 48.8, 65.2, 100.8, 110.9, 115.7, 127.2, 128.0, 128.5, 129.1, 130.1, 136.4, 137.6, 143.2, 151.1, 155.7, 162.6. MS (ES+): m/z (%) 229.1 (69), 91.1 (100).
3.1.11. 3-Benzyl-1-[2-(3,5-dimethylphenoxy)ethyl]uracil (14)
Was synthesized in a similar manner as 4 to give 14 (1.2 g, 3.42 mmol, 90%) as white crystals, mp: 78–79.5 °C, R f 0.62 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.32 (6H, s, CH3), 4.11 (2H, t, J = 4.3 Hz, CH2), 4.20 (2H, t, J = 4.2 Hz, CH2), 5.16 (2H, s, CH2Ph), 5.78 (1H, d, J = 7.8 Hz, H-5), 6.52 (2H, s, H-2′, H-6′), 6.67 (1H, s, H-4′), 7.29–7.36 (5H, m, C6H5), 7.50 (1H, d, J = 7 Hz, H-6). 13C NMR (CDCl3): δ 21.0, 43.9, 48.8, 65.1, 100.9, 111.8, 122.9, 127.2, 128.0, 136.4, 139.1, 143.2, 151.1, 157.6, 162.6. MS (ES+): m/z (%) 229.1 (74), 91.1 (100).
3.1.12. 3-(2-Methylbenzyl)-1-[2-(4-methylphenoxy)ethyl]uracil (15)
Was synthesized in a similar manner as 4 to give 15 (1.05 g, 3.00 mmol, 92%) as white crystals, mp: 107–108 °C, R f 0.57 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.34 (3H, s, CH3), 2.49 (3H, s, CH3), 4.12 (2H, t, J = 4.3 Hz, CH2), 4.20 (2H, t, J = 4.3 Hz, CH2), 5.17 (2H, s, CH2Ar), 5.82 (1H, d, J = 8 Hz, H-5), 6.81 (2H, d, J = 8.4 Hz, H-3′, H-5′), 7.03–7.05 (1H, m, aromatic H), 7.12–7.21 (5H, m, H-2′, H-6′, aromatic H), 7.37 (1H, d, J = 8 Hz, H-6). 13C NMR (CDCl3): δ 18.9, 20.1, 41.4, 48.9, 65.3, 100.7, 114.0, 125.4, 125.7, 126.7, 129.7, 129.9, 130.5, 134.1, 135.4, 143.4, 151.2, 155.5, 162.7. MS (ES+): m/z (%) 243.0 (74), 139.2 (100), 105.3 (42).
3.1.13. 3-(3-Methylbenzyl)-1-[2-(4-methylphenoxy)ethyl]uracil (16)
Was synthesized in a similar manner as 4 to give 16 (1.0 g, 2.85 mmol, 88%) as white crystals, mp: 102–103 °C, R f 0.63 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.32 (3H, s, CH3), 2.35 (3H, s, CH3), 4.11 (2H, t, J = 4.4 Hz, CH2), 4.20 (2H, t, J = 4.4 Hz, CH2), 5.12 (2H, s, CH2Ar), 5.77 (1H, d, J = 7.9 Hz, H-5), 6.77 (2H, d, J = 8.7 Hz, H-3′, H-5′), 7.10 (2H, d, J = 8.3 Hz, H-2′, H-6′), 7.22 (1H, t, J = 7.5 Hz, H-5″), 7.28–7.33 (4H, m, H-2″, H-4″, H-6″, H-6). 13C NMR (CDCl3): δ 20.1, 21.0, 43.9, 48.8, 65.3, 100.9, 113.9, 125.5, 127.9, 129.2, 129.7, 130.4, 136.3, 137.6, 143.1, 151.1, 155.4, 162.6. MS (ES+): m/z (%) 243.0 (74), 105.3 (100).
3.1.14. 3-(4-Methylbenzyl)-1-[2-(4-methylphenoxy)ethyl]uracil (17)
Was synthesized in a similar manner as 4 to give 17 (1.05 g, 3.00 mmol, 84%) as white crystals, mp: 124–125 °C, R f 0.63 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.32 (3H, s, CH3), 2.35 (3H, s, CH3), 4.11 (2H, t, J = 4.3 Hz, CH2), 4.19 (2H, t, J = 4.5 Hz, CH2), 5.12 (2H, s, CH2Ar), 5.76 (1H, d, J = 7.9 Hz, H-5), 6.78 (2H, d, J = 8.4 Hz, H-3′, H-5′), 7.10 (2H, d, J = 8.3 Hz, H-2′, H-6′), 7.14 (2H, d, J = 7.7 Hz, H-3″, H-5″), 7.30 (1H, d, J = 7.8 Hz, H-6), 7.41 (2H, d, J = 7.7 Hz, H-2″, H-6″). 13C NMR (CDCl3): δ 20.1, 20.8, 43.6, 48.8, 65.3, 100.9, 113.9, 125.5, 128.6, 128.7, 129.7, 130.4, 133.5, 136.9, 143.1, 151.1, 155.4, 162.6. MS (ES+): m/z (%) 243.0 (78), 139.1 (100), 105.3 (33).
3.1.15. 1-[2-(4-Methylphenoxy)ethyl]-3-(2,5-dimethylbenzyl)uracil (18)
Was synthesized in a similar manner as 4 to give 18 (1.2 g, 3.29 mmol, 83%) as white crystals, mp: 143–144 °C, R f 0.64 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.22 (3H, s, CH3), 2.33 (3H, s, CH3), 2.44 (3H, s, CH3), 4.14 (2H, t, J = 4.5 Hz, CH2), 4.20 (2H, t, J = 4.7 Hz, CH2), 5.13 (2H, s, CH2Ar), 5.83 (1H, d, J = 7.8 Hz, H-5), 6.79–6.83 (3H, m, H-3′, H-5′, H-6″), 6.97 (1H, d, J = 7.4 Hz, H-4″), 7.06–7.13 (3H, m, H-2′, H-6′, H-3″), 7.39 (1H, d, J = 7.8 Hz, H-6). 13C NMR (CDCl3): δ 18.5, 20.1, 20.7, 41.4, 48.9, 65.4, 100.8, 113.9, 126.1, 127.4, 129.7, 129.8, 130.5, 132.3, 133.8, 135.0, 143.3, 151.1, 155.5, 162.8. MS (ES+): m/z (%) 257.1 (100), 139.1 (99), 119.2 (49).
3.1.16. 1-[2-(4-Methylphenoxy)ethyl]-3-(3,5-dimethylbenzyl)uracil (19)
Was synthesized in a similar manner as 4 to give 19 (1.07 g, 2.94 mmol, 80%) as white crystals, mp: 126–127 °C, R f 0.66 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.31 (6H, s, CH3), 2.32 (3H, s, CH3), 4.12 (2H, t, J = 4.2 Hz, CH2), 4.20 (2H, t, J = 4.4 Hz, CH2), 5.09 (2H, s, CH2Ar), 5.78 (1H, d, J = 7.9 Hz, H-5), 6.78 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.92 (1H, s, H-4″), 7.09–7.11 (4H, m, H-2′, H-6′, H-2″, H-6″), 7.31 (1H, d, J = 8 Hz, H-6). 13C NMR (CDCl3): δ 20.1, 20.9, 43.8, 48.8, 65.3, 100.9, 113.9, 126.2, 128.9, 129.7, 130.4, 133.5, 136.2, 137.5, 143.1, 151.1, 155.4, 162.7. MS (ES+): m/z (%) 257.1 (92), 119.1 (100).
3.1.17. 1-[2-(4-Methylphenoxy)ethyl]-3-(2,4,6-trimethylbenzyl)-uracil (20)
Was synthesized in a similar manner as 4 to give 20 (1.15 g, 3.04 mmol, 83%) as white crystals, mp: 142–142.5 °C, R f 0.67 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.26 (3H, s, CH3), 2.33 (9H, s, CH3), 4.08 (2H, t, J = 4.4 Hz, CH2), 4.14 (2H, t, J = 4.4 Hz, CH2), 5.15 (2H, s, CH2Ar), 5.74 (1H, d, J = 7.8 Hz, H-5), 6.78 (2H, t, J = 9 Hz, H-3′, H-5′), 6.83 (2H, s, H-3″, H-5″), 7.25 (2H, d, J = 9 Hz, H-2′, H-6′), 7.29 (1H, d, J = 7.8 Hz, H-6). 13C NMR (CDCl3): δ 19.8, 20.5, 39.6, 48.7, 65.5, 100.7, 115.3, 126.0, 128.9, 129.1, 129.5, 136.2, 137.0, 142.9, 150.8, 156.2, 162.7. MS (ES+): m/z (%) 271.1 (100), 132.1 (55).
3.1.18. 3-Benzyl-1-[2-(4-methylphenoxy)ethyl]thymine (21)
A suspension of 1.2 g (4.61 mmol) of 1-[2-(4-methylphenoxy)ethyl]thymine15 and 0.8 g (5.79 mmol) of К2СО3 in DMF (8 mL) was stirred at 80 °С for 1 h. Benzyl chloride (0.5 mL, 4.34 mmol) was added and the mixture stirred at the same temperature for 24 h. The reaction mixture was cooled to room temperature, poured into cold H2O (250 mL) and kept at 4 °С for 10 h. The resulting precipitate was filtered and recrystallized from mixture of acetone (25 mL) and H2O (6 mL) to give large colorless crystals which were filtered and air-dried to give 21 (1.4 g, 87 %), mp: 143–144 °С, R f 0.73 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 1.97 (3Н, s, СН3), 2.32 (3H, s, CH3), 4.10 (2H, t, J = 4.5 Hz, CH2), 4.20 (2H, t, J = 4.6 Hz, CH2), 5.17 (2H, s, CH2Ph), 5.76 (1H, d, J = 7.9 Hz, H-5), 6.78 (2H, d, J = 8.6 Hz, H-3′, H-5′), 7.10 (2H, d, J = 8.5 Hz, H-2′, H-6′), 7.16 (1H, s, H-6), 7.26–7.35 (3H, m, aromatic H), 7.51–7.53 (2H, m, aromatic H). 13C NMR (CDCl3): δ 12.6, 20.1, 44.2, 48.6, 65.5, 109.0, 113.9, 127.1, 128.0, 128.7, 129.7, 130.4, 136.6, 139.3, 151.2, 155.5, 163.3. MS (ES+): m/z (%) 243.1 (100), 91.1 (92).
3.1.19. 1-[2-(4-Methylphenoxy)ethyl]-3-(2-phenylethyl)uracil (22)
A suspension of 0.8 g (3.25 mmol) of 1-[2-(4-methylphenoxy)ethyl]uracil (1)15 and 0.55 g (3.98 mmol) of К2СО3 in DMF (8 mL) was stirred at 80 °С for 1 h. 2-Phenyl chloroethane (0.5 mL, 3.98 mmol) was added and the mixture was stirred at the same temperature for 2 h and then refluxed for an additional 4 h. The reaction mixture was cooled to room temperature, poured into cold H2O (150 mL) and kept at 4 °С for 10 h. The aqueous layer was decanted and the solid residue recrystallized from a mixture of acetone (25 mL) and H2O (8 mL). The precipitate was filtered and air dried to give 22 (0.4 g, 35 %) as small white crystals, mp: 86–87 °С, R f 0.65 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.32 (3H, s, CH3), 2.96 (2H, t, J = 5.8 Hz, CH2), 4.11 (2H, t, J = 4.1 Hz, CH2), 4.16–4.22 (4H, m, CH2), 5.75 (1H, d, J = 7.9 Hz, H-5), 6.80 (2H, d, J = 8.6 Hz, H-3′, H-5′), 7.12 (2H, d, J = 8.4 Hz, H-2′, H-6′), 7.25–7.33 (6H, m, C6H5, H-6). 13C NMR (CDCl3): δ 20.8, 33,2 42.0, 48.9, 65.2, 100.8, 113.9, 126.0, 128.0, 128.6, 129.7, 130.4, 138.1, 143.1, 150.9, 155.4, 162.5. MS (ES+): m/z (%) 243.1 (78), 139.1 (100).
3.1.20. 1-[2-(4-Methylphenoxy)ethyl]-3-(1-phenylethyl)uracil (23)
A suspension of 0.9 g (3.55 mmol) of 1-[2-(4-methylphenoxy)ethyl]uracil (1)15 and 0.6 g (4.83 mmol) of К2СО3 in DMF (8 mL) was stirred at 80 °С for 1 h. 1-Phenylchloroethane (0.6 g, 4.27 mmol) was added and the mixture stirred at the same temperature for 4 h. The reaction mixture was cooled to room temperature, poured into cold H2O (250 mL) and kept at 4 °С for 10 h. The aqueous layer was decanted and the solid residue recrystallized from mixture of acetone (30 mL) and H2O (10 mL). The precipitate was filtered and air-dried to give 23 (0.87 g, 70 %) as small lamellar white crystals, mp: 103–105 °С, R f 0.57 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): δ 1.90 (3Н, d, J = 7.1 Hz, СН3), 2.33 (3Н, s, СН3), 4.04–4.18 (4Н, m, СН2), 5.74 (1Н, d, J = 7.9 Hz, Н-5), 6.35 (1Н, q, J = 7.1, PhСН), 6.77 (2H, d, J = 8.5 Hz, H-3′, H-5′), 7.11 (2Н, d, J = 8.4 Hz, H-2′, H-6′), 7.27–7.36 (5Н, m, С6Н5), 7.44 (1Н, d, J = 7.5 Hz, Н-6). 13C NMR (CDCl3): δ 15.3, 20.1, 48.7, 50.0, 65.4, 100.9, 113.9, 126.6, 126.8, 127.7, 129.7, 130.4, 139.9, 143.1, 150.5, 155.5, 163.0. MS (ES+): m/z (%) 243.1 (24), 139.1 (100).
3.1.21. 3-Benzyl-1-[2-(2-naphthyloxy)ethyl]uracil (25)
A suspension of 1.0 g (3.54 mmol) of 1-[2-(2-naphthyloxy)ethyl]uracil (24)15 and К2СО3 (0.6 g, 4.34 mmol) in DMF (8 mL) was stirred at 80 °С for 1 h. Benzyl chloride (0.45 mL, 3.91 mmol) was added and the mixture stirred at the same temperature for 24 h. The reaction mixture was cooled to room temperature, poured into cold water (250 mL) and kept at 4 °С for 10 h. The solids were filtered, dried and recrystallized from a mixture of toluene (10 mL) and hexanes (5 mL) and air-dried to give 25 (1.1 g, 80 %) as white crystals, mp: 88–90 °С, R f 0.65 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3): 4.16 (2H, t, J = 4.5 Hz, CH2), 4.32 (2H, t, J = 4.5 Hz, CH2), 5.17 (2H, s, CH2Ph), 5.79 (1H, d, J = 7.9 Hz, H-5), 7.10–7.13 (2H, m, aromatic H), 7.27–7.7.35 (5H, m, aromatic H), 7.38–7.42 (1H, m, aromatic H), 7.47–7.53 (3H, m, aromatic H, H-6), 7.73–7.82 (3H, m, aromatic H). 13C NMR (CDCl3): 44.0, 48,7, 65.2, 101.0, 106.6, 117.9, 123.7, 126.3, 126.4, 127.2, 127.3, 128.0, 128.6, 128.9, 129.3, 134.0, 136.4, 143.0, 151.2, 155.4, 162.6. MS (ES+): m/z (%) 229.0 (100), 91.1 (82).
3.1.22. 3-Benzyl-1-[2-(4-methylphenoxy)ethyl]quinazoline-2,4(1H,3H)-dione (27)
A suspension of 26 16 (0.55 g, 2.18 mmol) and К2СО3 (0.4 g, 2.89 mmol) of in DMF (8 mL) was stirred at 80 °С for 1 h. 1-Bromo-2-(4-methylphenoxy)ethane (0.56 g, 2.60 mmol) was added and the mixture stirred at the same temperature for 24 h. The reaction mixture was cooled to room temperature, poured into cold water (250 mL) and kept at 4 °С for 10 h. The aqueous layer was decanted and the residue crystallized from mixture of acetone (30 mL) and H2O (10 mL). The precipitate was filtered and air dried to give 27 (0.75 g, 89 %) as colorless crystals, mp: 137–138 °C, R f 0.84 (hexane:ethyl acetate, 1:1); 1H NMR (CDCl3): δ 2.30 (3H, s, CH3), 4.31 (2H, t, J = 5.7 Hz, CH2), 4.54 (2H, t, J = 5.8 Hz, CH2), 5.32 (2H, s, CH2Ph), 6.77 (2H, d, J = 8.6 Hz, H-3′, H-5′), 7.07 (2H, J = 8.5 Hz, H-2′, H-6′), 7.25–7.37 (5H, m, C6H5), 7.49 (1H, d, J = 8.4 Hz, quinazoline H), 7.58 (1H, d, J = 7 Hz, quinazoline H), 7.68 (1H, dt, J = 7.1 and 1.5 Hz, quinazoline H), 8.27 (1H, dd, J = 7.9 and 1.5 Hz, quinazoline H). 13C NMR (CDCl3): δ 20.1, 43.1, 44.6, 64.9, 113.8, 114.0, 115.2, 122.6, 127.2, 128.1, 128.6, 128.7, 129.5, 130.1, 134.5, 136.6, 140.0, 150.7, 155.7, 161.3. MS (ES+): m/z (%) 279.2 (96), 91.1 (100).
3.1.23. 1-Cinnamyluracil (28)
A mixture of uracil (3.0 g, 26.76 mmol) and ammonium chloride 0.3 g (5.6 mmol) in hexamethyldisilazane (15 mL) was refluxed for 10 hr with exclusion of moisture until a clear solution was obtained. The excess silylating agent was removed in vacuum, the residual clear oil of 2,4-bis(trimethylsilyloxy)pyrimidine was dissolved in 50 mL of dry 1,2-dichloroethane (distilled with P2O5), was added cinnamyl bromide (4.2 mL, 28.35 mmol) to the solution and the mixture was refluxed for 20 h under N2. The reaction mixture was treated with isopropanol (10 mL), and the resulting sediment filtered, washed with ethylacetate/hexane (1:1) and dried at room temperature to give 28 (5.5 g, 90%) as small crystals which were recrystallized from iPrOH-DMF-water (2:2:1); mp: 199–200 °C (199–200 °C lit.18), Rf 0.56 (ethylacetate/hexane 1:1); 1H NMR (CDCl3), δ: 4.45 (2H, m, NCH2), 5.61 (1H, dd, J = 8.0 and 2.3, H-5), 6.32 (1H, dt, J = 15.9 and 6.0, CH–), 6.55 (1H, d, J = 16.0, PhCH ), 7.24 (1H, m, aromatic Н), 7.32 (2H, t, J = 7.6, aromatic Н), 7.43 (2H, d, J = 7.3, aromatic Н), 7.64 (1H, d, J = 7.9, H-6), 11.32 (1H, s, H-3). 13C NMR (CDCl3): 52.2, 104.7, 127.6, 129.8, 131.3, 132.0, 135.9, 139.3, 148.7, 154.3, 167.2.
3.1.24. 3-Benzyl-1-cinnamyluracil (29)
A suspension of compound 28 (1.0 g, 4.38 mmol) and К2СО3 (0.8 g, 7.79 mmol) in DMF (15 ml) was stirred at 80 °С for 1 h. Benzyl chloride (0.55 mL, 4.78 mmol) was added and the mixture stirred at the same temperature for 8 h. The reaction mixture was cooled to room temperature, poured into cold H2O (250 mL) and kept at 4 °С for 24 h. The aqueous layer was decanted and the solid residue recrystallized from mixture of acetone-H2O (2:1). The precipitate was filtered and air-dried to give 29 (1.09 g, 3.42 mmol, 78 %) as small needle white crystals, mp: 97–99 °C, R f 0.61 (hexane/ethyl acetate, 1:1); 1H NMR (DMSO-d 6), δ: 4.51 (2H, dd, J = 6.6 and 1.4 Hz, NCH2), 5.18 (2H, s, CH2Ph), 5.79 (1H, d, J = 7.9 Hz, H-5), 6.22 (1H, dt, J = 15.9 and 6.5 Hz, CH–), 6.62 (1H, d, J = 15.8 Hz, PhCH ), 7.18 (1Н, d, J = 7.9 Hz, aromatic H), 7.27–7.41 (8H, m, aromatic H, H-6), 7.52–7.54 (2H, m, aromatic H). 13C NMR (DMSO-d 6): 44.0, 50.4, 101.6, 122.0, 126.2, 127.2, 128.0, 128.3, 128.6, 134.6, 135.2, 136.5, 141.2, 151.1, 162.5. MS (ES+): m/z (%) 318.1 (38), 227.1 (66), 115.1 (100).
3.1.25. 1-Cinnamyl-3-(3,5-dimethylbenzyl)uracil (30)
Was synthesized in a similar manner as 29 to give 30 (1.7 g, 4.91 mmol, 75%) as white crystals, mp: 144.5–146 °C, R f 0.58 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3), δ: 2.27 (6H, s, CH3), 4.49 (2H, dd, J = 6.7 and 1.25 Hz, NCH2), 5.07 (2H, s, CH2Ph), 5.76 (1H, d, J = 7.94 Hz, H-5), 6.19 (1H, dt, J = 15.88 and 6.53 Hz, CH–), 6.59 (1H, d, J = 15.88 Hz, PhCH ), 6.88 (2H, s, aromatic H), 7.16 (1Н, d, J = 7.94 Hz, aromatic H), 7.24–7.37 (6H, m, aromatic H, Н-6). 13C NMR (CDCl3): 21.3, 44.4, 50.8, 102.2, 122.5, 126.7, 126.8, 128.5, 128.8, 129.4, 135.1, 135.7, 136.7, 138.0, 141.5, 151.6, 163.0. MS (ES+): m/z 346 (19), 227 (49), 115 (100).
3.1.26. 1-Cinnamyl-3-(3,5-difluorobenzyl)uracil (31)
Was synthesized in a similar manner as 29 to give 31 (1.65 g, 4.66 mmol, 71%) as white crystals, mp: 77–79 °C, R f 0.44 (hexane/ethyl acetate, 1:1); 1H NMR (DMSO-d 6), δ: 4.54 (2H, d, J = 5.9, NCH2), 5.02 (2H, s, CH2), 5.83 (1H, d, J = 7.8, H-5), 6.37 (1H, dt, J = 15.9 and 6.0, CH–), 6.58 (1H, d, J = 16.0, PhCH ), 6.95–7.00 м (2H, aromatic Н), 7.10 (1H, dt, J = 9.4 and 2.3, aromatic Н), 7.23–7.27 м (1H, aromatic Н), 7.33 (2H, t, J = 7.5, aromatic Н), 7.43 (2H, d, J = 7.3, aromatic Н), 7.78 (1H, d, J = 7.9, H-6). 13C NMR (DMSO-d 6): 43.0, 50.2, 100.5, 102.7 (t, J = 25.6 Hz), 110.5 (dd, J = 18.4 and 6.9 Hz), 124.0, 126.5, 128.0, 128.7, 132.7, 135.9, 141.7, 144.4, 151.1, 161.1, 163.5. MS (ES+): m/z (%) 354 (15), 239 (88), 115 (100).
3.1.27. 1-Cinnamyl-3-(3,5-dichlorobenzyl)uracil (32)
Was synthesized in a similar manner as 29 to give 32 (2.0 g, 5.16 mmol, 78%) as white crystals, mp: 113–114 °C, R f 0.59 (hexane/ethyl acetate, 1:1); 1H NMR (CDCl3), δ: 4.52 (2H, dd, J = 6.7 and 1.25 Hz, NCH2), 5.06 (2H, s, CH2Ph), 5.79 (1H, d, J = 7.94 Hz, H-5), 6.20 (1H, dt, J = 15.88 and 6.7 Hz, CH–), 6.62 (1H, d, J = 15.88 Hz, PhCH ), 7.21 (1Н, d, J = 7.94 Hz, aromatic H), 7.25–7.39 (8H, m, aromatic H, Н-6). 13C NMR (CDCl3): 43.6, 51.0, 102.1, 122.1, 126.7, 127.5, 128.0, 128.6, 128.8, 135.0, 135.5, 135.6, 140.0, 141.9, 151.4, 162.7. MS (ES+): m/z (%) 386 (9), 271 (50), 115 (100).
3.1.28. 1-Cinnamyl-3-(3,5-dibromobenzyl)uracil (33)
Was synthesized in a similar manner as 29 to give 33 (2.55 g, 5.36 mmol, 81%) as white crystals, mp: 124–126 °C, R f 0.60 (hexane/ethyl acetate, 1:1); 1H NMR (DMSO-d 6), δ: 4.53 (2H, d, J = 5.7, NCH2), 4.99 (2H, s, CH2), 5.82 (1H, d, J = 7.9, H-5), 6.35 (1H, dt, J = 15.9 and 5.9, CH–), 6.57 (1H, d, J = 16.0, PhCH ), 7.22–7.24 (1H, m, aromatic Н), 7.32 (2H, t, J = 7.5, aromatic Н), 7.41 (2H, s, H-2′, H-6′), 7.42 (1H, s, Н-4′), 7.50 (2H, s, aromatic Н), 7.68 (1H, s, aromatic Н), 7.76 (1H, d, J = 7.8, H-6). 13C NMR (DMSO-d 6): 50.2, 100.6, 122.4, 123.9, 126.5, 127.9, 128.6, 129.7, 132.3, 132.7, 135.9, 141.8, 144.4, 151.1, 162.5. MS (ES+): m/z 361 (35), 227 (21), 115 (100).
3.1.29. 3-(3-Bromo-5-methylbenzyl)-1-cinnamyluracil (34)
Was synthesized in a similar manner as 29 to give 34 (2.1 g, 5.11 mmol, 77%) as white crystals, mp: 134–135.5 °C, R f 0.65 (hexane/ethyl acetate, 1:1); 1H NMR (DMSO-d 6), δ: 2.23 (3H, s, CH3), 4.52 (2H, d, J = 5.9, NCH2), 4.95 (2H, s, CH2), 5.81 (1H, d, J = 7.9, H-5), 6.35 (1H, dt, J = 15.9 and 6.0, CH–), 6.55 (1H, d, J = 16.0, PhCH ), 7.09 (1H, s, aromatic Н), 7.23–7.26 (3H, m, Н-2′, H-4′, H-6′), 7.32 (2H, t, J = 7.5, aromatic Н), 7.42 (2H, d, J = 7.7, aromatic Н), 7.75 (1H, d, J = 7.8, H-6). 13C NMR (DMSO-d 6): 23.9, 46.3, 103.9, 124.7, 127.3, 129.8, 130.8, 131.3, 132.0, 133.9, 136.1, 139.3, 143.0, 143.7, 147.6, 154.4, 165.8. MS (ES+): m/z 227 (34), 115 (100).
3.1.30. 1-Cinnamyl-3-(1-naphthylmethyl)uracil (35)
Was synthesized in a similar manner as 29 to give 35 (1.75 g, 4.75 mmol, 72%), mp: 151–152.5 °C, R f 0.59 (hexane/ethyl acetate, 1:1); 1H NMR (DMSO-d 6), δ: 4.57 (2H, d, J = 5.3, NCH2), 5.53 (2H, s, CH2Naph), 5.90 (1H, d, J = 7.8, H-5), 6.40 (1H, dt, J = 16.0 and 5.9, CH–), 6.61 (1H, d, J = 16.0, PhCH ), 7.12 (1H, d, J = 7.1, aromatic Н), 7.25–7.31 (1H, m, aromatic Н), 7.34 (2H, t, J = 7.4, aromatic Н), 7.39–7.46 (3H, m, aromatic Н), 7.55–7.62 (2H, m, aromatic Н), 7.82–7.85 (2H, m, aromatic Н, H-6), 7.95–7.97 (1H, m, aromatic Н), 8.23 (1H, d, J = 8.2, aromatic Н). 13C NMR (DMSO-d 6): 41.5, 50.2, 100.6, 122.4, 123.1, 124.1, 125.4, 125.9, 126.3, 126.5, 127.3, 128.0, 128.6, 128.7, 130.5, 132.1, 132.7, 133.3, 136.0, 144.3, 151.2, 162.6. MS (ES+): m/z 256 (40), 227 (45), 115 (100).
4. Biological evaluation
4.1. Antiviral assays
The methodology of the anti-HIV assays in MT-4 cell cultures was as follows: virus stocks were titrated in MT-4 cells and expressed as the 50% cell culture infective dose (CCID50). MT-4 cells were suspended in culture medium at 1 × 105 cells/ml and infected with HIV at a multiplicity of infection of 0.02. Immediately after viral infection, 100 μl of the cell suspension was placed in each well of a flat-bottomed microtiter tray containing various concentrations of the test compounds. After 4 days of incubation at 37 °C, the number of viable cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. The selection and characterization of mutant virus strains have been performed previously.
4.2. Antiviral activity assays other than HIV
The compounds were evaluated against the following viruses: herpes simplex virus type 1 (HSV-1) strain KOS, Thymidine kinase-deficient (TK−) HSV-1 KOS strain resistant to ACV (ACVr), herpes simplex virus type 2 (HSV-2) strains Lyons and G, varicella-zoster virus (VZV) strain Oka, TK− VZV strain 07−1, human cytomegalovirus (HCMV) strains AD-169 and Davis, vaccinia virus Lederle strain, respiratory syncytial virus (RSV) strain Long, vesicular stomatitis virus (VSV), Coxsackie B4, Parainfluenza 3, Influenza virus A (subtypes H1N1, H3N2), influenza virus B, Reovirus-1, Sindbis and Punta Toro. The antiviral assays were based on inhibition of virus-induced cytopathicity or plaque formation in human embryonic lung (HEL) fibroblasts, African green monkey cells (Vero), human epithelial cells (HeLa) or Madin-Darby canine kidney cells (MDCK). Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) or with 20 plaque forming units (PFU) (VZV) in the presence of varying concentrations of the test compounds. Viral cytopathicity or plaque formation was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 or compound concentration required to reduce virus-induced cytopathogenicity or viral plaque formation by 50%.
4.3. Reverse transcriptase plasmids, RT expression and purification
Plasmid encoding p66 subunits of the wild-type HIV-1 reverse transcriptase (RT) or mutant forms (L100I, K103N, V106A, Y181C, Y188L, G190A and K103N/Y181C) were described previously.14, 30 A similar plasmid encoding wild-type p51-subunit was a kind gift of Professor S. Le Grice.31 RTs were expressed in Rosetta (DE3) E. coli strain as individual subunits, RT-harboring cells were mixed together and the p66/p51 heterodimeric RTs were purified using standard Ni-NTA agarose procedure.31
4.4. RT enzyme assay
The RT assays using activated DNA were performed as follows: the standard reaction mixture (20 μl) contained 0.75 μg of activated DNA, 0.05 μg p66/p66 RT, 3 μM dATP, 30 μM of dCTP, dGTP and dTTP, 1 μCi [α-32P]dATP in a Tris–HCl buffer (50 mM, pH 8.1) containing also 10 mM MgCl2, and 0.2 M KCl. The test compounds were dissolved in DMSO and added to both assays to a final 10% DMSO concentration. The reaction mixtures were incubated for 30 min at 37 °C, and applied onto Whatman 3MM filters. After drying on air the filters were washed 2× with 10% trichloroacetic acid, then 2× with 5% trichloroacetic acid, once with ethanol and then air-dried. Radioactivity was determined in a Perkin Elmer Tri-Carb 2810 TR liquid scintillation counter.
Acknowledgments
This work was supported by Ministry of Education and Science of Russian Federation (state contract 16.512.11.2233), by the Russian Foundation For Basic Research (Grant 10-04-00056a), by the Presidium of the Russian Academy of Sciences (Programme ‘Molecular and Cellular Biology’) and by the K.U. Leuven (GOA no. 10/014). A.I. and V.V.-E. were supported by Grant of President of Russian Federation (МК-5035.2011.4). The authors like to thank Kartik Temburnikar (UMBC), Lizette van Berckelaer, Kristien Minner, Kristien Erven and Kris Uyttersprot (Rega) for their dedicated technical assistance.
Footnotes
Supplementary data (NMR spectra for all of the synthesized compounds) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2012.12.027.
Supplementary data
NMR spectra for all of the synthesized compounds.
References and notes
- 1.UNAIDS, http://www.unaids.org/en/.
- 2.Jonckheere H., Anne J., De Clercq E. Med. Res. Rev. 2000;20:129. doi: 10.1002/(sici)1098-1128(200003)20:2<129::aid-med2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 3.Pauwels R. Curr. Opin. Pharmacol. 2004;4:437. doi: 10.1016/j.coph.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Merluzzi V.J., Hargrave K.D., Labadia M., Grozinger K., Skoog M., Wu J.C., Shih C.K., Eckner K., Hattox S., Adams J., et al. Science. 1990;250:1411. doi: 10.1126/science.1701568. [DOI] [PubMed] [Google Scholar]
- 5.Dueweke T.J., Poppe S.M., Romero D.L., Swaney S.M., So A.G., Downey K.M., Althaus I.W., Reusser F., Busso M., Resnick L., et al. Antimicrob. Agents Chemother. 1993;37:1127. doi: 10.1128/aac.37.5.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young S.D., Britcher S.F., Tran L.O., Payne L.S., Lumma W.C., Lyle T.A., Huff J.R., Anderson P.S., Olsen D.B., Carroll S.S., et al. Antimicrob. Agents Chemother. 1995;39:2602. doi: 10.1128/aac.39.12.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sweeney Z.K., Klumpp K. Curr. Opin. Drug Discov. Devel. 2008;11:458. [PubMed] [Google Scholar]
- 8.Andries K., Azijn H., Thielemans T., Ludovici D., Kukla M., Heeres J., Janssen P., De Corte B., Vingerhoets J., Pauwels R., de Bethune M.P. Antimicrob. Agents Chemother. 2004;48:4680. doi: 10.1128/AAC.48.12.4680-4686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mordant C., Schmitt B., Pasquier E., Demestre C., Queguiner L., Masungi C., Peeters A., Smeulders L., Bettens E., Hertogs K., Heeres J., Lewi P., Guillemont J. Eur. J. Med. Chem. 2007;42:567. doi: 10.1016/j.ejmech.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 10.Tucker T.J., Sisko J.T., Tynebor R.M., Williams T.M., Felock P.J., Flynn J.A., Lai M.T., Liang Y., McGaughey G., Liu M., Miller M., Moyer G., Munshi V., Perlow-Poehnelt R., Prasad S., Reid J.C., Sanchez R., Torrent M., Vacca J.P., Wan B.L., Yan Y. J. Med. Chem. 2008;51:6503. doi: 10.1021/jm800856c. [DOI] [PubMed] [Google Scholar]
- 11.Ren J., Chamberlain P.P., Stamp A., Short S.A., Weaver K.L., Romines K.R., Hazen R., Freeman A., Ferris R.G., Andrews C.W., Boone L., Chan J.H., Stammers D.K. J. Med. Chem. 2008;51:5000. doi: 10.1021/jm8004493. [DOI] [PubMed] [Google Scholar]
- 12.Romines K.R., Freeman G.A., Schaller L.T., Cowan J.R., Gonzales S.S., Tidwell J.H., Andrews C.W., 3rd, Stammers D.K., Hazen R.J., Ferris R.G., Short S.A., Chan J.H., Boone L.R. J. Med. Chem. 2006;49:727. doi: 10.1021/jm050670l. [DOI] [PubMed] [Google Scholar]
- 13.Sweeney Z.K., Harris S.F., Arora S.F., Javanbakht H., Li Y., Fretland J., Davidson J.P., Billedeau J.R., Gleason S.K., Hirschfeld D., Kennedy-Smith J.J., Mirzadegan T., Roetz R., Smith M., Sperry S., Suh J.M., Wu J., Tsing S., Villasenor A.G., Paul A., Su G., Heilek G., Hang J.Q., Zhou A.S., Jernelius J.A., Zhang F.J., Klumpp K. J. Med. Chem. 2008;51:7449. doi: 10.1021/jm800527x. [DOI] [PubMed] [Google Scholar]
- 14.Novikov M.S., Ivanova O.N., Ivanov A.V., Ozerov A.A., Valuev-Elliston V.T., Temburnikar K., Gurskaya G.V., Kochetkov S.N., Pannecouque C., Balzarini J., Seley-Radtke K.L. Bioorg. Med. Chem. 2011;19:5794. doi: 10.1016/j.bmc.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novikov M.S., Ozerov A.A. Chem. Heterocycl. Compd. 2005;69:905. [Google Scholar]
- 16.Kirincich S.J., Xiang J., Green N., Tam S., Yang H.Y., Shim J., Shen M.W., Clark J.D., McKew J.C. Bioorg. Med. Chem. 2009;17:4383. doi: 10.1016/j.bmc.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 17.Goux C., Sigismondi S., Sinou D., Perez M., Moreno-Manas M., Pleixats R., Villaroya M. Tetrahedron. 1996;52:9521. [Google Scholar]
- 18.Moreno-Manas M., Pleixats R., Villaroya M. Tetrahedron. 1993;49:1457. [Google Scholar]
- 19.Solani Rad M.N., Khalafi-Nezhad A., Behrouz S., Faghihi M.A., Zare A., Parhami A. Tetrahedron. 2008;64:1778–1785. [Google Scholar]
- 20.Malik V., Singh P., Kumar S. Tetrahedron. 2005;61:4009. [Google Scholar]
- 21.Malik V., Singh P., Kumar S. Tetrahedron. 2006;62:5944. [Google Scholar]
- 22.Skaric V., Skaric D., Cuzmek A. Perkin Trans. 1984;1:2221. [Google Scholar]
- 23.Maguire A.R., Hladezuk I., Ford A. Carbohydr. Res. 2002;337:369. doi: 10.1016/s0008-6215(01)00325-1. [DOI] [PubMed] [Google Scholar]
- 24.Bollini M., Domaoal R.A., Thakur V.V., Gallardo-Macias R., Spasov K.A., Anderson K.S., Jorgensen W.L. J. Med. Chem. 2011;54:8582. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su D.S., Lim J.J., Tinney E., Wan B.L., Young M.B., Anderson K.D., Rudd D., Munshi V., Bahnck C., Felock P.J., Lu M., Lai M.T., Touch S., Moyer G., DiStefano D.J., Flynn J.A., Liang Y., Sanchez R., Perlow-Poehnelt R., Miller M., Vacca J.P., Williams T.M., Anthony N.J. J. Med. Chem. 2009;52:7163. doi: 10.1021/jm901230r. [DOI] [PubMed] [Google Scholar]
- 26.Buckheit R.W., Jr., Watson K., Fliakas-Boltz V., Russell J., Loftus T.L., Osterling M.C., Turpin J.A., Pallansch L.A., White E.L., Lee J.W., Lee S.H., Oh J.W., Kwon H.S., Chung S.G., Cho E.H. Antimicrob. Agents Chemother. 2001;45:393. doi: 10.1128/AAC.45.2.393-400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buckheit R.W., Jr., Hartman T.L., Watson K.M., Kwon H.S., Lee S.H., Lee J.W., Kang D.W., Chung S.G., Cho E.H. Antivir. Chem. Chemother. 2007;18:259. doi: 10.1177/095632020701800502. [DOI] [PubMed] [Google Scholar]
- 28.Trott O., Olson A.J.J. Comput. Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kroemer R.T., Vulpetti A., McDonald J.J., Rohrer D.C., Trosset J.Y., Giordanetto F., Cotesta S., McMartin C., Kihlen M., Stouten P.F.J. Chem. Inform. Comput. Sci. 2004;44:871. doi: 10.1021/ci049970m. [DOI] [PubMed] [Google Scholar]
- 30.Pokholok D.K., Gudima S.O., Yesipov D.S., Dobrynin V.N., Rechinsky V.O., Kochetkov S.N. FEBS Lett. 1993;325:237. doi: 10.1016/0014-5793(93)81080-j. [DOI] [PubMed] [Google Scholar]
- 31.Le Grice S.F., Grüninger-Leitch F. Eur. J. Biochem. 1990;187:307. doi: 10.1111/j.1432-1033.1990.tb15306.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NMR spectra for all of the synthesized compounds.