

Abstract
CD8+ T-cell memory to viruses is stable in the absence but volatile in the presence of other infections. Apoptotic events that occur early in acute infections delete pre-existing memory T cells, leaving the host with reduced memory (except for cross-reactive responses) to previously encountered viruses. Apoptotic events also silence the acute immune response, leaving the host with a residual population of memory T cells. Persistent infections can induce apoptotic deletions of memory T cells that are specific to the persisting virus and to previously encountered pathogens.
Abbreviations: AICD, activation-induced cell death; FasL, Fas ligand; IFN, interferon; IL, interleukin; LCMV, lymphocytic choriomeningitis; TCR, T-cell receptor; TNF, tumor necrosis factor
Introduction
Viral infections are potent stimulators of CD8+ T cells, which recognize viral peptides in association with Class 1 MHC molecules on virus-infected antigen-presenting cells (APCs). CD8+ T-cell stimulation is so profound that the CD8+/CD4+ T-cell ratio sometimes shifts from about 1:2 to 3:1 but, thereafter, returns to normal in the memory state (Figure 1 ). The proliferating cells are mostly specific to viral peptides, but their fate and the fate of bystander CD8+ T cells are highly regulated by apoptotic events that come in two waves during acute infections. The first wave is associated with a transient lymphopenia, occurring before the development of the splenomegaly and lymphadenopathy characteristic of the developing T-cell response [1]. The second wave occurs as the immune response silences and enters the memory state [2]. Recent work has correlated the degree of apoptotic events to deletions and preservations of the memory pool.
Figure 1.
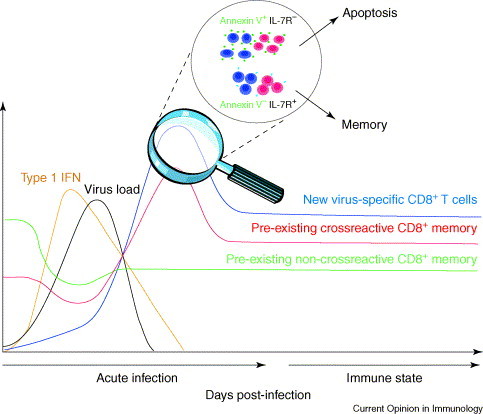
CD8+ T-cell proliferation and apoptosis during acute viral infection. This graft reflects data generated in the LCMV system, showing the magnitude of type 1 IFN, virus load, and frequencies of cross-reactive and non-cross-reactive memory T cells as a new T-cell response is generated. Pre-existing memory cells are driven into apoptosis, but cross-reactive cells ultimately expand and are preserved or increased in memory. Virus-specific T cells undergo apoptosis during the silencing phase, but those expressing IL-7Rα and not reacting with Annexin V survive this process and enter the long-term memory pool.
Virus-induced lymphopenia and memory CD8+ T-cell loss
Virus-induced lymphopenia has been observed in many severe acute viral infections in humans, such as influenza, measles, West Nile, Ebola, Lassa fever, lymphocytic choriomeningitis (LCMV) and, most recently, severe acute respiratory syndrome (SARS) corona viruses 1., 3.••, 4., 5., but common mechanisms have only recently been proposed. Infection of mice with LCMV and other viruses, as well as bacteria such as Listeria monocytogenes, causes a reduction of lymphocytes early in infection 1., 2., 6.•, 7.. Memory CD8+ T cells are particularly affected and express apoptotic markers, including Annexin V and TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling) stain positivity. There are probably several mechanisms for this apoptotic loss of CD8+ T cells, but a common mechanism might be provided by type 1 interferons (IFNs). CD8+ T-cell apoptosis was elicited by the IFN-inducer poly I:C, and the early apoptosis occurring during LCMV infection of mice correlated with the type 1 IFN response [1]. Notably, mice lacking type 1 IFN receptors resisted the CD8+ T-cell apoptosis, induced by either poly I:C or by LCMV. It was not clear whether IFN stimulated apoptosis directly or indirectly, either by altering the susceptibility of CD8+ T cells to apoptotic stimuli or by inducing other cytokines. IFN can, for example, induce other pro-apoptotic proteins, such as TNF (tumor necrosis factor) family member TRAIL (TNF-related apoptosis-inducing ligand) [8], and Jiang et al. discussed preliminary data indicating that this apoptosis is reduced in TRAIL-deficient mice [6•]. This early apoptosis does not seem to be regulated greatly by Fas, FasL, perforin, Bcl-2, Bcl-XL, or IFN γ, and thus, further characterization of this event is necessary.
The induction of a lymphopenic state by irradiation or cytotoxic drugs can lead to enhanced immune responses, by making room in lymphoid organs for T cells to seed and develop [3••]. Cyclophosphamide, for example, enhances T-cell responses to herpes simplex virus type 1 infections in mice [9]. It is possible, therefore, that this virus-induced early loss of CD8+ T cells might facilitate the development of a more vigorous virus-specific CD8+ T-cell response. Consistent with this hypothesis, younger mice have been observed to experience greater initial lymphopenia [10], yet generate stronger virus-specific responses than older mice 11., 12.. Adoptive transfer studies have indicated that the environment of aged mice inhibits the apoptosis of young T cells [10]. T cells in aged mice also express higher levels of Bcl-2 and resist poly I:C-induced and interleukin (IL)-15-induced memory cell division [13•].
CD8+ T-cell memory to viruses is normally relatively stable, as there is a continuous IL-15-dependent turnover of memory CD8+ T cells, which maintains their numbers, probably by equally balancing division with apoptosis 14., 15.. This has been referred to as ‘basal proliferation’, in contrast to the ‘acute homeostatic proliferation’ that occurs when lymphocytes divide to fill up lymphopenic environments. The stability of memory is disrupted by infections with unrelated viruses or bacteria, or by exposure to bacterial superantigens, all of which lead to a reduction in frequencies of pre-existing memory cells 16., 17.•, 18.•, 19., 20.. We propose two models to explain this loss of memory. One is a passive competition model, whereby newly generated memory cells, at termination of infection, compete with pre-existing memory cells for survival niches. Such a model appears to be supported by a study showing that inclusion of an IL-15 gene into Mycobacterium bovis enhances memory T-cell frequencies to the Mycobacterium, but decreases previously generated memory T cells that are specific to L. monocytogenes [21•]. An active model of memory-cell decay proposes that some mechanism kills off the resident memory cells, thereby enhancing the survival of the newly emerging memory cells. Recent studies have demonstrated that LCMV infection causes substantial reductions in the frequencies of CD8+ T cells that are specific to a heterologous virus as early as day two post-infection, and that these frequencies do not recover as the infection resolves [20]. Thus, the long-term loss in memory might, in large part, be a consequence of the early lymphopenia and apoptotic loss in memory CD8+ T cells.
The failure to recover memory after the occurrence of virus-induced lymphopenia is probably due to the inability of CD8+ T cells that are not specific to the virus to compete with the proliferation of T cells that are specific. Additionally, it may reflect the poor ability of virus-specific memory cells to repopulate lymphopenic environments in general [3••]. Lymphopenic conditions induce some T cells to proliferate and fill-up available space; this ‘acute homeostatic proliferation’ is primarily dependent on IL-7, and somewhat on IL-15 [15]. Type 1 IFN induces the division of memory phenotype CD8+ T cells through the action of IL-15 [22]. This division, however, might not be just a response to IL-15 but, instead, may be driven by this tendency of T cells to divide in lymphopenic environments, in this case created by IFN 1., 3.••. The T cells that proliferate in the most lymphopenic environments seem to be those with the highest affinity for self-antigens and, thus, may not be the same T cells that have expanded and entered the memory pool in response to foreign antigens 23., 24.. An analysis of the repopulation of lymphopenic environments with adoptively transferred splenocytes, containing clearly defined virus-specific memory cells demonstrated, in every incidence, that the bona fide memory cells divided less frequently and were diluted out by other, presumably self-reactive T cells [3••]. Memory CD8+ cells express higher levels of Bcl-2 and resist irradiation-induced apoptosis [25], but when time is allowed for the repopulation of the irradiated environment, other cells proliferate more than the virus-specific memory cells and dilute that population proportionally [3••]. Thus, a permanent loss of memory cells is observed under conditions of lymphopenia, even when there is not competition by the T cells responding to viral antigens during an acute infection.
Preservation of memory after infections can occur if the memory cells cross-react with antigens that are encoded by the infecting virus [17•] (Figure 1). LCMV and Pichinde virus (PV) encode epitopes that share six out of eight amino acids, and T-cell responses to these epitopes are enriched rather than deleted in mice that are sequentially infected by these viruses. A similar phenomenon of deletion of non-cross-reactive but enrichment of cross-reactive T cells is seen with sequential bacterial infections [26•]. Thus, T-cell cross-reactivity acts in opposition to the forces driving the deletion in memory. This leads us to question whether T-cell receptor (TCR) engagement inhibits the apoptotic loss of memory CD8+ T cells during the lymphopenic phase of infection. One study examining the proliferation of LCMV-specific transgenic T cells in LCMV-infected mice argued that antigen engagement blocks the early apoptosis, although it was unclear whether there was a transient apoptotic loss before a rapid antigen-specific proliferation [6•]. T cells that are specific to the cross-reactive epitope between LCMV and PV were deleted, just like those that are specific to other epitopes at the early stages of infection, and thereafter began to proliferate [20].
CD8+ T-cell apoptosis during silencing of the immune response
The reduction of T-cell number from the spleen and lymph nodes as the immune responses silences at the end of infection is, in part, due to apoptosis [2] and, in part, due to dissemination of the T cells out of the lymphoid organs and into the peripheral tissue [27]. The level of apoptosis in splenocytes was high during LCMV infection, as judged by an altered mitochondrial membrane potential (Δψm) [28] and by reactivity with Annexin V, often on more than 50% of the CD8+ cells [29•]. The spontaneous apoptosis could not be prevented by transgenic expression of Bcl-2 or Bcl-XL 2., 30., yet Bcl-2 was shown to be elevated in memory cells [25]. Silencing was reduced in mice lacking CD43, as the CD43−/− T cells demonstrated increased Bcl-2 and decreased apoptosis [31]. It seems, therefore, that anti-apoptotic Bcl-2 family members cannot prevent immune silencing, but might still be helpful for memory-cell survival. Expression of a Bcl-2 family pro-apoptotic molecule, BNIP3, was found to be elevated in the apoptotic T cells [32].
An analysis of cell-surface antigen expression, predictive of survival of spleen CD8+ T cells into the memory state revealed that IL-7 receptor (R) α-expressing and IL-7Rα-sorted CD8+ T cells gave rise to memory when transferred into naïve hosts [33••]. Cells with high IL-7Rα-expression had higher levels of the anti-apoptotic molecules Bcl-2 and Bcl-XL than those that did not express IL-7Rα (Figure 1). Using similar strategies, we have shown that Annexin-V (−) but not Annexin-V (+) T cells gives rise to LCMV-specific memory and that the expression of IL-7Rα is much higher on Annexin-V (−) than Annexin-V (+) cells (XZ Wang, MA Brehm and RM Welsh, unpublished data). The factor(s) that cause some cells to express IL-7Rα and others not to remains unclear, as does the mechanism of apoptosis during immune silencing conditions. No singular pro-apoptotic event, such as that mediated by FasL or TNF, has been shown to mediate this process. Silencing was delayed in mice lacking perforin or IFNγ, but so was clearance of virus 34., 35.. It has been hypothesized that immune silencing might be a consequence of a pre-determined ‘programmed contraction’ event [36•].
Silencing of the T-cell response to infection is also associated with the dissemination of T cells into the periphery, where memory T cells can reside at high frequencies [27]. Of note, throughout the infection of mice with LCMV, a much lower level of T-cell apoptosis was seen in peripheral tissue, such as the lung, fat and peritoneal cavity, than in the spleen and lymph nodes [29•]. T cells from the spleen of LCMV-infected mice expressed more Fas and Fas ligand (FasL), and were more susceptible to TCR-mediated activation-induced cell death (AICD) than those in the periphery, but the tissue-dependent differences in spontaneous in vivo apoptosis levels during immune silencing were also seen in mice deficient in Fas and FasL expression. This indicates that T cells in the secondary lymphoid organs are more susceptible to both Fas-dependent and Fas-independent mechanisms of apoptosis [29•]. Interestingly, IL-7Rα is expressed on a much higher proportion of peripheral than lymphoid organ T cells (XZ Wang, MA Brehm, and RM Welsh, unpublished observations). Mixture experiments have demonstrated that the CD8+ T-cell expression of Fas and FasL, and susceptibility to AICD was dependent on the tissue-specific leukocyte environment, indicating that peripheral tissues create environments to preserve the survival of memory cells [29•]. These tissue-dependent differences may reflect the fact that many cytokines and cell adhesion molecules can influence CD8+ T-cell apoptosis and the expression of pro-apoptotic molecules 29.•, 37., 38..
Antigen-dependent exhaustion of T-cell responses during acute and persistent infections
So far, the focus has been on apoptosis and loss of CD8+ T cells in the absence of available antigen to signal the TCR. The presence of antigen, however, can drive T cells into clonal exhaustion, which may be due either to a complete loss of T cells or to a loss in T-cell function [39]. Inoculation of adult mice with high doses of LCMV can result in persistent infections with weak T-cell responses that differ, depending on the epitope. T cells that are specific for the NP396 epitope are completely lost from the spleen, presumably by apoptotic mechanisms, but T cells with other specificities, such as GP33, undergo a predictable loss of function in the sequence of cytotoxicity and IL-2 production, TNFα production and then IFNγ production 40.•, 41.•, 42., 43.•• (Figure 2 ). This differential effect between NP396- and GP33-specific T cells may be explained by overstimulation of the T cells with NP396, which is derived from the most highly expressed protein and has a high affinity to the presenting MHC. Differences in the apoptosis levels of epitope-specific CD8+ T cells can be seen early during infection and can be imprinted such that the differences can be detected even in the memory state after acute infection, when antigen is mostly cleared. As early as five days after acute LCMV infection, NP396-specific T cells had more profound increases in Δψm than GP33-specific T cells [28], and higher proportions of NP396-specific than GP33-specific CD8+ T cells bound Annexin V at day 7, 9, 12, 65, and 128 post-infection [29•], suggesting that the apoptotic properties of these antigen-specific T cells were forever altered by their initial antigenic exposure. Kinetic studies have also demonstrated differences in proliferation rates of various LCMV epitope-specific T cells, although not necessarily correlating with an apoptotic phenotype [44]; this does, however, support the idea that T cells that are specific to different epitopes might behave in distinct ways.
Figure 2.
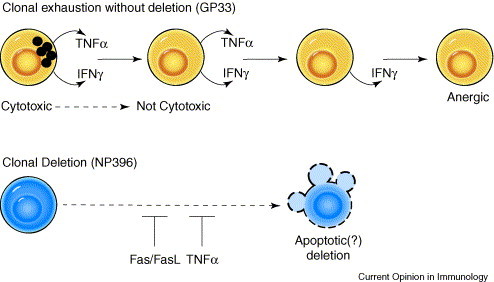
Progression of clonal exhaustion during persistent infection. This figure, based on persistent LCMV infections, induced by infecting adult mice with high doses of widely disseminating virus, depicts an epitope-specificity-based functional exhaustion or complete deletion of T cells. Prior to deletion, T cells lose functions in a prescribed order, as depicted for GP33-specific T cells. NP396-specific T cells will also lose function, but depicted here is their propensity to be completely eliminated, presumably by apoptotic mechanisms, which are, in part, controlled by Fas/FasL and TNFα.
The clonal elimination under conditions of high antigen load during persistent infection has long been thought to be mediated by some form of AICD but has never been clarified because it occurred in mice defective in AICD functions. Important new kinetic studies, examining the frequencies of LCMV NP396-specific T cells by MHC-tetramers at different time periods after persistent infection, have indicated that the elimination of these T cells is substantially delayed, although not prevented, in FasL- or TNFR1-deficient mice, arguing for an AICD effect that might entail either of at least two pro-apoptotic mechanisms [43••] (Figure 2).
Persistent infections, therefore, cause a continual evolution of the function and numbers of T cells that are specific for the infecting virus, but what is the impact of a persistent infection on memory T cells that are not specific to the persisting virus? New studies have shown that persistent infections of mice with mouse γ herpes virus or LCMV can cause substantial deletions in CD8+ memory T cells that are specific to influenza, PV, vesicular stomatitis, or vaccinia viruses 18.•, 20.. Adoptive transfer of CFSE (carboxyfluorescein diacetate succinimidyl ester)-labeled splenocytes into mice that are persistently infected as adults with LCMV revealed a very low level of IL-15-mediated division and a substantial loss in frequency of donor memory T cells, specific to a heterologous virus. Thus, attrition of memory to unrelated pathogens can be a continuous process during persistent infections.
Conclusions
This review has highlighted recent work demonstrating the way that CD8+ T-cell memory is modulated through apoptotic processes during viral infections. Memory T-cell loss can be driven non-specifically as a consequence of virus-induced cytokines, or specifically through excess antigenic stimulation. This volatility of the stability of CD8+ T-cell memory should be taken into consideration in the design of new vaccines.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
-
•
of special interest
-
••
of outstanding interest
Acknowledgements
This work was supported by US National Institutes of Health research grants AI17672 and AR35506. We thank Susan Stepp and Michael Brehm for help with the graphics.
Contributor Information
Raymond M Welsh, Email: Raymond.welsh@umassmed.edu.
Kapil Bahl, Email: 2kapil.bahl@umassmed.edu.
Xiaoting Z Wang, Email: xiaoting.wang@umassmed.edu.
References
- 1.McNally J.M, Zarozinski C.C, Lin M.Y, Brehm M.A, Chen H.D, Welsh R.M. Attrition of bystander CD8 T cells during virus-induced T cell and interferon responses. J Virol. 2001;75:5965–5976. doi: 10.1128/JVI.75.13.5965-5976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Razvi E.S, Jiang Z, Woda B.A, Welsh R.M. Lymphocyte apoptosis during the silencing of the immune response to acute viral infections in normal, lpr and Bcl-2-transgenic mice. Am J Pathol. 1995;147:79–91. [PMC free article] [PubMed] [Google Scholar]
- 3.••.Peacock C.D, Kim S.-K, Welsh R.M. Attrition of virus-specific memory CD8(+) T cells during reconstitution of lymphopenic environments. J Immunol. 2003;171:655–663. doi: 10.4049/jimmunol.171.2.655. [DOI] [PubMed] [Google Scholar]; This paper shows that bona fide virus-specific memory T cells are diluted out under conditions of homeostatic reconstitution of lymphopenic environments, caused by genetic deficiency, irradiation, cytokine stimulation or virus infection.
- 4.Wong R.S, Wu A, To K.F, Lee N, Lam C.W, Wong C.K, Chan P.K, Ng M.H, Yu L.M, Hui D.S. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ. 2003;326:1358–1362. doi: 10.1136/bmj.326.7403.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nabeshima S, Murata M, Kikuchi K, Ikematsu H, Kashiwagi S, Hayashi J. A reduction in the number of peripheral CD28+CD8+T cells in the acute phase of influenza. Clin Exp Immunol. 2002;128:339–346. doi: 10.1046/j.1365-2249.2002.01819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.•.Jiang J, Lau L.L, Shen H. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J Immunol. 2003;171:4352–4358. doi: 10.4049/jimmunol.171.8.4352. [DOI] [PubMed] [Google Scholar]; The authors demonstrate apoptotic deletion of CD8+ T cells as a consequence of infection and argue that TCR stimulation can prevent this deletion.
- 7.Merrick J.C, Edelson B.T, Bhardwaj V, Swanson P.E, Unanue E.R. Lymphocyte apoptosis during early phase of Listeria infection in mice. Am J Pathol. 1997;151:785–792. [PMC free article] [PubMed] [Google Scholar]
- 8.Sato K, Hida S, Takayanagi H, Yokochi T, Kayagaki N, Takeda K, Yagita H, Okumura K, Tanaka N, Taniguchi T, Ogasawara K. Antiviral response by natural killer cells through TRAIL gene induction by IFN-alpha/beta. Eur J Immunol. 2001;31:3138–3146. doi: 10.1002/1521-4141(200111)31:11<3138::aid-immu3138>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 9.Pfizenmaier K, Jung H, Starzinski-Powitz A, Rollinghoff M, Wagner H. The role of T cells in anti-herpes simplex virus immunity. I. Induction of antigen-specific cytotoxic T lymphocytes. J Immunol. 1977;119:939–944. [PubMed] [Google Scholar]
- 10.Jiang J, Anaraki F, Blank K.J, Murasko D.M. Cuttine edge: T cells from aged mice are resistant to depletion early during virus infection. J Immunol. 2003;171:3353–3357. doi: 10.4049/jimmunol.171.7.3353. [DOI] [PubMed] [Google Scholar]
- 11.Po J.L, Gardner E.M, Anaraki F, Katsikis P.D, Murasko D.M. Age-associated decrease in virus-specific CD8+ T lymphocytes during primary influenza infection. Mech Ageing Dev. 2002;123:1167–1181. doi: 10.1016/s0047-6374(02)00010-6. [DOI] [PubMed] [Google Scholar]
- 12.Kapasi Z.F, Murali-Krishna K, McRae M.L, Ahmed R. Defective generation but normal maintenance of memory T cells in old mice. Eur J Immunol. 2002;32:1567–1573. doi: 10.1002/1521-4141(200206)32:6<1567::AID-IMMU1567>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 13.•.Zhang X, Fujii H, Kishimoto H, LeRoy E, Surh C.D, Sprent J. Aging leads to disturbed homeostasis of memory phenotype CD8(+) cells. J Exp Med. 2002;195:283–293. doi: 10.1084/jem.20011267. [DOI] [PMC free article] [PubMed] [Google Scholar]; This shows that the effects of IL-15 and poly I:C on CD8+ T-cell homeostasis can be regulated by the age of the host.
- 14.Becker T.C, Wherry E.J, Boone D, Murali-Krishna K, Antia R, Ma A, Ahmed R. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. 2002;195:1541–1548. doi: 10.1084/jem.20020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldrath A.W, Sivakumar P.V, Glaccum M, Kennedy M.K, Bevan M.J, Benoist C, Mathis D, Butz E.A. Cytokine requirements for acute and basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med. 2002;195:1515–1522. doi: 10.1084/jem.20020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selin L.K, Lin M.Y, Kraemer K.A, Schneck J.P, Pardoll D, Varga S.M, Santolucito P.A, Pinto A.K, Welsh R.M. Attrition of T cell memory:selective loss of lymphocytic choriomeningitis virus (LCMV) epitope-specific memory CD8 T cells following infections with heterologous viruses. Immunity. 1999;11:733–742. doi: 10.1016/s1074-7613(00)80147-8. [DOI] [PubMed] [Google Scholar]
- 17.•.Brehm M.A, Pinto A.K, Daniels K.A, Schneck J.P, Welsh R.M, Selin L.K. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat Immunol. 2002;3:627–634. doi: 10.1038/ni806. [DOI] [PubMed] [Google Scholar]; This paper shows that, while non-cross-reactive memory CD8+ T cells are lost after a virus infection, cross-reactive T cells are enriched.
- 18.•.Liu H, Andreansky S, Diaz G, Turner S.J, Wodarz D, Doherty P.C. Quantitative analysis of long-term virus-specific CD8+-T-cell memory in mice challenged with unrelated pathogens. J Virol. 2003;77:7756–7763. doi: 10.1128/JVI.77.14.7756-7763.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrate that a persistent infection can drive a loss in memory to an unrelated virus.
- 19.Huang C.C, Shah S, Nguyen P, Altman J.D, Blackman M.A. Bacterial superantigen exposure after resolution of influenza virus infection perturbs the virus-specific memory CD8(+)-T-cell repertoire. J Virol. 2002;76:6852–6856. doi: 10.1128/JVI.76.13.6852-6856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim S.K, Welsh R.M. Comprehensive early and lasting loss of memory CD8 T cells and functional memory during acute and persistent viral infections. J Immunol. 2004;172:3139–3150. doi: 10.4049/jimmunol.172.5.3139. [DOI] [PubMed] [Google Scholar]
- 21.•.Chapdelaine Y, Smith D.K, Pedras-Vasconcelos J.A, Krishnan L, Sad S. Increased CD8+ T cell memory to concurrent infection at the expense of increased erosion of pre-existing memory: the paradoxical role of IL-15. J Immunol. 2003;171:5454–5460. doi: 10.4049/jimmunol.171.10.5454. [DOI] [PubMed] [Google Scholar]; Here, incorporation of IL-15 into a pathogen is shown to enhance memory to it but cause greater attrition of memory to a previously encountered pathogen, possibly supporting a competition model for the deletion of memory.
- 22.Zhang X, Sun S, Hwang I, Tough D.F, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 1998;8:591–599. doi: 10.1016/s1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- 23.Kieper W.C, Burghardt J.T, Surh C.D. A role for TCR affinity in regulating naive T cell homeostasis. J Immunol. 2004;172:40–44. doi: 10.4049/jimmunol.172.1.40. [DOI] [PubMed] [Google Scholar]
- 24.Kassiotis G, Zamoyska R, Stockinger B. Involvement of avidity for major histocompatibility complex in homeostasis of naive and memory T cells. J Exp Med. 2003;197:1007–1016. doi: 10.1084/jem.20021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grayson J.M, Harrington L.E, Lanier J.G, Wherry E.J, Ahmed R. Differential sensitivity of naive and memory CD8+ T cells to apoptosis in vivo. J Immunol. 2002;169:3760–3770. doi: 10.4049/jimmunol.169.7.3760. [DOI] [PubMed] [Google Scholar]
- 26.•.Smith D.K, Dudani R, Pedras-Vasconcelos J.A, Chapdelaine Y, van Faassen H, Sad S. Cross-reactive antigen is required to prevent erosion of established T cell memory and tumor immunity: a heterologous bacterial model of attrition. J Immunol. 2002;169:1197–1206. doi: 10.4049/jimmunol.169.3.1197. [DOI] [PubMed] [Google Scholar]; Cross-reactivity in antibacterial T-cell responses can prevent the memory loss that otherwise occurs against non-cross-reactive epitopes.
- 27.Masopust D, Vezys V, Marzo A.L, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 28.Grayson J.M, Laniewski N.G, Lanier J.G, Ahmed R. Mitochondrial potential and reactive oxygen intermediates in antigen-specific CD8+ T cells during viral infection. J Immunol. 2003;170:4745–4751. doi: 10.4049/jimmunol.170.9.4745. [DOI] [PubMed] [Google Scholar]
- 29.•.Wang X.Z, Stepp S.E, Brehm M.A, Chen H.D, Selin L.K, Welsh R.M. Virus-specific CD8 T cells in peripheral tissues are more resistant to apoptosis than those in lymphoid organs. Immunity. 2003;18:631–642. doi: 10.1016/s1074-7613(03)00116-x. [DOI] [PubMed] [Google Scholar]; This paper demonstrates that CD8+ T cells in the periphery are more resistant to apoptosis than those in secondary lymphoid organs, offering a potential explanation for the survival of T cells in the periphery after clearance of infection.
- 30.Petschner F, Zimmerman C, Strasser A, Grillot D, Nunez G, Pircher H. Constitutive expression of Bcl-xL or Bcl-2 prevents peptide antigen-induced T cell deletion but does not influence T cell homeostasis after a viral infection. Eur J Immunol. 1998;28:560–569. doi: 10.1002/(SICI)1521-4141(199802)28:02<560::AID-IMMU560>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 31.Onami T.M, Harrington L.E, Williams M.A, Galvan M, Larsen C.P, Pearson T.C, Manjunath N, Baum L.G, Pearce B.D, Ahmed R. Dynamic regulation of T cell immunity by CD43. J Immunol. 2002;168:6022–6031. doi: 10.4049/jimmunol.168.12.6022. [DOI] [PubMed] [Google Scholar]
- 32.Wan J, Martinvalet D, Ji X, Lois C, Kaech S.M, Von Andrian U.H, Lieberman J, Ahmed R, Manjunath N. The Bcl-2 family pro-apoptotic molecule, BNIP3 regulates activation-induced cell death of effector cytotoxic T lymphocytes. Immunology. 2003;110:10–17. doi: 10.1046/j.1365-2567.2003.01710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.••.Kaech S.M, Tan J.T, Wherry E.J, Konieczny B.T, Surh C.D, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]; This shows that IL-7Rα expression defines a subset of activated CD8+ T cells that survive to enter the memory pool.
- 34.Lohman B.L, Welsh R.M. Apoptotic regulation of T cells and absence of immune deficiency in virus-infected IFN-gamma receptor knock-out mice. J Virol. 1998;72:7815–7821. doi: 10.1128/jvi.72.10.7815-7821.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Badovinac V.P, Tvinnereim A.R, Harty J.T. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 36.•.Badovinac V.P, Porter B.B, Harty J.T. Programmed contraction of CD8(+) T cells after infection. Nat Immunol. 2002;3:619–626. doi: 10.1038/ni804. [DOI] [PubMed] [Google Scholar]; The authors argue that the silencing of the T-cell response at the resolution of infection may be a consequence of a pre-determined programmed contraction event.
- 37.Genestier L, Kasibhatla S, Brunner T, Green D.R. Transforming growth factor beta1 inhibits Fas ligand expression and subsequent activation-induced cell death in T cells via downregulation of c-Myc. J Exp Med. 1999;189:231–239. doi: 10.1084/jem.189.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S.W, Park Y, Yoo J.K, Choi S.Y, Sung Y.C. Inhibition of TCR-induced CD8 T cell death by IL-12: regulation of Fas ligand and cellular FLIP expression and caspase activation by IL-12. J Immunol. 2003;170:2456–2460. doi: 10.4049/jimmunol.170.5.2456. [DOI] [PubMed] [Google Scholar]
- 39.Moskophidis D, Lechner F, Pircher H, Zinkernagel R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 40.•.Fuller M.J, Zajac A.J. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol. 2003;170:477–486. doi: 10.4049/jimmunol.170.1.477. [DOI] [PubMed] [Google Scholar]; See annotation for [41•].
- 41.•.Wherry E.J, Blattman J.N, Murali-Krishna K, van der M.R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper, together with [40•], explains how there is a progression in loss of function of T cells during persistent infection.
- 42.van der Most R.G, Murali-Krishna K, Lanier J.G, Wherry E.J, Puglielli M.T, Blattman J.N, Sette A, Ahmed R. Changing immunodominance patterns in antiviral CD8 T-cell responses after loss of epitope presentation or chronic antigenic stimulation. Virology. 2003;315:93–102. doi: 10.1016/j.virol.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 43.••.Zhou S, Ou R, Huang L, Moskophidis D. Critical role for perforin-, Fas/FasL-, and TNFR1-mediated cytotoxic pathways in down-regulation of antigen-specific T cells during persistent viral infection. J Virol. 2002;76:829–840. doi: 10.1128/JVI.76.2.829-840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a kinetic study, demonstrating that the elimination of viral epitope-specific T cells under conditions of persistent infections is dependent, in part, on Fas/FasL and TNF, arguing for a role of apoptosis in this process.
- 44.De Boer R.J, Homann D, Perelson A.S. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J Immunol. 2003;171:3928–3935. doi: 10.4049/jimmunol.171.8.3928. [DOI] [PubMed] [Google Scholar]