

Graphical abstract

Keywords: Cysteine proteinase, Antiviral, Picornavirus, Halopyridinyl ester
Abstract
The related 3C and 3C-like proteinase (3Cpro and 3CLpro) of picornaviruses and coronaviruses, respectively, are good drug targets. As part of an effort to generate broad-spectrum inhibitors of these enzymes, we screened a library of inhibitors based on a halopyridinyl ester from a previous study of the severe acute respiratory syndrome (SARS) 3CL proteinase against Hepatitis A virus (HAV) 3Cpro. Three of the compounds, which also had furan rings, inhibited the cleavage activity of HAV 3Cpro with Kics of 120–240 nM. HPLC-based assays revealed that the inhibitors were slowly hydrolyzed by both HAV 3Cpro and SARS 3CLpro, confirming the identity of the expected products. Mass spectrometric analyses indicated that this hydrolysis proceeded via an acyl-enzyme intermediate. Modeling studies indicated that the halopyridinyl moiety of the inhibitor fits tightly into the S1-binding pocket, consistent with the lack of tolerance of the inhibitors to modification in this portion of the molecule. These compounds are among the most potent non-peptidic inhibitors reported to date against a 3Cpro.
1. Introduction
Regulated proteolysis is a critical feature in the processing of viral polyproteins and is essential for their replication. As such, proteinases constitute an important target for novel antivirals. The main proteinases responsible for this processing in picornaviruses and coronaviruses are the 3C and 3C-like proteinases (3Cpro and 3CLpro), respectively.1 Picornaviruses include important human pathogens such as human rhinovirus (HRV), poliovirus (PV), and hepatitis A virus (HAV), as well as significant insect, plant and agricultural pathogens, such as foot-and-mouth disease virus (FMDV). Coronaviruses include the etiological agent of the severe acute respiratory syndrome (SARS) outbreak in 2002. Given the significance of these viral pathogens, intense efforts are underway to develop novel antiviral compounds that target 3Cpros and 3CLpros.
The crystal structure of HAV 3Cpro was the first solved of the picornaviral proteinases.2 It has an overall fold resembling the two-domain β-barrel fold of chymotrypsin but with a cysteine in the active site typical of the 3C proteinases.3 The active site consists of a long groove between the two β-barrel domains and the enzyme’s specificity is determined by the residues that line this groove. In the central portion of the groove between the two domains, lie the conserved residues of a catalytic triad: the nucleophilic Cys172; the general acid-base catalyst His44; and Asp84, that forms a hydrogen bond to His44.4 This catalytic triad is consistent with other 3Cpro structures identified to date including those of PV, HRV, and FMDV.5, 6, 7 The coronaviral 3CLpros have a similar two-domain β-barrel fold with a third α-helical domain that plays a role in dimerization and contributes to the active site pocket.8 Coronaviruses have an active site Cys–His catalytic dyad9 but for both types of proteinases the catalytic mechanism is similar. Following binding of a substrate, the latter’s carbonyl carbon atom of the scissile peptide bond is subject to nucleophilic attack by the sulfur atom of the active site Cys. Formation of a tetrahedral intermediate is stabilized by residues of the electrophilic oxyanion hole, and loss of the amide leads to formation of an acyl enzyme intermediate.4 The catalysis is complete following deacylation and restoration of the active site.
With some exceptions, such as peptidyl aldehydes10 and peptidyl fluoromethyl ketones,11 many of the inhibitors of HAV 3Cpro that have been developed to date are not particularly potent in their initial binding to the enzyme. Keto-glutamine analogues have been well studied as potential inhibitors. Such analogues with a phthalhydrazide moiety display initial competitive (reversible) inhibition of HAV 3Cpro with the best having a K i of 9 μM.12 Over a longer period (hours), they then alkylate the active site cysteine thiol with concomitant loss of the phthalhydrazide moiety.13 Such compounds were improved through cyclization of the glutamine side chain, resulting in inhibitors with IC50s in the low micromolar range.14 Lall et al.15 also investigated interaction of β-lactones with HAV 3C and found a reversible inhibitor having a K i of 9 μM. Interestingly, the enantiomeric compound was an irreversible inhibitor with a k inact/K i = 3800 M−1 min−1.4
Given the similarity of the active site architectures of 3Cpro and 3CLpro, it is expected that these proteinases will be inhibited by similar classes of compounds. Indeed, it may be possible to generate broad-spectrum inhibitors of these enzymes. Screening of a 50,000-member library of small molecules revealed a pyridinyl thiophene ester, 1 (Table 1 ), that inhibited SARS 3CLpro with an IC50 of 0.5 μM.16 This compound also inhibited HAV 3Cpro with an essentially identical IC50. More recently, a library of 82 heteroaromatic esters based on 1 was created and screened against SARS 3CLpro.17 The most potent ester 24 (Table 2 ) inhibited the proteinase with an IC50 of 50 nM. Mass spectrometric analysis suggested a mechanism of inhibition involving acylation of the enzyme by the furoyl group. Slow reactivation of the enzyme further suggested that the acyl-enzyme was subject to deacylation, and that the enzyme catalyzed the slow hydrolysis of the ester.
Table 1.
Percentage of inhibition of HAV 3Cpro for preliminary analysis of inhibitor library at various concentrations of inhibitors
| Compound | Structure | % 10 μM | % 1 μM | % 0.25 μM | Compound | Structure | % 10 μM | % 1 μM | % 0.25 μM |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 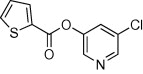 |
93 | ND | ND | 43 | 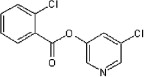 |
>90 | 67 | 23 |
| 2 | 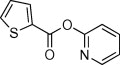 |
89 | 8 | ND | 44 | See Table 2 | 54 | 69 | 53 |
| 3 |  |
83 | 21 | ND | 45 | See Table 2 | >90 | >90 | 50 |
| 4 | 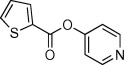 |
<10 | 10 | ND | 46 | 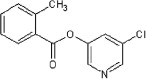 |
82 | ND | ND |
| 5 | 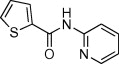 |
<10 | ND | ND | 47 | 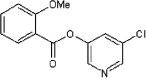 |
85 | 57 | <10 |
| 6 | 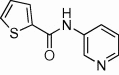 |
<10 | ND | ND | 48 | 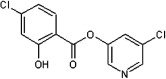 |
>90 | 59 | 10 |
| 7 |  |
<10 | ND | ND | 49 | 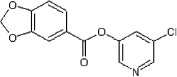 |
>90 | 11 | ND |
| 8 | 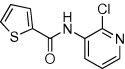 |
<10 | ND | ND | 50 | 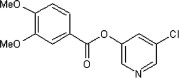 |
59 | ND | ND |
| 9 | 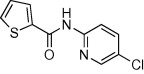 |
<10 | ND | ND | 51 | 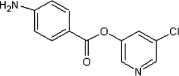 |
>90 | 26 | ND |
| 10 | 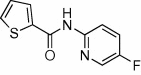 |
<10 | ND | ND | 52 | 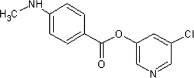 |
82 | <10 | ND |
| 11 | 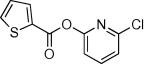 |
16 | ND | ND | 53 | 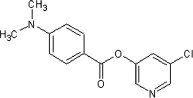 |
>90 | >90 | 20 |
| 12 |  |
91 | <10 | ND | 54 | 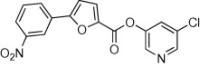 |
NDa | ND | ND |
| 13 | 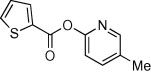 |
12 | ND | ND | 55 | 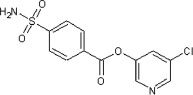 |
64 | 57 | <10 |
| 14 | 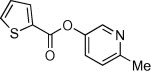 |
23 | ND | ND | 56 | 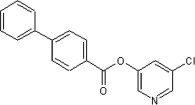 |
39 | 23 | ND |
| 15 | 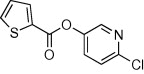 |
25 | ND | ND | 57 | 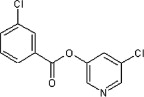 |
29 | 50 | ND |
| 16 | 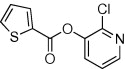 |
11 | ND | ND | 58 |  |
63 | 32 | ND |
| 17 |  |
<10 | <10 | ND | 59 |  |
<10 | ND | ND |
| 18 |  |
14 | ND | ND | 60 | 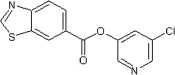 |
74 | 54 | 27 |
| 19 | 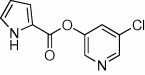 |
92 | <10 | <10 | 61 | 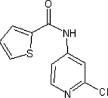 |
19 | ND | ND |
| 20 | See Table 2 | >90 | >90 | 49 | 62 | 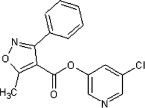 |
<10 | ND | <10 |
| 21 | 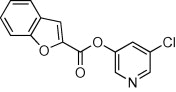 |
>90 | >90 | <10 | 63 | 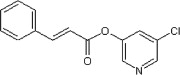 |
>90 | 53 | ND |
| 22 | 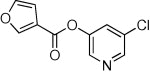 |
>90 | ND | ND | 64 | 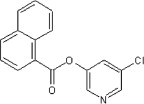 |
>90 | 30 | ND |
| 23 | 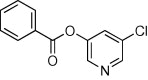 |
>90 | >90 | <10 | 65 | 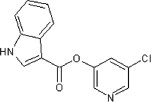 |
13 | ND | ND |
| 24 | See Table 2 | ND | >90 | 87 | 66 | 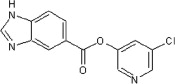 |
82 | 75 | 32 |
| 25 | 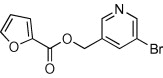 |
67 | ND | ND | 67 | 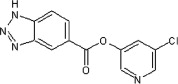 |
>90 | ND | ND |
| 26 | 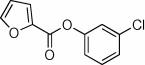 |
11 | ND | ND | 68 | 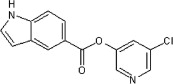 |
87 | 60 | 22 |
| 27 | 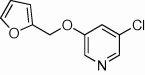 |
<10 | ND | ND | 69 |  |
>90 | 27 | ND |
| 28 |  |
>90 | ND | ND | 70 |  |
75 | ND | ND |
| 29 |  |
<10 | ND | ND | 71 |  |
81 | 32 | ND |
| 30 |  |
10 | ND | ND | 72 |  |
>90 | 45 | ND |
| 31 |  |
80 | 58 | ND | 73 | 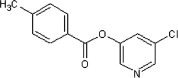 |
77 | 70 | ND |
| 32 | 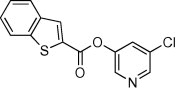 |
>90 | 47 | ND | 74 | 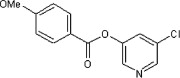 |
85 | ND | ND |
| 33 | 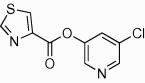 |
>90 | 85 | 37 | 75 | 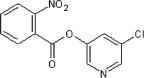 |
>90 | 24 | ND |
| 34 | 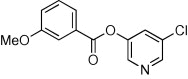 |
>90 | 50 | ND | 76 | 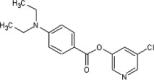 |
64 | ND | ND |
| 35 | 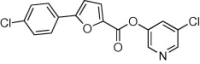 |
>90 | ND | 36 | 77 | 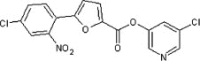 |
40 | ND | ND |
| 36 | See Table 2 | >90 | ND | 54 | 78 | 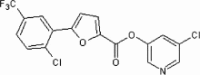 |
76 | 36 | ND |
| 37 | See Table 2 | 83 | >90 | 43 | 79 | 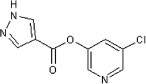 |
56 | 21 | ND |
| 38 |  |
<10 | ND | ND | 80 | 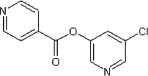 |
37 | 34 | ND |
| 39 | 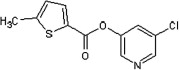 |
>90 | ND | 24 | 81 | 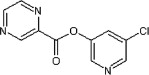 |
22 | ND | ND |
| 40 | 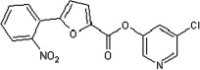 |
37 | ND | ND | 82 | 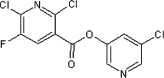 |
44 | 19 | ND |
| 41 | 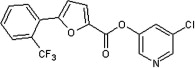 |
>90 | ND | <10 | 83 | 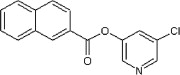 |
23 | 35 | ND |
| 42 | 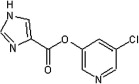 |
33 | 47 | 34 |
ND, not determined.
Not soluble.
Table 2.
Kinetic parameters of a series of pyridinyl analogue inhibitors of HAV 3Cpro and SARS 3CLpro and half-life of pyridinyl analogues in buffer
| Compound | Structure | IC50 (nM) | Km (nM) | kcat (s−1) × 10−5 | kcat/Km (M−1s−1) × 103 | Half-life (s−1) × 10−3 |
|---|---|---|---|---|---|---|
| HAV 3Cpro | ||||||
| 20 | 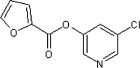 |
338 ± 2 | 240 ± 30 | 110 ± 6 | 4.6 ± 0.6 | 1.2 ± 0.2 |
| 37 |  |
140 ± 20 | 180 ± 30 | 25 ± 8 | 1.4 ± 0.5 | 4.9 ± 0.6 |
| 24 | 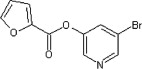 |
53 ± 2 | 120 ± 20 | 119 ± 8 | 10 ± 2 | 1.1 ± 0.3 |
| 45 | 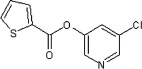 |
470 ± 60 | ND | ND | ND | ND |
| 36 | 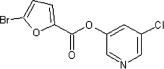 |
1200 ± 130 | ND | ND | ND | ND |
| 44 | 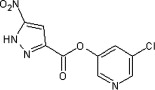 |
250 ± 50 | ND | ND | ND | ND |
| SARS 3CLpro | ||||||
| 24 | ND | 26 ± 7 | 17 ± 6 | 7 ± 3 | ||
ND, not determined.
Herein, we investigate the ability of the heteroaromatic esters to inhibit HAV 3Cpro. The inhibitors were screened against HAV 3Cpro using a fluorescent peptide substrate and the steady-state inhibition parameters of the most potent inhibitors were evaluated. The mode of action of the inhibitors was further investigated using an HPLC-based kinetic assay and mass spectrometric analyses. The results of these studies were compared to the inhibition of SARS 3CLpro by the heteroaromatic esters as well as to the previously described HAV 3Cpro inhibitors.
2. Results
2.1. Initial screen
The library of 82 pyridinyl esters (Scheme 1 ) that yielded potent inhibitors of SARS 3CLpro 17 (a coronoviral proteinase) was screened against HAV 3Cpro (a picornaviral enzyme) using a microplate assay. Of the 82 compounds, 49 completely inhibited the proteinase’s activity when tested at 10 μM, 10 showed complete inhibition at 1 μM, and none showed complete inhibition at 0.25 μM (Table 1). For six compounds (Table 2) that inhibited the proteinase activity to the greatest extent at 1 μM, IC50 values were assessed. The IC50 experiments were performed as described for the initial screening except that the substrate at a concentration of 20 μM was used. As summarized in Table 2, the six compounds had IC50s ranging from 50 nM to 1.2 μM. The lowest value, for 24, is close to the theoretical minimal value that can be determined in this experiment (i.e., 50% of the concentration of the enzyme).
Scheme 1.

Synthesis of a library of 3-chloropyridinyl esters by Method A or B.
2.2. Determination of kinetic parameters
In studies using sufficient pyridinyl ester to completely inhibit the initial hydrolysis of the fluorogenic peptide by HAV 3Cpro, peptide hydrolysis was sometimes observed after incubating the reaction mixture for ∼4 min. This suggested that HAV 3Cpro might catalyze the slow hydrolysis of the ester inhibitor. To investigate this possibility, we developed an HPLC-based assay to monitor inhibitor hydrolysis by HAV 3Cpro and SARS 3CLpro. Reactions were performed using a concentration of pyridinyl ester high enough to ensure enzyme saturation. HPLC analysis of the reaction mixture demonstrated that the expected hydrolysis products could be observed in a time-dependent manner concomitant with the disappearance of the ester (Fig. 1 ) and that this hydrolysis was dependent on the enzyme. For each of the tested esters, product identification was confirmed when possible by comparison to known standards. The assay was performed at several concentrations of ester to confirm saturation of the proteinase with the ester and enzymatic hydrolysis rates were corrected for the non-enzymatic hydrolysis of the inhibitor.
Figure 1.

Hydrolysis of 24 by HAV 3Cpro. The reaction mixture contained 50 μM inhibitor and 2 μM 3Cpro. It was subjected to HPLC analysis after 0, 17, 35, and 52 min. Traces were recorded at (A) 252 nm and (B) 287 nm. Peaks corresponding to each of 24, furoic acid, and 3-bromo-5-hydroxypyridine (3B5H) are labeled. Over the time course of this experiment, non-enzymatic hydrolysis of the ester was negligible (data not shown).
The calculated and deduced steady-state kinetic parameters for three of the best inhibitors are summarized in Table 2. The k cat values were calculated from the hydrolysis rates at saturating ester concentrations. The K m values were taken to be the equivalent of the determined K ic values, which is the case for substrates that are slowly turned over (e.g., Seah et al.).18 Among the three inhibitors tested with HAV 3Cpro, the two with the same acyl group, 24 and 20, had very similar k cat values. Equivalent studies using SARS 3CLpro and the most potent inhibitor, 24, demonstrated that this proteinase also catalyzed ester hydrolysis with parameters similar to those observed for HAV 3Cpro.
2.3. MS analyses of inhibitor–proteinase complex
The kinetic data indicate that HAV 3Cpro catalyzes the slow hydrolysis of the pyridinyl ester inhibitors. To investigate whether hydrolysis proceeds via an acyl-enzyme intermediate, samples of proteinase were incubated for 10 min with 100 μM 24 and the resulting sample was analyzed by mass spectrometry. As shown in Figure 2 , incubation of HAV 3Cpro resulted in a mass increase of 100 ± 2.5 Da. This is consistent with the covalent attachment of the furoyl moiety (M W 95 Da) and the departure of the 3-bromo-5-hydroxypyridine as the leaving group. In samples that were incubated for both 2 and 10 min, only the acylated form of the proteinase was detected.
Figure 2.

(A) Mass spectrum of wild-type HAV 3Cpro (M+ 23875.08 Da). (B) Mass spectrum of the complex of 3Cpro and inhibitor 24 following 2 min of incubation (M+ 23974.38 Da).
2.4. Docking
To gain insight into the interactions between the inhibitors and HAV 3Cpro prior to acylation, compound 24 was computer-modeled into the active site pocket of the enzyme. From the 20 docked complexes obtained using the program AUTODOCK, the best binding mode as judged from binding energies and allowable bond lengths and angles is one in which the halopyridine ring occupies the S1-binding pocket (Fig. 3 a). More specifically, the nitrogen of the halopyridinyl ring is hydrogen bonded to the Nε2 of His191 (3.0 Å) and the halogen atom points out toward solvent. The carbonyl oxygen of the ester is located in the oxyanion hole of the enzyme, forming two hydrogen bonds each with the N of Gly170 and Cys172 at distances of 3.1 and 2.7 Å, respectively.
Figure 3.

Possible prehydrolysis-binding modes of 24 to (a) HAV 3Cpro and (b) SARS 3CLpro. Protein surfaces are colored according to electrostatic potentials with red and blue indicating negative and positive potentials, respectively. The carbon atoms of 24 and the amino acid residues are gray and purple, respectively. The nitrogen, oxygen and sulfur atoms are colored blue, red, and orange, respectively. Hydrogen bonds are displayed by dashed lines.
3. Discussion
In this study, a targeted library of compounds was screened to identify several potent inhibitors of the picornaviral HAV 3Cpro. The six most potent inhibitors all contain an ester bond connecting a halopyridine ring to a 5-membered ring (Table 2). The most potent of these, 24, competitively inhibited the hydrolysis of a peptidyl substrate with a K ic (120 nM) that is among the lowest for an inhibitor of a 3Cpro enzyme reported to date. HPLC and mass spectrometric analysis demonstrated that the esters are slow substrates of HAV 3Cpro, with hydrolysis proceeding via an acyl-enzyme intermediate. Consistent with a mechanism involving rate-limiting deacylation, the two compounds possessing the same acylating group, 20 and 24, had very similar k cat values.
Interestingly, the halopyridinyl esters are better substrates for HAV 3Cpro than the peptide substrates characterized to date. For example, the k cat/K m value for 37 was 1400 ± 500 M−1 s−1 versus 800 ± 100 M−1 s−1 for Ac-ELRTQSFS-NH2.19 This is due to the enzyme’s exceptionally low K m values for the halopyridinyl esters (e.g., 120 ± 20 nM for 24), that were considerably lower than for good peptide substrates (2.1 ± 0.5 mM for Ac-ELRTQSFS-NH2). By contrast, the enzyme turns over peptide substrates much more efficiently (k cat = 1.8 ± 0.1 s−1 for Ac-ELRTQSFS-NH2 20 versus 25 ± 8 × 10−5 s−1 for 24). Importantly, the specificity of the viral proteinase for its natural polypeptide substrate has yet to be determined. The presence of an ester linkage is a critical feature for effective inhibition of 3Cpro by the tested small molecules. For example, compounds lacking the ester bond, such as 29, 30, and 61, are poor inhibitors suggesting that these compounds do not interact with the proteinase particularly strongly.
Comparison of the inhibition characteristics of the esters reveals that there is more flexibility in the cyclic acid moiety of the ester than in the pyridinyl alcohol. For example, 60, 66, 68 and 78 moderately inhibited HAV 3Cpro at concentrations of 1 μM despite the different sizes of the cyclic acid moiety. While the cyclic acid moiety is subject to some constraints, as illustrated by compounds 65, 69, and 78, these constraints are difficult to deduce from the current data set. The spacing between the ester and alcohol or acid moiety was also more critical for the alcohol than the acid. Compound 37 was a good inhibitor despite possessing two additional carbon atoms (–CH CH–) on the acid side of the ester with respect to 20. By contrast, the insertion of a single additional carbon atom (–CH2–) on the alcohol side of the ester of the potent inhibitor 24 yielded 25, a poor inhibitor. Finally, the presence and position of the halogen on the pyridine ring were important determinants of inhibition as was the position of the ring nitrogen with respect to the ester bond. Thus, compounds containing 2- or 4-pyridinyls (2, 4, 11 and 13) were poor inhibitors. Similarly, compounds lacking the halogen substituent (3 vs 45) or in which this substituent is ortho or para to the ester group (15 and 16 vs 45) were also poor inhibitors. Overall, these results indicate that the halopyridine ring is a particularly important determinant of inhibition.
The model of 24 docked to HAV 3Cpro is consistent with the structure–activity relationship of the inhibitors. In particular, the model indicates that the halopyridinyl ring of 24 essentially fills the S1 pocket of the enzyme with the pyridine nitrogen atom forming a hydrogen bond to Nε2 of His191 in this pocket (Fig. 3a) in contrast to the considerable space in the enzyme’s active site around the furan ring. Although the modeled HAV 3Cpro-24 complex does not clarify the role of the halogen group on the pyridine ring as this group, it is possible that the halogen influences the electronic characteristics and/or the solvation of the pyridinyl ring so as to enhance inhibition.
The modeled HAV 3Cpro-24 complex is also consistent with the hydrolysis of these ester compounds via an acyl-enzyme intermediate in two aspects. First, the distance between the Sγ of Cys172 and the carbonyl carbon in 24 is 3.4 Å. Second, these two atoms are almost co-planar with the imidazole ring of His44. Therefore, His44 is in an ideal position to have its Nε2 acting as a general base. During normal enzymatic hydrolysis of peptidyl substrates, the scissile peptide bond is expected to be in line with the spatial orientation between the Cys and His catalytic pair in the active site. However, the modeled HAV 3Cpro-24 complex shows a rather different direction for the central scissile ester bond of compound 24, primarily due to the predicted strong propensity of the bromopyridinyl ring to bind inside the S1 pocket. Interestingly, a similar binding mode was predicted for the complexes between SARS 3CLpro and a group of pyridinyl ester-based inhibitors.21
Considering the similarities of picornaviral 3Cpro and coronaviral 3CLpro, it is not surprising that the library of pyridinyl esters yielded similar potent inhibitors of the HAV and SARS proteinases.17 In particular, both enzymes are catalytically driven by a Cys–His pair and have a high specificity of Gln as the P1 residue. This specificity reflects their similar S1 pockets, especially the histidine at its base (His163 and His191 of SARS 3CLpro and HAV 3Cpro, respectively) which is an important determinant of P1 specificity in both enzymes.4, 22 Consistent with these similarities, the predicted binding mode of halopyridinyl esters to SARS 3CLpro 17 is very similar to that predicted for HAV 3Cpro, with the halopyridinyl moiety occupying the S1 pocket and the ring of the acid moiety extending into the S2 pocket.
4. Conclusion
We have screened a library of 82 non-peptidic inhibitors, including 49 not previously described, against HAV 3Cpro and have characterized in more detail several of the most potent of these inhibitors. Some of these inhibitors had been previously shown to potently inhibit the related SARS 3CLpro. The ability of the halopyridinyl esters to inhibit two such divergent 3Cpros strongly suggests that these compounds could be used to inhibit the related proteinases of other coronaviruses and picornaviruses such as FMDV and HRV. These inhibitors might be improved by increasing their specificity, presumably by targeting the S2 pocket better, and further slowing their enzymatic hydrolysis. Indeed, given the correlation between turnover of the esters (k cat) and non-enzymatic stability (half-life), decreasing enzymatic hydrolysis might improve their stability in aqueous solution. Finally, given the slightly different substrate-binding pockets of the different 3Cpros, a comparison of the inhibition of related proteinases by a series of these halopyridinyl esters might provide insight into the mode of substrate binding and how the physicochemical properties of the substrate-binding pocket determine the excellent specificity of these enzymes.
5. Experimental
5.1. Chemicals and reagents
Factor Xa was purchased from Hematologic Technologies Inc. (Essex Junction, USA). Peptides were synthesized at the Nucleic Acid Protein Service Unit of the University of British Columbia and confirmed by mass spectrometric analysis. All other chemicals were of analytical grade and used without further purification.
5.2. Inhibitor synthesis
Two general procedures were used to prepare pyridinyl esters. In method A, the following compounds were added to a solution of carboxylic acid (0.5 mmol, 1.0 equiv) in DMF (2 mL) at rt: EDCI (97 mg, 0.5 mmol, 1.0 equiv), HOBt (68 mg, 0.5 mmol, 1.0 equiv), DIPEA (90 μL, 0.5 mmol, 1.0 equiv), and 5-chloro pyridinol (65 mg, 0.5 mmol, 1.0 equiv). After 24 h of stirring, the solvent was removed in vacuo to afford the crude mixture.
In method B, the following compounds were added to a solution of carboxylic acid (1 mmol, 1.0 equiv) in DCM (5 mL) at rt: thionyl chloride (0.4 mL, 2.6 equiv) and a catalytic amount of DMF (2 drops). After 20 h of stirring, the solvent was removed in vacuo to afford the acyl chloride product. A solution of the acyl chloride in DCM (5 mL) was added dropwise to a solution of 5-chloro pyridinol (130 mg, 1 mmol, 1.0 equiv) and pyridine (0.09 mL, 1.1 equiv) in DCM (5 mL) at 0 °C. After 3 h of stirring, the solvent was removed in vacuo to afford the crude mixture.
Crude mixtures were purified using an 1100 HPLC coupled with an ES-MSD Agilent 1956B with positive ion detection. The HPLC was fitted with a semi-preparative column, Zorbax RX-C8 (9.4 × 250 mm, 5 μM) equipped with a guard column. The column was operated at a flow rate of 3 mL/min. Compounds were eluted using a linear gradient of 35–100% acetonitrile in 0.05% formic acid/H2O over 20 min, followed by 100% acetonitrile in 0.05% formic acid/H2O (2 min) and a final return to 35% acetonitrile in 0.05% formic acid/H2O in 0.5 min. The quality of selected purified samples was confirmed by re-injection of the samples on an analytic column (:Zorbax RX-C18, 4.6 × 150 mm, 5 μM) operated at a flow rate of 0.7 mL/min using the above-described linear gradient.
5.2.1. 5-Chloropyridin-3-yl 5-bromofuran-2-carboxylate (36)
Method B. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.52 (d, 1H, J = 2.1 Hz), 8.47 (d, 1H, J = 2.3 Hz), 7.68 (dd, 1H, J = 2.3, 2.1 Hz), 7.38 (d, 1H, J = 3.7 Hz), 6.59 (d, 1H, J = 3.6 Hz); HRMS [EI] Calcd for C10H5ClNO3Br (M+) 302.9112, found 302.9114.
5.2.2. (E)-5-Chloropyridin-3-yl 3-(furan-2-yl)acrylate (37)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.47 (dd, 1H, J = 2.1, 0.4 Hz), 8.41 (dd, 1H, J = 2.3, 0.4 Hz), 7.63 (d, 1H, J = 15.5 Hz), 7.60 (t, 1H, J = 2.2 Hz), 7.57 (ddd, 1H, J = 2.3, 1.2, 0.5 Hz), 6.72 (d, 1H, J = 3.4 Hz), 6.51 (dd, 1H, J = 3.4, 1.8 Hz), 6.46 (d, 1H, J = 15.6 Hz); HRMS [EI] Calcd for C12H8ClNO3 (M+) 249.0193, found 249.0194.
5.2.3. 5-Chloropyridin-3-yl 3-methylthiophene-2-carboxylate (38)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.50 (d, 1H, J = 2.3, 0.5 Hz), 8.47 (dd, 1H, J = 2.3, 0.5 Hz), 7.69 (dd, 1H, J = 2.3, 2.3 Hz), 7.57 (d, 1H, J = 5.0 Hz), 7.03 (d, 1H, J = 5.0 Hz), 2.63 (s, 3H); HRMS [EI] Calcd for C11H8ClNO2S (M+) 252.9964, found 252.9962.
5.2.4. 5-Chloropyridin-3-yl 5-methylthiophene-2-carboxylate (39)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.49 (d, 1H, J = 2.0 Hz), 8.46 (d, 1H, J = 2.4 Hz), 7.83 (d, 1H, J = 3.8 Hz), 7.68 (d, 1H, J = 2.3, 2.0 Hz), 6.88 (dd, 1H, J = 3.9, 1.0 Hz), 2.60 (s, 3H); HRMS [EI] Calcd for C11H8ClNO2S (M+) 252.9964, found 252.9954.
5.2.5. 5-Chloropyridin-3-yl 5-(2-nitrophenyl)furan-2-carboxylate (40)
Method A. A slightly brown solid. 1H NMR (CDCl3, 500 MHz) δ 8.52 (d, 1H, J = 2.2 Hz), 8.49 (dd, 1H, J = 2.3, 0.3 Hz), 7.87 (t, 1H, J = 1.2 Hz), 7.86 (t, 1H, J = 1.2 Hz), 7.72–7.69 (m, 2H), 7.59 (ddd, 1H, J = 8.2, 7.6, 1.5 Hz), 7.51 (d, 1H, J = 3.8 Hz), 6.82 (d, 1H, J = 3.7 Hz); HRMS [EI] Calcd for C16H9ClN2O5 (M+) 344.0200, found 344.0197.
5.2.6. 5-Chloropyridin-3-yl 5-(2-(trifluoromethyl)phenyl)furan-2-carboxylate (41)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.53 (d, 1H, J = 2.3 Hz), 8.51 (d, 1H, J = 2.3 Hz), 8.08–8.06 (m, 1H), 8.04–8.01 (m, 1H), 7.72 (dd, 1H, J = 2.2, 2.2 Hz), 7.68–7.58 (m, 2H), 7.54 (d, 1H, J = 3.7 Hz), 6.96 (d, 1H, J = 3.7 Hz); HRMS [EI] Calcd for C17H9ClNO3F3 (M+) 367.0223, found 367.0223.
5.2.7. 5-Chloropyridin-3-yl 1H-imidazole-4-carboxylate (42)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.51 (d, 1H, J = 2.2 Hz), 8.48 (d, 1H, J = 2.3 Hz), 7.99–7.98 (m, 1H), 7.85–7.85 (m, 1H), 7.69 (t, 1H, J = 2.2 Hz); HRMS [EI] Calcd for C9H6ClN3O2 (M+) 223.0149, found 223.0150.
5.2.8. 5-Chloropyridin-3-yl 2-chlorobenzoate (43)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.53 (d, 1H, J = 2.1 Hz), 8.50 (d, 1H, J = 2.3 Hz), 8.06 (ddd, 1H, J = 7.9, 1.9, 1.0 Hz), 7.72 (dd, 1H, J = 2.2, 2.2 Hz), 7.57–7.54 (m, 2H), 7.46–7.40 (m, 1H); HRMS [EI] Calcd for C12H7NO2Cl2 (M+) 266.9854, found 266.9857.
5.2.9. 5-Chloropyridin-3-yl 5-nitro-1H-pyrazole-3-carboxylate (44)
Method B. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.60 (dd, 1H, J = 2.1, 0.5 Hz), 8.52 (dd, 1H, J = 2.4, 0.5 Hz), 7.72 (t, 1H, J = 2.1 Hz), 7.64 (s, 1H); HRMS [EI] Calcd for C9H5ClN4O4 (M+) 267.9999, found 267.9992.
5.2.10. 5-Chloropyridin-3-yl thiophene-2-carboxylate (45)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.51 (d, 1H, J = 2.1 Hz), 8.48 (d, 1H, J = 2.1 Hz), 8.02 (dd, 1H, J = 3.9, 1.3 Hz), 7.74 (dd, 1H, J = 5.0, 1.2 Hz), 7.69 (dd, 1H, J = 2.1, 2.1 Hz), 7.22 (dd, 1H, J = 5.0, 3.9 Hz); HRMS [EI] Calcd for C10H6O2NSCl (M+) 238.9808, found 238.9809.
5.2.11. 5-Chloropyridin-3-yl 2-methylbenzoate (46)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.56–8.42 (m, 2H), 8.16 (dd, 1H, J = 8.3, 1.5 Hz), 7.68 (dd, 1H, J = 2.0, 2.0 Hz), 7.53 (dt, 1H, J = 7.5, 1.4 Hz), 7.39–7.32 (m, 2H), 2.68 (s, 3H); HRMS [EI] Calcd for C13H10NO2Cl (M+) 247.0400, found 247.0396.
5.2.12. 5-Chloropyridin-3-yl 2-methoxybenzoate (47)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.50 (d, 1H, J = 2.0 Hz), 8.48 (d, 1H, J = 2.3 Hz), 8.03 (dd, 1H, J = 8.0, 1.8 Hz), 7.75 (dd, 1H, J = 2.2, 2.2 Hz), 7.60 (ddd, 1H, J = 8.8, 7.5, 1.8 Hz), 7.11–7.04 (m, 2H), 3.96 (s, 3H); HRMS [EI] Calcd. for C13H10NO3Cl (M+) 263.0349, found 263.0350.
5.2.13. 5-Chloropyridin-3-yl 4-chloro-2-hydroxybenzoate (48)
Method B. A white solid. 1H NMR (CDCl3, 300 MHz) δ 10.3 (s, 1H), 8.56 (dd, 1H, J = 2.1, 0.4 Hz), 8.48 (dd, 1H, J = 2.4, 0.4 Hz), 7.98 (dd, 1H, J = 8.6, 0.3 Hz), 7.67 (dd, 1H, J = 2.4, 2.1 Hz), 7.10 (dd, 1H, J = 2.0, 0.3 Hz), 6.99 (dd, 1H, J = 8.6, 2.0 Hz); HRMS [EI] Calcd for C12H7Cl2NO3 (M+) 282.9803, found 282.9805.
5.2.14. 5-Chloropyridin-3-yl benzo[d][1,3]dioxole-5-carboxylate (49)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.50 (d, 1H, J = 2.1 Hz), 8.45 (d, 1H, J = 2.3 Hz), 7.82 (dd, 1H, J = 8.2, 1.7 Hz), 7.67 (dd, 1H, J = 2.2, 2.2 Hz), 7.59 (d, 1H, J = 1.7 Hz), 6.93 (d, 1H, J = 8.2 Hz), 6.11 (s, 2H); HRMS [EI] Calcd for C13H8NO4Cl (M+) 277.0142, found 277.0140.
5.2.15. 5-Chloropyridin-3-yl 3,4-dimethoxybenzoate (50)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.51 (d, 1H, J = 2.1 Hz), 8.47 (d, 1H, J = 2.1 Hz), 7.86 (dd, 1H, J = 8.4, 2.1 Hz), 7.67 (dd, 1H, J = 2.3, 2.1 Hz), 7.64 (d, 1H, J = 2.1 Hz), 6.98 (d, 1H, J = 8.4 Hz), 3.99 (s, 3H), 3.997 (s, 3H); HRMS [EI] Calcd for C14H12NO4Cl (M+) 293.0455, found 293.0454.
5.2.16. 5-Chloropyridin-3-yl 4-aminobenzoate (51)
Method B. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.48 (d, 1H, J = 2.1 Hz), 8.44 (d, 1H, J = 2.3 Hz), 8.00 (d, 2H, J = 8.7 Hz), 7.67 (dd, 1H, J = 2.2, 2.2 Hz), 6.71 (d, 2H, J = 8.7 Hz), 4.50–3.60 (br s, 2H); HRMS [EI] Calcd for C12H9N2O2Cl (M+) 248.0353, found 248.0352.
5.2.17. 5-Chloropyridin-3-yl 4-(methylamino)benzoate (52)
Method B. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.47 (dd, 1H, J = 2.1, 0.5 Hz), 8.45 (dd, 1H, J = 2.4, 0.5 Hz), 8.01 (d, 2H, J = 9.0 Hz), 7.67 (dd, 1H, J = 2.2, 2.2 Hz), 6.62 (d, 2H, J = 9.0 Hz), 2.95 (s, 3H); HRMS [EI] Calcd for C13H11N2O2Cl (M+) 262.0509, found 262.0513.
5.2.18. 5-Chloropyridin-3-yl 4-(dimethylamino)benzoate (53)
A slightly yellow solid. 1H NMR (CDCl3, 500 MHz) δ 8.47 (d, 1H, J = 2.0 Hz), 8.45 (d, 1H, J = 2.3 Hz), 8.04 (d, 2H, J = 9.2 Hz), 7.67 (dd, 1H, J = 2.2, 2.2 Hz), 6.71 (d, 2H, J = 9.2 Hz), 3.10 (s, 6H); HRMS [EI] Calcd for C14H13N2O2Cl (M+) 276.0666, found 276.0670.
5.2.19. 5-Chloropyridin-3-yl 5-(3-nitrophenyl)furan-2-carboxylate (54)
Method B. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.65 (ddd, 1H, J = 2.2, 1.7, 0.5 Hz), 8.56–8.50 (m, 2H), 8.26 (ddd, 1H, J = 8.2, 2.2, 1.0 Hz), 8.17 (ddd, 1H, J = 7.8, 1.7, 1.0 Hz), 7.72 (t, 1H, J = 2.2 Hz), 7.68 (ddd, 1H, J = 8.3, 7.9, 0.5 Hz), 7.55 (d, 1H, J = 3.8 Hz), 7.03 (d, 1H, J = 3.7 Hz); HRMS [EI] Calcd for C16H9ClN2O5 (M+) 344.0200, found 344.0200.
5.2.20. 5-Chloropyridin-3-yl 4-sulfamoylbenzoate (55)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.55 (dd, 1H, J = 2.1, 0.5 Hz), 8.50 (dd, 1H, J = 2.4, 0.5 Hz), 8.35 (d, 2H, J = 8.8 Hz), 8.10 (d, 2H, J = 8.9 Hz), 7.71 (dd, 1H, J = 2.4, 2.1 Hz), 4.92–4.87 (br, 2H); HRMS [EI] Calcd for C12H9N2O4ClS (M+) 311.9972, found 311.9978.
5.2.21. 5-Chloropyridin-3-yl biphenyl-4-carboxylate (56)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.53 (dd, 1H, J = 2.1, 0.5 Hz), 8.51 (dd, 1H, J = 2.3, 0.5 Hz), 8.27 (d, 2H, J = 8.7 Hz), 7.77 (d, 2H, J = 8.7 Hz), 7.72 (dd, 1H, J = 2.2, 2.2 Hz), 7.67 (dd, 2H, J = 8.4, 1.4 Hz), 7.54–7.43 (m, 3H); HRMS [EI] Calcd. for C18H12NO2Cl (M+) 309.0557, found 309.0556.
5.2.22. 5-Chloropyridin-3-yl 3-chlorobenzoate (57)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.53 (dd, 1H, J = 2.0, 0.5 Hz), 8.48 (dd, 1H, J = 2.4, 0.5 Hz), 8.18 (ddd, 1H, J = 2.1, 1.7, 0.5 Hz), 8.08 (ddd, 1H, J = 7.8, 1.7, 1.1 Hz), 7.69 (dd, 1H, J = 2.4, 2.1 Hz), 7.66 (ddd, 1H, J = 8.0, 2.1, 1.1 Hz), 7.50 (ddd, 1H, J = 8.0, 7.8, 0.5 Hz); HRMS [EI] Calcd for C12H7NO2Cl2 (M+) 266.9854, found 266.9858.
5.2.23. 5-Chloropyridin-3-yl 3-methylbenzoate (58)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.51 (dd, 1H, J = 2.0, 0.5 Hz), 8.47 (dd, 1H, J = 2.3, 0.5 Hz), 8.02–8.00 (m, 1H), 8.00–7.98 (m, 1H), 7.68 (dd, 1H, J = 2.3, 2.1 Hz), 7.50 (dddd, 1H, J = 7.6, 2.4, 1.3, 0.6 Hz), 7.43 (ddd, 1H, J = 8.0, 7.6, 0.6 Hz), 2.47 (s, 3H); HRMS [EI] Calcd for C13H10NO2Cl (M+) 247.0400, found 247.0398.
5.2.24. 5-Chloropyridin-3-yl 4-hydroxy-7-(trifluoromethyl)quinoline-3-carboxylate (59)
Method B. A yellow solid. 1H NMR (CD3OD, 300 MHz) δ 9.03 (s, 1H), 8.55 (d, 1H, J = 2.1 Hz), 8.50 (d, 1H, J = 2.4 Hz), 8.36 (d, 1H, J = 8.4 Hz), 8.06–8.04 (m, 1H), 8.01 (dd, 1H, J = 2.3, 2.2 Hz), 7.74 (dd, 1H, J = 8.5, 1.4 Hz), 4.00 (s, 1H); HRMS [EI] Calcd for C16H8ClN2O3F3 (M+) 368.0176, found 368.0176.
5.2.25. 5-Chloropyridin-3-yl benzo[d]thiazole-6-carboxylate (60)
Method B. A white solid. 1H NMR (CDCl3, 300 MHz) δ 9.24 (s, 1H), 8.88 (dd, 1H, J = 1.6, 0.6 Hz), 8.54 (dd, 1H, J = 2.1, 0.4 Hz), 8.52 (dd, 1H, J = 2.4, 0.4 Hz), 8.35 (dd, 1H, J = 8.6, 1.7 Hz), 8.28 (dd, 1H, J = 8.7, 0.6 Hz), 7.73 (t, 1H, J = 2.2 Hz); HRMS [EI] Calcd for C13H7N2O2ClS (M+) 289.9917, found 289.9907.
5.2.26. N-(6-Chloropyridin-3-yl)thiophene-2-carboxamide (61)
Method B. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.47 (dd, 1H, J = 2.8, 0.6 Hz), 8.26 (ddd, 1H, J = 8.6, 2.8, 0.3 Hz), 7.71–7.68 (br s, 1H), 7.68 (dd, 1H, J = 3.8, 1.2 Hz), 7.62 (dd, 1H, J = 5.0, 1.2 Hz), 7.36 (dt, 1H, J = 8.7, 0.5 Hz), 7.17 (dd, 1H, J = 5.0, 3.8 Hz); HRMS [EI] Calcd for C10H7N2OSCl (M+) 237.9968, found 237.9966.
5.2.27. 5-Chloropyridin-3-yl 5-methyl-3-phenylisoxazole-4-carboxylate (62)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 8.47 (d, 1H, J = 2.0 Hz), 8.28 (d, 1H, J = 2.4 Hz), 7.67- 7.64(m, 2H), 7.58 (dd, 1H, J = 2.4, 2.0 Hz), 7.54–7.46 (m, 3H), 2.87 (s, 3H); HRMS [EI] Calcd for C16H11ClN2O3 (M+) 314.0458, found 314.0458.
5.2.28. 5-Chloropyridin-3-yl cinnamate (63)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.49 (dd, 1H, J = 2.1, 0.4 Hz), 8.43 (dd, 1H, J = 2.3, 0.4 Hz), 7.92 (d, 1H, J = 16.1 Hz), 7.65 (t, 1H, J = 2.2 Hz), 7.63–7.59 (m, 2H), 7.47–7.44 (m, 3H,), 6.62 (d, 1H, J = 16.0 Hz); HRMS [EI] Calcd for C14H10NO2Cl (M+) 259.0400, found 259.0406.
5.2.29. 5-Chloropyridin-3-yl 1-naphthoate (64)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 9.02 (ddd, 1H, J = 8.7, 1.8, 0.8 Hz), 8.55 (dd, 1H, J = 4.0, 0.5 Hz), 8.55 (t, 1H, J = 0.5 Hz), 8.51 (d, 1H, J = 1.3 Hz), 8.50 (d, 1H, J = 1.3 Hz), 8.18–8.15 (m, 1H), 7.96 (ddt, 1H, J = 8.2, 1.5, 0.7 Hz), 7.76 (dd, 1H, J = 2.3, 2.1 Hz), 7.69 (ddd, 1H, J = 8.4, 6.8, 1.5 Hz), 7.63–7.59 (m, 1H); HRMS [EI] Calcd for C16H10NO2Cl (M+) 283.0400, found 283.0402.
5.2.30. 5-Chloropyridin-3-yl 1H-indole-3-carboxylate (65)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.82–8.66 (br s, 1H), 8.51 (dd, 1H, J = 2.3, 0.4 Hz), 8.50 (dd, 1H, J = 2.1, 0.4 Hz), 8.22 (dddd, 1H, J = 6.1, 3.2, 0.8, 0.8 Hz), 8.13 (d, 1H, J = 3.1 Hz), 7.74 (t, 1H, J = 2.2 Hz), 7.49 (ddd, 1H, J = 6.0, 3.3, 0.8 Hz), 7.35 (dd, 2H, J = 6.1, 3.2 Hz); HRMS [EI] Calcd for C14H9N2O2Cl (M+) 272.0353, found 272.0349.
5.2.31. 5-Chloropyridin-3-yl 1H-benzo[d]imidazole-5-carboxylate (66)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.60–8.58 (m, 1H), 8.52 (d, 1H, J = 2.1 Hz), 8.51 (d, 1H, J = 2.3 Hz), 8.25 (s, 1H), 8.17 (dd, 1H, J = 8.5, 1.7 Hz), 7.77 (d, 1H, J = 8.5 Hz), 7.73 (t, 1H, J = 2.0 Hz); HRMS [EI] Calcd for C13H8N3O2Cl (M+) 273.0305, found 273.0303.
5.2.32. 5-Chloropyridin-3-yl 1H-benzo[d][1,2,3]triazole-5-carboxylate (67)
Method A. A white solid. 1H NMR (CD3OD, 300 MHz) δ 8.85 (dd, 1H, J = 1.4, 0.7 Hz), 8.55 (d, 1H, J = 2.3 Hz), 8.53 (d, 1H, J = 2.1 Hz), 8.29 (ddd, 1H, J = 8.7, 1.5, 0.6 Hz), 7.99 (s, 1H), 7.98 (ddd, 1H, J = 4.9, 1.4, 0.6 Hz); HRMS [EI] Calcd for C12H7N4O2Cl (M+) 274.0258, found 274.0260.
5.2.33. 5-Chloropyridin-3-yl 1H-indole-5-carboxylate (68)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.60–8.58 (m, 1H), 8.50 (d, 2H, J = 2.2 Hz), 8.48–8.40 (br s, 1H), 8.04 (dd, 1H, J = 8.7, 1.7 Hz), 7.51 (t, 1H, J = 2.2 Hz), 7.50 (dt, 1H, J = 8.6, 0.7 Hz), 7.34 (dd, 1H, J = 3.4, 2.5 Hz), 6.73 (ddd, 1H, J = 3.3, 2.1, 1.0 Hz); HRMS [EI] Calcd for C14H9N2O2Cl (M+) 272.0353, found 272.0354.
5.2.34. 5-Chloropyridin-3-yl 3-acetoxybenzoate (69)
Method B. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.52 (d, 1H, J = 2.1 Hz), 8.43 (d, 1H, J = 2.4 Hz), 8.21 (dd, 1H, J = 7.9, 1.8 Hz), 7.69 (ddd, 1H, J = 8.0, 7.5, 1.7 Hz), 7.64 (t, 1H, J = 2.2 Hz), 7.42 (dt, 1H, J = 7.8, 1.2 Hz), 7.21 (dd, 1H, J = 8.1, 1.2 Hz), 2.33 (s, 3H); HRMS [ES] Calcd for C14H11ClNO4 (MH+) 292.0371, found 292.0369.
5.2.35. 5-Chloropyridin-3-yl 2-oxo-2H-chromene-3-carboxylate (70)
Method B. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.76 (s, 1H), 8.53 (d, 1H, J = 2.0 Hz), 8.50 (d, 1H, J = 2.2 Hz), 7.78–7.74 (m, 1H), 7.72 (t, 1H, J = 2.2 Hz), 7.70 (dd, 1H, J = 7.7, 1.4 Hz), 7.46–7.38 (m, 2H); HRMS [EI] Calcd for C15H8NO4Cl (M+) 301.0142, found 301.0141.
5.2.36. 5-Chloropyridin-3-yl cyclohexanecarboxylate (71)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.45 (d, 1H, J = 2.1 Hz), 8.31 (d, 1H, J = 2.3 Hz), 7.53 (dd, 1H, J = 2.3, 2.1 Hz), 2.59 (tt, 1H, J = 11.0, 3.6 Hz), 2.12–2.02 (m, 2H), 1.90–1.75 (m, 2H), 1.75–1.50 (m, 3H), 1.44–1.26 (m, 3H); HRMS [EI] Calcd for C12H14NO2Cl (M+) 239.0713, found 239.0717.
5.2.37. 5-Chloropyridin-3-yl 4-fluorobenzoate (72)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.52 (d, 1H, J = 2.0 Hz), 8.47 (d, 1H, J = 2.3 Hz), 8.22 (dd, 2H, J = 9.1, 5.3 Hz), 7.68 (dd, 1H, J = 2.3, 2.0 Hz), 7.22 (dd, 2H, J = 9.0, 8.3 Hz)); HRMS [EI] Calcd for C12H7NO2ClF (M+) 251.0149, found 251.0145.
5.2.38. 5-Chloropyridin-3-yl 4-methylbenzoate (73)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.54–8.45 (m, 2H), 8.08 (d, 2H, J = 8.4 Hz), 7.68 (dd, 1H, J = 2.2, 2.2 Hz), 7.34 (d, 2H, J = 8.3 Hz), 2.47 (s, 3H); HRMS [EI] Calcd for C13H10NO2Cl (M+) 247.0400, found 247.0403.
5.2.39. 5-Chloropyridin-3-yl 4-methoxybenzoate (74)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.50 (d, 1H, J = 2.0 Hz), 8.46 (d, 1H, J = 2.3 Hz), 8.15 (d, 2H, J = 8.7 Hz), 7.68 (dd, 1H, J = 2.3, 2.0 Hz), 7.01 (d, 2H, J = 8.8 Hz), 3.92 (s, 3H); HRMS [EI] Calcd for C13H10NO3Cl (M+) 263.0349, found 263.0351.
5.2.40. 5-Chloropyridin-3-yl 2-nitrobenzoate (75)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.68–8.45 (m, 2H), 8.10 (dd, 1H, J = 7.6, 1.7 Hz), 7.91–7.76 (m, 3H), 7.75–7.72 (m, 1H); HRMS [EI] Calcd for C12H7N2O4Cl (M+) 278.0094, found 278.0095.
5.2.41. 5-Chloropyridin-3-yl 4-(diethylamino)benzoate (76)
Method B. A yellow solid. 1H NMR (CDCl3, 500 MHz) δ 8.46 (d, 1H, J = 2.1 Hz), 8.44 (d, 1H, J = 2.3 Hz), 8.01 (d, 2H, J = 9.2 Hz), 7.66 (dd, 1H, J = 2.2, 2.2 Hz), 6.68 (d, 2H, J = 9.2 Hz), 3.46 (q, 4H, J = 7.2 Hz), 1.24 (t, 6H, J = 7.1 Hz); HRMS [EI] Calcd for C16H17N2O2Cl (M+) 304.0979, found 304.0978.
5.2.42. 5-Chloropyridin-3-yl 5-(4-chloro-2-nitrophenyl)furan-2-carboxylate (77)
Method A. A slightly yellow solid. 1H NMR (CDCl3, 500 MHz) δ 8.53 (d, 1H, J = 2.0 Hz), 8.49 (d, 1H, J = 2.4 Hz), 7.85 (d, 1H, J = 2.1 Hz), 7.83 (d, 1H, J = 8.6 Hz), 7.70 (dd, 1H, J = 2.4, 2.1 Hz), 7.67 (dd, 1H, J = 8.4, 2.1 Hz), 7.50 (d, 1H, J = 3.8 Hz), 6.82 (d, 1H, J = 3.7 Hz); HRMS [EI] Calcd for C16H8Cl2N2O5 (M+) 377.9810, found 377.9807.
5.2.43. 5-Chloropyridin-3-yl 5-(2-chloro-5-(trifluoromethyl)phenyl)furan-2-carboxylate (78)
Method A. A yellow solid. 1H NMR (CDCl3, 500 MHz) δ 8.54 (dd, 1H, J = 2.0, 0.5 Hz), 8.52 (dd, 1H, J = 2.3, 0.5 Hz), 8.28–8.26 (m, 1H), 7.72 (t, 1H, J = 2.2 Hz), 7.67–7.64 (m, 1H), 7.61–7.58 (m, 1H), 7.56 (d, 1H, J = 3.8 Hz), 7.40 (d, 1H, J = 3.8 Hz); HRMS [EI] Calcd for C17H8Cl2NO3F3 (M+) 402.9833, found 400.9823.
5.2.44. 5-Chloropyridin-3-yl 1H-pyrazole-4-carboxylate (79)
Method A. A white solid. 1H NMR (CDCl3, 500 MHz) δ 10.6–10.4 (br s, 1H), 8.51 (d, 1H, J = 2.1 Hz), 8.46 (d, 1H, J = 2.4 Hz), 8.24 (s, 2H), 7.67 (t, 1H, J = 2.2 Hz); HRMS [EI] Calcd for C9H6ClN3O2 (M+) 223.0149, found 223.0149.
5.2.45. 5-Chloropyridin-3-yl isonicotinate (80)
Method B. A white solid. 1H NMR (CD3OD, 300 MHz) δ 8.84 (d, 2H, J = 6.2 Hz), 8.54 (d, 2H, J = 2.2Hz), 8.11 (d, 2H, J = 6.2 Hz), 7.99 (t, 1H, J = 2.2 Hz); HRMS [EI] Calcd for C11H7N2O2Cl (M+) 234.0196, found 234.0191.
5.2.46. 5-Chloropyridin-3-yl pyrazine-2-carboxylate (81)
Method B. A white solid. 1H NMR (CDCl3, 300 MHz) δ 9.48 (d, 1H, J = 1.5 Hz), 8.90 (d, 1H, J = 2.5 Hz), 8.84 (dd, 1H, J = 2.4, 1.5 Hz), 8.57 (d, 1H, J = 2.0 Hz), 8.55 (d, 1H, J = 2.3 Hz), 7.74 (t, 1H, J = 2.2 Hz); HRMS [EI] Calcd for C10H6N3O2Cl (M+) 235.0149, found 235.0149.
5.2.47. 5-Chloropyridin-3-yl 2,6-dichloro-5-fluoronicotinate (82)
Method B. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.57 (dd, 1H, J = 2.0, 0.4 Hz), 8.50 (dd, 1H, J = 2.4, 0.4 Hz), 8.21 (d, 1H, J H–F = 7.1 Hz), 7.71 (t, 1H, J = 2.2 Hz); HRMS [EI] Calcd for C11H4Cl3N2O2F (M+) 319.9323, found 319.9327.
5.2.48. 5-Chloropyridin-3-yl 2-naphthoate (83)
Method A. A white solid. 1H NMR (CDCl3, 300 MHz) δ 8.80–8.78 (m, 1H), 8.54 (d, 2H, J = 2.2 Hz), 8.17 (dd, 1H, J = 8.6, 1.8 Hz), 8.04–7.92 (m, 3H), 7.75 (t, 1H, J = 2.2 Hz), 7.70–7.58 (m, 2H); HRMS [EI] Calcd for C16H10NO2Cl (M+) 283.0400, found 283.0405.
5.3. Protein expression and purification
The HAV 3Cpro (C24S) was produced heterologously using Escherichia coli BL21(DE3) pLysS containing pHAV-3CEX.20 Substitution of the non-essential surface cysteine residue in the C24S variant prevents intermolecular disulfide bond formation. Freshly transformed cells were grown overnight at 30 °C in LB broth supplemented with 100 μg/mL ampicillin and 25 μg/mL chloramphenicol, and used to inoculate (1:200) one litre of the same medium. The 1-L culture was grown at 37 °C to an optical density at 600 nm of approximately 0.6 whereupon heterologous gene expression was induced by adding 0.25 mM IPTG. The cells were incubated for a further 6 h at 30 °C, harvested by centrifugation, washed using 20 mM potassium phosphate, pH 6.5, containing 1 mM EDTA and 2 mM DTT, and then frozen at −80 °C until further use.
To purify HAV 3Cpro, the frozen cells were resuspended in 20 mL of 20 mM potassium phosphate, 1 mM EDTA, 2 mM DTT, pH 6.5 and disrupted using a french press operated at 20,000 psi. Cell debris was removed by centrifugation (37,000g for 30 min) and the supernatant was passed through a 45 μm filter. The filtered cell extracted was loaded onto a MonoS 10/10 column pre-equilibrated with 20 mM potassium phosphate, 0.5 mM EDTA, pH 6.5 and operated at 3.5 mL/min an ÄKTA Explorer (GE Healthcare). The proteinase was eluted using a gradient of 80–280 mM NaCl in 96 mL of the equilibration buffer. Eight milliliters of fractions were collected. Those containing the proteinase, as judged from SDS–PAGE, were combined and concentrated using a stirred cell concentrator equipped with a YM10 membrane (Amicon, Etobicoke, ON, Canada). The protein solution was concentrated to 3.0 mg/mL and frozen as beads in liquid nitrogen. Sixty milligrams of 3Cpro was typically obtained from one liter of cell culture.
The His-tagged SARS 3CLpro was produced and purified as described previously16 except that the His-tag was removed. To remove the His-tag, the protein was exchanged three times into 50 mM Tris pH 8.0 and then amended with 100 mM NaCl and 1 mM Ca2+. The His-tag was removed proteolytically using Factor Xa:3CL (0.002:1, w:w) overnight at 23 °C. The concentrated protein was loaded onto a MonoQ 10/10 column pre-equilibrated with 20 mM Tris, pH 7.5. The protein was eluted with a gradient of 0–150 mM NaCl over 100 mL of the equilibration buffer. The fractions containing proteinase were combined, exchanged into 20 mM Tris, pH 7.5, concentrated to 21 mg/mL, and then frozen as beads in liquid nitrogen. A yield of 50 mg of 3CLpro was obtained from 4 L of cell culture.
5.4. Fluorogenic peptide assay
The steady-state proteolytic activities of HAV 3Cpro and SARS 3CLpro were measured using the fluorogenic peptide substrates Dabcyl-GLRTQSND(edans)G and Abz-SVTLQSGY(NO2)R, respectively. Standard assays were performed using 0.1 M potassium phosphate, pH 7.5, 2 mM EDTA containing 0.1 μM proteinase and 10–75 μM peptide at 37 °C. Initial inhibitor screening and IC50 determination for HAV 3Cpro were done using a 96-well plate and a Victor2 fluorescence plate reader (PerkinElmer, Woodbridge, ON, Canada). Substrate (10 μM) and inhibitor (0.25–10 μM) were prewarmed for three minutes in black 96-well microplates in a total volume of 200 μl before the reaction was started with the addition of enzyme (λ ex = 355 nm, λ em = 460 nm). Determination of K i was done using a Varian Eclipse Fluorescence Spectrophotometer (Varian, Cary, NC, USA). The assay was performed using a 100 μL quartz cuvette (λ ex = 340 nm, λ em = 490 nm for SARS 3CLpro; λ ex = 320 nm, λ em = 420 nm for SARS 3CLpro). Initial rates were calculated using the first 3 min of the progress curve and were corrected for the inner filter effect as described by Liu et al.,23 using SND(edans)G or Abz-SVTLQ as appropriate. Steady-state equations were fitted to the initial-rate data using the least-squares and dynamic weighting options of LEONORA.24
5.5. Pyridinyl ester hydrolysis assay
Rates of pyridinyl ester hydrolysis were determined by monitoring product formation using an HPLC. Assays were performed at 37 °C in HPLC vials containing 400 μL of 0.1 M potassium phosphate, pH 7.5, 2 mM EDTA, 2 μM proteinase, and 10–75 μM pyridinyl ester. Reactions were initiated with the addition of the ester. Aliquots of 50 μL were withdrawn and analyzed using a Waters 2695 separation module equipped with Phenomenex prodigy 10 μ OD-prep column and a Waters 2996 photodiode array detector. The column was operated at a flow rate of 1 mL/min and developed using the following three-step profile: 1 mL 5% acetonitrile in 0.05% formic acid/H2O; 10 mL linear gradient of 5–100% acetonitrile in 0.05% formic acid/H2O; and 1 mL 100% acetonitrile. Rates were calculated over the first hour of the reaction. Enzymatic rates were corrected for non-enzymatic hydrolysis.
5.6. Mass spectrometry of enzyme-inhibitor complexes
Samples of HAV 3Cpro were prepared by reacting 50 μM enzyme with 100 μM pyridinyl ester in 100 mM potassium phosphate buffer, pH 7.5, 2 mM EDTA at 37 °C for 2 or 10 min. Samples were quenched with 25% acetic acid and submitted to NAPS for MALDI-TOF analysis.
5.7. Molecular docking
Automated molecular docking was performed using the program AutoDock 3.0.5 and a recent structural model of HAV 3Cpro (PDB: 2CXV)4 as the target. The 3D structures of ester compounds were constructed using Sybyl 7.1 (Tripos Inc., St. Louis, USA). Inhibitor-enzyme interactions were evaluated using the Lamarckian genetic algorithm (LGA). Binding energies between the compounds and the protein were evaluated using a grid map with 60 × 60 × 60 points spaced at 0.375 Å generated using the AUTOGRID program. Default values of the docking parameters were used except for the following: step sizes for translation (0.2 Å), orientation (5°), and torsion (5°); and the number of generations (37,000); energy evaluations (1,500,000), and docking runs (20). The docked inhibitor–enzyme complexes were ranked according to the lowest predicted binding energies and the conformity to ideal geometry of the docked structures.
Acknowledgments
Financial support from the Natural Sciences and Engineering Research Council of Canada (Discovery grants to L.D.E. and J.C.V.), the Canada Research Chairs Program (CRC in Protein Structure & Function and CRC in Bioorganic & Medicinal Chemistry), and the Alberta Heritage Foundation for Medical Research (AHFMR) is gratefully acknowledged. C.H. is the recipient of an NSERC fellowship. J.Y. is the recipient of AHFMR and Killam Foundation fellowships. We thank Suzanne Perry for help with mass spectrometry and Geoff Horsman for helpful discussions.
References and notes
- 1.Tong L. Chem. Rev. 2002;102:4609. doi: 10.1021/cr010184f. [DOI] [PubMed] [Google Scholar]
- 2.Allaire M., Chernaia M.M., Malcolm B.A., James M.N. Nature. 1994;369:72. doi: 10.1038/369072a0. [DOI] [PubMed] [Google Scholar]
- 3.Bergmann E.M., Mosimann S.C., Chernaia M.M., Malcolm B.A., James M.N. J. Virol. 1997;71:2436. doi: 10.1128/jvi.71.3.2436-2448.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin J., Bergmann E.M., Cherney M.M., Lall M.S., Jain R.P., Vederas J.C., James M.N. J. Mol. Biol. 2005;354:854. doi: 10.1016/j.jmb.2005.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosimann S.C., Cherney M.M., Sia S., Plotch S., James M.N. J. Mol. Biol. 1997;273:1032. doi: 10.1006/jmbi.1997.1306. [DOI] [PubMed] [Google Scholar]
- 6.Matthews D.A., Smith W.W., Ferre R.A., Condon B., Budahazi G., Sisson W., Villafranca J.E., Janson C.A., McElroy H.E., Gribskov C.L., et al. Cell. 1994;77:761. doi: 10.1016/0092-8674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 7.Birtley J.R., Knox S.R., Jaulent A.M., Brick P., Leatherbarrow R.J., Curry S. J. Biol. Chem. 2005;280:11520. doi: 10.1074/jbc.M413254200. [DOI] [PubMed] [Google Scholar]
- 8.Shi J., Wei Z., Song J. J. Biol. Chem. 2004;279:24765. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. PNAS. 2003;100:13190. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malcolm B.A., Lowe C., Shechosky S., McKay R.T., Yang C.C., Shah V.J., Simon R.J., Vederas J.C., Santi D.V. Biochemistry. 1995;34:8172. doi: 10.1021/bi00025a024. [DOI] [PubMed] [Google Scholar]
- 11.Morris T.S., Frormann S., Shechosky S., Lowe C., Lall M.S., Gauss-Muller V., Purcell R.H., Emerson S.U., Vederas J.C., Malcolm B.A. Bioorg. Med. Chem. 1997;5:797. doi: 10.1016/s0968-0896(97)88649-x. [DOI] [PubMed] [Google Scholar]
- 12.Ramtohul Y.K., James M.N.G., Vederas J.C. J. Org. Chem. 2002;67:3169. doi: 10.1021/jo0157831. [DOI] [PubMed] [Google Scholar]
- 13.Yin J., Cherney M.M., Bergmann E.M., Zhang J., Huitema C., Pettersson H., Eltis L.D., Vederas J.C., James M.N. J. Mol. Biol. 2006;361:673. doi: 10.1016/j.jmb.2006.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain R.P., Vederas J.C. Bioorg. Med. Chem. Lett. 2004;14:3655. doi: 10.1016/j.bmcl.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 15.Lall M.S., Ramtohul Y.K., James M.N.G., Vederas J.C. J. Org. Chem. 2002;67:1536. doi: 10.1021/jo0109016. [DOI] [PubMed] [Google Scholar]
- 16.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J., Pettersson H.I., Huitema C., Niu C., Yin J., James M.N., Eltis L.D., Vederas J.C. J. Med. Chem. 2007;50:1850. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- 18.Seah S.Y., Labbe G., Nerdinger S., Johnson M.R., Snieckus V., Eltis L.D. J. Biol. Chem. 2000;275:15701. doi: 10.1074/jbc.275.21.15701. [DOI] [PubMed] [Google Scholar]
- 19.Jewell D.A., Swietnicki W., Dunn B.M., Malcolm B.A. Biochemistry. 1992;31:7862. doi: 10.1021/bi00149a017. [DOI] [PubMed] [Google Scholar]
- 20.Malcolm B.A., Chin S.M., Jewell D.A., Stratton-Thomas J.R., Thudium K.B., Ralston R., Rosenberg S. Biochemistry. 1992;31:3358. doi: 10.1021/bi00128a008. [DOI] [PubMed] [Google Scholar]
- 21.Niu C., Yin J., Zhang J., Vederas J.C., James M.N. Bioorg. Med. Chem. 2007;16:293. doi: 10.1016/j.bmc.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee T.W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N. J. Mol. Biol. 2005;353:1137. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y., Kati W., Chen C.M., Tripathi R., Molla A., Kohlbrenner W. Anal. Biochem. 1999;267:331. doi: 10.1006/abio.1998.3014. [DOI] [PubMed] [Google Scholar]
- 24.Cornish-Bowden A. Oxford University Press; Oxford, New York: 1995. Analysis of Enzyme Kinetic Data. [Google Scholar]