

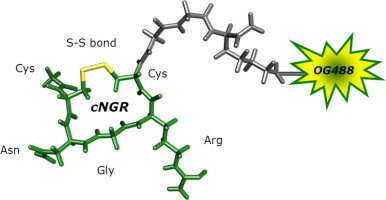
Graphical abstract
Keywords: Angiogenesis, cNGR, Multimerization, Quantum dots, Molecular imaging
Abstract
Angiogenesis is a multi-step process regulated by pro- and anti-angiogenic factors. Inhibition of angiogenesis is a potential anti cancer treatment strategy that is now investigated clinically. In addition, advances in the understanding of the angiogenic process have led to the development of new angiogenesis therapies for ischemic heart disease.
Currently, researchers search for objective measures that indicate pharmacological responses to pro- and anti-angiogenic drugs and therefore, there is a great interest in techniques to visualize angiogenesis noninvasively. As CD13 is selectively expressed in angiogenic blood vessels, it can serve as a target for molecular imaging tracers to noninvasively visualize angiogenic processes in animal models and patients. Here, an overview on the currently used CD13 targeted molecular imaging probes for noninvasive visualization of angiogenesis is given.
1. Introduction
Angiogenesis, the formation of new blood vessels from existing ones, plays an essential role in embryonic development. However, once the vascular network is fully deployed, angiogenesis is only triggered locally and transiently under specific conditions. Well-known pathological conditions in which angiogenesis plays a role are myocardial infarction and cancer.1 Currently, a potential pathological role of angiogenesis in atherosclerotic plaques is under investigation.2, 3, 4, 5, 6, 7
In solid tumors, angiogenesis causes the growth of new capillaries that are necessary for the continuous supply of oxygen and nutrients to the tumor tissue. This allows the tumor to grow beyond a certain size and to metastasize.
In myocardial infarction (MI) however, angiogenesis plays a much more benign role, allowing neovascularization following myocardial ischemia. For this reason, therapeutic myocardial angiogenesis is being explored as a therapeutic option in ischemic heart disease.8
Neovascularization of atherosclerotic plaques contributes to the development of the plaques, increasing the risk of rupture. The clinical complications of atherosclerosis, for example, MI and stroke, are mainly caused by thrombus formation, which results from rupture of an atherosclerotic plaque and exposure of tissue factor to the blood. An atherosclerotic plaque is formed when platelets adhere to subendothelial collagen. Tissue factor captures factor VII(a) from the circulation and initiates coagulation and fibrin formation to stabilize the primary platelet plug.9 The formed blood clot can then either locally occlude the blood vessel or embolize and occlude a narrower vessel downstream. In both cases this manifests itself in the clinical symptoms of MI or stroke. It is thought that intraplaque microvessels contribute to the unstable plaque phenotype and are mainly derived from the vasa vasorum by angiogenic sprouting, followed by ingrowth into the lesion.4, 10, 11 Recently, Rademakers et al. demonstrated that plaque-associated vasa vasorum were angiogenically active and, albeit poorly, perfused.7 Moreover, plaque-associated vasa vasorum showed increased permeability, reduced blood flow, and increased leukocyte adhesion and extravasation. These characteristics could contribute to plaque progression and destabilization. Imaging of plaque-angiogenesis could ascertain the exact importance of intraplaque angiogenesis in the prediction of plaque growth and rupture.
Angiogenesis involves a complex interplay of growth factors, adhesion molecules, and the extracellular matrix. Activated endothelial cells present in angiogenic blood vessels express markers that are minimally expressed or completely absent in normal blood vessels. These biomarkers include integrins αvβ3 and αvβ5,12, 13 certain receptors for vascular growth factors,14, 15 and matrix metalloproteases (MMPs).16, 17 Aminopeptidase N (APN), also called CD13 receptor, is a zinc dependent membrane-bound ectopeptidase, which has been identified as an important regulator of endothelial morphogenesis during angiogenesis.18
CD13 binds peptides and proteins through exposed tripeptide asparagine-glycine-arginine (NGR) amino acid residues. This motif emerged as a recurring sequence during phage display experiments directed against α5β1 integrin, and also surfaced in phage display experiments against αvβ3 integrin.19 As CD13 is selectively expressed in angiogenesis, it can serve as a target for molecular imaging tracers to noninvasively visualize angiogenic processes in vivo. Noninvasive determination of CD13 expression potentially can be used to monitor treatment response to pro- or antiangiogenic drugs in tumors or ischemic heart disease, respectively.
In this paper, we discuss the target, CD13, the discovery of its peptide ligand, the NGR motif, the NGR-isoDGR molecular switch, and the design and development of NGR-based imaging probes. An overview of imaging studies with these cyclic NGR-based tracers in cancer and cardiovascular disease is given.
2. Discovery of the NGR motif
In their search for α5β1-binding ligands, Koivunen et al. found that besides various RGD-containing peptides, the NGR-containing peptide NGRAHA represented an α5β1-binding sequence.20 In addition, they isolated 8 NGR-containing peptides upon screening of cyclic peptide phage libraries under similar experimental conditions.21 In vivo selection of phage display libraries was used to isolate peptides that home specifically to tumor blood vessels. The CNGRCVSGCAGRC-phage demonstrated to target tumor vasculature in nude mice bearing human MDA-MB-435 breast carcinoma.22 The linear NGRAHA- and cyclic CVLNGRMEC-peptide, previously identified in vitro, also selectively localized to tumors.22 Co-injection of the CNGRCVSGCAGRC-, NGRAHA- or CVLNGRMEC-phage with the CNGRC peptide (NGR-2C; Cys1–Cys5 disulfide), inhibited the accumulation of these phages in breast cancer xenografts, indicating that NGR-2C is a functional tumor targeting peptide.
Another tumor targeting peptide discovered by phage display-based studies was ACDCRGDCFC (RGD-4C), which binds to αvβ3 and αvβ5 integrins expressed on sprouting endothelial cells.22, 23 NGR-containing peptides can bind to integrins, although with lower affinity than RGD peptides.24, 25 In cross-inhibition experiments tumor homing of the RGD-4C-phage could not be inhibited by free NGR-2C peptide, whereas free RGD-4C peptide could only partially compete with NGR-2C-phage for tumor homing.22 These results suggested that NGR-2C and RGD-4C bind to distinct receptors.
Soon, it was found that CD13 was a receptor for NGR-containing peptides.26 Although CD13 appeared to be the major target of NGR containing peptides, there are large differences in NGR binding among CD13 isoforms. NGR peptides preferentially bind to CD13 on endothelial cells of small angiogenic vessels and to a much lesser extent on CD13 positive large blood vessels or CD13 expressing macrophages.27 This may be due to the differential utilization of O-glycosylation sites among CD13 isoforms or to conformational changes induced in some CD13 isoforms due to complexation with the proangiogenic protein galectin-3 or other unidentified compounds.19
3. Structure of aminopeptidase N (CD13)
CD13 (EC.3.4.11.2) is a broad specificity aminopeptidase with a preference for neutral amino acids. CD13 occurs as a soluble and a membrane-bound form and multiple distinct glycoforms have been reported. The structure of the 967 amino acids long human CD13 protein has not yet been resolved. However, the membrane-bound form of the protein is known to occur as a non-covalent homodimer with a relative molecular mass of 160 kDa. Each of the monomers consists of a short N-terminal cytoplasmatic domain, a single transmembrane domain and a large, glycosylated extracellular domain that contains the aminopeptidase active site.28 In addition to the membrane-bound form, soluble CD13 has also been reported in both plasma and urine.29
4. Function of amino peptidase N (CD13)
Many functions have been ascribed to CD13, which can be divided in three groups: enzymatic cleavage of peptides, endocytosis or signal transduction. These were recently reviewed by Mina-Osorio.30
CD13 is a peptidase that removes N-terminal amino acids from oligopeptides, with the exception of peptides that contain Pro in the penultimate position. Its amino acid substrate order preference has been determined to be Ala > Phe > Tyr > Leu > Arg > Thr > Trp > Lys > Ser > Asp > His > Val. Known peptide substrates of CD13 include the enkephalin, kinin, and hemorphin neuropeptides, the vasoactive angiotensins, the immunomodulatory tetrapeptide tuftsin, and the chemotactic peptides monocyte chemotactic protein 1, and N-formyl-methionyl-leucyl-phenylalanine.29
In addition, human CD13 is a viral receptor for a number of viruses, including human coronavirus 229E and possibly also human cytomegalovirus (HCMV). This function was shown to be independent from its catalytic function, since neither active site mutations nor active site inhibitors affected virus binding to CD13.
The importance of CD13 in angiogenesis was only discovered after it had been identified as the main target of NGR. Studies in CD13-null mice showed that these mice suffered from decreased retinal neovascularization under hypoxic conditions and showed a reduced angiogenic response to growth factors.31
CD13 expression is strongly correlated with the invasive capacity of a number of tumor cell types. It plays a role in cell differentiation, motility, proliferation and apoptosis. Membrane-bound CD13 is involved in phagocytosis and adhesion.
Finally, CD13 is one of the main targets of ezetimibe, a clinically used drug against hypercholesterolaemia.30
5. The NGR/isoDGR motif as a molecular switch
Quite remarkably, the NGR motif was discovered during a phage display search for integrin binders, even though the NGR sequence had little or no integrin binding affinity. More recently it was observed that the asparagine residue in NGR can spontaneously convert into the isoaspartate resulting in isoDGR. IsoDGR mimics the RGD motif, which is a well-known integrin binding motif.32
The asparagine to isoaspartate conversion is a known protein aging reaction. The first step for this conversion is the nucleophilic attack of the peptide bond nitrogen of the adjacent amino on the asparagine side chain carbonyl. This leads to the formation of a cyclic succinimide intermediate and the loss of one molecule of ammonia. The succinimide intermediate can undergo hydrolytic ring opening at either carbonyl in the imide ring, giving rise to aspartate or isoaspartate. Isomerization also occurs during this reaction, but only to a limited extent. The major products of the reaction are l-aspartate and l-isoaspartate, typically in a 1:3 ratio (Scheme 1 ).
Scheme 1.
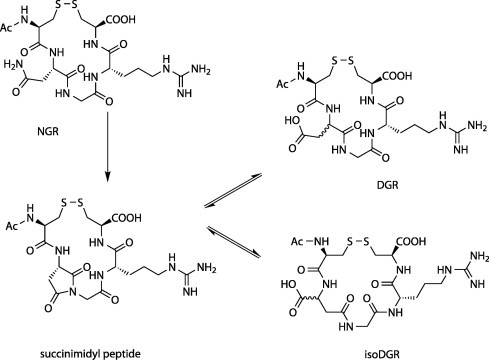
The NGR-isoDGR switch. The cNGR moiety can spontaneously deamidate into a succinimide containing intermediate. Hydrolysis of the succinimide results in the formation of isoDGR or DGR, with limited racemization.
Asparagine to isoaspartate conversion causes quite large local changes in a protein, since it causes both a change in the charge state (from 0 to −1) and a change in the protein backbone (conversion of an α-amino acid into a longer β-amino acid). Generally, conversion of asparagine to aspartate can take hours, days or years and depends on a number of factors, including protein microenvironment, pH, ionic strength and temperature. The reaction rates of the NGR to isoDGR reaction vary as well, but are typically quite rapid (hours in vitro). While most protein aging reactions cause some loss of function, NGR to isoDGR may actually have gain-of-function effects. In fibronectin, four NGR motif containing sequences have been identified that function as latent αvβ3 integrin binding sites.
Molecular modeling has shown that isoDGR neatly docks onto the same site of αvβ3 integrin as RGD, albeit in an inverted orientation.33 It was also shown that isoDGR containing peptides can bind the α5β1 integrin as well and that the α5β1/αvβ3 selectivity depends on the flanking residues of the NGR/isoDGR motif.34
6. Design and synthesis of NGR-based imaging agents
Magnetic Resonance Imaging (MRI) is a powerful, non-invasive technique for the visualization of soft tissue anatomy and the diagnosis of diseases. To enhance the contrast in T1-weighted MR images, gadolinium(III) (Gd(III)) chelates are commonly used as MRI contrast agents.35 Several (patho)fysiological processes, such as angiogenesis, are difficult to image with MRI. Therefore, target-specific MRI contrast agents designed to bind to markers that are (over)expressed by cells involved in these processes could drastically improve the imaging angiogenesis due to accumulation of contrast agents around these cells.
6.1. Monomeric and monomodal cyclicNGR peptide
To explore the possibilities and efficacies of achieving multimodal and multivalent cyclicNGR constructs, several approaches and synthetic routes have been explored. Initially, fluorescent monomodal constructs were prepared for optical imaging that consisted of a cyclicNGR (cNGR) moiety to which a Gly-Gly-Lys(N ε-Oregon Green 488) sequence was added C-terminally. Therefore, Ac-Cys(4MeBzl)-Asn(Xanthyl)-Gly-Arg(Tosyl)-Cys-(4MeBzl)-Gly-Gly-Lys(Fmoc) was synthesized by tBoc (tert-butyloxycarbonyl) solid-phase peptide synthesis (SPPS).36 On resin, the Fmoc (9-fluorenylmethyloxycarbonyl) protecting group of lysine was removed and subsequently Oregon Green 488 (OG488) was coupled to the lysine ε-amino group. After deprotection and cleavage of the peptide from the resin, the peptide was cyclized by the formation of an internal disulfide bond by oxidative folding. Unconjugated cNGR was used as control compound. This compound was synthesized analogously, though it was acetylated at the lysine ε-amino group; Ac-Cys-Asn-Gly-Arg-Cys-Gly-Gly-Lys-(Ac)-NH2.
6.2. Monomeric bimodal cNGR peptide
To gain more insight into the targeting process of cNGR, a bimodal fluorescence and MRI CD13-specific probe, based on OG488, gadolinium(III) diethylenetriaminepentaacetic acid (Gd(III)DTPA), and cNGR, has been synthesized.37 Highly efficient, chemoselective reaction procedures were used for the synthesis of this bimodal target-specific contrast agent, which allows imaging with both MRI and optical methods. The groundbreaking native chemical ligation (NCL) technologies enabled the formation of a peptide bond between the NGR peptide and the cysteine-functionalized DTPA chelator.38, 39, 40 Subsequently, the free thiol of the introduced cysteine was used for conjugation to the maleimide of OG488. Removal of the acetamidomethyl (Acm)-protecting groups and subsequent oxidative folding resulted in the DTPA- and OG488-conjugated cyclicNGR peptide (Scheme 2, Scheme 3 ).
Scheme 2.
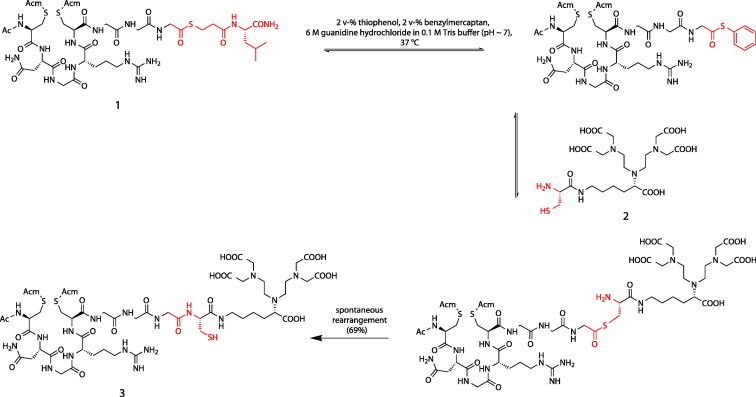
DTPA conjugation of NGR peptide using NCL, resulting in NAcC(Acm)NGRC(Acm)GGC-DTPA (3).
Scheme 3.
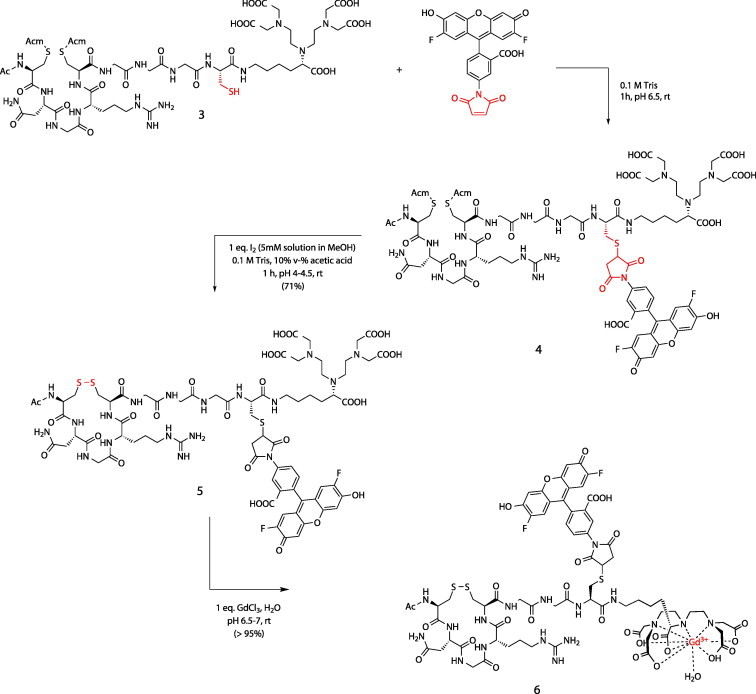
Coupling of OG488 to NGR peptide and subsequent oxidative folding of the peptide. Chelation of DTPA with Gd(III) resulted in the final product: Ac-cNGR-GGC(OG488)-Gd(III)DTPA (6).
6.3. Tetrameric monomodal cNGR-containing MRI contrast agent
Multivalent structures have become a new focus of investigation in drug design and chemical biology. A multimeric compound could theoretically bind multivalently to targets and thus more avidly to the target cell. By immobilizing multiple cyclic NGR moieties and MRI labels on one single carrier, the binding of cNGR to CD13 may be improved through cooperative binding (multivalency),41 resulting in a stronger accumulation of MRI contrast agent around regions of angiogenesis. Next to targeting, sensitivity is the other major challenge for molecular MR imaging. One way to increase the contrast is through the connection of multiple Gd(III) chelates to one carrier.35, 42
A next generation of constructs consisted of a tetrameric avidin core bound to four identical peptide based molecules consisting of a cNGR part, a biotin and a DTPA moiety. The idea was to both increase the binding avidity to CD13 by cooperative binding and to increase imaging sensitivity by locally increasing the Gd(III) (gadolinium) chelate concentration. The synthesis of these constructs is shown in Scheme 4, Scheme 5 .
Scheme 4.
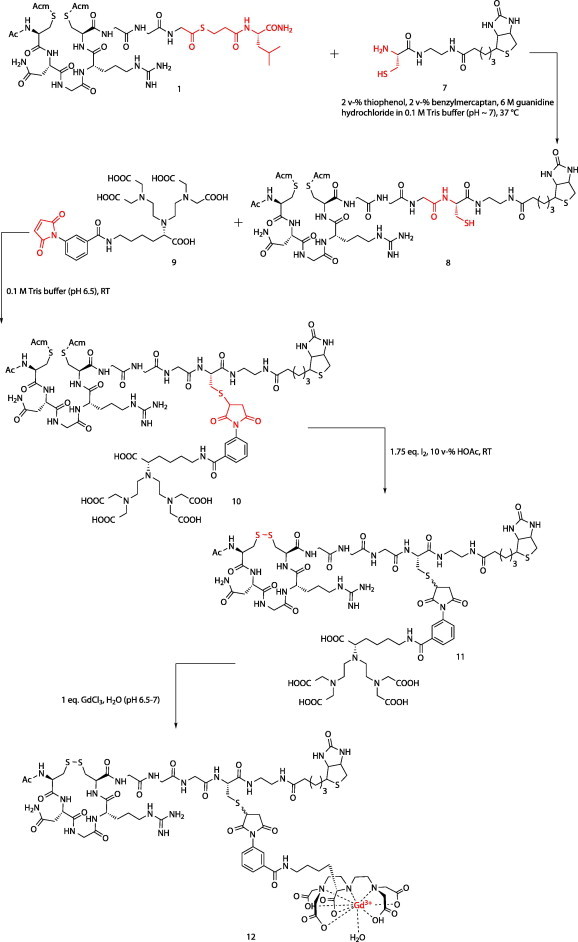
Synthesis of biotinylated Gd(III)DTPA-cNGR using native chemical ligation (NCL) techniques.
Scheme 5.
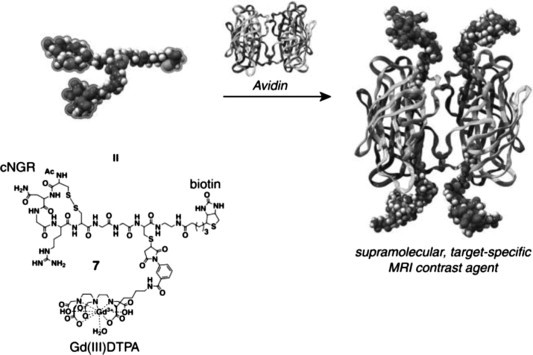
Synthesis of a multivalent target-specific MRI contrast agent based on biotinylated Gd(III)DTPA-cNGR (12) and avidin.
Briefly, the synthesis involved the tBoc SPPS of a bis-S-Acm protected thioester precursor which was coupled to a cysteine functionalized biotin by native chemical ligation. Next, the single free thiol at the ligation site was reacted with a maleimide functionalized DTPA molecule. At this stage, the S-Acm deprotection and simultaneous disulfide bond formation could be safely performed without risking disulfide shuffling or transthioesterification. Finally, Gd(III) was chelated and the fragments were mixed with avidin, creating the tetravalent constructs.43
6.4. Multivalent multimodal cNGR imaging agents
In addition, a number of modified cNGR nanoparticle-based contrast agents for angiogenesis imaging were prepared and investigated, at the same time evaluating nanoparticles as a universal scaffold for targeted molecular imaging. These multivalent cNGR coated Quantum Dots (QDs) were composed of a CdSe core with a ZnS shell and covered with polyethyleneglycol-2000. Each QD (λ em = 585 nm or λ em = 525 nm) contained about 10 surface-bound streptavidins, which allowed 30 biotinylated compounds to bind on average to each QD. Quantum Dots will not likely be applied to clinical use, as they accumulate in the spleen and liver,44 but constitute a facile, and sensitive toolbox to test combinations of target peptides and imaging modalities, allowing multimodal detection in animal models, as well as cellular localization with use of multiphoton laserscanning microscopy (Fig. 1 ). In case of optimal combinations of targeting and imaging moieties, tailored organic synthesis of contrast agent without QDs will yield imaging agents suitable for clinical translation. The modular nature of these multivalent constructs allows the rational design of targeting agents and imaging modalities. For instance, by changing the ratio of targeting and imaging moieties, constructs can be skewed towards multivalent binding to low affinity targets or multivalent detection of low abundance targets.
Figure 1.
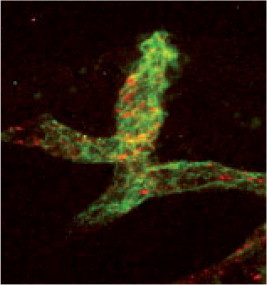
Ex vivo two-photon laser scanning microscopy (TPLSM) image obtained in an intact LS174T tumor. Colocalization of cNGR-pQDs (red) with tumor endothelial cells (green) is shown. This indicates contrast agent binding to activated endothelial cells.
As a proof of concept of this approach, biotinylated cNGR peptide and biotin-functionalized dendritic DTPA constructs were developed, and tested in combination on a QD-scaffold. First, QDs (λ em = 585 nm or λ em = 525 nm) loaded with nothing but cNGR peptide were prepared. Therefore, Ac-Cys(4MeBzl)-Asn(Xanthyl)-Gly-Arg(Tosyl)-Cys(4MeBzl)-Gly-Gly-Lys(Fmoc) was synthesized by tBoc SPPS as described above (monomeric and monovalent cNGR peptide). After on resin removal of the Fmoc protecting group of lysine, biotin-succinimidyl ester was coupled to the lysine-Nε group. Subsequently, the peptide was simultaneously deprotected and cleaved from the resin, purified, and oxidatively folded to generate the internal disulfide bridge.
cNGR coated QDs were generated by mixing QD-streptavidin with a sixfold molar excess of cNGR-biotin, ensuring multivalent loading of each QD with cNGR.
6.5. Synthesis of the biotin-poly(lysine) dendritic DTPA wedge
The poly(lysine) dendritic DTPA wedges were obtained using manual solid-phase peptide synthesis.45 In the first steps of SPPS the focal point of the wedge is defined, whereas in the final coupling steps the periphery is determined and an additional functionality can be introduced at the focal point.
To obtain DTPA-functionalized wedges, 1st and 2nd generation poly(lysine) dendritic wedges with thioester-functionalities at their focal point and sulfhydryl groups along their periphery were synthesized. A glycine spacer consisting of 4 glycine residues was introduced between the thioester at the focal point and the poly(lysine) dendritic wedge to create space for coupling of the wedges to their ligands. The 1st and 2nd generation wedges were used without further purification and were functionalized with DTPA using maleimide-functionalized DTPA. For formation of DTPA-Gd(III) complexes, 4 or 8 equiv GdCl3 were added to the wedges at pH 6.5–7.
6.6. cNGR-containing quantum dots conjugated with DTPA wedge
As QDs enabled binding of both multiple cNGR peptides and multiple DTPA moieties, the next generation of molecular imaging tracers contained not only cNGR peptides, but also DTPA-Gd(III) complexes.
Hence, the poly(lysine) dendritic DTPA wedge (8 moieties, 2nd generation) with a thioester-functionality at its focal point was coupled to a cysteine functionalized biotin by native chemical ligation. Subsequently, QDs (λ em = 585 nm), biotin-cNGR and biotin-poly(lysine) dendritic wedge (8 Gd(III)DTPA moieties) were mixed in a molar ratio of 1:6:24, resulting in a maximum of 192 Gd(III)DTPA and 6 cNGR peptides per QD (Scheme 6, Scheme 7 ).
Scheme 6.
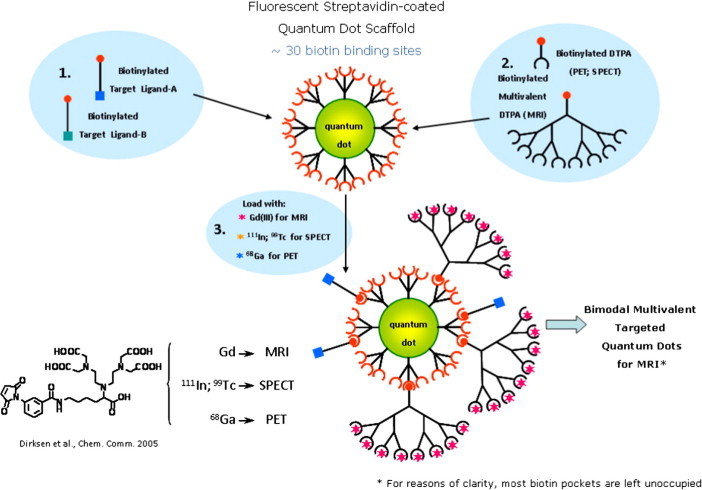
General synthetic strategy for the preparation of nanoparticle based imaging agents. The nanoparticles used allow for the modular construction of multivalent agents. Depending on the metal ion used, the same scaffold and synthetic approach can be used for the synthesis of MRI, PET or SPECT agents.
Scheme 7.
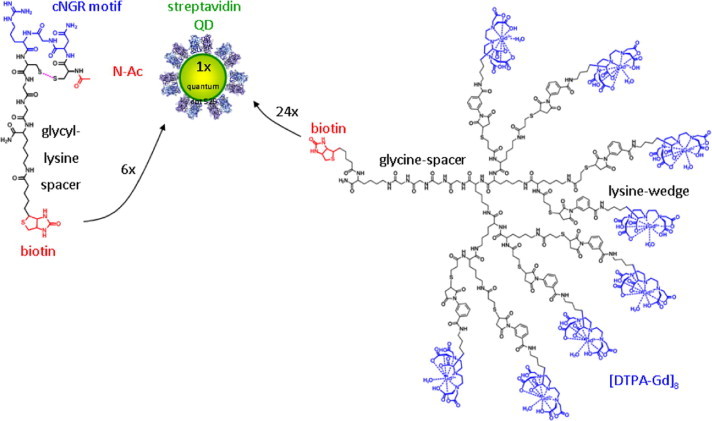
Synthesis of cNGR/Gd(III)-DTPA loaded quantum dots. The cNGR moieties allow for the multivalent targeting of CD13 expressing cells, while coupling with multiple dendritic constructs, each containing 8 Gd(III) ions per molecule allows for locally high concentrations of Gd(III), which is optimal for MRI.
7. Imaging of cardiac angiogenesis with NGR-based constructs
The utility of cNGR-OG488 and cNGR–QDs as CD13-specific targeted tracers for the visualization of angiogenesis, was investigated in in vivo and ex vivo studies.27, 46 Myocardial infarction (MI) was induced in 10 to 12-week-old male mice by ligation of the left coronary artery.47 Sham surgery consisted of the same procedure without tying the ligature.
Both the multimeric bimodal Quantum Dots and the monomeric monomodal agent could successfully be used to identify areas of the heart that had suffered hypoxia and as a result developed angiogenic vessels due to induced myocardial infarction in mice. In comparing the small fluorescent probe and the multivalent QD conjugates, it was found that the latter showed both a brighter fluorescence signal and a more persistent signal. While CD13 was found to be expressed on vessels in healthy myocardium and also on larger vessels, the cNGR-based probes were found to bind preferentially to CD13 positive small vessels in the angiogenic infarction area. The spontaneous neovascularization through angiogenesis and arteriogenesis likely determines prognosis in patients with coronary artery disease, making this a promising new tool in the cardiovascular imaging toolbox. While the Cd and Se containing QDs used in this study are likely too toxic for human use, the QD scaffolds may be replaced by other nanoparticles or peptide-like scaffolds, especially since the fluorescent properties of QDs are no longer required following validation of the nanoparticle–biomarker probe conjugate.
8. Imaging of tumor angiogenesis with NGR-based constructs
Angiogenesis is an important process for tumors to grow and metastasize. Therefore, we studied the possible use of cNGR peptides conjugated to paramagnetic quantum dots for tumor imaging. The cNGR–QDs mentioned above were designed for bimodal imaging by in vivo MRI and ex vivo two-photon laser scanning microscopy (TPLSM), thereby allowing validation of the results. QDs conjugated with only biotin-poly(lysine) dendritic DTPA wedges and no biotin-cNGR peptides were used as a control.
Swiss nu/nu mice with subcutaneously growing LST174 tumors (human colorectal adenocarcinoma) were studied by in vivo MRI and ex vivo TPLSM. It was found that the cNGR-conjugated QDs gave a threefold higher quantitative MRI contrast in the tumor rim, that is, the tumor region with the highest angiogenic activity, compared with non-cNGR-conjugated QDs (Fig. 2 ). In addition, the cNGR-conjugated QDs were barely detectable in muscle tissue, indicating the high specificity for angiogenic vessels compared to regular blood vessels. Finally, TPLSM showed colocalization of cNGR-conjugated QDs with endothelial cells in the tumor vasculature, validating that the binding was specific.48
Figure 2.
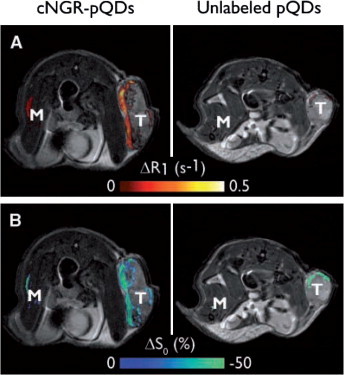
T2-weighted anatomic images with color overlay of ΔR1 (A) and ΔS0 (B) for tumor (T) and muscle (M) tissue of mice injected with cNGR-labeled or unlabeled pQDs (n = 7 for both groups). Changes in R1 were most pronounced at the tumor rim for cNGR-pQDs. Although an R1 increase in the tumor rim was also observed for unlabeled pQDs, the average response was threefold lower when compared with cNGR-pQDs, indicating a high specificity of cNGR for angiogenic tumor endothelium. This is further supported by the low changes in R1 found in muscle tissue. Changes in S0 (B) colocalized almost completely with changes in R1 (A).
This initial study was followed up by a more in depth study of the pharmacokinetic and tumor imaging properties of these cNGR–QD conjugates. In comparing two pharmacokinetic models for the distribution of cNGR–QDs, it was found that a two compartment model fitted best with the observed data. This two compartment model comprised the blood space and endothelial cell surface. Addition of a third extravascular–extracellular compartment revealed an irrelevantly low extravasation parameter and was therefore neglected. Interestingly, some accumulation of cNGR negative QDs in tumor tissue was also observed, probably due to easier extravasation and the presence of large intercellular spaces in tumor tissue.49
9. Radiolabeled cNGR probes
MRI offers good depth tissue penetration and good resolution. In addition, anatomical information can be coregistered with functional and molecular information within a single imaging method. A major disadvantage of MRI compared to radiotracer techniques such as positron emission tomography (PET) and single photon emission computed tomography (SPECT), is its lower sensitivity for the detection of targeted agents.
Recently, Chen et al. synthesized a monomeric and a dimeric NGR-containing peptide (NGR1 and NGR2, resp.) and conjugated these with the chelator 1,4,7,10-tetraazadodecane-N,N′,N″,N‴-tetraacetic acid (DOTA).50 The resulting peptides were radiolabeled with the PET isotope 64Cu (t 1/2 = 12.7 h; β+ 655 keV, 17.8%) and evaluated in vitro and in vivo.
In vitro experiments demonstrated that both 64Cu-DOTA−NGR1 and 64Cu-DOTA−NGR2 are stable enough in phosphate-buffered saline (PBS) at room temperature and mouse serum at 37 °C for 24 h. More than 94% of 64Cu-DOTA−NGR1 or 64Cu-DOTA−NGR2 remained intact after 24 h of incubation in mouse serum at 37 °C.
In order to select appropriate tumor cell lines for targeting CD13 receptor, the CD13 expression levels in human fibrosarcoma HT-1080 cells and human colon adenocarcinoma HT-29 cells were investigated. Both Western blot analysis and immunofluorescence staining showed that CD13 receptors are highly overexpressed in HT-1080 cells but not in HT-29 cells. The binding affinity of 64Cu-DOTA−NGR2 to HT-1080 cells was measured to be within the low nanomolar range and about twofold higher than that of 64Cu-DOTA−NGR1 (1.27 ± 0.25 nM and 0.62 ± 0.29 nM, respectively). Subsequent in vivo studies confirmed that 64Cu-DOTA−NGR2 showed higher tumor uptake and better tumor retention than 64Cu-DOTA−NGR1, presumably due to the bivalency effect and increase in apparent molecular size. 64Cu-DOTA−NGR2 is a suitable PET probe for noninvasive detection of CD13 receptor expression in vivo.
10. Conclusions
Molecular imaging advances not only through the continuing improvement in imaging equipment and technology, but also through the design of powerful probes with optimal in vivo biodistribution and imaging characteristics. Synthetic access to cNGR-containing mono-, bi-, and multimodal imaging agents for visualization of CD13 expression in tumors and cardiovascular diseases was established.
Further preclinical research of the presented molecular imaging tracers includes early noninvasive evaluation of pro- and anti-angiogenic therapy in myocardial infarction and solid tumors, respectively. Future clinical applications would be patient stratification and development of individualized therapy.
11. Materials and methods
11.1. Solvents and starting materials
Unless stated otherwise, all reagents and solvents were purchased from commercial sources and used without further purification.
11.2. Instrumentation
1H NMR and 13C NMR spectra were recorded at 298 K on a Varian Unity Inova 500 MHz spectrometer at 499.86 and 125.70 MHz, respectively. Chemical shifts are given in parts per million (ppm). Reversed phase high pressure liquid chromatography (RP HPLC) was performed on a Varian Pro Star HPLC system coupled to a UV–Vis detector probing at 214 nm using a VydacTM protein & peptide C18 column. Electrospray ionization mass spectrometry (ES-MS) was performed on a Perkin Elmer PE SCIEX Turbo Ionspray. To determine T1 and T2 relaxation times, a 2D mixed dual echo sequence was performed (voxel size 10 × 10 × 8 mm). The gadolinium content was determined by means of inductively coupled plasma atomic emission spectroscopy (ICP-AES) on a Leeman Labs Echelle spectrometer.
11.3. Synthesis
11.3.1. Ac-C(Acm)NGRC(Acm)GG-mpa-l-NH2 (1)
Manual solid phase peptide synthesis (SPPS) using the in situ neutralization/2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) activation procedure for Boc chemistry on an MBHA resin, as described earlier by Schnölzer et al.,36 was applied to synthesize a peptide containing the target-specific NGR sequence and a C-terminal thioester. ES-MS calcd for C39H67N14O14S3 ([M+H]+): 1050.4, found 1050.2.
11.3.2. C-DTPA (2)
The cysteine-functionalized DTPA synthon 2 was synthesized according to a literature procedure.40
11.3.3. Ac-C(Acm)NGRC(Acm)GGC-DTPA (3)
30.6 mg (0.029 mmol) of 1 and 1.5 equiv (24.7 mg, 0.043 mmol) of 2 were dissolved in 1 mL of 6 M Guanidine, 0.07 M Tris (aq). To this solution 20 μL (2 v-%) of thiophenol and 20 μL (2 v-%) of benzyl mercaptan were added. The pH was adjusted to pH 7.5 by the addition of small aliquots of 0.5 M NaOH (aq). The reaction was continued for 2 h at 37 °C. The reaction mixture was filtered and the product was purified by preparative RP HPLC over a C18 column (gradient: 0–22% MeCN in H2O, 0.1% TFA in 90 min). Freeze drying rendered 28.2 mg (0.020 mmol, 69.3%) of 3 as a fluffy white powder: ES-MS calcd for C51H86N18O22S3 ([M+H]+): 1399.5, found 1399.4.
11.3.4. Ac-C(Acm)NGRC(Acm)GGC(OG488)-DTPA (4)
25.8 mg (0.018 mmol) of 3 was dissolved in 1 mL 0.1 M Tris (aq, pH 6.92). The pH of the solution was adjusted to pH 6.5 by the addition of small aliquots of 0.5 M NaOH (aq) and subsequently added to 6.7 mg (0.014 mmol) of malOG488. The reaction mixture was shaken until all malOG488 dissolved and the reaction was continued for 2 h at room temperature. The reaction was monitored by reversed phase HPLC over a C18 column (gradient: 0–60% MeCN in H2O, 0.1% TFA in 30 min) and showed that the reaction went to completion. The reaction mixture was used for the next reaction step without intermediate purification. ES-MS calcd for C75H97F2N19O29S3 ([M+H]+): 1862.6, found 1862.8.
11.3.5. Ac-CNGRCGGC(OG488)-DTPA, 1–5 disulfide (5)
The reaction mixture was diluted ∼30 times by adding 27 mL of 0.1 M Tris (aq, pH 6.92) and 3 mL (i.e., 10 v-%) of acetic acid. Subsequently, 3.72 mL of a solution of 5 mM I2 in MeOH (0.018 mmol of I2) was added and the reaction was continued for 1 h at room temperature. The product was purified by preparative RP HPLC over a C18 column (gradient: 0–18% MeCN in H2O, 0.1% TFA in 5 min, then 18–36% MeCN in H2O, 0.1% TFA in 90 min). After freeze drying, 17.9 mg (0.010 mmol, 71.4%) of 5 was obtained as an orange-colored fluffy powder. ES-MS calcd for C69H85F2N17O27S3 ([M+H]+): 1718.5, found 1718.7.
11.3.6. Ac-CNGRCGGC(OG488)-Gd(III)DTPA, 1–5 disulfide (6)
17 mg (0.010 mmol) of 5 was dissolved in 10 mL of H2O. The pH was adjusted to pH 7 by adding small aliquots of 0.5% NH4OH (aq). To this solution was added 1 mL of a 10 mM solution of GdCl3 in H2O (1 equiv, 0.010 mmol). This was done in a stepwise manner and monitored by ES-MS to ensure full complexation, thus avoiding the addition of excess of GdCl3. Freeze drying rendered 6 as an orange powder. ES-MS calcd For C69H82F2GdN17O27S3 ([M+H]+): 1873.4, found 1873.7. ICP-AES gadolinium content: 94%.
11.3.7. C-Biotin (7)
C-Biotin was synthesized according to a literature procedure.40
11.3.8. Ac-C(Acm)NGRC(Acm)GGC-Biotin (8)
59.4 mg (0.057 mmol) of 1 and 1.1 equiv (24.6 mg, 0.066 mmol) of 7 were dissolved in 1 mL of 6 M Guanidine in 0.07 M Tris (aq). To this solution 20 μL (2 v-%) of thiophenol and 20 μL (2 v-%) of benzylmercaptan were added. The pH was adjusted to pH ∼7 by the addition of small aliquots of 0.5 M NaOH (aq). The reaction was continued for 2 h at 37 °C. The reaction mixture was filtered and the product was purified by preparative RP HPLC over a C18 column (gradient: 7–27% MeCN in H2O, 0.1% TFA in 90 min). Freeze drying rendered 57.6 mg (0.047 mmol, 83%) of 8 as a fluffy white powder: ESI-MS calcd for C45H76N18O14S4 ([M+H]+): 1221.5, found 1221.3.
11.3.9. malDTPA (9)
The synthesis of 9 will be published elsewhere.
11.3.10. Ac-C(Acm)NGRC(Acm)GGC(DTPA)-Biotin (10)
45.0 mg (0.0369 mmol) of 8 was dissolved in 1 mL 0.1 M Tris (aq, pH 6.9). This solution was added to 24.5 mg (0.0369 mmol) of 9. The pH of the solution was adjusted to pH 6.5 by the addition of small aliquots of 0.5 M NaOH (aq) and the reaction was continued for 2 h at room temperature. The reaction was monitored until completion by analytical RP HPLC over a C18 column (gradient: 0–67% MeCN in H2O, 0.1% TFA in 30 min). The reaction mixture was used for the next reaction step without intermediate purification. ESI-MS calcd for C74H113N23O27S4 ([M+H]+): 1884.7, found 1885.0.
11.3.11. Ac-CNGRCGGC(DTPA)-Biotin, 1–5 disulfide (11)
The reaction mixture was diluted ∼30 times by adding 40.5 mL of 0.1 M Tris (aq, pH 6.9) and 4.5 mL (i.e., 10 v-%) of acetic acid. Subsequently, 870 μL of a 0.075 M solution of I2 in MeOH (0.064 mmol of I2, 1.75 equiv) were added and the reaction was continued for 1 h at room temperature. The product was purified by preparative RP HPLC over a C18 column (gradient: 10–30% MeCN in H2O, 0.1% TFA in 90 min). Freeze drying rendered 17.2 mg (9.9 μmol, 27%) of 11 as a fluffy white powder. ESI-MS calcd For C68H101N21O25S4 ([M+H]+): 1740.6, found 1740.9.
11.3.12. Ac-CNGRCGGC(Gd(III)DTPA)-Biotin, 1–5 disulfide (12)
14.6 mg (8.4 μmol) of 11 was dissolved in 5 mL of H2O. The pH was adjusted to pH 7 by adding small aliquots of 0.5% NH4OH (aq). To this solution was added 0.5 mL of a 16.8 mM (8.4 μmol) GdCl3 solution in H2O. This was done in a stepwise manner and monitored by ESI-MS to ensure full complexation, avoiding the addition of excess GdCl3. Freeze drying rendered 12 in quantitative yield (>99%). ESI-MS calcd for C68H98GdN21O25S4 ([M−H]−): 1893.5, found 1893.8. ICP-AES gadolinium content: 81%.
Acknowledgments
This research was in part performed within the framework of CTMM, the Center for Translational Molecular Medicine (www.ctmm.nl), project EMINENCE (grant 01C-204).
(Part of) this work was financially supported by Cyttron II (LSH framework: FES0908).
References and notes
- 1.Brack S.S., Dinkelborg L.M., Neri D. Eur. J. Nucl. Med. Mol. Imaging. 2004;31:1327. doi: 10.1007/s00259-004-1648-0. [DOI] [PubMed] [Google Scholar]
- 2.Kolodgie F.D., Gold H.K., Burke A.P., Fowler D.R., Kruth H.S., Weber D.K., Farb A., Guerrero L.J., Hayase M., Kutys R., Narula J., Finn A.V., Virmani R.N. Engl. J. Med. 2003;349:2316. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 3.Fleiner M., Kummer M., Mirlacher M., Sauter G., Cathomas G., Krapf R., Biedermann B.C. Circulation. 2004;110:2843. doi: 10.1161/01.CIR.0000146787.16297.E8. [DOI] [PubMed] [Google Scholar]
- 4.Jeziorska M., Woolley D.E. J. Pathol. 1999;188:189. doi: 10.1002/(SICI)1096-9896(199906)188:2<189::AID-PATH336>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 5.Moreno P.R., Purushothaman K.R., Fuster V., Echeverri D., Truszczynska H., Sharma S.K., Badimon J.J., O’Connor W.N. Circulation. 2004;110:2032. doi: 10.1161/01.CIR.0000143233.87854.23. [DOI] [PubMed] [Google Scholar]
- 6.Hellings W.E., Peeters W., Moll F.L., Piers S.R., van Setten J., Van der Spek P.J., de Vries J.P., Seldenrijk K.A., De Bruin P.C., Vink A., Velema E., de Kleijn D.P., Pasterkamp G. Circulation. 1941;2010:121. doi: 10.1161/CIRCULATIONAHA.109.887497. [DOI] [PubMed] [Google Scholar]
- 7.Rademakers T., Douma K., Hackeng T.M., Post M.J., Sluimer J.C., Daemen M.J., Biessen E.A., Heeneman S., van Zandvoort M.A. Arterioscler. Thromb. Vasc. Biol. 2013;33:249. doi: 10.1161/ATVBAHA.112.300087. [DOI] [PubMed] [Google Scholar]
- 8.Higuchi T., Wester H.J., Schwaiger M. Eur. J. Nucl. Med. Mol. Imaging. 2007;34:S9. doi: 10.1007/s00259-007-0436-z. [DOI] [PubMed] [Google Scholar]
- 9.Seré K.M., Hackeng T.M. Semin. Vasc. Med. 2003;3:3. doi: 10.1055/s-2003-38328. [DOI] [PubMed] [Google Scholar]
- 10.de Boer O.J., van der Wal A.C., Teeling P., Becker A.E. Cardiovasc. Res. 1999;41:443. doi: 10.1016/s0008-6363(98)00255-7. [DOI] [PubMed] [Google Scholar]
- 11.Kumamoto M., Nakashima Y., Sueishi K. Hum. Pathol. 1995;26:450. doi: 10.1016/0046-8177(95)90148-5. [DOI] [PubMed] [Google Scholar]
- 12.Friedlander M., Brooks P.C., Shaffer R.W., Kincaid C.M., Varner J.A., Cheresh D.A. Science. 1995;270:1500. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- 13.Hammes H.P., Brownlee M., Jonczyk A., Sutter A., Preissner K.T. Nat. Med. 1996;2:529. doi: 10.1038/nm0596-529. [DOI] [PubMed] [Google Scholar]
- 14.Korpelainen E.I., Alitalo K. Curr. Opin. Cell Biol. 1998;10:159. doi: 10.1016/s0955-0674(98)80137-3. [DOI] [PubMed] [Google Scholar]
- 15.Lappi D.A. Semin. Cancer Biol. 1995;6:279. doi: 10.1006/scbi.1995.0036. [DOI] [PubMed] [Google Scholar]
- 16.Hass T.L., Davis S.J., Madri J.A. J. Biol. Chem. 1998;273:3604. doi: 10.1074/jbc.273.6.3604. [DOI] [PubMed] [Google Scholar]
- 17.Vu T.H., Shipley J.M., Bergers G., Berger J.E., Helms J.A., Hanahan D., Shapiro S.D., Senior R.M., Werb Z. Cell. 1998;93:411. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhagwat S.V., Lahdenranta J., Giordano R., Arap W., Pasqualini R., Shapiro L.H. Blood. 2001;97:652. doi: 10.1182/blood.v97.3.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corti A., Curnis F., Arap W., Pasqualini R. Blood. 2008;112:2628. doi: 10.1182/blood-2008-04-150862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koivunen E., Gay D.A., Ruoslahti E. J. Biol. Chem. 1993;268:20205. [PubMed] [Google Scholar]
- 21.Koivunen E., Wang B., Ruoslahti E. J. Cell. Biol. 1994;124:373. doi: 10.1083/jcb.124.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arap W., Pasqualini R., Ruoslahti E. Science. 1998;279:377. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 23.Pasqualini R., Koivunen E., Ruoslahti E. Nat. Biotechnol. 1997;15:542. doi: 10.1038/nbt0697-542. [DOI] [PubMed] [Google Scholar]
- 24.Koivunen E., Wang B., Ruoslahti E. Bio/Technology. 1995;13:265. doi: 10.1038/nbt0395-265. [DOI] [PubMed] [Google Scholar]
- 25.Healy J.M., Murayama O., Maeda T., Yoshino K., Sekiguchi K., Kikuchi M. Biochemistry. 1995;34:3948. doi: 10.1021/bi00012a012. [DOI] [PubMed] [Google Scholar]
- 26.Pasqualini R., Koivunen E., Kain R., Lahdenranta J., Sakamoto M., Stryhn A., Ashmun R.A., Shapiro L.H., Arap W., Ruoslahti E. Cancer Res. 2000;60:722. [PMC free article] [PubMed] [Google Scholar]
- 27.Buehler A., van Zandvoort M.A., Stelt B.J., Hackeng T.M., Schrans-Stassen B.H., Bennaghmouch A., Hofstra L., Cleutjens J.P., Duijvestijn A., Smeets M.B., de Kleijn D.P., Post M.J., de Muinck E.D. Arterioscler. Thromb. Vasc. Biol. 2006;26:2681. doi: 10.1161/01.ATV.0000245807.65714.0b. [DOI] [PubMed] [Google Scholar]
- 28.Sjöström H., Norén O., Olsen J. Adv Exp Med Biol. 2002;477:25. doi: 10.1007/0-306-46826-3_2. [DOI] [PubMed] [Google Scholar]
- 29.Bauvois B., Dauzonne D. Med. Res. Rev. 2006;26:88. doi: 10.1002/med.20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mina-Osorio P. Trends Mol. Med. 2008;14:361. doi: 10.1016/j.molmed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rangel R., Sun Y., Guzman-Rojas L., Ozawa M.G., Sun J., Giordano R.J., Van Pelt C.S., Tinkey P.T., Behringer R.R., Sidman R.L., Arap W., Pasqualini R. Natl. Acad. Sci. U.S.A. 2007;104:4588. doi: 10.1073/pnas.0611653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curnis F., Sacchi A., Gasparri A., Longhi R., Bachi A., Doglioni C., Bordignon C., Traversari C., Rizzardi G.P., Corti A. Cancer Res. 2008;68:7073. doi: 10.1158/0008-5472.CAN-08-1272. [DOI] [PubMed] [Google Scholar]
- 33.Corti A., Curnis F. J. Cell Sci. 2011;124:515. doi: 10.1242/jcs.077172. [DOI] [PubMed] [Google Scholar]
- 34.Frank A.O., Otto E., Mas-Moruno C., Schiller H.B., Marinelli L., Cosconati S., Bochen A., Vossmeyer D., Zahn G., Stragies R., Novellino E., Kessler H. Angew. Chem., Int. Ed. 2010;49:9278. doi: 10.1002/anie.201004363. [DOI] [PubMed] [Google Scholar]
- 35.Caravan P., Ellison J.J., McMurry T.J., Lauffer R.B. Chem. Rev. 1999;99:2293. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 36.Schnölzer M., Alewood P., Jones A., Alewood D., Kent S.B.H. Int. J. Pept. Protein Res. 1992;40:180. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 37.Dirksen A., Langereis S., de Waal B.F., van Genderen M.H., Meijer E.W., de Lussanet Q.G., Hackeng T.M. Org. Lett. 2004;6:4857. doi: 10.1021/ol048084u. [DOI] [PubMed] [Google Scholar]
- 38.Dawson P.E., Muir T.W., Clark-Lewis I., Kent S.B. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 39.Hackeng T.M., Griffin J.H., Dawson P.E. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10068. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tolbert J.T., Wong C.H. J. Am. Chem. Soc. 2000;122:5421. [Google Scholar]
- 41.Mammen M., Choi S.K., Whitesides G.M. Angew. Chem., Int. Ed. 1998;37:2754. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. For example: [DOI] [PubMed] [Google Scholar]
- 42.Jacques V., Desreux J.F. Top. Curr. Chem. 2002;221:123. [Google Scholar]
- 43.Dirksen A., Langereis S., de Waal B.F., van Genderen M.H., Hackeng T.M., Meijer E.W. Chem. Commun. (Camb). 2005;22:2811. doi: 10.1039/b502347e. [DOI] [PubMed] [Google Scholar]
- 44.Douma K., Prinzen L., Slaaf D.W., Reutelingsperger C.P., Biessen E.A., Hackeng T.M., Post M.J., van Zandvoort M.A. Small. 2009;5:544. doi: 10.1002/smll.200801079. [DOI] [PubMed] [Google Scholar]
- 45.Dirksen A., Meijer E.W., Adriaens W., Hackeng T.M. Chem. Commun. (Camb). 2006;15:1667. doi: 10.1039/b600286b. [DOI] [PubMed] [Google Scholar]
- 46.Oostendorp M., Douma K., Wagenaar A., Slenter J.M., Hackeng T.M., van Zandvoort M.A., Post M.J., Backes W.H. Circulation. 2010;121:775. doi: 10.1161/CIRCULATIONAHA.109.889451. [DOI] [PubMed] [Google Scholar]
- 47.Lutgens E., Daemen M.J., de Muinck E.D., Debets J., Leenders P., Smits J.F. Cardiovasc. Res. 1999;41:586. doi: 10.1016/s0008-6363(98)00216-8. [DOI] [PubMed] [Google Scholar]
- 48.Oostendorp M., Douma K., Hackeng T.M., Dirksen A., Post M.J., van Zandvoort M.A., Backes W.H. Cancer Res. 2008;68:7676. doi: 10.1158/0008-5472.CAN-08-0689. [DOI] [PubMed] [Google Scholar]
- 49.Oostendorp M., Douma K., Hackeng T.M., van Zandvoort M.A., Post M.J., Backes W.H. Contrast Media Mol. Imaging. 2010;5:9. doi: 10.1002/cmmi.361. [DOI] [PubMed] [Google Scholar]
- 50.Chen K., Ma W., Li G., Wang J., Yang W., Yap L.P., Hughes L.D., Park R., Conti P.S. Mol. Pharm. 2013;10:417. doi: 10.1021/mp3005676. [DOI] [PubMed] [Google Scholar]