

Graphical abstract
A comprehensive overview on methods applied for syntheses of β-amino-α-trifluoromethyl alcohols, including stereocontrolled variants, is presented. In addition, reported cases of the exploration of β-amino-α-trifluoromethyl alcohols for the preparation of trifluoromethylated peptidomimetics and other biologically active, fluorinated compounds are discussed. Attractive opportunities for their applications as organocatalysts are also presented.
Keywords: Organic synthesis, Amino alcohols, Fluorinated compounds, Trifluoromethyl alcohols, Stereocontrolled processes, Biological activity
Abstract
A comprehensive overview on methods applied for syntheses of β-amino-α-trifluoromethyl alcohols, including stereocontrolled variants, is presented. In addition, reported cases of the exploration of β-amino-α-trifluoromethyl alcohols for the preparation of trifluoromethylated peptidomimetics and other biologically active, fluorinated compounds are discussed. Attractive opportunities for their applications as organocatalysts are also presented.
1. Introduction
Amino alcohols form a group of difunctional compounds with great importance as building blocks in organic synthesis and their diverse applications as pharmaceuticals and catalysts are widely known [1]. Of special interest are β-amino alcohols [1f], and many of them display significant biological activity (e.g. ephedrine, quinidine, terbutaline, etc.). On the other hand, some of them are known as efficient catalysts for stereocontrolled syntheses (e.g. 3-aminoborneols, phenylalaninols, prolinols, etc.) [1a–e,h] and as building blocks for the preparation of optically active heterocycles [1g].
In the recent three decades, a rapid development of fluorinated organic compounds was observed [2a], and special attention is focused on the preparation and practical exploration of trifluoromethylated systems. The incorporation of the CF3 group results in relevant modification of physico-chemical and biological properties of organic compounds [2b–f]. The current development of methods for enantioselective trifluoromethylations opens an attractive access to enantiopure compounds, which are of particular interest for further applications [3].
It seems obvious that β-amino-α-trifluoromethyl alcohols, which combine the important structural features of the compounds mentioned above, are especially attractive as unique building blocks, catalysts, and potentially, as pharmaceuticals. In spite of the fact that there are numerous papers related to the chemistry of β-amino-α-trifluoromethyl alcohols, their chemistry has never been summarized in the form of a review. Therefore, the aim of the present paper is the presentation of reported methods, which were developed for their preparation as well as their most relevant applications in the field of modern organic synthesis.
2. Synthetic approaches to β-amino-α-trifluoromethyl alcohols
2.1. Reduction of cyanohydrines derived from trifluoromethylketones and fluoral
The conversion of hexafluoroacetone (1a) into its cyanohydrine 2a was achieved by treatment of 1a with potassium or sodium cyanide in THF and subsequent acidification of the solution containing the obtained salt [4] (Scheme 1 ).
Scheme 1.

The same method was used for the formation of corresponding cyanohydrines starting with other trifluoromethyl ketones [4], [5] or fluoral [6]. In recent papers, analogous conversions were reported to occur efficiently by using trimethylsilyl cyanide (Me3SiCN, TMS-CN) instead of the poisonous alkalimetal cyanides [7].
The reduction of the cyano group in cyanohydrines of type 2 offers a convenient route to the desired β-amino-α-trifluoromethyl alcohols. In the case of 2a, hydrogenolysis over Raney-Ni under high-pressure led to β-amino alcohol 3a in 75% yield [4e] (Scheme 1).
Cyanohydrine 2b, derived from α,α,α-trifluoroacetophenone (1b) by treatment with TMS-CN, was converted to 3b by reduction with LiAlH4 in ether [7a] (Scheme 2 ). This two-step protocol was successfully applied for the conversion of trifluoromethyl ketones of type 1c into the corresponding racemic 1-amino-2-(trifluoromethyl)propan-2-ol 3c [7d] (Scheme 2). The partially deuterated derivatives (e.g. R = CH2D, CHD2) were converted into various amides, which were subsequently tested as glucocorticoid agonists.
Scheme 2.

Cyanosilylation of the α,β-unsaturated trifluoromethyl ketone 4 (R = Et) gave the desired 1,2-adduct 5 in high yield, in the presence of catalytic amounts of Et3N, whereas under acidic conditions the 1,4-adduct was obtained [7b,c] (Scheme 3 ). Attempted diastereoselective cyanosilylations of ketones of type 4 bearing an enantiopure residue R (e.g. (−)-menthyl) resulted in poor diastereoselectivity [7c]. The silylated cyanohydrine 5 (R = Et), upon treatment with LiAlH4 in ether, yielded the β-amino alcohol 6 (R = Et).
Scheme 3.

2.2. Henry reaction and subsequent reduction of β-nitro alcohols
The base-catalyzed aldol-type addition of α-CH nitroalkanes with carbonyl compounds is known as Henry reaction [8]. Depending on reaction conditions and the substitution pattern, the primary β-nitro alcohol can be isolated or transferred into the α,β-unsaturated nitro compound. The initially formed β-nitro alcohols are convenient precursors for the preparation of β-amino alcohols.
As a result of the electron-withdrawing properties of the trifluoromethyl group, trifluoromethylketones and fluoral are suitable reaction partners for nitroalkanes. In a pioneering work with fluorinated aldehydes, fluoral hydrate (7) was reacted with nitromethane in the presence of K2CO3 to give the trifluoromethyl β-nitro alcohol 8 in fair yield [9] (Scheme 4 ). The subsequent reduction with Raney-Ni led to the β-amino alcohol 9a. In contrast to chlorinated analogues, no reductive replacement of F-atoms by H-atoms was observed. More complex nitroalkanes were used in the reaction with 7 under the same conditions leading, after reduction, to β-amino alcohols of type 9 bearing amino acid side chain residues [10].
Scheme 4.

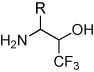
In the described reactions, fluoral hydrate can be replaced by its ethyl hemiacetal 10 [11]. In the case of 2-methyl-1-nitropropane, a mixture of four stereoisomeric nitroalcohols (threo + erythro-11) was formed [12]. After fractional crystallization from pentane, the racemic threo-11 was isolated and reduced with Raney-Ni/H2 in isopropanol. Finally, the resolution of the racemic threo-9b was achieved by crystallization with (−)-tartaric acid in ethanol (Scheme 5 ).
Scheme 5.
The free trifluoroacetone underwent smoothly the reaction with excess nitromethane in the presence of catalytic amounts of Et2NH or K2CO3 yielding 1-nitro-2-trifluoromethylpropan-2-ol [13]. The latter was converted into the corresponding β-amino alcohol upon reduction with Raney-Ni/H2 at 5 bar [14].
A series of substituted α,α,α-trifluoroacetophenones 12 was used in the Henry reaction with nitromethane (cat. K2CO3), and the in situ generated β-nitro alcohols 13 were reduced with H2 over Pd/charcoal under high-pressure to afford the desired β-amino alcohols 14 in good yields [15] (Scheme 6 ).
Scheme 6.

In a recent paper, a chiral version of the Henry reaction was reported, in which α,α,α-trifluoroacetophenones and aliphatic trifluoromethyl ketones reacted with nitromethane in the presence of a chiral lanthanide(III)–‘(S)-binolam’ complex to give optically active products with ee-values of 67–98% [16]. Selected enantiomerically enriched β-nitro alcohols (80–98% ee) were reduced with NiCl2/NaBH4 in methanol. The obtained β-amino alcohols showed ee-values of 72–97%. In the case of the optically active form of 3b (94.5% ee), single crystals suitable for the X-ray crystal-structure determination were isolated and the absolute (S)-(+)-configuration was established for this enantiomer.
2.3. Reduction of α-aminoalkyl trifluoromethyl ketones
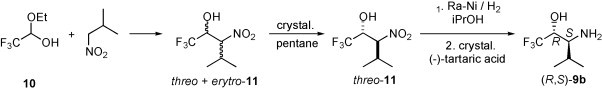
A general method for the preparation of α-amino alcohols is the reduction of the corresponding α-amino ketones [17]. Therefore, the reduction of α-aminoalkyl trifluoromethyl ketones is a convenient approach to β-amino-α-trifluoromethyl alcohols. A general method for the synthesis of the trifluoromethyl ketones is the Dakin–West reaction [18], in which an α-amino acid derivative, by treatment with trifluoroacetic acid anhydride (TFAA), is converted in a multi-step reaction into the desired α-aminoalkyl trifluoromethyl ketone. The alkyl residue is determined by the amino acid used. For example, a series of N-benzyl-N-(methoxycarbonyl)-α-amino acids 15a, was transformed to the trifluoromethyl ketones 16a [19] (Scheme 7 ).
Scheme 7.
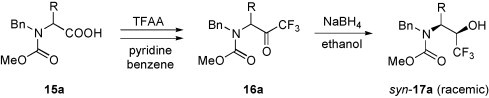
Further conversion of 16a into the amino alcohol syn-17a occurred diastereoselectively by the reduction with NaBH4 in ethanol. In some instances, however, the reduction was accompanied by the formation of the corresponding 1,3-oxazolidin-2-one. The same conversion was achieved by treatment of syn-17a with NaH in THF.
A modified version of the presented transformation started with an N-benzoyl α-amino acid 15b, which in the presence of acetic acid anhydride (Ac2O) underwent cyclization to give the 2,4-disubstituted 1,3-oxazol-5(4H)-one 18. The latter compound was trifluoroacetylated at C(4) and subsequent treatment of the resulting 19 with oxalic acid resulted in the formation of N-benzoyl-α-aminoalkyl trifluoromethyl ketone 16b, which was finally reduced to give the β-N-benzoylamino alcohol 17b [20] (Scheme 8 ). Based on the 19F NMR spectra, mixtures of two diasteroisomers of 17b were obtained. Without separation, these mixtures were hydrolyzed with HCl in water/ethanol to give the desired amino alcohols as hydrochlorides 20.
Scheme 8.

An efficient method for the preparation of β-amino-α-trifluoromethyl alcohols is the nucleophilic trifluoromethylation of α-amino aldehydes with (trifluoromethyl)trimethyl silane (TMS-CF3, see Section 2.5). The addition occurred diastereoselectively in favour of the formation of the anti-isomer. In order to obtain the syn-isomer, a two-step procedure was developed [21]. For example, the N,N-dibenzyl derivative anti-21a with R = Me was oxidized under Swern conditions to give the corresponding ketone 22, which then was diastereoselectively converted to syn-21a (Scheme 9 ).
Scheme 9.

In a recent report, the reduction of ethyl 2-dibenzylamino-4,4,4-trifluoro-3-oxobutanoate with KBH4 was described to yield the corresponding 2-amino-3-hydroxybutanoate in a diastereoselective manner [22].
Another convenient approach to α-aminoalkyl trifluoromethyl ketones was developed via the regioselective nucleophilic ring opening of 2-ethoxy-2-trifluoromethyloxiranes with secondary amines [23] (Scheme 10 ). In the studied series of aminoketones 22b, the reduction with NaBH4 occurred with very high stereoselectivity in favour of the syn-isomers. However, replacement of the secondary amines by dimethylaluminium benzylamide (Me2AlNHBn) and subsequent reduction of the crude aminoketone at −78 °C resulted in the formation of anti-21c as the major product [24]. The authors claimed that in this case an aluminium complex is formed as the intermediate and it governs the inversed stereochemical course of the reaction. On the one hand, the reaction of racemic epoxide 23 with enantiopure dimethylaluminium (R)-α-methylbenzylamide led to 1:1 mixtures of the diastereoisomeric products [24]. The analogous reaction carried out with enantiomerically enriched oxirane 23 (R = Ph) afforded optically active N-benzylated amino alcohols of type 21, which were smoothly debenzylated (H2, Pd(OH)2/C) yielding the optically active 3-amino-1,1,1-trifluoro-3-phenylpropan-2-ol [24].
Scheme 10.

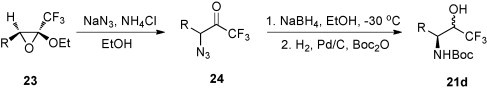
In another study, the ring opening of trifluoromethyloxiranes 23 was performed with sodium azide in ethanol in the presence of ammonium chloride [25] (Scheme 11 ). Treatment of the crude product with NaBH4 and subsequent hydrogenolysis of the azido group, followed by Boc-protection, led to a mixture of diastereoisomers of 21d in favour of the syn-isomer. The reversed sequence of the reduction steps, i.e., reduction of the azido group and Boc-protection, followed by reduction of the keto function with LiAlH4, or NaBH4 yielded a mixture of 21d, in which the anti-isomer was predominant [25].
Scheme 11.
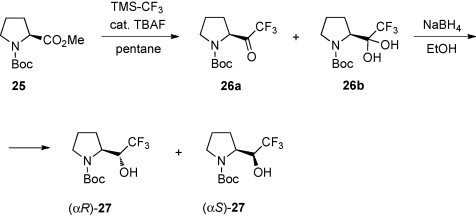
Amino alcohols derived from proline, i.e., prolinols, are of particular interest [26]. However, only little is known about fluorinated representatives. In a recent paper, an elegant approach to trifluoromethyl derivatives was reported [27]. In this case, the trifluoromethyl ketone 26a, required for the next step, was obtained from N-Boc-proline methyl ester (25) via substitution of MeO by CF3 in the reaction with (trifluoromethyl)trimethylsilane (CF3SiMe3, TMS-CF3) [28] (Scheme 12 ). The mixture of ketone 26a and its hydrate 26b was reduced with NaBH4 in ethanol at room temperature to give an 85:15 mixture of two diastereoisomers 27. Carrying out the reduction at 0 °C, the diastereoselectivity increased to 96:4.
Scheme 12.
Another approach to proline-based trifluoromethyl ketones, which potentially can be used for the preparation of the corresponding amino alcohols, is the reaction of N-protected proline with TFAA in pyridine in the presence of 4-(dimethylamino)pyridine (DMAP, ‘Steglich Base’), and subsequent hydrolysis with 10% HCl solution [29].
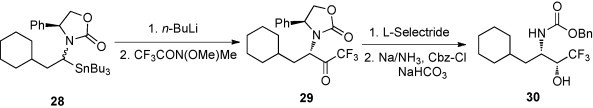
The diastereoisomeric mixture of the organostannane 28 was transmetallated with n-BuLi leading to the lithium compound, which in solution existed predominantly as the ‘trans’ isomer [30] (Scheme 13 ). Treatment of this intermediate with the Weinreb-amide of trifluoroacetic acid led to the protected α-aminoalkyl trifluoromethyl ketone 29 diastereoselectively with retention of the configuration. Subsequent diastereoselective reduction of the keto group with l-selectride, followed by reductive deprotection and in situ carboxybenzylation of the amino group, gave the 2-protected β-amino-α-trifluoromethyl alcohol 30 with >95% de.
Scheme 13.
2.4. Ring opening of trifluoromethyloxiranes with N-nucleophiles
It is well established that oxiranes easily undergo ring-opening reactions with primary and secondary amines [31]. The reactions of trifluoromethyloxirane (31) with ammonia or diethylamine yield the trifluoromethyl amino alcohols 9 and 3-diethylamino-1,1,1-trifluoropropan-2-ol, respectively [32]. The ring cleavage occurred regioselectively. Secondary amines of type 32 smoothly react with commercially available trifluoromethyloxirane (31) yielding the desired amino alcohols 33 [33]. The ring opening of the oxirane proceeds in the presence of catalytic amounts of ytterbium(III) triflate in acetonitrile with complete regioselectivity (Scheme 14 ). The biologically most active examples are also prepared in the enantiomerically pure (R)-(+) form.
Scheme 14.

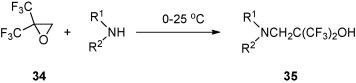
It was also reported that 31 and ammonium hydroxide at ambient temperature undergo a threefold reaction to give tris(3,3,3-trifluoro-2-hydroxypropyl)amine [34]. Under similar conditions, the reaction of 2,2-bis(trifluoromethyl)oxirane (34) and ammonia or ammonium hydroxide yield β-amino-α,α-bis(trifluoromethyl)ethanol as the major product [35]. As a minor product, the secondary amine with two 2-hydroxy-3,3,3,3′,3′,3′-hexafluoroisopropyl residues is formed (ratio 5:1). The latter is the only product when the reaction is carried out at 60 °C. The reactions of 34 with dimethylamine, aniline and 4-fluoroaniline, respectively, occurred already at ambient temperature in the absence of any solvent, and lead to the corresponding N-substituted β-amino-α,α-bis(trifluoromethyl)ethanols 35 in 66–83% yield [35] (Scheme 15 ).
Scheme 15.
In situ generated 34 (2 mol.-equiv.) reacts at room temperature with 1 mol.-equiv. of (R,R)-N,N′-dimethylcyclohexane-1,2-diamine (36) yielding a mixture of the mono-substituted product 37 (major) and the disubstituted 38 (minor) one. Separation of both amino alcohols was achieved by fractional sublimation followed by recrystallization. Restricted rotation (fluxionality) in 38, resulting from the intramolecular O–H⋯N hydrogen bonding, was observed in both 1H and 19F NMR spectra at room temperature [36] (Scheme 16 ).
Scheme 16.

The optically active oxirane (S)-31 is an attractive building block for the preparation of chiral organofluorine compounds. As an example, initial methylation of enantiomerically enriched (S)-31 (75% ee) by treatment with n-BuLi and methyl iodide followed by aminolysis of the in situ generated (S)-39 with (S)-α-methylbenzylamine led to a 9:1 mixture of diastereoisomers with (S,S)-40 as the major product [37] (Scheme 17 ). An analogous procedure was reported as an excellent method for the preparation of a series of optically active (>90% ee) amino alcohols derived from piperidine, starting with (S)-39 (optical purity >99% ee) [38].
Scheme 17.

Similarly, the in situ obtained (S)-39 was trapped with benzophenone and the modified oxirane was cleaved with (R)-α-methylbenzylamine [39]. The expected structure of the major diastereoisomer was confirmed by X-ray crystallography.
The oxirane (S)-31 with high ee-value (96%) was prepared from 1,1,1-trifluoro-3-bromopropanone in a two-step procedure via enantioselective borane-mediated reduction with β-chlorodiisopinocampheylborane (DIP-Cl) [40]. The reactions with potassium bis(trimethylsilyl)amide or diethylamine afforded the optically active amino alcohol (S)-9 (95% ee) and (S)-3-(diethylamino)-1,1,1-trifluoropropan-2-ol (96% ee) [40]. An alternative approach to (S)-9 was the ring opening of (S)-31 with sodium azide in ethanol and subsequent reduction with hydrogen over Pd/C. Also in this case, both reaction steps occurred with high stereoselectivity.
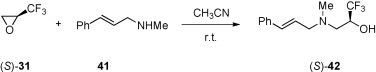
The enantiomerically pure oxirane (S)-31 reacts easily with N-methylallylamine (41) in acetonitrile solution without a catalyst to give stereoselectively the ‘allylic’ amino alcohol (S)-42 in 90% yield (Scheme 18 ) [41].
Scheme 18.
The presence of the p-tolylsulfinyl methyl auxiliary attached to C(2) of trifluoromethyloxirane governs efficiently the regio- and stereoselective opening of the three-membered ring. The oxiranes are available by treatment of the corresponding trifluoromethylketones with diazomethane at room temperature [42]. Depending on the solvent, the ratio of oxirane and the isomeric methyl vinyl ether varied from 1:1 (in benzene) to 63:1 (in ethanol) and, in all cases, the (S,R)-diastereoisomeric oxirane was formed in high excess (>97:3).
The reactions of oxirane (S,R)-43 with benzyl- or dibenzylamine were carried out at room temperature in THF and the corresponding amino alcohols (S,R)-44 were obtained in high yields [42] (Scheme 19 ). Upon treatment with typical agents, the sulfinyl auxiliary was removed and diverse derivatives of the amino trifluoromethyl alcohols 45–47 were obtained.
Scheme 19.

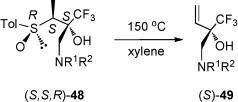
Analogous conversions with higher substituted sulfinylmethyl residues (CHMe, CHPr, CHPh instead of CH2) were reported by the same research group [43] and reviewed in a recent survey [44]. In addition to conversions presented in Scheme 19, (S,S,R)-48 yielded, after heating in boiling xylene, the optically active vinyl-substituted amino alcohol (S)-49 in 82% yield [43] (Scheme 20 ).
Scheme 20.
2.5. Nucleophilic trifluoromethylation of α-amino aldehydes and ketones
The nucleophilic addition of (trifluoromethyl)trimethylsilane (TMS-CF3, Ruppert–Prakash reagent [45]) to a carbonyl group followed by desilylation of the initial adduct is a powerful and clean method for the synthesis of α-trifluoromethyl alcohols. In a typical procedure, anhydrous ethers are used as solvents, and the fluoride ion is used as a catalyst. However, recent studies showed that the reaction can be performed without fluoride in DMF/K2CO3 [46a] or in DMSO only [46b].
The reaction of TMS-CF3 with α-amino aldehydes offers an attractive approach to β-amino-α-trifluoromethyl alcohols. For example, the N-protected α-amino aldehydes 51, which were prepared by reduction of the Weinreb-amides 50, were treated with TMS-CF3 in the presence of tetrabutylammonium fluoride (TBAF) and, after acidic hydrolysis, amino alcohols 52 were obtained in 40–50% yield [47] (Scheme 21 ). The same reaction sequence was applied in the case of Boc-protected l-phenylalanine [11a]. The obtained trifluoromethylated amino alcohol was identified as a mixture of the two diastereoisomers.
Scheme 21.

A two-step procedure for the synthesis of another series of N-protected α-amino aldehydes is outlined in Scheme 22 [48]. The N-protected α-amino acid ester 53 was reduced to the alcohol and the latter was selectively oxidized to 54. The addition of TMS-CF3 was reported to occur diastereoselectively and the amino alcohol 55 was finally obtained, after reduction/deprotection, as a 4.5:1 mixture of the diastereoisomers in favour of 55.
Scheme 22.

A series of enantiomerically pure N,N-dibenzyl-α-amino aldehydes 56 (R = alkyl or aryl) was used for the reaction with TMS-CF3 to examine the influence of the substituent R on the stereochemical outcome of the reaction [21] (Scheme 23 ). Whereas in the case of alkyl groups the ratio of anti/syn-21a was between 84:16 and 62:38, the phenylglycinal derivative 56 (R = Ph) gave a 46:54 mixture of the anti/syn-isomers.
Scheme 23.

The pure anti-21a was modified by the replacement of the N-benzyl groups by Me2, Et2, –(CH2)4– and –(CH2)5–, respectively, in order to obtain a series of compounds which were tested as catalysts of enantioselective Reformatzky reactions [49]. Starting with l-isoleucine, the N,N-dimethylisoleucine methyl ester was prepared in a two-step procedure. Subsequent reduction and Swern oxidation, followed by treatment with TMS-CF3 and desilylation afforded the anti- and syn-isomers of type 21a in 37 and 7% yield, respectively.
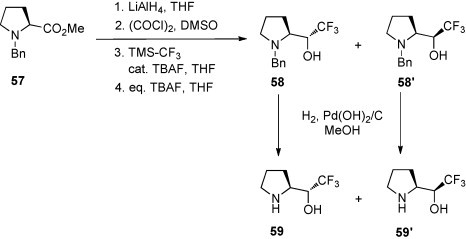
The analogous procedure starting with methyl N-benzyl l-prolinate (57) yielded a mixture of comparable amounts of the isomeric amino alcohols 58/58′, which, after separation, were debenzylated to give 59 and 59′ [49] (Scheme 24 ). Finally, these amino alcohols were converted into the N-methyl derivatives.
Scheme 24.
The N-Boc-protected prolinal reacts under typical conditions (TBAF, dry THF, 0 °C to room temp.) with TMS-CF3 yielding a mixture (ratio 1:1.2) of the corresponding diastereoisomeric amino alcohols 27 (Scheme 12), analogous to 58 and 58′, which by treatment with trifluoroacetic acid in CH2Cl2 were converted into 59 and 59′ [50].
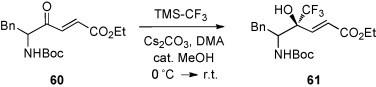
In contrast to α-amino aldehydes, reactions of TMS-CF3 with α-amino ketones were only scarcely studied. In a very recent report, compound 60, derived from 5-amino-4-oxohex-2-enoate, was described to react with TMS-CF3 in N,N-dimethyl acetamide (DMA) in the presence of alkali metal carbonates. Under these conditions the desilylation occurred spontaneously, and the desired trifluoromethylated product 61 was obtained in fair yield [51] (Scheme 25 ). It is worth mentioning that the reaction proceeded chemoselectively at the keto group. Starting with enantiomerically pure 60, a mixture of syn and anti-diastereoisomers was isolated, and the de values for 61 varied between 61 and 83% depending on the reaction conditions. The presence of a chiral additive (BINOL) did not effect the de value.
Scheme 25.
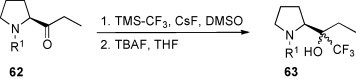
Ketones 62 (R1 = Bz or Boc), derived from proline, reacted with TMS-CF3 in DMSO in the presence of CsF at room temperature yielding, after desilylation with TBAF in THF, mixtures of diastereoisomers of the corresponding trifluoromethylated prolinol 63 [52] (Scheme 26 ). The ratios of the diastereoisomers were determined by 19F NMR spectroscopy to ca. 1:1.
Scheme 26.
2.6. Nucleophilic trifluoromethylation of α-iminocarbonyl derivatives
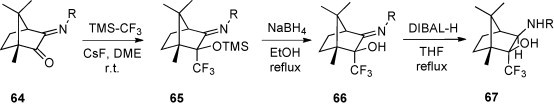
Reactions of TMS-CF3 with α-imino ketones offer a straightforward access to the corresponding trifluoromethylated amino alcohols. However, similarly to α-amino ketones, this class of substrates was explored to a limited extend, only. Monoimines 64, derived from camphorquinone, are easily available and stable under standard conditions. Nucleophilic trifluoromethylations of 64 occur smoothly at room temperature in dimethoxyethane (DME) in a chemo- and diastereoselective manner. The 19F NMR spectra of the crude product confirmed the formation of a single diastereoisomer 65 in all cases studied [53] (Scheme 27 ). Desilylation leading to the imino alcohols 66 was achieved by treatment with NaBH4 in boiling ethanol. Under these conditions, the C N bond was not reduced and only the reduction with DIBAL-H led to the exo,exo-isomer of the α-trifluoromethylated amino alcohols 67 in a diastereoselective manner. The studies were carried out with monoimines 64 bearing an alkyl or aryl residue at the N-atom.
Scheme 27.
Imino ketones of type 68, which are quite labile compounds obtained from 2-aryl-2-oxoethanol hydrates (arylglyoxal hydrates) and primary amines, reacted easily with TMS-CF3 with complete chemoselectivity to afford the O-silylated α-trifluoromethyl alcohols 69 in high yields [54] (Scheme 28 ). Typically, the reaction was performed in DME/CsF, but comparable yields were obtained in DMF/K2CO3 or in DMSO without any additive. In contrast to adducts 65, treatment of 69 with NaBH4 in ethanol at room temperature led directly to the amino alcohol 70.
Scheme 28.

Compounds 71, i.e. α-imino lactones, contain the structure fragment of an α-imino carbonyl derivative. They reacted with TMS-CF3 in THF at 0 °C in the presence of an activator, and the corresponding products 72 were obtained exclusively [55] (Scheme 29 ). The latter were transformed to amino alcohols 73 by treatment with LiBH4 in THF. Subsequent hydrogenolysis, Boc-protection, and acidic deprotection led to the hydrochlorides of the anti-diastereoisomers 74 in a diastereoselective manner. In an alternative approach, imino lactones 71 were stereoselectively hydrogenated and N-benzylated to give the corresponding cis-disubstituted α-amino lactones 75. Trifluoromethylation followed by treatment with NaBH4 in methanol/water gave the diastereoisomers 76, which finally were converted to syn-74.
Scheme 29.

The enatiomerically pure β-amino alcohol 77 derived from (+)-pulegon is a useful auxiliary for a highly diastereoselective multi-step procedure applied for the preparation of α-hydroxy aldehydes [56]. Condensation of 77 with glyoxal derivatives (R = alkyl, aryl) led to the intermediate products 78, which, upon treatment with TMS-CF3 and TBAF, were converted into trifluoromethyl alcohols 79 with diastereoselectivities between 11:1 and >50:1 [57] (Scheme 30 ).
Scheme 30.
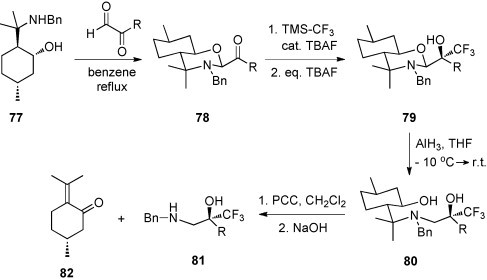
Selective reduction with AlH3 in THF resulted in the cleavage of the C,O bond of the aminoacetal to give 80. Finally, removal of the auxiliary by treatment with pyridinium chlorochromate (PCC) followed by NaOH afforded the N-benzylamino alcohols 81 in excellent yields [57].
2.7. Petasis reaction of 2-hydroxy-3,3,3-trifluoropropanal
An attractive multi-step approach to diverse β-amino-α-trifluoromethyl alcohols offers the Petasis reaction [58] with 2-hydroxy-3,3,3-trifluoropropanal (83), boronic acids and secondary amines [59] (Scheme 31 ). The key compound 83 was synthesised via nucleophilic addition of TMS-CF3 to cinnamic aldehyde, subsequent ozonolysis and typical workup with dimethyl sulfide. Remarkably, the Petasis reaction leading to 84 occurred with high diastereoselectivity.
Scheme 31.

In the extension of the study, enantiomerically enriched (R)- and (S)-83 were prepared and used for the reaction with dibenzylamine and 2-bromo-2-phenylvinyl boronic acid. The best result with almost complete diastereoselectivity was obtained in a 1:4 mixture of methanol and dichloromethane without chromatographic purification of the intermediate hydroxyaldehyde 83, leading to the product 84 (R1 = R2 = Bn, R3 = Ph(Br)C CH2) with ee-values up to 92%.
2.8. Miscellaneous reactions
Some less general methods for the preparation of β-amino-α-trifluoromethyl alcohols from CF3-containing starting materials are also known. In one case, ethyl trifluoroacetyl acetate (85) was converted into the O-protected 4,4,4-trifluoro-3-hydroxybutanoic acid 86 [60] (Scheme 32 ). This intermediate was reacted with diphenyl phosphorazidate (DPPA) in the presence of Et3N and benzyl alcohol yielding the protected amino alcohol 87.
Scheme 32.

The presented protocol was used for the conversion of 2-alkyl-4,4,4-trifluoro-3-hydroxybutanoates, which are accessible via LDA-mediated aldol addition of a carboxylic acid with fluoral.
In the second case, the conversion of the unsaturated ether 88 to the amino alcohol 91 was achieved via initial Sharpless dihydroxylation to give 89, transformation to the azide 90, reduction of the azido group, and in situ Boc-protection of the amino group [61] (Scheme 33 ). Compound 91 was further transformed into the amino alcohol 92 and finally to the trifluoromethylated α-amino acid 93.
Scheme 33.

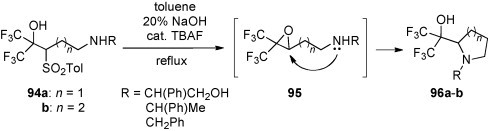
Heating of bis(trifluoromethyl)carbinols 94a–b in a two-phase system of toluene and 20% aqueous NaOH, in the presence of tetrabutylammonium fluoride (TBAF) as the catalyst, was reported to yield pyrrolidines 96a (n = 1) and piperidines 96b (n = 2) in satisfactory to good yields (50–90%) [62] (Scheme 34 ). The reaction course was explained via initial formation of oxiranes 95 and subsequent cyclization by intramolecular nucleophilic substitution. The analogous protocol applied for the attempted synthesis of the corresponding seven-membered azepine derived amino alcohol (n = 3) failed. In an additional experiment, the appearance of the intermediate oxirane derivative 95 was convincingly evidenced. Thus, the reaction follows a two-step mechanism via oxirane formation/oxirane ring-opening sequence.
Scheme 34.
Enantiomerically enriched (R)-(−)-3-amino-1,1,1-trifluoro-2-phenylpropan-2-ol (3b) was prepared via a 3-step synthesis starting with (R)-(−)-2-hydroxy-3,3,3-trifluoro-2-phenylpropanoic acid (97) (Scheme 35 ), and its optical purity was proved by NMR spectroscopy with a chiral europium complex [63].
Scheme 35.

The optically active diol (S)-100 can be prepared from 3,3,3-trifluoropropene by osmium-catalyzed asymmetric dihydroxylation in the presence of phthalazine and pyrimidine ligands [64]. Subsequent reaction of (S)-100 with sulfuryl chloride in the presence of imidazole leads to 1,3,2-dioxathiolane 101, which upon treatment with benzylamine and in situ performed acidic hydrolysis provides enantioselectively amino alcohol (S)-102 in 75% yield (Scheme 36 ). The same method was applied to produce the corresponding CCl3-analogue.
Scheme 36.

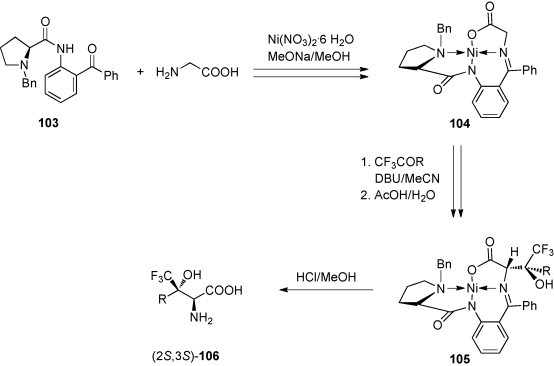
A highly diastereoselective asymmetric aldol reaction was elaborated for the synthesis of trifluoromethylated threonine and analogous amino acids [65]. The monochiral Schiff base–Ni(II) complex 104, which is conveniently accessible from (S)-ortho-[N-(N-benzylprolyl)amino]benzophenone (103) and glycine, reacts with trifluoromethylketones in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to yield the aldol adduct 105 with high to excellent diastereoselectivity (de 95 to >98%). Hydrolysis leads to the amino acids (2S,3S)-106 and the chiral auxiliary 103 in very good chemical yields (Scheme 37 ).
Scheme 37.
3. Applications of β-amino-α-trifluoromethyl alcohols in organic synthesis
3.1. Transformations of β-amino-α-trifluoromethyl alcohols into trifluoromethylated carboxylic acids and heterocycles
Diverse transformations of amino alcohols described in Section 2 are known and they lead to either linear or cyclic products. Thus, in the case of the protected amino alcohol 91, dehydroxylation by the Barton-McCombie method and debenzylation afforded amino alcohol 92, which, after oxidation with Jones reagent, gave the enantiomerically pure α-amino acid derivative 93 [61] (Scheme 33).
Mesylation of 61 to give 107 and subsequent treatment with dimethylzinc or methyl magnesium chloride in the presence of copper(I) or copper(II) salts led to the α-methyl-β,γ-unsaturated esters (Z)- and (E)-108 [51] (Scheme 38 ). The major product (Z)-108 was formed with lower diastereoselectivity (20–86% de) then the minor (E)-configured product (88–99% de). Analogous alkylations were carried out with other zinc-organic compounds.
Scheme 38.

Compound 109, i.e. ethyl 2-dibenzylamino-4,4,4-trifluoro-3-hydroxybutanoate, after tosylation and base-induced elimination was converted into (Z)-but-2-enoate 110 [22] (Scheme 39 ). Debenzylation of 109 followed by twofold benzoylation and elimination according to Scheme 39 afforded the corresponding N-benzoyl derivative of type 110, which was used for the cyclopropanation with diazomethane. On the other hand, the attempted cyclopropanation with dimethyl sulfoxonium methylide unexpectedly gave a 4,5-dihydro-4-(2,2,2-trifluoroethyl)-1,3-oxazole-4-carboxylate [22].
Scheme 39.

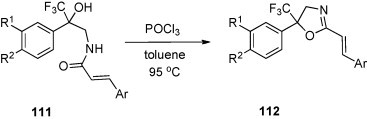
A series of 4,5-dihydro-1,3-oxazoles of type 112 was obtained from amino alcohols 14 (Scheme 6) via cinnamoylation to 111 and subsequent treatment with POCl3 in toluene at 95 °C [15] (Scheme 40 ).
Scheme 40.
Numerous conversions of α-trifluoromethylated β-amino alcohols into the corresponding 1,3-oxazolidin-2-ones 113 were reported. They were performed via carbonylation with phosgene [24], [66], triphosgene [21], and carbonyldiimidazole [51], respectively, or via base-induced 1,5-cyclization of the corresponding N-Boc-protected derivatives [19], [25], [55] (Scheme 41 ). Irrespective of the applied method, the anti-amino alcohol led to the cis-4,5-disubstituted product and the syn-isomer to the trans-disubstituted oxazolidinone.
Scheme 41.

The carbonylation of the trifluoromethylated amino alcohol 67a with the bornane skeleton by treatment with phosgene, leading to the oxazolidinone 114, proved the exo orientation of both the hydroxyl and amino groups [53] (Scheme 42 ).
Scheme 42.

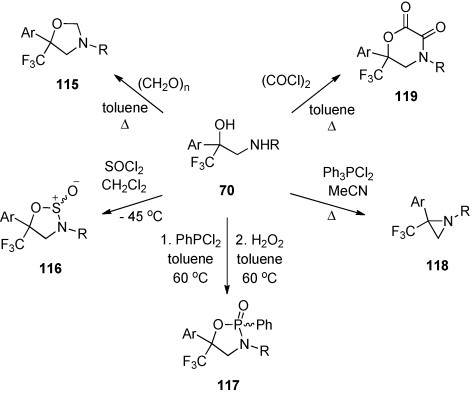
Cyclization of amino alcohols 70 (Scheme 28) with formaldehyde, thionylchloride or dichlorophenyl phosphane led to the formation of trifluoromethylated derivatives of 1,3-oxazolidine 115, 1,2,3-oxathiazolidine 116, and 1,3,2-oxazaphospholidine 117, respectively [66] (Scheme 43 ). On the other hand, treatment of 70 with Ph3PCl2 in boiling acetonitrile in the presence of triethylamine gave the corresponding aziridines 118. Finally, the reaction of 70 with oxalyl chloride afforded the 1,4-oxazidine-2,3-dione 119.
Scheme 43.
The unsaturated amino alcohols 6a were hydrolyzed and the in situ formed γ-amino-β-hydroxyalkyl trifluoromethyl carbonyl compounds 120 underwent spontaneously cyclization and dehydration leading to 3-trifluoromethyl pyrrols 121 [7e] (Scheme 44 ).
Scheme 44.

3.2. Preparation of peptidomimetics and other biologically active compounds
It is a well-established fact that the trifluoromethyl group can significantly enhance the biological activity of organic compounds and, therefore, trifluoromethylated products are of high interest in life sciences [67]. In general, the trifluoromethyl group is recognized as an important pharmacophore displaying a key role in modulation of biological activity of the studied compounds, e.g. in binding ability and agonist activity of a glucocorticoid receptor ligand [68]. With regard to the electrostatic properties, the trifluoromethyl group is evaluated as an equivalent of the phenyl group [69]. Therefore, trifluoromethylated β-amino alcohols can be considered as compounds, which structurally mimic α-hydroxy-β-phenylethylamine, well known as β-agonist and/or β-antagonist.
For these reasons, β-amino-α-trifluoromethyl alcohols are very attractive building blocks for the synthesis of biologically active compounds. To the best of our knowledge, there are only few examples known, e.g. 3-amino-1,1,1-trifluoro-2-phenylpropan-2-ol, which showed biological activity without chemical modification [7a]. Another example offers a series of N-[3-(1,1,2,2-tetrafluoroethoxy)benzyl]-N-(3-phenoxyphenyl)trifluoro-3-aminopropan-2-ols of type 33 (Scheme 14), which potently and reversibly inhibit cholesteryl ester transfer protein (CETP) [33]. In most cases, the amino alcohols were converted into amides and eventually oxidized to the trifluoromethylated ketones.
A large series of carboxamides and a sulfonamide of type 122 and 123, respectively, derived from amino alcohols 3c (Scheme 2), were tested as non-steroidal glucocorticoid agonists [7d]. In some cases, aryl analogues of type 124 were also investigated.
A mixture of the diastereoisomeric amino alcohols 59 and 59′ (Scheme 24) was treated with a sulfonyl chloride derived from isatin, and the diastereoisomeric sulfonamides 125 obtained thereby were evaluated for their caspase-inhibition potency. Unfortunately, the results obtained in this case were not encouraging [50].

The unsaturated protected amino alcohol 126 derived from 6 (Scheme 3) was hydrolyzed to the corresponding aldehyde, which then was oxidized and deprotected yielding finally 3-trifluoromethyl-4-amino-3-hydroxybutanoic acid (127) as a racemate [7c] (Scheme 45 ).
Scheme 45.

Both, amino alcohols 6 and carboxylic acid 127, were resolved to give the pure enantiomers. The carboxylic acids 127 are recognized as β-hydroxy-β-trifluoromethyl derivatives of γ-aminobutyric acid (GABA).
The syn/anti-amino alcohols 21d (R = PhCH2CH2) (Scheme 11) were separated and, after deprotection, converted into mixtures of diastereoisomeric dipeptide-like products 128 [25]. Chromatographic separation led to all four diastereoisomers in pure from, which were tested as inhibitors of human leucocyte elastase.

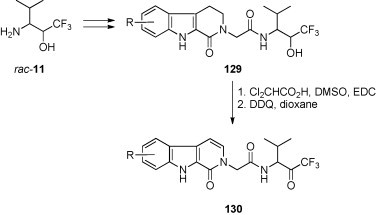
Trifluoromethyl ketones of type 130 containing the β-carbolinone skeleton were tested as potential non-peptidic inhibitors of human leucocyte elastase [70]. The key step of their synthesis was the oxidation of the secondary alcohol fragment in 129 originating from the starting trifluoromethylated amino alcohol (Scheme 46 ). This oxidation was performed using a modified Pfitzner-Moffatt protocol. Finally, dehydrogenation with dichlorodicyanobenzoquinone (DDQ) led to the target molecules.
Scheme 46.
An analogous strategy with oxidation of a α-trifluoromethyl alcohol fragment leading to the trifluoroacetyl group was described in numerous reports on preparations of trifluoroacetyl peptidomimetics. They were tested as renin inhibitors [60], inhibitors of human leucocyte elastase [11], [12], metallo-β-lactamase inhibitors [47b], serine protease inhibitors [10], inhibitors of the human cytomegalovirus protease [11b], human plasma kallikrein [48], and SARS-CoV 3CL protease [11d].
In another paper, peptidyl trifluoromethyl ketones were investigated as inhibitors of porcine pancreatic elastase, human neutrophil elastase, and rat and human neutrophil cathepsin G [20]. The oxidiations of the α-trifluoromethyl alcohol residue to the trifluoroacetyl function were carried out by using different protocols, such as Dess–Martin periodinane oxidation, Swern and Pfitzner-Moffatt reactions as well as potassium permanganate oxidation under basic aqueous conditions. The incorporation of the α-trifluoromethyl-β-amino alcohol into the peptidomimetics was also performed by using solid-phase methodology [11b,c].
3.3. Application in stereocontrolled synthesis
The enantiomerically pure β-amino-α-trifluoromethyl alcohols of type (S)-131 turned out to be more efficient ligands for the enantioselective addition of diethylzinc to benzaldehyde than the non-fluorinated analogue [38], [71]. The ee-values increased up to >90% (ca. 45% with the non-fluorinated analogue) with increasing concentration of the ligand.

By treatment with bis(iodomethyl)zinc (ICH2)2Zn in dichloromethane solution (–5 °C, 1 h) (Simmons–Smith reaction), the ‘allylic’ amino alcohol (S)-42b (Scheme 18) easily undergoes cyclopropanation to give 132 in 86% yield and with very high diastereoselectivity (>99%). Acceleration of the reaction rate results from the electron-withdrawing effect of the trifluoromethylated amino alcohol auxiliary [41].
A series of differently substituted α-trifluoromethyl-β-amino alcohols 133–134 was applied as chiral ligands in enantioselective additions of a Reformatzky reagent to benzaldehyde and in diethylzinc additions to N-(diphenylphosphinoyl)imine of benzaldehyde [49].

In the first reaction, the ee-values varied between 39 and 86%, and the best result (chemical yield 89%) was obtained with 133a (R1 = R2 = Et). The reactions with the two diastereoisomers of 134 led to enantiomeric products with comparable yields and ee-values. In the diethylzinc addition to the imine bond, the results were even better, e.g. with 133c (R = t-Bu), ee-values of 95% were obtained.
Complexes of amino alcohol 3a (Scheme 1) with Cu2+, Ni2+, and Co2+ were obtained and their stoichiometry was determined as 2:1 [4e].
The chiral fluorinated diamino-diol 38 (Scheme 16) reacts smoothly as a proligand with Zr(CH2Ph)4 and Ti(OiPr)4 forming the corresponding (dialkoxy)zirconium and (dialkoxy)titanium complexes. The cationic zirconium complex was tested as a new, efficient catalyst for the regioselective polymerization of 1-hexene [36].
4. Conclusions
The results summerized in the present review evidence that β-amino-α-trifloromethyl alcohols form an important subclass of vicinal amino alcohols. The presence of the trifluomethyl group enables their exploration in the synthesis of more complex molecules with special properties. On the other hand, both, the amino and the hydroxy group can easily be transformed to other functional groups. Of special importance is the fact that many of the trifluoromethylated alcohols can be prepared in enantio- and diastereomerically pure from. For that reason, they are promising substances for stereocontrolled reactions. However, their exploration in this part of organic synthesis is limited to a few cases only. Similarly, syntheses of new heterocycles via selective reactions of the OH or NH2 group are almost unknown.
The incorporation of β-amino-α-trifluoromethyl alcohols into the backbone of peptidomimetics, and eventually further oxidation of the secondary alcohol function to the trifluoroacetyl group, results in a remarkable increase of the biological activities of the obtained products.
The presented review supplements the survey on the synthesis and biological activity of structurally similar β-amino-β-fluoroalkyl alcohols published in 1999 [72].
Acknowledgments
The authors thank the Polish Ministry for Science and Higher Education for a grant (Grant # N N204 130335) (G.M. and E.O.), and financial support by F. Hoffmann-La Roche AG, Basel, is gratefully acknowledged (H.H.).
Contributor Information
Grzegorz Mlostoń, Email: gmloston@uni.lodz.pl.
Heinz Heimgartner, Email: heimgart@oci.uzh.ch.
References
- 1.(a) Kukharev B.F., Stankevich V.K., Klimenko G.R. Russ. Chem. Rev. (Engl. Transl.) 1995;64:523–540. [Google Scholar]; (b) Ager D.J., Prakash I., Schaad D.R. Chem. Rev. 1996;96:835–875. doi: 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]; (c) Periasamy M. Pure Appl. Chem. 1996;68:663–666. [Google Scholar]; (d) Senenayake C.H. Aldrichim. Acta. 1998;31:3–15. [Google Scholar]; (e) Fache F., Schulz E., Tommasino M.L., Lemaire M. Chem. Rev. 2000;100:2159–2232. doi: 10.1021/cr9902897. [DOI] [PubMed] [Google Scholar]; (f) Bergmeier S.C. Tetrahedron. 2000;56:2561–2576. [Google Scholar]; (g) Anakabe E., Badia D., Carillo L., Reyes E., Vicario J.L. In: Attanasi O.A., Spinelli D., editors. vol. 6. Italian Society of Chemistry; Rome, Italy: 2002. pp. 270–311. (Targets in Heterocyclic Systems-Chemistry and Properties). [Google Scholar]; (h) Zaidlewicz M., Sokol W., Wolan A., Cytarska J., Tafelska-Kaczmarek A., Dzielendziak A., Prewysz-Kwinto A. Pure Appl. Chem. 2003;75:1349–1355. [Google Scholar]; (i) Lait S.M., Rankic D.A., Keay B.A. Chem. Rev. 2007;107:767–796. doi: 10.1021/cr050065q. [DOI] [PubMed] [Google Scholar]
- 2.(a) Z.-P. Liu (Guest Ed.) Curr. Org. Chem. 2010;14:888–999. [Google Scholar]; (b) Olah G.A., Chambers R.D., Prakash G.K.S., editors. Synthetic Fluorine Chemistry. J. Wiley & Sons; New York: 1992. [Google Scholar]; (c) Chambers R.D. Blackwell; Oxford: 2004. Fluorine in Organic Chemistry. [Google Scholar]; (d) Kirsch P. Wiley-VCH; Weinheim: 2004. Modern Fluoroorganic Chemistry. [Google Scholar]; (e) Bégué J.-P., Bonnet-Delpon D. J. Wiley & Sons; Hoboken, NJ: 2008. Bioorganic and Medicinal Chemistry of Fluorine. [Google Scholar]; (f) S.E. Lopez (Guest Ed.), Curr. Org. Synth., 7, in press.
- 3.(a) Kawai H., Kusuda A., Nakamura S., Shiro M., Shibata N. Angew. Chem. Int. Ed. 2009;48:6324–6327. doi: 10.1002/anie.200902457. [DOI] [PubMed] [Google Scholar]; (b) Ma J.-A., Cahard D. Chem. Rev. 2008;108:PR1–PR43. doi: 10.1021/cr800221v. [DOI] [PubMed] [Google Scholar]; (c) Shibata N., Mizuta S., Kawai H. Tetrahedron: Asymmetry. 2008;19:2633–2644. [Google Scholar]; (d) Billard T., Langlois B.R. Eur. J. Org. Chem. 2007:891–897. [Google Scholar]
- 4.(a) Darrall R.A., Smith F., Stacey M., Tatlow J.C. J. Chem. Soc. 1951:2329–2332. [Google Scholar]; (b) Mill T., Rodin J.O., Silverstein R.M., Woolf C. J. Org. Chem. 1964;29:3715–3716. [Google Scholar]; (c) Zeifman Y.V., Postovoi S.A., Aerov A.F., Mysov E.I. J. Fluorine Chem. 1993;65:189–194. [Google Scholar]; (d) Sutin L., Andersson S., Bergquist L., Castro V.M., Danielsson E., James S., Hendriksson M., Johansson L., Kaiser C., Flyren K., Williams M. Bioorg. Med. Chem. Lett. 2007;17:4837–4840. doi: 10.1016/j.bmcl.2007.06.054. [DOI] [PubMed] [Google Scholar]; (e) Chang I.-S., Wills C.J. Can. J. Chem. 1977;55:2465–2472. [Google Scholar]
- 5.(a) Burdon J., McLaughlin V.C.R., Tatlow J. J. Chem. Soc. 1960:3184–3188. [Google Scholar]; (b) Usui S., Tsuboya S., Umezawa Y., Hazama K., Okamura M. Bull. Chem. Soc. Jpn. 2009;82:254–260. [Google Scholar]
- 6.Kitazume T. J. Fluorine Chem. 1987;35:287–294. [Google Scholar]
- 7.(a) Choudhury-Mukherjee I., Schenck H.A., Cechova S., Pajewski T.N., Kapur J., Ellena J., Cafiso D.S., Brown M.L. J. Med. Chem. 2003;46:2494–2501. doi: 10.1021/jm020546r. [DOI] [PubMed] [Google Scholar]; (b) Kruchok I.S., Gerus I.I., Kukhar V.P. Tetrahedron. 2000;56:6533–6539. [Google Scholar]; (c) Shaitanova E.N., Gerus I.I., Belik M.Y., Kukhar V.P. Tetrahedron: Asymmetry. 2007;18:192–198. [Google Scholar]; (d) Clackers M., Coe D.M., Demaine D.A., Hardy G.W., Humphreys D., Inglis G.G.A., Jonston M.J., Jones H.D., House D., Loiseau R., Minick D.J., Skone P.A., Uings I., McLay I.M., Macdonald S.J.F. Bioorg. Med. Chem. Lett. 2007;17:4737–4745. doi: 10.1016/j.bmcl.2007.06.066. [DOI] [PubMed] [Google Scholar]; (e) Shaitanova E.N., Gerus I.I., Kukhar V.P. Tetrahedron Lett. 2008;49:1184–1187. [Google Scholar]
- 8.(a) Hass H.B., Riley E.F. Chem. Rev. 1943;32:373–430. [Google Scholar]; (b) Rosini G. Compr. Org. Synth. 1991;2:321–340. [Google Scholar]
- 9.Cook D.J., Pierce O.R., McBee E.T. J. Am. Chem. Soc. 1954;76:83–87. [Google Scholar]
- 10.Imperiali B., Abeles R.H. Tetrahedron Lett. 1986;27:135–138. [Google Scholar]
- 11.(a) Skiles J.W., Fuchs V., Miao C., Sorcek R., Grotzinger K.G., Mauldin S.C., Vitous J., Mui P.W., Jacober S., Chow G., Matteo M., Skoog M., Weldon S.M., Possanza G., Keirns J., Letts G., Rosenthal A.R. J. Med. Chem. 1992;35:641–662. doi: 10.1021/jm00082a005. [DOI] [PubMed] [Google Scholar]; (b) Ogilvie W., Bailey M., Poupart M.A., Abraham A., Bhavsar A., Bonneau P., Bordeleau J., Bousquet Y., Chabot C., Duceppe J.-S., Fazal G., Goulet S., Grand-Maître Ch., Guse I., Halmos T., Lavallee P., Leach M., Malenfant E., O’Meara J., Plante R., Plouffe C., Poirier M., Soucy F., Yoakim Ch., Déziel R. J. Med. Chem. 1997;40:4113–4135. doi: 10.1021/jm970104t. [DOI] [PubMed] [Google Scholar]; (c) Poupart M.-A., Fazal G., Goulet S., Mar L.T. J. Org. Chem. 1999;64:1356–1361. [Google Scholar]; (d) Shao Y.-M., Yang W.B., Kuo T.-H., Tsai K.-C., Lin C.-H., Yang A.-S., Liang P.-H., Wong C.-H. Bioorg. Med. Chem. 2008;16:4652–4660. doi: 10.1016/j.bmc.2008.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veale C.A., Bernstein P.R., Bohnert C.M., Brown F.J., Bryant C., Damewood J.R., Jr., Earley R., Feeney S.W., Edwards P.D., Gomes B., Hulsizer J.M., Kosmider B.J., Krell R.D., Moore G., Salcedo T.W., Shaw A., Silberstein D.S., Steelman G.B., Stein M., Strimpler A., Thomas R.M., Vacek E.P., Williams J.C., Wolanin D.J., Woolson S. J. Med. Chem. 1997;40:3173–3181. doi: 10.1021/jm970250z. [DOI] [PubMed] [Google Scholar]
- 13.(a) McBee E.T., Hathaway C.E., Roberts C.W. J. Am. Soc. 1956;78:4053–4057. [Google Scholar]; (b) Schechter H., Ley D.E., Roberson E.B., Jr. J. Am. Chem. Soc. 1956;78:4984–4991. [Google Scholar]
- 14.Gassen K.-R., Kirmse W. Chem. Ber. 1986;119:2233–2248. [Google Scholar]
- 15.Poszáwácz L., Simig G. J. Heterocycl. Chem. 2000;37:343–348. [Google Scholar]
- 16.Tur F., Saá J.M. Org. Lett. 2007;9:5079–5082. doi: 10.1021/ol702434t. [DOI] [PubMed] [Google Scholar]
- 17.Tramontani M. Synthesis. 1982:605–644. [Google Scholar]
- 18.Buchanan G.L. Chem. Soc. Rev. 1988;17:91–109. [Google Scholar]
- 19.Kawase M., Hirabayashi M., Kumakura H., Saito S., Yamamoto K. Chem. Pharm. Bull. 2000;48:114–119. doi: 10.1248/cpb.48.114. [DOI] [PubMed] [Google Scholar]
- 20.Peet N.P., Burkhart J.P., Angelastro M.R., Giroux E.L., Mehdi S., Bey P., Kolb M., Neises B., Schirlin D. J. Med. Chem. 1990;33:394–407. doi: 10.1021/jm00163a063. [DOI] [PubMed] [Google Scholar]
- 21.Andrés J.M., Pedrosa R., Pérez-Encabo A. Eur. J. Org. Chem. 2004:1558–1566. [Google Scholar]
- 22.Wan W., Gao Y., Jiang W., Hao J. J. Fluorine Chem. 2008;129:510–514. [Google Scholar]
- 23.Bégué J.P., Bonnet-Delpon D., Sdassi H. Tetrahedron Lett. 1992;33:1882–1892. [Google Scholar]
- 24.Abouabdellah A., Bégué J.-P., Bonnet-Delpon D., Kornilov A., Rodrigues I., Richard C. J. Org. Chem. 1998;63:6529–6534. [Google Scholar]
- 25.Bégué J.-P., Bonnet-Delpon D., Fischer-Durand N. Tetrahedron: Asymmetry. 1994;5:1099–1110. [Google Scholar]
- 26.(a) Enders D., Klatt M. Synthesis. 1996:1403–1418. [Google Scholar]; (b) Palomo C., Mielgo A. Angew. Chem. Int. Ed. 2006;45:7876–7880. doi: 10.1002/anie.200602943. [DOI] [PubMed] [Google Scholar]
- 27.Funabiki K., Shibata A., Iwata H., Hatano K., Kubota Y., Komura K., Ebihara M., Matusi M. J. Org. Chem. 2008;73:4694–4697. doi: 10.1021/jo8004952. [DOI] [PubMed] [Google Scholar]
- 28.Wiedemann J., Heiner T., Mlostoń G., Prakash G.K.S., Olah G.A. Angew. Chem. Int. Ed. 1998;37:820–821. doi: 10.1002/(SICI)1521-3773(19980403)37:6<820::AID-ANIE820>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 29.(a) Kawase M., Miyamae H., Narita M., Kurihara T. Tetrahedron Lett. 1993;34:859–862. [Google Scholar]; (b) Kawase M., Miyamae H., Kurihara T. Chem. Pharm. Bull. 1998;46:749–756. [Google Scholar]
- 30.Tomoyasu T., Tomooka K., Nakai T. Synlett. 1998:1147–1149. [Google Scholar]
- 31.(a) Yudin A.K., editor. Aziridines and Epoxides in Organic Synthesis. Wiley-VCH; Weinheim: 2006. [Google Scholar]; (b) Dake G. In: Katritzky A.R., Ramsden C.A., Scriven E.F.V., Taylor R.J.K., editors. vol. 1. Elsevier; 2008. Oxiranes and Oxirenes; pp. 174–217. (Comprehensive Heterocyclic Chemistry III). (Chapter 1.03) [Google Scholar]
- 32.McBee E.T., Hathaway C.E., Roberts C.W. J. Am. Chem. Soc. 1956;78:3851–3854. [Google Scholar]
- 33.Reinhard E.J., Wang J.L., Durley R.C., Fobian Y.M., Grapperhaus M.L., Hickory B.S., Massa M.A., Norton M.B., Promo M.A., Tollefson M.B., Vernier W.F., Connolly D.T., Witherbee B.J., Melton M.A., Regina K.J., Smith M.E., Sikorski J.A. J. Med. Chem. 2003;46:2152–2168. doi: 10.1021/jm020528+. [DOI] [PubMed] [Google Scholar]
- 34.Voronkov M.G., Baryshok V.P., Tandura S.N., Vitkovskii V.Y., D’yakov V.M., Pestunovich V.A. Zh. Obsch. Khim. 1978;48:2238–2245. (J. Gen. Chem. USSR, 48, 1978, 2033) [Google Scholar]
- 35.Petrov V.A. Synthesis. 2002:2225–2231. [Google Scholar]
- 36.Kirillov E., Lavanant L., Thomas Ch., Roisnel T., Chi Y., Carpentier J.-F. Chem. Eur. J. 2007;13:923–935. doi: 10.1002/chem.200600895. [DOI] [PubMed] [Google Scholar]
- 37.Yamauchi Y., Kawate T., Katagiri T., Uneyama K. Tetrahedron. 2003;59:9839–9847. [Google Scholar]
- 38.Harada A., Fujiwara Y., Katagiri T. Tetrahedron: Asymmetry. 2008;19:1210–1214. [Google Scholar]
- 39.Yamauchi Y., Katagiri T., Uneyama K. Org. Lett. 2002;4:173–176. doi: 10.1021/ol016829f. [DOI] [PubMed] [Google Scholar]
- 40.Ramachandran P.V., Gong B., Brown H.C. J. Org. Chem. 1995;60:41–46. [Google Scholar]
- 41.Katagiri T., Iguchi N., Kawate T., Takahashi S., Uneyama K. Tetrahedron: Asymmetry. 2006;17:1157–1160. [Google Scholar]
- 42.Bravo P., Farina A., Frigerio M., Meille S.V., Viani F. Tetrahedron: Asymmetry. 1994;5:987–1004. [Google Scholar]
- 43.Arnone A., Bravo P., Frigerio M., Viani F., Soloshonok V.A. Tetrahedron. 1998;54:11841–11860. [Google Scholar]
- 44.Sorochinsky A.E., Soloshonok V.A. J. Fluorine Chem. 2010;131:127–139. [Google Scholar]
- 45.(a) Prakash G.K.S., Yudin A.K. Chem. Rev. 1997;97:757–786. doi: 10.1021/cr9408991. [DOI] [PubMed] [Google Scholar]; (b) Prakash G.K.S., Mandal M. J. Fluorine Chem. 2001;112:123–131. [Google Scholar]; (c) Olah G.A., Prakash G.K.S., Wang Q., Li X.-Y. In: Paquette L.A., editor. vol. 7. J. Wiley & Sons; New York: 1995. pp. 5165–5166. (Encyclopedia of Reagents in Organic Synthesis). [Google Scholar]; (d) Bastos R.S. Synlett. 2008:1425–1426. [Google Scholar]
- 46.(a) Prakash G.K.S., Panja C., Yaghoo H., Surampudi V., Kultyshev R., Mandal M., Rasul G., Mathew T., Olah G.A. J. Org. Chem. 2006;71:112–114. doi: 10.1021/jo060835d. [DOI] [PubMed] [Google Scholar]; (b) Iwanami K., Oriyama T. Synlett. 2006:112–114. [Google Scholar]
- 47.(a) Walter M.W., Felici A., Galleni M., Soto R.P., Adlington R.M., Baldwin J.E., Frère J.-M., Gololobov M., Schofield C.J. Bioorg. Med. Chem. Lett. 1996;6:2455–2458. [Google Scholar]; (b) Walter M.W., Adlington R.M., Baldwin J.E., Schofield C.J. Tetrahedron. 1997;53:7275–7290. [Google Scholar]
- 48.Garrett G.S., McPhail S.J., Tornheim K., Correa P.E., McIver J.M. Bioorg. Med. Chem. Lett. 1999;9:301–306. doi: 10.1016/s0960-894x(98)00562-9. [DOI] [PubMed] [Google Scholar]
- 49.Xu X.-H., Qiu X.-L., Qing F.-L. Tetrahedron. 2008;64:7353–7361. [Google Scholar]
- 50.Podichetty A.K., Wagner S., Faust A., Schäfers M., Schober O., Kopka K., Haufe G. Future Med. Chem. 2009;1:969–989. doi: 10.4155/fmc.09.66. [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi K., Narumi T., Oishi S., Ohno H., Fujii N. J. Org. Chem. 2009;74:4626–4629. doi: 10.1021/jo9005602. [DOI] [PubMed] [Google Scholar]
- 52.Mlostoń G., Gębicki K., Kołodziej B. XXII Conference on Advances in Organic Synthesis; Karpacz, Poland; 2009. Book of Abstracts, P07. [Google Scholar]
- 53.Obijalska E., Mlostoń G., Linden A., Heimgartner H. Tetrahedron: Asymmetry. 2008;19:1676–1683. [Google Scholar]
- 54.G. Mlostoń, E. Obijalska, A. Tafelska-Kaczmarek, M. Zaidlewicz, J. Fluorine Chem., submitted for publication.
- 55.Fustero S., Albert L., Aceña J.L., Sanz-Cervera J.F., Asensio A. Org. Lett. 2008;10:605–608. doi: 10.1021/ol702947n. [DOI] [PubMed] [Google Scholar]
- 56.(a) He X.-C., Eliel E.L. Tetrahedron. 1987;43:4979–4987. [Google Scholar]; (b) Eliel E.L., He X.-C. J. Org. Chem. 1990;55:2114–2119. [Google Scholar]
- 57.Pedrosa R., Sayalero S., Vicente M., Maestro A. J. Org. Chem. 2006;71:2177–2180. doi: 10.1021/jo052458v. [DOI] [PubMed] [Google Scholar]
- 58.(a) Petasis N.A., Akritopoulou I. Tetrahedron Lett. 1993;34:583–586. [Google Scholar]; (b) Petasis N.A., Zavialov I.A. J. Am. Chem. Soc. 1997;119:445–446. [Google Scholar]; (c) Petasis N.A., Zavialov I.A. J. Am. Chem. 1998;120:11798–11799. [Google Scholar]
- 59.Prakash G.K.S., Mandal M., Schweizer S., Petasis N.A., Olah G.A. Org. Lett. 2000;2:3173–3176. doi: 10.1021/ol000195f. [DOI] [PubMed] [Google Scholar]
- 60.Patel D.V., Rielly-Gauvin K., Ryono D.E. Tetrahedron Lett. 1988;29:4665–4668. [Google Scholar]
- 61.Jiang Z.-X., Qin Y.-Y., Qing F.L. J. Org. Chem. 2003;68:7544–7547. doi: 10.1021/jo0344384. [DOI] [PubMed] [Google Scholar]
- 62.Taguchi T., Suda Y., Hamochi M., Fujino Y., Itaka Y. Chem. Lett. 1991:1425–1428. [Google Scholar]
- 63.Mioskowski C., Solladié G. Tetrahedron. 1973;29:3669–3674. (cf. data given in [16]) [Google Scholar]
- 64.Vanhessche K.P.M., Sharpless K.B. Chem. Eur. J. 1997;4:517–522. [Google Scholar]
- 65.(a) Soloshonok V.A., Avilov D.V., Kukhar V.P. Tetrahedron: Asymmetry. 1996;7:1547–1550. [Google Scholar]; (b) Soloshonok V.A., Kukhar V.P., Galushko S.V., Svistunova N.Y., Avilov D.V., Kuz’mina N.A., Raevski N.I., Struchkov Y.T., Pysarevski A.P., Belkova Y.N. J. Chem. Soc., Perkin Trans. 1993;1:3143–3155. [Google Scholar]
- 66.E. Obijalska, G. Mlostoń, H. Heimgartner, Helv. Chim. Acta, submitted for publication.
- 67.Maienfisch P., Hall R.G. Chimia. 2004;58:93–99. [Google Scholar]
- 68.Betageri R., Zhang Y., Zindell R.M., Kuzmich D., Kirrane T.M., Bentzien J., Cardozo M., Capolino A.J., Fadra T.N., Nelson R.M., Paw Z., Shih D.-T., Shih Ch.-K., Zuvela-Jelaska L., Nabozny G., Thomson D.S. Bioorg. Med. Chem. Lett. 2005;15:4761–4769. doi: 10.1016/j.bmcl.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 69.T. Katagari, K. Uneyama, Functional Group Transformations at α-Carbon to Trifluoromethyl Group, in Fluorine Containing Synthons, in: V.A., Soloshonok (Ed.), ACS Symposium Series, 2005, vol. 911, Chapter 18, pp. 318–341.
- 70.Veale C.A., Damewood J.R., Jr., Steelman G.B., Bryant C., Gomes B., Williams J. J. Med. Chem. 1995;38:86–97. doi: 10.1021/jm00001a014. [DOI] [PubMed] [Google Scholar]
- 71.Katagiri T., Fujiwara Y., Takahashi S., Ozaki N., Uneyama K. Chem. Commun. 2002:986–987. doi: 10.1039/b201238c. [DOI] [PubMed] [Google Scholar]
- 72.Bravo P., Crucianelli M., Ono T., Zanda M. J. Fluorine Chem. 1999;97:27–49. [Google Scholar]