

Abstract
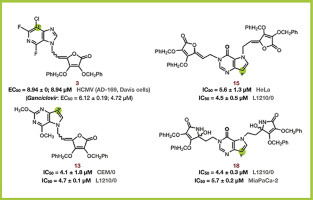
Keeping the potential synergy of biological activity of synthetic anomalous derivatives of deazapurines and l-ascorbic acid (l-AA) in mind, we have synthesized new 3-, 7- and 9-deazapurine derivatives of l-ascorbic (1–4, 8–10, 13–15) and imino-l-ascorbic acid (5–7, 11, 12, 16–19). These novel compounds were evaluated for their cytostatic and antiviral activity in vitro against a panel of human malignant tumour cell lines and normal murine fibroblasts (3T3). Among all evaluated compounds, the 9-deazapurine derivative of l-AA (13) exerted the most potent inhibitory activity on the growth of CEM/0 cells (IC50 = 4.1 ± 1.8 μM) and strong antiproliferative effect against L1210/0 (IC50 = 4.7 ± 0.1 μM) while the 9-deazahypoxanthine derivative of l-AA (15) showed the best effect against HeLa cells (IC50 = 5.6 ± 1.3 μM) and prominent effect on L1210/0 (IC50 = 4.5 ± 0.5 μM). Furthermore, the 9-deazapurine derivative disubstituted with two imino-l-AA moieties (18) showed the best activity against L1210/0 tumour cells (IC50 = 4.4 ± 0.3 μM) and the most pronounced antiproliferative effects against MiaPaCa-2 cells (IC50 = 5.7 ± 0.2 μM). All these compounds showed selective cytostatic effect on tumour cell lines in comparison with embryonal murine fibroblasts (3T3). When evaluating their antiviral activity, the 3-deazapurine derivative of l-AA (3) exhibited the highest activity against both laboratory-adapted strains of human cytomegalovirus (HCMV) (AD-169 and Davis) with EC50 values comparable to those of the well-known anti-HCMV drug ganciclovir and without cytotoxic effects on normal human embryonal lung (HEL) cells.
Keywords: 3-Deazapurines, 7-Deazapurines, 9-Deazapurines, l-Ascorbic acid, Imino-l-ascorbic acid, Cytostatic activity, Antiviral activity
Graphical abstract
Highlights
-
•
Deazapurine derivatives of l-ascorbic and imino-l-ascorbic acid were synthesized.
-
•
Compound 13 exerted strong activity on the growth of CEM/0 and L1210/0 cells.
-
•
Compound 15 showed the best cytostatic effect against HeLa cells.
-
•
Compound 18 showed the best cytostatic activity against L1210/0 and MiaPaCa-2.
-
•
Compound 3 exhibited highest effect against HCMV comparable to ganciclovir.
1. Introduction
Numerous nucleoside derivatives are being used in treatment of viral, tumoural, bacterial as well as other diseases. Purine nucleoside antiviral drugs carbovir, didanosine and its prodrug 2′,3′-dideoxyadenosine are currently used in treatment of human immunodeficiency virus (HIV) caused disease (AIDS), while acyclovir, ganciclovir [1], penciclovir [2] and (S)-9-(2,3-dihydroxypropyl)adenine ((S)-DHPA) [3] are used as nucleoside antiherpetic drugs. Moreover, 6-mercaptopurine and thioguanine have been used as antitumoural drugs, azathioprine as an immunosuppressive drug used in organ transplant recipients and cladiribine in treatment of systemic mastocytosis [1].
With the aim to synthesize better and selective substances, modifications in the sugar moiety or its analogue replacement and alterations in nucleoside bases were investigated synthetically and biologically. Thus, 3-deazaadenosine, a potent inhibitor of S-adenosylhomocysteine hydrolase and an attractive target for the design of antiviral drugs, has shown significant activity against HSV-1, HIV and oncogenic DNA viruses [4]. 3-Deazaadenine derivatives of cyclopentyl adenine drugs aristeromycin and neoplanocin A retained their strong activity against HSV-1, vaccinia virus and hepatitis C virus (HCV) with lower cytotoxicity [5], [6], [7], [8]. Also, 3-deazaguanosine has shown a wide spectrum of activity against DNA and RNA viruses as well as antitumoural activity [1]. C-2 and C-3 halogenated 3-deazapurine derivatives showed anti-HBV activity, a broad spectrum of antitumour properties [9] and strong ability of subtracting Mycobacterium tuberculosis adenosine kinase [4]. C-7 fluorinated and C-2′ methylated derivatives of the naturally isolated antibiotic tubercidin (7-deazaadenosine) have shown strong inhibition of NS5B, a nonstructural hepatitis C protein, while C-7 halogenated also exhibited substantial activity against human cytomegalovirus (HCMV) [10], [11], [12]. Acyclic derivatives of sangivamycin (7-deazapurine broad spectrum antibiotic) showed the same antiviral activity against herpes simplex virus 1 (HSV-1) and HCMV as the reference drug ganciclovir, without cytotoxic effects. In addition, compound BMK-Y101 and its prodrug are preclinical candidates for the treatment of hepatocellular carcinoma [12]. Furthermore, C-2 fluorinated, C-7 alkylated and 2′,3′-dideoxy derivatives of 9-deazaadenosine have shown a broad spectrum of antitumoural activities with lower cytotoxicity in relation to 9-deazaadenosine [13], [14].
Our research group has synthesized numerous nucleoside derivatives of l-ascorbic acid which have demonstrated a broad spectrum of antiproliferative activity, as well anti-HCMV, anti-VZV and anti-CMV activity at micromolar concentrations [15], [16], [17], [18], [19]. Alterations of the sugar moiety in natural or synthetic nucleosides with l-ascorbic acid succeeded from its antioxidant properties and antitumoural properties of its stearate and palmitate derivatives [20], [21], [22], [23]. These findings prompt us to synthesize new types of variously halogenated 3-deazapurine, 7-deazapurine and alkylated 9-deazapurine derivatives of l-ascorbic or imino-l-ascorbic acid (Fig. 1 ) with a primary aim to evaluate their antiviral potency and cytotoxic activity.
Fig. 1.
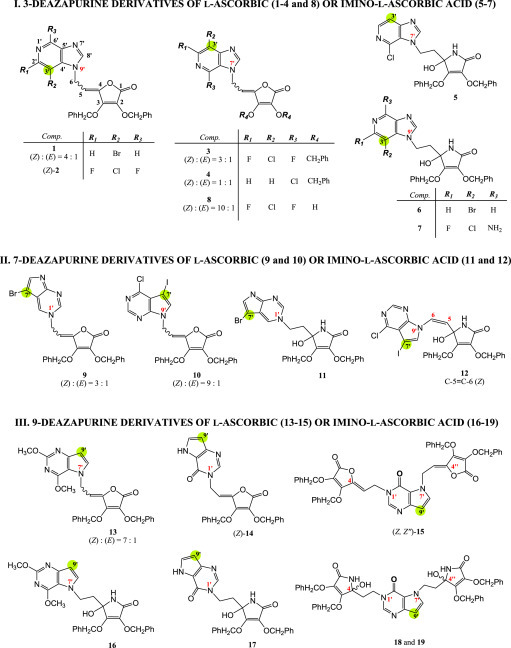
Structures of novel halogenated 3-, 7- and 9-deazapurine derivatives of l-ascorbic (1–4, 8–10 and 13–15) or imino-l-ascorbic acid (5–7, 11, 12 and 16–19).
2. Results and discussion
2.1. Chemistry
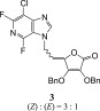
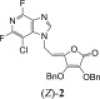
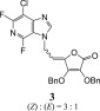
The 3-deazapurine derivatives of l-ascorbic acid (1–4) were prepared under modified Vorbrüggen reaction conditions (Scheme 1 ). The 3-deazapurine bases (3-DP) (A, B and C) were silylated with either 1,1,1,3,3,3-hexamethyldisilazane (HMDS) and catalytic amount of ammonium-sulphate (2,6-difluoro-3-chloro-3-deazapurine, B or 6-chloro-3-deazapurine, C) or bis(trimethylsilyl)acetamide (BSA) (3-bromo-3-deazapurine, A). Condensation reaction in situ of silylated 3-deazapurine bases (A, B and C) and 5,6-di-O-acetyl-2,3-di-O-benzyl-l-ascorbic acid (dAdBAA, synthesized as previously described [15]) with Friedel–Crafts catalyst, trimethylsilyltrifluoromethane sulphonate (TMSOTf) afforded the new 3-deazapurine derivatives of l-ascorbic acid (1–4, Scheme 1) containing ethylenic spacer between two moieties. 2,6-Difluoro-3-chloro-3-deazapurine base (B) was synthesized in six steps as previously described by Liu, M.-C. et al. [9] while 3-bromo-3-deazapurine base (A) was synthesized by cyclization reaction of 3,4-diamino-5-bromopyridine with diethoxymethyl acetate (DEMA). 3-Bromo-3-deazapurine derivative of l-AA (1) was isolated as N-9 regioisomer and as a mixture of Z and E isomers in the ratio 4:1, while reaction between 3-deazapurine base B and dAdBAA resulted in two regioisomers, N-9 as a Z isomer (2) and N-7 as a mixture of Z and E isomers (3) in the ratio 3:1. 6-Chloro-3-deazapurine derivative of l-AA (4) was isolated as N-7 regioisomer in equal Z and E isomer amounts. Ammonolysis of compounds 1, 2 and 4 in dioxane and methanol gave compounds 5–7, bearing a hydroxy group at the C-4 of now imino-l-ascorbic acid (imino-l-AA). Also, substitution of the fluorine atom with amino group at the C-6 position of the 3-deazapurine base occurred in the synthesis of 3-deazapurine derivative of imino-l-AA (7). Compound 4 underwent debenzylation with boron-trichloride (1 M in dichloromethane) which yielded the compound 8, 3-deazapurine derivative of l-AA with free hydroxy groups at the C-2 and C-3 positions of l-AA, as mixture of Z and E isomers in the ratio 10:1.
Scheme 1.
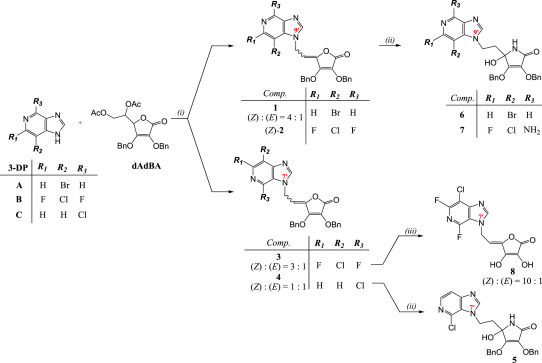
Synthesis of novel halogenated 3-deazapurine derivatives of l-ascorbic (1–4, 8) or imino-l-ascorbic acid (5–7). Reagents and conditions: (i) a) HMDS/(NH4)2SO4/Ar/reflux/24 h or BSA/reflux/1.5 h; b) TMSOTf/C2H4Cl2 or CH3CN/Ar/55–60 °C/24 h; (ii) NH4OH/MeOH/1,4-dioxane/0 °C/r. t./24 h; (iii) BCl3/CH2Cl2/Ar/−78 °C/r. t./24 h.
The 7-bromo-7-deazapurine derivative of l-AA (9) was prepared in a similar manner as compounds 2–4. Condensation reaction of 7-deazapurine base (7-DP) D and dAdBAA in Vorbrüggen reaction conditions gave N-1 regioisomer of compound 9 (Z:E = 3:1) (Scheme 2 ). 6-Chloro-7-iodo-7-deazapurine derivative of l-AA (10) was not isolated under Vorbrüggen conditions, but was successfully obtained via reaction under Mitsunobu reaction conditions. Reaction between 7-deazapurine base E and 6-hydroxy-2,3-di-O-benzyl-4,5-didehydro-l-ascorbic acid (HdBAA) with triphenylphosphine (PPh3) and diethyl azodicarboxylate (DEAD) afforded compound 10, 6-chloro-7-iodo-7-deazapurine substituted with 4,5-didehydro-5,6-dideoxy-l-ascorbic acid at N-9 position in the isomer ratio Z:E = 9:1 (Scheme 2). Ammonolysis of compounds 9 and 10 in similar procedures as for the synthesis of 3-deazapurine derivatives of imino-l-AA (5–7) gave 7-deazapurine derivatives of imino-l-AA 11 and 12, bearing the amino group at the C-4 chiral position of the lactame moiety (Scheme 2). As opposed to the structure of analogue derivatives of deazapurines and imino-l-AA described in this paper (5–7, 11 and 16–19) which contain ethyl spacer between deazapurine and lactame moieties, compound 12 contains ethylenic spacer between those two moieties with the double bond between C-5 and C-6 atoms. Structural difference of the products 11 and 12 may be explained by the mechanism from which these lactams arise. The mechanism of the formation of the lactams from l-ascorbic acid involves nucleophilic attack of the ammonia to the carbonyl group at C-1 of the lactone moiety with concomitant lactone ring opening and formation of the keto-enolic tautomers that subsequently close to form lactams [24]. Thus, our findings of the two structural different compounds 11 and 12 may be in accordance to that mechanism; the keto tautomer undergoes the nucleophilic attack of the amino group, followed by ring closure and formation of the lactame derivative 11, whereas the enolic tautomer undergoes the same nucleophilic attack followed by a consecutive shift of the double bond and formation of imino-l-AA derivative 12.
Scheme 2.
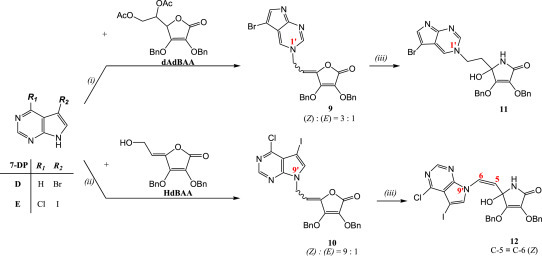
Synthesis of novel halogenated 7-deazapurine derivatives of l-ascorbic (9, 10) or imino-l-ascorbic acid (11, 12). Reagents and conditions: (i) a) HMDS/(NH4)2SO4/Ar/reflux/24 h; b) TMSOTf/C2H4Cl2/Ar/55–60 °C/24 h; (ii) DEAD/PPh3/THF/Ar/−50 to −40 °C/3 h/r. t./24 h/40 °C/24 h; (iii) NH4OH/MeOH/1,4-dioxane/0 °C/r. t./24 h.
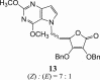
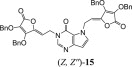
The 9-deazapurine derivatives of l-AA (13–15) were prepared under Mitsunobu reaction conditions, via similar synthetic method for obtaining 7-deazapurine derivative of l-AA (11) (Scheme 3 ). Reaction of 2,6-dimethoxy-9-deazapurine base (F) and HdBAA gave N-7 regioisomer 13 in equal isomer ratio (Z:E = 1:1), while reaction of 9-deazahypoxanthine base (G) and HdBAA yielded two condensated derivatives, 9-deazahypoxanthine monosubstituted with 4,5-didehydro-5,6-dideoxy-l-ascorbic acid at position N-1 (Z-14) and 9-deazahypoxanthine disubstituted with l-AA (15) at positions N-1 and N-7 (Z, Z″ related to both double bonds in 15). Ammonolysis of compound 13 produced 2,6-dimethoxy-9-deazapurine derivative of imino-l-AA (16) while ammonolysis of 14 gave the 9-deazahypoxanthine derivative of imino-l-AA (17) (Scheme 3). When subdued to ammonia, the disubstituted 9-deazahypoxanthine derivative of l-AA (15) yielded two derivatives of 9-deazahypoxanthine and imino-l-AA (18 and 19) that presumably are in diasteroisomeric relation (Scheme 3).
Scheme 3.
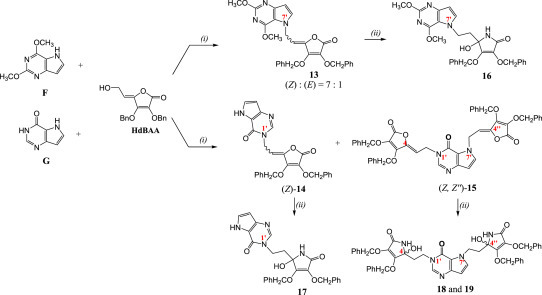
Synthesis of novel alkylated 9-deazapurine derivatives of l-ascorbic (13–15) or imino-l-ascorbic acid (16–19). Reagents and conditions: (i) DEAD/PPh3/THF/Ar/−50 to −40 °C/3 h/r. t./24 h/40 °C/24 h; (ii) NH4OH/MeOH/1,4-dioxane/0 °C/r. t./24 h.
2.2. Structural and conformational properties
The chemical identities of deazapurine derivatives of l-ascorbic acid (1–4, 8–10, 13–15) and imino-l-ascorbic acid (5–7, 11, 12, 16–19) were confirmed by 1H, 13C, 15N, 19F and 2D NMR measurements, as well as mass spectrometry spectra analysis (HPLC/MS/MS and MALDI-TOF/TOF). NMR chemical shifts are reported in Table 1 (1H), Experimental section (1H and 13C) and Supplementary data 1 and 2 (19F and 15N).
Table 1.
1H NMR chemical shifts (δ/ppm)a and H–H coupling constants (J/Hz) of compounds 1–19 (for numeration of atoms in molecular skeleton cf. Fig. 1).
| Comp. | H-2′ | H-3′ | H-6′ | H-8′ | H-9′ | OCH2-2 | OCH2-3 | Ph | OH-4 | H-5 | H-6 | NH (imino-l-AA) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Z)-1 | 8.44 (s, 1H) |
– | 8.93 (s, 1H) |
8.49 (s, 1H) |
– | 5.14 (s, 2H) |
5.30 (s, 2H) |
7.24–7.50 (m, 10H) |
– | 5.67 (t, 3J = 6.5, 1H) |
5.39 (d, 3J = 6.5, 2H) |
– |
| (Z)-2 | – | – | – | 8.54 (s, 1H) |
– | 5.15 (s, 2H) |
5.30 (s, 2H) |
7.28–7.44 (m, 10H) |
– | 5.70 (t, 3J = 6.6, 1H) |
5.39 (d, 3J = 6.6, 2H) |
– |
| (Z)-3 | – | – | – | 8.71 (s, 1H) |
– | 5.16 (s, 2H) |
5.31 (s, 2H) |
7.28–7.46 (m, 10H) |
– | 5.70 (t, 3J = 7.0, 1H) |
5.25 (d, 3J = 7.0, 2H) |
– |
| (E)-3 | – | – | – | 8.58 (s, 1H) |
– | 5.19 (s, 2H) |
5.40 (s, 2H) |
7.28–7.46 (m, 10H) |
– | 6.01 (t, 3J = 7.5, 1H) |
5.32 (d, 3J = 7.5, 2H) |
– |
| (Z)-4 | 8.14* (d, 3J = 5.5, 1H) /8.15* (d, 3J = 5.5, 1H) |
7.70* (d, 3J = 5.5, 1H) /7.72* (d, 3J = 5.5, 1H) |
– | 8.56 (s, 1H) |
– | 5.15 (s, 2H) |
5.03 (s, 2H) |
7.3–7.5 (m, 10H) |
– | 5.67 (t, 3J = 6.5, 1H) |
5.40 (d, 3J = 6.5, 2H) |
– |
| (E)-4 | 8.14* (d, 3J = 5.5, 1H) /8.15* (d, 3J = 5.5, 1H) |
7.70* (d, 3J = 5.5, 1H) /7.72* (d, 3J = 5.5, 1H) |
– | 8.43 (s, 1H) |
– | 5.21 (s, 2H) |
5.40 (s, 2H) |
7.3–7.5 (m, 10H) |
– | 5.97 (t, 3J = 7.2, 1H) |
5.47 (d, 3J = 7.2, 2H) |
– |
| 5 | 8.12 (d, 3J = 5.5, 1H) |
7.69 (d, 3J = 5.5, 1H) |
– | 8.39 (s, 1H) |
– | 5.00 (d, 2J = 11.2, 1H), 5.06 (d, 2J = 11.2, 1H) |
5.01 (d, 2J = 12.0, 1H), 5.14 (d, 2J = 12.0, 1H) |
7.2–7.4 (m, 10H) |
6.26 (s, 1H) |
2.22 (t, 3J = 7.6, 2H) |
4.45 (m, 2H) |
8.28 (s, 1H) |
| 6 | 8.42 (s, 1H) |
– | 8.91 (s, 1H) |
8.30 (s, 1H) |
– | 5.00 (d, 2J = 11.1, 1H), 5.07 (d, 2J = 11.1, 1H) |
5.02 (d, 2J = 12.0, 1H), 5.13 (d, 2J = 12.0, 1H) |
7.18–7.50 (m, 10H) |
6.30 (s, 1H) |
2.22 (t, 3J = 7.4, 2H) |
4.43 (m, 2H) |
8.27 (s, 1H) |
| 7b | – | – | – | 7.92 (s, 1H) |
– | 5.00 (d, 2J = 11.2, 1H), 5.06 (d, 2J = 11.2, 1H) |
5.00 (d, 2J = 11.8, 1H), 5.13 (d, 2J = 11.8, 1H) |
7.22–7.43 (m, 10H) |
6.25 (s, 1H) |
2.18 (t, 3J = 7.5, 2H) |
4.30 (m, 2H) |
8.25 (s, 1H) |
| 8 | – | – | – | 8.74 (s, 1H) |
– | – | – | – | – | 5.54 (t, 3J = 7.2, 1H) |
5.22 (d, 3J = 7.2, 2H) |
– |
| (Z)-9 | 8.74 (d, 2J = 1.9, 1H) |
– | 8.92 (d, 2J = 1.9, 1H) |
7.31–7.47 (m, 11H)c |
– | 5.16 (s, 2H) |
5.31 (s, 2H) |
7.31–7.47 (m, 11H)c |
– | 5.85 (t, 3J = 7.1, 1H) |
5.27 (d, 2J = 7.1, 2H) |
– |
| 10 | 8.73 (s, 1H) |
– | – | 8.50 (s, 1H) |
– | 5.04 (s, 2H) |
5.31 (s, 2H) |
7.22–7.42 (m, 10H) |
– | 6.68–6.72 (m, 1H) |
4.83 (d, 2J = 6.0, 2H) |
– |
| 11 | 8.75 (m, 1H) |
– | 8.94 (m, 1H) |
7.2–7.5 (m, 11H)c |
– | 4.99 (s, 2H) |
5.02 (d, 2J = 11.9, 1H), 5.11 (d, 2J = 11.9, 1H) |
7.2–7.5 (m, 11H)c |
6.25 (s, 1H) |
2.21 (m, 1H), 2.40 (m, 1H) |
4.44 (m, 2H) |
8.30 (s, 1H) |
| (E)-12 | 8.73 (s, 1H) |
– | – | 8.50 (s, 1H) |
– | 5.01 (s, 2H) |
5.15 (d, 2J = 12.2, 1H), 5.19 (d, 2J = 12.2, 1H) |
7.25–7.45 (m, 10H) |
6.72 (d, 4J = 0.7, 1H) |
6.40 (dd, 3J = 14.3, 4J = 0.7, 1H) |
7.62 (d, 3J = 14.3, 1H) |
8.46 (s, 1H) |
| (Z)-13d | – | – | – | 7.58 (d, 3J = 3.0, 1H) |
6.34 d, 3J = 3.0 1H) |
5.16 (s, 2H) |
5.29 (s, 2H) |
7.22–7.50 (m, 10H) |
– | 5.47 (t, 3J = 7.4, 1H) |
5.10 (d, 3J = 7.4, 2H) |
– |
| (Z)-14e | 8.10 (s, 1H) |
– | – | 7.38 (m, 1H) |
6.37 (dd, 3J = 2.7, 1.9, 1H) |
5.14 (s, 2H) |
5.29 (s, 2H) |
7.25–7.45 (m, 10H) |
– | 5.54 (t, 3J = 6.9, 1H) |
4.82 (d, 3J = 6.9, 2H) |
– |
| 16f | – | – | – | 7.41 (d, 3J = 3.0, 1H) |
6.31 (d, 3J = 3.0, 1H) |
4.97 (s, 2H) |
4.94 (d, 2J = 11.9, 1H), 5.11 (d, 2J = 11.9, 1H) |
7.20–7.40 (m, 10H) |
6.17 (s, 1H) |
2.12 (m, 2H) |
4.16 (t, 3J = 7.5, 2H) |
8.18 (s, 1H) |
| 17g | 7.93 (s, 1H) |
– | – | 7.37 (m, 1H) |
6.36 (d, 3J = 2.3, 1H) |
5.02 (s, 2H) |
5.01 (d, 2J = 11.8, 1H), 5.13 (d, 2J = 11.8, 1H) |
7.20–7.45 (m, 10H) |
6.16 (s, 1H) |
2.08 (m, 2H) |
3.95 (m, 2H) |
8.23 (s, 1H) |
| Comp. | H-2′ | H-3′ | H-6′ | H-8′ | H-9′ | OCH2-2 /OCH2-2″ |
OCH2-3 /OCH2-3″ |
Ph/Ph″ | OH-4/ OH-4″ |
H-5/ H-5″ |
H-6/ H-6″ |
NH/NH″ (imino-l-AA) |
| (Z, Z″)-15 | 8.10 (s, 1H) |
– | – | 7.46 (d, 3J = 3.0, 1H) |
6.36 (d, 3J = 3.0, 1H) |
5.13 (s, 2H), 5.14 (s, 2H)* /5.13 (s, 2H), 5.14 (s, 2H)* |
5.29 (s, 2H)/ 5.29 (s, 2H) |
7.2–7.5 (m, 10H)/ 7.2–7.5 (m, 10H) |
– | 5.56 (t, 3J = 7.1, 1H), /5.53 (t, 3J = 6.8, 1H) |
5.24 (d, 3J = 7.1, 2H), /4.79 (d, 3J = 6.8, 2H) |
– |
| 18 | 7.91 (s, 1H) |
– | – | 7.26 (d, 3J = 2.9, 1H) |
6.30 (d, 3J = 2.9, 1H) |
5.01 (s, 2H)/ 5.01 (s, 2H) |
4.95–5.15 (m, 2H) /4.95–5.15 (m, 2H) |
7.20–7.40 (m, 10H)/ 7.20–7.40 (m, 10H) |
6.15 (s, 1H)/ 6.17 (s, 1H) |
2.18 (m, 2H)/ 2.07 (m, 2H) |
4.31 (m, 2H)/ 3.92 (t, 3J = 7.2, 2H) |
8.15 (s, 1H)/ 8.24 (s, 1H) |
| 19 | 7.91 (s, 1H) |
– | – | 7.27 (d, 3J = 2.9, 1H) |
6.30 (d, 3J = 2.9, 1H) |
5.01 (s, 2H)/ 5.01 (s, 2H) |
4.95–5.20 (m, 2H) /4.95–5.20 (m, 2H) |
7.20–7.50 (m, 10H)/ 7.20–7.50 (m, 10H) |
6.14 (s, 1H)/ 6.16 (s, 1H) |
2.17 (m, 2H)/ 2.07 (m, 2H) |
4.31 (m, 2H)/ 3.91 (t, 3J = 7.3, 2H) |
8.15 (s, 1H)/ 8.24 (s, 1H) |
1H NMR chemical shifts are determined in reference to chemical shift of the solvent DMSO-d6, δ (DMSO) = 2.50 ppm.
δ (NH2-6′) = 6.84 ppm (s, 2H).
Chemical shifts of H-8′ in 9 and 11 are overlapped with phenyl protons in the range δ 7.2–7.5 ppm (11H).
δ (OCH3-2′) = 3.84 (s, 3H), δ (OCH3-6′) = 3.94 (s, 3H).
δ (NH-7′) = 12.11 (s, 1H).
δ (OCH3-2′) = 3.83 (s, 3H), δ (OCH3-6′) = 3.91 (s, 3H).
δ (NH-7′) = 12.10 (s, 1H). *Chemical shifts could not be unequivocally assigned.
1H decoupled 13C NMR spectra showed C–F coupling constants that enabled straightforward identification of fluorinated carbon atoms and their neighbours. Notably, C-4′ and C-5′ displayed different multiplicity pattern due to the long-range coupling constants with two fluorine atoms in 2 and 3; C-4′ is coupled to both fluorine atoms over three bonds (3 J = 4–12 Hz), whereas C-5′ showed coupling constants over two (2 J = 27–34 Hz) and four bonds (4 J = 3–4 Hz). Furthermore, 2D 19F–13C NMR spectra allowed unambiguous assignment of C-2′ and C-6′ in difluorinated deazapurines 2 and 3 (Fig. 2 ).
Fig. 2.
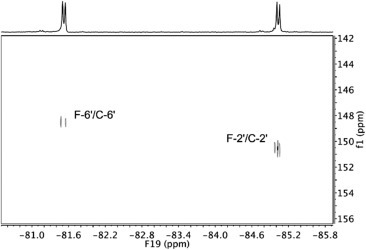
19F–13C HSQC NMR spectrum of 2 in DMSO-d6 showed correlation signals for fluorine and carbon atoms coupled across a single bond.
N-7 and N-9 regioisomers 2 and 3 were distinguished on the basis of heteronuclear 1H–13C and 1H–15N correlation signals in 2D HMBC spectra. H-6 methylene protons in 2 (δ 5.39 ppm) showed correlation signal with C-4′ (δ 141.67 ppm), which suggested that benzyl-protected ascorbic acid is attached to N-9 position of deazapurine scaffold. 15N chemical shifts of N-7 and N-9 in deazapurine 2 were observed at δ 240 and 163 ppm, respectively. In contrast, perusal of 2D NMR spectra of 3 showed a correlation signal between H-6 methylene protons (δ 5.25 ppm) and C-5′ (δ 116.37 ppm) indicating that the reaction proceeded at the N-7 position of the deazapurine moiety. 15N chemical shifts in deazapurine 3 corroborated that ascorbic acid is attached at N-7 position. N-7 and N-9 were observed at δ 158 and 243 ppm, respectively. Distinct shielding of C-4′ and C-5′ as well as N-7 and N-9 atoms in regioisomeric pair of 2/3 facilitated the assessment of structural features of the remaining regioisomers (Fig. 3 ).
Fig. 3.
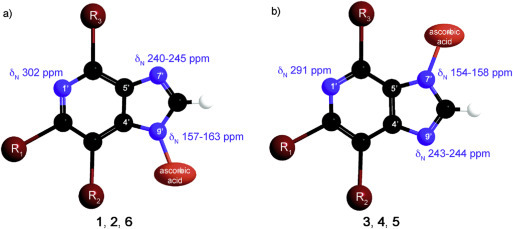
The characteristic 15N chemical shifts for N-9′ (a) and N-7′ regioisomers (b) in compounds 1–6.
Vicinal coupling constant 3 J H5H6 = 14.3 Hz suggested an E-configuration along C4 C5 double bond in compound 12. Some NMR spectra exhibited two sets of signals, which were attributed to Z- and E-isomers. The configurations along C4 C5 double bond in 1–4 and 13–15 were evaluated through long-range 3 J C13H11 coupling constants, which were determined by J-HMBC NMR experiments. Small values between 2 and 3 Hz are in agreement with Z-configuration along C4 C5 double bond, whereas 3 J C13H11 between 7 and 8 Hz correspond to E-isomers (Table 2 ). Conformational study of compounds 2 and 4 showed only trivial NOESY cross-peaks. Consequently, no particular conformational preferences could be established for these compounds.
Table 2.
1H–13C coupling constants (JC3H5, JC3″H5″/Hz) and isomer ratio (Z:E) for compounds 1–4 and 13–15.
|
1 |
2 |
3 |
4 |
13 |
14 |
15 |
|||
|---|---|---|---|---|---|---|---|---|---|
| Z | Z | Z | E | Z | E | Z | Z | Z, Z″ | |
| J | 2.0 | 2.5 | 2.6 | 7.3 | 2.0 | 7.0 | 2.0 | 3.0 | 3.0, 3.0 |
| Isomer ratio (Z:E) | 4:1 | 9.5:0.5 | 3:1 | 1:1 | 7:1 | 9.5:0.5 | – | ||
2.3. Biological results
2.3.1. Cytostatic activity
Newly synthesized 3-, 7- and 9-deazapurine derivatives of l-ascorbic (1–4, 8–10 and 13–15) and imino-l-ascorbic acid (5–7, 12 and 16–19) have been evaluated in vitro for their cytostatic effects on malignant human tumour cell lines including cervical carcinoma (HeLa), colorectal adenocarcinoma (SW620), murine leukemia (L1210/0), acute lymphoblastic leukemia (CEM/0), pancreatic carcinoma (MiaPaCa-2) and hepatocellular carcinoma (HepG2) as well as their cytotoxic effects on embryonal murine fibroblasts (3T3) (Table 3 and Supplementary data 3). Compound 9 was tested on pancreatic adenocarcinoma cells, CFPAC-1.
Table 3.
Inhibitory effects of selected 3- and 9-deazapurine derivatives of l-ascorbic (1, 13 and 15) or imino-l-ascorbic acid (17–19) on the growth of malignant cell lines (HeLa, SW260, L1210/0, CEM/0, MiaPaCa-2 and HepG2) as well as their cytotoxic effects on embryonal murine fibroblasts (3T3).
| Comp. | IC50a |
||||||
|---|---|---|---|---|---|---|---|
| HeLa | SW620 | L1210/0 | CEM/0 | MiaPaCa-2 | HepG2 | 3T3 | |
 |
15 ± 0 | 32.9 ± 0.1 | 6.8 ± 0.1 | 5.7 ± 1.4 | 22.6 ± 0.1 | 46.9 ± 0.4 | 26.4 ± 0.1 |
 |
12 ± 4 | 47.1 ± 0.1 | 4.7 ± 0.1 | 4.1 ± 1.8 | 33.9 ± 0.1 | >100 | >100 |
 |
5.6 ± 1.3 | 47.2 ± 0.1 | 4.5 ± 0.5 | 19 ± 12 | 26.2 ± 0.1 | >100 | >100 |
 |
92 ± 3 | 90.7 ± 0.1 | 107 ± 6 | 87 ± 0 | 36.6 ± 0.1 | >100 | >100 |
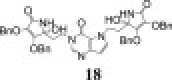 |
17 ± 2 | 69.9 ± 0.1 | 4.4 ± 0.3 | 15 ± 4 | 5.7 ± 0.2 | >100 | >100 |
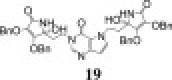 |
18 ± 2 | 30.5 ± 0.1 | 7.6 ± 3.8 | 4.9 ± 0.3 | 4.4 ± 0.1 | 52 ± 0.1 | 63.5 ± 0.1 |
| 5-Fluorouracil | 66.5 ± 0.01 | 0.79 ± 0.7 | n. d.b | n. d. | 11.67 ± 0.53 | 55.2 ± 15 | 28.3 ± 0.01 |
Bolded values emphasize the most pronunced biological activities (results).
Inhibitory concentration (IC50) on tumour and normal fibroblast cell growth.
Not determined.
Among the tested compounds that exhibited the most prominent antitumour activities, only compounds 1 and 19 showed low cytotoxic effects on fibroblasts 3T3 (Table 3). Several deazapurine derivatives of l-AA and imino-l-AA expressed antiproliferative activity against L1210/0 cell lines in the micromolar concentration range: 3-bromo-3-deazapurine derivative of l-AA (1) (IC50 = 6.8 ± 0.1 μM), 2,6-dimethoxy-9-deazapurine derivative of l-AA (13) (IC50 = 4.7 ± 0.1 μM) and 9-deazahypoxanthine derivatives (15, 18, 19) (IC50 = 4.5 ± 0.5; 4.4 ± 0.3 and 7.6 ± 3.8 μM, respectively). Compounds 13, 15 and 18 were not cytotoxic for 3T3 cells. 3-Deazapurine derivative of l-AA (1), as well as 9-deazapurine derivatives of l-AA (13) and imino-l-AA (19) showed the most pronounced activity against CEM/0 cell lines (IC50 = 5.7 ± 1.4; 4.1 ± 1.8 and 4.9 ± 0.3 μM, respectively) while amongst them, only 2,6-dimethoxy-9-deazapurine derivative of l-AA (13) did not exhibit cytotoxic effects on fibroblasts 3T3. N-1 regioisomers of 7-bromo-7-deazapurine derivative of l-AA (9) and 9-deazahypoxanthine derivative of l-AA (14) have shown moderate effects against L1210/0 (IC50 = 15 ± 0; 28 ± 5 μM, respectively) and against the CEM/0 cell line (IC50 = 12 ± 0; 16 ± 2 μM, respectively) without cytotoxic effects on 3T3 cells (Supplementary data 3). The 9-deazahypoxanthine derivative disubstituted with l-AA (15) exhibited marked effects against the HeLa cell line with an IC50 value of 5.6 ± 1.3 μM without cytotoxic effects on embryonal murine fibroblasts. 3-Deazapurine derivative of l-AA (1), 7-deazapurine derivative of l-AA (9) and the 9-deazapurine derivatives of l-AA (13) and of imino-l-AA (18, 19) expressed moderate activity against HeLa cells where compounds 13 and 18 exerted no cytotoxic effects on 3T3 cells.
Compounds 1, 6, 12, 16 and 19 exerted weak and nonselective antiproliferative activity against all tested malignant cell lines and moderate or low cytotoxic effects on 3T3 cells (Supplementary data 3), while amongst them, the 3-bromo-3-deazapurine derivative of l-AA (1) exhibited moderate activity against HeLa cells.
The 9-deazapurine derivatives of l-AA (13) and the imino-l-AA derivatives (15 and 18), which exhibited the most prominent antitumoural activities, will serve as model molecules for developing more selective and more active antitumoural substrates in this class of compounds.
2.3.2. Antiviral activity
The newly synthesized 3-, 7- and 9-deazapurine derivatives of l-ascorbic (1–4, 8–10, 13–15) and of imino-l-ascorbic acid (5–7, 12, 16–19) were evaluated in vitro against numerous pathogenic viruses:human immunodeficiency virus type 1 and 2 (HIV-1, HIV-2) in human T-lymphocyte cells (CEM); human cytomegalovirus (HCMV), Varicella-zoster virus (VZV), herpes simplex virus type 1 and 2 (HSV-1 i 2), vaccinia virus (VV), vesicular stomatitis virus (VSV) and adenovirus-2 in human embryonic HEL cells; vesicular stomatitis, Coxsackie B4 and respiratory syncytial virus in human HeLa cells; para-influenza, reovirus-1, Sindibis virus, Coxsackie B4, Punta Toro virus in Vero cells (African green monkey cells); feline Corona virus (feline infectious Peritonitis virus, FIVP) and herpes virus in Crandell-Rees feline kidney cells (CRFK); influenza A, subtype H1N1 and influenza B subtype H3N2 in Madin Darby canine kidney cell lines (MDCK).
2,6-Difluoro-3-chloro-3-deazapurine derivative of l-ascorbic acid (3) exhibited substantial effects against HCMV replication with EC50 value of 8.94 μM against both viral strains (AD-169 and Davis) (Table 4 ). The EC50's for compound 3 were in the same order as those found for the reference anti-HCMV drug ganciclovir and without cytotoxic effects on normal human embryonal lung (HEL) cells. The removal of benzyl protection groups of C-2 and C-3 hydroxyl groups of l-AA moiety in compound 8 resulted in the imino-l-AA analogue derivative 7, which lost anti-HCMV activity compared to the most active compound 3. The 2,6-dimethoxy-9-deazapurine derivative of l-AA (13) exhibited lower EC50 values against both HCMV strains (EC50 = 4.99 μM for AD-169 strain and EC50 = 1.99 for Davis strain) but also cytotoxic effects on HEL cells (CC50 = 15.4 μM).
Table 4.
Antiviral activity of selected 3-deazapurine (2 and 3) and 9-deazapurine (13) derivatives of l-AA against human cytomegalovirus (HCMV) and their cytotoxicity (MCC, CC50) on human embryonic lung cells (HEL).
| Comp. | Cytotoxicity |
EC50 (μM)c |
||
|---|---|---|---|---|
| MCCa | CC50b | HCMV |
||
| AD-169 cell line | Davis cell line | |||
 |
>100 | ≥100 | 44.7 ± 0 | 44.7 |
 |
≥100 | >100 | 8.94 ± 0 | 8.94 |
 |
≥20 | 36.6 ± 30 | 4.99 ± 4.52 | 1.99 ± 0.28 |
| Ganciclovir | >350 | 328.5 ± 92.6 | 6.12 ± 0.19 | 4.72 |
| Cidofovir | >300 | 153 | 0.61 ± 0.13 | 0.51 |
Bolded values emphasize the most pronunced biological activities (results).
Minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology.
Cytotoxic concentration required to reduce cell growth by 50%.
Effective concentration required to reduce virus plaque formation by 50%. Virus input was 100 plaque forming units (PFU).
The 9-deazahypoxanthine derivative substituted with two l-AA moieties (15) exerted very weak activity against both strains of VZV (TK+ and TK−) (EC50 = 13.8; 7.6 μM, respectively) with cytotoxic effect on HEL cells (CC50 = 20.0 μM) (Table 5 ).
Table 5.
Antiviral activity of selected 3-deazapurine (2 and 3) and 9-deazapurine (15) derivatives of l-AA against viruses: para-influenza-3, reovirus-1, Sindibis, Coxsackie B4 and Punta Toro virus and their cytotoxic effect (MCC) on African green monkey kidney epithelial cells (Vero).
| Comp. | MCCb (μM) | EC50a (μM) |
||||
|---|---|---|---|---|---|---|
| Para-influenza-3 virus | Reovirus-1 | Sindbis virus | Coxsackie B4 virus | Punta Toro virus | ||
 |
≥20 | >20 | >20 | >20 | 14.5 | 12 |
 |
20 | >20 | >20 | >20 | >20 | 12 |
 |
≥20 | >20 | >20 | >20 | >20 | 6.7 ± 3.1 |
| DS-5000 (μg/ml) | >100 | >100 | >100 | >100 | >100 | >100 |
| DS-10000 (μg/ml) | >100 | ≥100 | >100 | 9.45 ± 0.78 | 34 ± 34 | 20 |
| (S)-DHPA | >250 | >250 | >250 | >250 | >250 | >250 |
| Ribavirin | >250 | 73.5 ± 54.4 | >250 | >250 | >250 | 150 ± 141 |
Bolded values emphasize the most pronunced biological activities (results).
Effective concentration required to reduce virus-induced cytopathogenicity by 50%.
Minimum cytotoxic concentration required to cause a microscopically detectable alteration of normal cell morphology.
The 2,6-difluoro-3-chloro-3-deazapurine derivatives of l-AA substituted at the N-9 position of the deazapurine moiety (2) and at the N-7 position (3) exhibited the same potency against Punta Toro virus (EC50 = 12 μM), but had strong cytotoxic effects on Vero cells (2: MCC ≥ 20 μM; 3: MCC = 20 μM). Besides that, compound 2 showed moderate activity against Coxsackie B4 virus in Vero cells with an EC50 value of 14.5 μM, while when tested on HeLa cells, it did not exhibit any activity against this virus (EC50 > 100 μM) (Table 5).
The 9-deazahypoxanthine derivative disubstituted with two l-AA moieties at the N-1 and N-7 positions (15) exhibited stronger activity against Punta Toro virus (EC50 = 6.7 ± 3.1 μM) compared to compounds 2 and 3. The effective concentration needed for reducing Punta Toro virus replication by 50% for compound 15 was 3- and 22-folds lower compared to, respectively, dexstran-sulphate (DS-10000) and ribavirin. However, compound 15 proved to be cytotoxic to Vero cells (MCC ≥ 20 μM) (Table 5).
2.3.3. Structure–activity relationship
Amongst the 3-deazapurine derivatives of l-AA (1–4, 8) and imino-l-AA (5–7), 2,6-difluoro-3-deazapurine derivative of l-ascorbic acid (3) showed important activity against human cytomegalovirus (AD-169 and Davis cell strain), at inhibitory concentrations similar as those for the reference anti-HCMV drug ganciclovir. Also, compound 3 did not show any cytostatic effects on normal human embryonal lung (HEL) cells, embryonal murine fibroblasts (3T3) nor against immortalized cells, i.e. cervical carcinoma (HeLa), Madin Darby canine kidney (MDCK), Crandell-Rees feline kidney cells (CRFK). However, removal of benzyl protection groups at C-2 and C-3 positions of l-AA moiety as in compound 8 and conversion of l-AA into imino-l-AA as in analogue 7, resulted in loss of anti-HCMV activity. Compound 3 and its N-9 regioisomer (2) have exhibited comparable effects against Punta Toro virus, but concomitantly showed cytostatic effects on the monkey cell line Vero. In addition, compound 2 exerted moderate activity against Coxsackie B4 virus in Vero cells and no cytostatic effects on cervical carcinoma cells (HeLa) used in the antiviral assays. The 7-deazapurine derivatives of l-AA (9, 10) and of imino-l-AA (12) showed no marked cytostatic nor antiviral activities. In contrast, the 9-deazapurine derivatives of l-ascorbic acid (13, 15) and of imino-l-ascorbic acid (18) exhibited marked effects against the murine leukemia cell line (L1210/0) at micromolar concentrations (IC50), while the 2,6-dimethoxy-9-deazapurine and l-ascorbic acid derivative (13) also displayed effects against CEM/0 cells. In addition, the 9-deazahypoxanthine derivative disubstituted with two l-ascorbic acid moieties (15) had strong activity against HeLa cells. Conversion of l-ascorbic acid moiety of compound 15 into imino-l-ascorbic acid, resulted in gaining compound 18 which showed activity against the pancreatic carcinoma cell line (MiaPaCa-2). The 9-deazapurine derivatives 13, 15 and 18 did not shown any cytotoxic effects on embryonal murine fibroblasts (3T3). Furthermore, the 9-deazahypoxanthine derivative disubstituted with two l-AA moieties at N-1 and N-7 positions (15) exhibited even stronger activity against Punta Toro virus than compounds 2 and 3, with 3- and 22-folds lower EC50's than dexstran-sulphate (DS-10000) and ribavirin, respectively. However compound 15 is also cytotoxic to uninfected Vero cells.
3. Experimental section
3.1. Materials and general methods
Commercially available chemicals were purchased from Sigma Aldrich (Germany) and Acros (Belgium) and where used without purification. All solvents used in synthesis were analytical grade purity and dried. Acetonitrile (CH3CN) was refluxed over calcium hydride (CaH2), distilled and stored over 3 Å molecular sieves. 1,2-Dichloroethane (C2H4Cl2) and dichloromethane (CH2Cl2) were distilled from phosphorus pentoxide (P2O5) and stored over 4 Å molecular sieves. 1,4-Dioxane was dried with sodium and also stored over 4 Å molecular sieves. Methanol (CH3OH) was stored over 3 Å molecular sieves without distillation. 1,4-Tetrahydrofuran (THF) was pre-dried over calcium hydride and refluxed over sodium benzophenone ketyl (sodium wire and benzophenone), distilled and stored over sodium. Melting points were determined on a Kofler micro hot-stage instrument (Reichter, Wien) and were uncorrected. Precoated Merck silica gel 60 F254 plates were used for thin-layer chromatography and spots were visualized by shortwave UV light (254 nm). Column chromatography was performed on Fluka silica gel (0.063–0.200 mm), with petroleum ether:ethylacetate, dichloromethane:methanol and dichloromethane as mobile phases. Additional purification by rechromatography afforded the analytical samples.
NMR spectroscopy 1H, 13C, 15N and 19F NMR spectra were recorded on a Varian Gemini 300 spectrometer (Institute Ruđer Bošković, Zagreb) and Varian NMR System 600 and Varian Unity Inova 300 and Agilent Technologies DD2 300 MHz NMR spectrometers (National Institute of Chemistry, Ljubljana, Slovenia). Samples were measured in DMSO-d 6 solutions at 25 °C in 5 mm NMR tubes. 1H and 13C NMR chemical shifts (δ) in ppm were referred to TMS (δ 0.0 ppm). 19F and 15N NMR chemical shifts were referenced externally with respect to trichlorofluoromethane (δ F 0.0 ppm) and ammonia (δ N 0.0 ppm). Individual resonances were assigned on the basis of their chemical shifts, signal intensities, multiplicity of resonances and H–H coupling constants. NOESY spectra were acquired with mixing times of 80 and 200 ms. 19F–13C NMR spectra utilized the one-bond coupling between fluorine and carbon (J = 240 Hz).
The electron impact mass spectra and the purity of compounds were assessed by using Agilent Technologies 6410 Triple Quad LC/MS instrument equipped with electrospray interface and triple quadrupole analyzer (LC-MS/MS) in positive instrument mode. High performance LC was performed on Agilent 1100 series system with UV detection (photodiode array detector) using Zorbax C18 reverse-phase analytical column (2.1 × 30 mm; 3.5 μm). All compounds used for biological evaluation showed >95% purity in HPLC-MS/MS system. Exact mass of compounds were analysed by using HRMS with MALDI-TOF/TOF analyser (4800 Plus MALDI-TOF/TOF, Applied Biosystems) in reflectron positive ion mode of the instrument. Elemental analyses indicated by the symbols of the elements were within ±0.4% of the theoretical values.
3.2. Synthesis
3.2.1. Procedure for the preparation of 3,4-di-O-benzyl-5-(9-(3-bromo-1H-imidazo[4,5-c]pyridine-9-yl)ethylidene)furan-2(5H)-one (1)
3-Bromo-3-deazapurine base (A, 426.0 mg; 2.15 mmol) was dissolved in N,O-bis(trimethylsylil)acetamide (BSA, 0.6 ml; 2.15 mmol) and the reaction mixture was refluxed at room temperature for 1 h and 30 min under inert atmosphere of argon. Excess BSA was evaporated under reduced pressure and yellow solid of silylated base was obtained. 6-Hydroxy-2,3-di-O-benzyl-4,5-didehydro-l-ascorbic acid (HdBAA, 622.7 mg; 1.43 mmol) and silylated base were dissolved in dry acetonitrile (20.0 ml) and stirred under argon atmosphere at room temperature. TMSOTf (1.80 ml, 4 × 0.45 ml) was then added to the reaction mixture and additionally stirred and heated at 55–70 °C overnight. After evaporating the solvent under reduced pressure, crude yellowish-green product was obtained and washed with CH2Cl2 and saturated solution of NaHCO3. Organic layer was dried over MgSO4 and concentrated under reduced pressure. Crude product mixture was purified by silica gel column chromatography (dichloromethane:methanol = 25:1) and product 1 (Z:E = 4:1; 14.7 mg; 1.32%) was obtained as yellow oil by rechromatography (petroleum ether:etylacetate = 1:1). MS (ESI): m/z = 518.2 ([M+H]+). Anal. Calcd. for C26H20BrN3O4 (517.06): C, 60.24; H, 3.89; N, 8.11. Found: C, 60.19; H, 3.88; N, 8.13.
(Z)-1: 1H NMR (DMSO-d 6): δ 5.14 (s, 2H, OCH 2-2), 5.30 (s, 2H, OCH 2-3), 5.39 (d, 2H, 3 J = 6.5 Hz, H-6), 5.67 (t, 1H, 3 J = 6.5 Hz, H-5), 7.24–7.50 (m, 10H, C6 H 5), 8.44 (s, 1H, H-2′), 8.49 (s, 1H, H-8′), 8.93 (s, 1H, H-6′) ppm.
(Z)-1: 13C NMR (DMSO-d 6): δ 40.96 (C-6), 72.97 (OCH2-3), 73.94 (OCH2-2), 100.96 (C-3′), 104.31 (C-5), 123.02 (C-2), 143.66 (C-2′), 127.8–128.9 (C 6H5), 135.33/135.64/135.72∗ (C-2a, C-3a), 135.33/135.64/135.72∗ (C-4′), 141.30 (C-6′), 141.98 (C-4), 142.10 (C-5′), 143.66 (C-2′), 147.86 (C-8′), 163.57 (C-1) ppm. *Could not be unequivocally assigned.
3.2.2. Method A. General method for the preparation of 3-deazapurine derivatives of 4,5-didehydro-5,6-dideoxy-l-ascorbic acid (2–4)
Suspension of anhydrous 3-deazapurine base (B or C, 1 eq.) and (NH4)2SO4 (0.1 eq.) in HMDS (25–30 ml) was refluxed overnight under inert atmosphere of argon. Excess HMDS was evaporated under reduced pressure (0.1 mmHg) and silylated base was obtained as a solid. 5,6-di-O-acetyl-2,3-di-O-benzyl-l-ascorbic acid (dAdBAA, 0.7 eq.) and silylated base were dissolved in dry 1,2-dichloroethane (20–25 ml) and stirred at room temperature under argon atmosphere. TMSOTf (1.5 eq.) was added to the reaction mixture and the reaction mixture was heated at 55–70 °C with stirring overnight. After evaporating the solvent under reduced pressure, crude product was obtained and washed with CH2Cl2 and saturated solution of NaHCO3. Organic layer was dried over MgSO4 and concentrated under reduced pressure. Crude product mixture was purified by silica gel column chromatography (petroleum ether:etylacetate) and by rechromatography (CH2Cl2 or petroleum ether:etylacetate). Compounds 2–4 were obtained as oils.
3.2.2.1. (3,4-di-O-benzyl-5-(9-(3-chloro-2,6-difluoro-1H-imidazo[4,5-c]pyridine-9-yl)ethylidene)furan-2(5H)-one (2) and 3,4-di-O-benzyl-5-(7-(3-chloro-2,6-difluoro-1H-imidazo[4,5-c]pyridine-7-yl)ethylidene)furan-2(5H)-one (3)
According to method A, mixture of 3-chloro-2,6-difluoro-3-deazapurine (B, 618.2 mg, 3.26 mmol) and dAdBAA (941.88 mg, 2.16 mmol) gave the crude product that was purified by silica gel column chromatography (petroleum ether:etylacetate = 3:1). Products 2 (Z; 174.0 mg; 31.60%) and 3 (Z:E = 3:1; 157.0 mg; 28.51%) were obtained as white oils by rechromatography (CH2Cl2). (Z)-2: MS (ESI): m/z = 510.1 ([M+H]+). Anal. Calcd. for C26H18ClF2N3O4 (509.1): C, 61.24; H, 3.56; N, 8.24. Found: C, 61.27; H, 3.56; N, 8.26. 3: MS (ESI): m/z = 510.1 ([M+H]+). Anal. Calcd. for C26H18ClF2N3O4 (509.1): C, 61.24; H, 3.56; N, 8.24. Found: C, 61.26; H, 3.56; N, 8.25.
(Z)-2: 1H NMR (DMSO-d 6): δ 5.15 (s, 2H, OCH 2-2), 5.30 (s, 2H, OCH 2-3), 5.39 (d, 2H, 3 J = 6.6 Hz, H-6), 5.70 (t, 1H, 3 J = 6.6 Hz, H-5), 7.28–7.44 (m, 10H, C6 H 5), 8.54 (s, 1H, H-8′) ppm.
(Z)-2: 13C NMR (DMSO-d 6): 41.43 (C-6), 73.03 (OCH2-3), 73.96 (OCH2-2), 95.49 (dd, 2 J CF = 40.4 Hz, 4 J CF = 8.0 Hz, C-3′), 103.77 (C-5), 123.12 (C-2), 126.06 (dd, 2 J CF = 33.5 Hz, 4 J CF = 3.3 Hz, C-5′), 127.93–128.81 (C 6H5), 135.31, 135.65 (C-2a, C-3a), 141.67 (dd, 3 J CF = 11.5 Hz, 3 J CF = 5.5 Hz, C-4′), 142.19 (C-4), 148.02 (C-3), 148.28 (dd, 1 J = 247.8 Hz, 3 J = 15.9 Hz, C-6′), 149.21 (C-8′), 150.46 (dd, 1 J CF = 229.6 Hz, 3 J CF = 15.4 Hz, C-2′), 163.49 (C-1) ppm.
(Z)-3: 1H NMR (DMSO-d 6): δ 5.16 (s, 2H, OCH 2-2), 5.25 (d, 2H, 3 J = 7.0 Hz, H-6), 5.31 (s, 2H, OCH 2-3), 5.70 (t, 1H, 3 J = 7.0 Hz, H-5), 7.28–7.46 (m, 10H, C6 H 5), 8.71 (s, 1H, H-8′) ppm.
(E)-3: 1H NMR (DMSO-d 6): δ 5.19 (s, 2H, OCH 2-2), 5.32 (d, 2H, 3 J = 7.5 Hz, H-6), 5.40 (s, 2H, OCH 2-3), 6.01 (t, 1H, 3 J = 7.5 Hz, H-5), 7.28–7.46 (m, 10H, C6 H 5), 8.58 (s, 1H, H-8′) ppm.
(Z)-3: 41.56 (C-6), 73.3 (OCH2-3), 73.99 (OCH2-2), 101.21 (dd, 2 J CF = 34.8 Hz, 4 J CF = 8.8 Hz, C-3′), 102.84 (C-5), 116.37 (dd, 2 J CF = 26.9 Hz, 4 J CF = 3.8 Hz, C-5′), 123.28 (C-2), 127.81–128.81 (C 6H5), 135.32, 135.67 (C-2a, C-3a), 142.80 (C-4), 143.62 (dd, 1 J = 242.0 Hz, 3 J = 17.0 Hz, C-6′), 147.97 (C-3), 149.70 (dd, 1 J CF = 229.6 Hz, 3 J CF = 14.3 Hz, C-2′), 151.02 (C-8′), 153.15 (dd, 3 J CF = 7.1 Hz, 3 J CF = 4.4 Hz, C-4′), 163.55 (C-1) ppm.
3.2.2.2. 3,4-bis(benzyloxy)-5-(2-(4-chloro-1H-imidazo[4,5-c]pyridine-1-yl)ethylidene)furan-2(5H)-one (4)
Suspension of anhydrous 6-chloro-3-deazapurine (C, 500.0 mg, 3.26 mmol) and dAdBAA (941.0 mg, 2.16 mmol) gave after reaction, according to method A, the crude product that was purified by silica gel column chromatography (petroleum ether:etylacetate = 2:1). Compound 4 (Z:E = 1:1; 255.8 mg; 25.08%) was obtained as white oil by rechromatoraphy (petroleum ether:etylacetate = 1:1). MS (ESI): m/z = 474.2 ([M+H]+). Anal. Calcd. for C26H20ClN3O4 (473.11): C, 65.89; H, 4.25; N, 8.87. Found: C, 65.93; H, 4.25; N, 8.90.
(Z)-4: 1H NMR (DMSO-d 6): δ 5.03 (s, 2H, OCH 2-3), 5.15 (s, 2H, OCH 2-2), 5.40 (d, 2H, 3 J = 6.5 Hz, H-6), 5.67 (t, 1H, 3 J = 6.5 Hz, H-5), 7.3–7.5 (m, 10H, C6 H 5), 7.70/7.72* (d, 1H, 3 J = 5.5 Hz, H-3′), 8.14/8.15* (d, 1H, 3 J = 6.5 Hz, H-2′), 8.56 (s, 1H, H-8′) ppm. *Chemical shifts could not be unequivocally assigned.
(Z)-4: 13C NMR (DMSO-d 6): 41.45 (C-6), 73.07 (OCH2-3), 74.01 (OCH2-2), 104.37 (C-5), 114.83/114.88* (C-3′), 123.13/124.67* (C-2), 126.29/126.45* (C-5′), 127.9–129.0 (C 6H5), 132.72/132.77* (C-6′), 135.4–135.7 (C-2a, C-3a), 140.75/140.77* (C-2′), 142.15 (C-4), 148.15 (C-3), 149.13 (C-8′), 150.99 (C-4′), 163.65/163.79* (C-1) ppm. *Chemical shifts between E and Z isomers could not be distinguished.
(E)-4: 1H NMR (DMSO-d 6): δ 5.21 (s, 2H, OCH 2-2), 5.40 (s, 2H, OCH 2-3), 5.47 (d, 2H, 3 J = 7.2 Hz, H-6), 5.97 (t, 1H, 3 J = 7.2 Hz, H-5), 7.3–7.5 (m, 10H, C6 H 5), 7.70/7.72* (d, 1H, 3 J = 5.5 Hz, H-3′), 8.14/8.15* (d, 1H, 3 J = 6.5 Hz, H-2′), 8.43 (s, 1H, H-8′) ppm. *Chemical shifts could not be unequivocally assigned.
(E)-4: 13C NMR (DMSO-d 6): 41.45 (C-6), 73.63 (OCH2-3), 74.01 (OCH2-2), 109.70 (C-5), 114.83/114.88* (C-3′), 123.13/124.67* (C-2), 126.29/126.45* (C-5′), 127.9–129.0 (C 6H5), 132.77 (C-6′), 135.4–135.7 (C-2a, C-3a), 140.75/140.77* (C-2′), 141.83 (C-4), 148.53 (C-8′), 148.94 (C-3), 150.99 (C-4′), 163.65/163.79* (C-1) ppm. *Chemical shifts between E and Z isomers could not be distinguished.
3.2.3. Method B. General procedure for the preparation of 3-deazapurine derivatives of imino-l-ascorbic acid (5–7)
Ammonia was induced in a stirred solution of compounds 1, 2 or 4 (0.11–0.25 mmol), in anhydrous methanol (5–10 ml) and 1,4-dioxane (5–10 ml) in ice-cold bath (0 °C). After saturation of ammonia, mixture was additionally stirred overnight at room temperature. Methanol, 1,4-dioxane and surplus ammonia were removed at reduced pressure. Purification of crude residue by silica gel column chromatography using dichloromethane:methanol = 10:1 gave the desired compounds 5–7, respectively as solids.
3.2.3.1. 3,4-di-O-benzyl-5-hydroxy-5-(7-(6-chloro-3H-imidazo[4,5-c]pyridine-7-yl)ethyl)-1H-pyrrolo-2(5H)-one (5)
According to procedure B, ammonolysis of compound 3 (96.2 mg; 0.20 mmol) gave crude residue which was purified by silica gel column chromatography (dichloromethane:methanol = 30:1) and gave compound 5 as beige solid (16.5 mg; 16.81%, m.p. = 216–218 °C). MS (ESI): m/z = 491.3 ([M+H]+). Anal. Calcd. for C26H23ClN4O4 (490.14): C, 63.61; H, 4.72; N, 11.41. Found: C, 63.69; H, 4.72; N, 11.42.
1H NMR (DMSO-d6): δ 2.22 (t, 2H, 3 J = 7.6 Hz, H-5), 4.45 (m, 2H, H-6), 5.00 (d, 1H, 2 J = 11.2 Hz, OCH 2-2), 5.01 (d, 1H, 2 J = 12.0 Hz, OCH 2-3), 5.06 (d, 1H, 2 J = 11.2 Hz, OCH 2-2), 5.14 (d, 1H, 2 J = 12.0 Hz, OCH 2-3), 6.26 (s, 1H, OH-4), 7.2–7.4 (m, 10H, C6 H 5), 7.69 (d, 1H, 3 J = 5.5 Hz, H-3′), 8.12 (d, 1H, 3 J = 5.5 Hz, H-2′), 8.28 (s, 1H, NHimino-l-AA), 8.39 (s, 1H, H-8′) ppm.
13C NMR (DMSO-d6): 38.60 (C-5), 41.64 (C-6), 72.10 (OCH2-3), 73.27 (OCH2-2), 81.74 (C-4), 114.79 (C-3′), 123.12 (C-2), 127.60 (C-5′), 127.6–128.7 (C 6H5), 136.23 (C-2a), 132.66 (C-6′), 136.70 (C-3a), 140.54 (C-2′), 149.03 (C-8′), 151.01 (C-4′), 153.23 (C-3), 167.96 (C-1) ppm.
3.2.3.2. 3,4-di-O-benzyl-5-(9-(3-bromo-1H-imidazo[4,5-c]pyridine-9-yl)ethyl)-5-hydroxy-1H-pyrrolo-2(5H)-one (6)
Ammonolysis of compound 1 (58.0 mg; 0.11 mmol) in accordance with procedure B, gave crude residue which was purified by silica gel column chromatography (dichloromethane:methanol = 10:1) and resulted in isolation of compound 6 in the form of transparent oil (9.7 mg; 16.47%). HRMS calcd. for C26H23BrN4O4: m/z = 535.0967 ([M+H]+). Found MS (TOF): m/z = 535.0962 ([M+H]+).
1H NMR (DMSO-d6): δ 2.22 (t, 2H, 3 J = 7.4 Hz, H-5), 4.43 (m, 2H, H-6), 5.00 (d, 1H, 2 J = 11.1 Hz, OCH 2-2), 5.02 (d, 1H, 2 J = 12.0 Hz, OCH 2-3), 5.07 (d, 1H, 2 J = 11.1 Hz, OCH 2-2), 5.13 (d, 1H, 2 J = 12.0 Hz, OCH 2-3), 6.30 (s, 1H, OH-4), 7.18–7.50 (m, 10H, C6 H 5), 8.27 (s, 1H, NHimino-l-AA), 8.30 (s, 1H, H-8′), 8.42 (s, 1H, H-2′), 8.91 (s, 1H, H-6′) ppm.
13C NMR (DMSO-d6): 38.41 (C-5), 41.09 (C-6), 72.05 (OCH2-3), 73.26 (OCH2-2), 81.72 (C-4), 100.85 (C-3′), 123.11 (C-2), 127.6–128.7 (C 6H5), 135.61 (C-4′), 136.25, 136.70 (C-2a, C-3a), 141.30 (C-6′), 142.22 (C-5′), 143.52 (C-2′), 147.85 (C-8′), 153.16 (C-3), 167.92 (C-1) ppm.
3.2.3.3. 5-(9-(6-amino-3-chloro-2-fluoro-1H-imidazo[4,5-c]pyridine-9-yl)ethyl)-3,4-di-O-benzyl-5-hydroxy-1H-pyrrolo-2(5H)-one (7)
In accordance to procedure B, ammonolysis of compound 2 (130.0 mg; 0.25 mmol) gave crude residue which was purified by silica gel column chromatography (dichloromethane:methanol = 10:1) which resulted in isolation of the compound 7 as white solid (24.3 mg; 18.55%; m.p. = 199–202 °C). MS (ESI): m/z = 524.2 ([M+H]+). Anal. Calcd. for C26H23ClFN5O4 (523.14): C, 59.60; H, 4.42; N, 13.37. Found: C, 59.71; H, 4.41; N, 13.35.
1H NMR (DMSO-d6): δ 2.18 (t, 2H, 3 J = 7.5 Hz, H-5), 4.30 (m, 2H, H-6), 5.00 (d, 1H, 2 J = 11.2 Hz, OCH 2-2), 5.00 (d, 1H, 2 J = 11.8 Hz, OCH 2-3), 5.06 (d, 1H, 2 J = 11.2 Hz, OCH 2-2), 5.13 (d, 1H, 2 J = 11.8 Hz, OCH 2-3), 6.25 (s, 1H, OH-4), 6.84 ppm (s, 2H, NH2-6′), 7.22–7.43 (m, 10H, C6 H 5), 7.92 (s, 1H, H-8′), 8.25 (s, 1H, NHimino-l-AA) ppm.
13C NMR (DMSO-d6): 38.55 (C-5), 41.05 (C-6), 72.03 (OCH2-3), 73.21 (OCH2-2), 81.64 (C-4), 82.67 (d, 2JCF = 44.0 Hz, C-3′), 123.06 (C-2), 126.02 (d, 4 J CF = 2.2, C-5′), 127.59–128.63 (C 6H5), 136.10 (d, 3 J CF = 7.1 Hz, C-4′), 136.20, 136.69 (C-2a, C-3a), 143.86 (C-8′), 148.48 (d, 3 J CF = 20.9 Hz, C-6′), 153.67 (d, 1 J CF = 219.2 Hz, C-2′), 153.13 (C-3), 167.88 (C-1) ppm.
3.2.4. Procedure for the preparation of 5-(2-(7-chloro-4,6-difluoro-3H-imidazo[4,5-c]pyridine-3-yl)ethylidene)-3,4-dihydroxyfuran-2(5H)-one (8)
To solution of compound 3 (117.3 mg; 0.23 mmol) in anhydrous dichloromethane (3 ml), BCl3 (0.76 ml; 0.76 mmol) was added dropwise in 10 portions at temperature −70 °C under argon atmosphere and mixture was stirred for 30 min. Temperature raised gradually to room temperature and reaction solution stirred overnight. Reaction was terminated by adding solution of dichloromethane:methanol in ratio 1:1 (10 ml) and concentrated by evaporation in vacuo. Purification of crude product mixture by silica gel column chromatography (CH2Cl2:MeOH = 5:1) gave of compound 8 as white oil (Z:E = 10:1; 15.3 mg; 20.18%). MS (ESI): m/z = 330.0 ([M+H]+). Anal. Calcd. for C12H6ClF2N3O4 (329.0): C, 43.72; H, 1.83; N, 12.75. Found: C, 43.77; H, 1.83; N, 12.75.
1H NMR (DMSO-d6): δ 5.22 (d, 2H, 3 J = 7.2 Hz, H-6), 5.54 (t, 2H, 3 J = 7.2 Hz, H-5), 8.74 (s, 1H, H-8′) ppm.
13C NMR (DMSO-d6): ≈42 (C-6), ≈99 (C-5), ≈116 (C-5′), ≈143 (C-4), ≈143 (C-3), ≈150 (C-8′) ppm. Chemical shifts of C-1, C-2, C-3′, C-2′, C-6′ carbons were not detectable because of the small quantity of the compound and it also decomposes over time.
3.2.5. Procedure for the preparation of 7-bromo-7-deazapurine derivative of 4,5-didehydro-5,6-dideoxy-l-ascorbic acid (3,4-di-O-benzyl-5-(1-(7-bromo-3H-pyrrolo[2,3-d]pyrimidine-1-yl)ethylidene)furan-2(5H)-one, 9)
Suspension of anhydrous 7-bromo-7-deazapurine base (D, 500.0 mg; 2.52 mmol) and (NH4)2SO4 (33.3 mg; 0.25 mmol) in HMDS (25 ml) was refluxed overnight under inert atmosphere of argon. Excess HMDS was evaporated in vacuo. Suspension of silylated base D and dAdBAA (727.2 mg; 1.67 mmol) in dry 1,2-dichloroethane (25 ml) was stirred under inert atmosphere of argon at room temperature. TMSOTf (1.5 eq.) was then added to the mixture and was heated at 55–70 °C and stirred overnight. Crude product was obtained after evaporating the solvent in vacuo, and extracted with CH2Cl2 and saturated solution of NaHCO3. Organic layer was dried over MgSO4 and concentrated in vacuo. Compound 9 (8.4 mg; 0.97%; m.p. = 123–125 °C) was obtained as greenish-yellow solid by purification of solid product by silica gel column chromatography (dichloromethane:methanol = 70:1) and rechromatography (dichloromethane:methanol = 50:1). HRMS Calcd. for C26H20BrN3O4: m/z = 518.0710 ([M+H]+). Found MS (TOF): m/z = 518.0704 ([M+H]+).
(Z)-9: 1H NMR (DMSO-d 6): δ 5.16 (s, 2H, OCH 2-2), 5.27 (d, 2H, 2 J = 7.1 Hz, H-6), 5.31 (s, 2H, OCH 2-3), 5.85 (t, 1H, 3 J = 7.1 Hz, H-5), 7.31–7.47* (m, 11H, C6 H 5, H-8′), 8.74 (d, 1H, 2 J = 1.9 Hz, H-2′), 8.92 (d, 1H, H-6′) ppm. *Chemical shift of H-8′ proton is overlapped with phenyl (C6 H 5) protons.
(Z)-9: 13C NMR (DMSO-d 6): δ 50.07 (C-6), 73.04 (OCH2-3), 74.05 (OCH2-2), 86.82 (C-7′), 102.51 (C-5), 119.20 (C-5′), 123.48 (C-2), 123.72 (C-8′), 127.9–128.8 (C 6H5), 135.31, 135.68 (C-2a, C-3a), 136.06 (C-6′), 141.90 (C-2′), 143.40 (C-4), 147.93 (C-3), 155.93 (C-4′), 163.58 (C-1) ppm.
3.2.6. Procedure for the preparation of 6-chloro-7-iodo-7-deazapurine derivative of 4,5-didehydro-5,6-dideoxy-l-ascorbic acid (3,4-di-O-benzyl-5-(9-(6-chloro-7-iodo-7H-pyrrolo[2,3-d]pyrimidine-9-yl)ethylidene)furan-2(5H)-one, 10)
Solution of triphenyl-phosphine (PPh3, 241.3 mg; 0.92 mmol), diethyl azodicarboxylate (DEAD, 0.18 ml; 0.92 mmol) in absolute dry tetrahydrofuran (THF, 3.8 ml) was stirred under inert argon atmosphere for one hour at −40 to −50 °C. Solution of 6-chloro-7-iodo-7-deazapurine (E, 140.0 mg; 0.50 mmol) and THF (1.5 ml) was then added to reaction mixture and stirred for another 1 h at −40 to −50 °C temperature, under inert atmosphere of argon. 6-Hydroxy-2,3-di-O-benzyl-4,5-didehydro-l-ascorbic acid (HdBAA, 185.4 mg; 0.55 mmol) was dissolved in absolute dry THF (1.8 ml) and added to the mixture which was then stirred at −40 to −50 °C which was risen gradually to room temperature and stirred overnight. Crude reaction product was obtained by evaporation of raw reaction mixture in vaccuo and was purified by to silica gel column chromatography (dichloromethane:methanol = 175:1) and rechromatoraphy (dichloromethane:methanol = 120:1) which resulted in gaining white powder of compound 10 (Z:E = 9:1; 93.0 mg; 30.95%; m.p. = 114–117 °C). MS (ESI): m/z = 599.1 ([M]+). Anal. Calcd. for C26H19ClIN3O4 (599.01): C, 52.06; H, 3.19; N, 7.01. Found: C, 52.11; H, 3.19; N, 7.00.
1H NMR (DMSO-d6): δ 4.83 (d, 2H, 2 J = 6.0 Hz, H-6), 5.04 (s, 2H, OCH 2-2), 5.31 (s, 2H, OCH 2-3), 6.68–6.72 (m, 1H, H-5), 7.22–7.42 (m, 10H, C6 H 5), 8.50 (s, 1H, H-8′), 8.73 (s, 1H, H-2′) ppm.
13C NMR (DMSO-d6): δ 69.15 (C-6), 71.05 (C-7′), 73.50′ (OCH2-3), 74.34 (OCH2-2), 113.54 (C-5), 123.01 (C-8′), 128.01–129.02 (C 6H5), 129.42 (C-2′), 135.58, 135.82 (C-2a, C-3a), 141.57 (C-4′), 151.99 (C-3), 153.81 (C-6′), 167.02 (C-1) ppm. *Chemical shifts of quarter carbons C-2, C-4 and C-5′ are not detectable in the spectrum.
3.2.7. Method C. General procedure for the preparation of 7-deazapurine derivatives of imino-l-ascorbic acid (11, 12)
Ammonia was induced in stirred solution of compounds 9 or 10 (0.13–0.14 mmol) and anhydrous methanol (2.0–2.25 ml) and dioxane (2.0–2.25 ml) in ice-cold bath (0 °C). After saturation with ammonia, mixture was stirred overnight at room temperature. Methanol, 1,4-dioxane and surplus ammonia were removed in vacuum. Purification of crude residue by silica gel column chromatography using dichloromethane:methanol = 10:1 gave compounds 11 and 12 as transparent oil and as white powder, respectively.
3.2.7.1. 3,4-di-O-benzyl-5-(2-(5-bromo-3H-pyrrolo[2,3-d]pyrimidine-ethyl)-5-hydroxy-1H-pyrrolo-2(5H)-one (11)
According to procedure C, ammonolysis of compound 9 (65.0 mg; 0.14 mmol) gave solid residue which was purified by silica gel column chromatography (dichloromethane:methanol = 10:1) which gave compound 11 as transparent oil (1.0 mg; 3.24%). MS (ESI): m/z = 535.2 ([M+H]+). Anal. Calcd. for C26H23BrN4O4 (534.09): C, 58.33; H, 4.33; N, 7.01. Found: C, 58.37; H, 4.32; N, 6.99.
1H NMR (DMSO-d6): δ 2.21 (m, 1H, H-5), 2.40 (m, 1H, H-5), 4.44 (m, 2H, H-6), 4.99 (s, 2H, OCH 2-2), 5.02 (d, 1H, 2 J = 11.9 Hz, OCH 2-3), 5.11 (d, 1H, 2 J = 11.9 Hz, OCH 2-3), 6.25 (s, 1H, OH-4), 7.2–7.5* (m, 11H, C6 H 5, H-8′), 8.30 (s, 1H, NHimino-l-AA), 8.75 (m, 1H, H-2′), 8.94 (m, 1H, H-6′) ppm. *Chemical shift of H-8′ proton is overlapped with phenyl (C6 H 5) protons.
13C NMR (DMSO-d6): δ 37.7 (C-5), 51.3 (C-6), 72.12 (OCH2-3), 73.37 (OCH2-2), 81.82 (C-4), 86.5 (C-7′), 121.5 (C-5′), 123.0 (C-8′), 123.25 (C-2), 127.6–128.4 (C 6H5), 136.13 (C-2a), 136.4 (C-6′), 136.71 (C-3a), 142.0 (C-2′), 153.21 (C-3), 155.5 (C-4′), 167.85 (C-1) ppm.
3.2.7.2. 3,4-di-O-benzyl-5-(9-(6-chloro-7-iodo-7H-pyrrolo[2,3-d]pyrimidine-9-yl)-vinyl)-5-hydroxy-1H-pyrrolo-2(5H)-one (12)
According to procedure C, ammonolysis of compound 10 (80.0 mg; 0.13 mmol) gave crude residue which was purified by silica gel column chromatography (dichloromethane:methanol = 40:1) and resulted in isolation of compound 12 as white powder (13.2 mg; 16.05%; m.p. = 165–168 °C). MS (ESI): m/z = 615.2 ([M+H]+). Anal. Calcd. for C26H20ClIN4O4 (614.02): C, 50.79; H, 3.28; N, 9.11. Found: C, 50.90; H, 3.28; N, 9.09.
(E)-12: 1H NMR (DMSO-d 6): δ 5.01 (s, 2H, OCH 2-2), 5.15 (d, 1H, 2 J = 12.2 Hz, OCH 2-3), 5.19 (d, 1H, 2 J = 12.2 Hz, OCH 2-3), 6.40 (dd, 1H, 3 J = 14.3 Hz, 4 J = 0.7 Hz, H-5), 6.72 (d, 1H, 4 J = 0.7 Hz, OH-4), 7.25–7.45 (m, 10H, C6 H 5), 7.62 (d, 1H, 3 J = 14.3 Hz, H-6), 8.46 (s, 1H, NHimino-l-AA), 8.50 (s, 1H, H-8′), 8.73 (s, 1H, H-2′) ppm.
(E)-12: 13C NMR (DMSO-d 6): δ 56.17 (C-7′), 72.02 (OCH2-3), 73.27 (OCH2-2), 81.23 (C-4), 116.90 (C-5′), 117.26 (C-5), 122.59 (C-6), 123.54 (C-2), 127.4–128.4 (C 6H5), 132.92 (C-8′), 136.38, 136.72 (C-2a, C-3a), 149.66 (C-4′), 151.32 (C-2′), 151.43 (C-6′), 153.50 (C-3), 167.74 (C-1) ppm.
3.2.8. Method D. General procedure for the preparation of 9-deazapurine derivatives of 4,5-didehydro-5,6-dideoxy-l-ascorbic acid (13–15)
Solution of triphenyl-phosphine (PPh3, 0.66 eq.), diethyl azodicarboxylate (DEAD, 0.66 eq.) in absolute dry tetrahydrofuran (THF, 4.0 ml) was stirred under inert argon atmosphere for one hour at −40 to −50 °C. Solution of 7-deazapurine bases (F or G, 1 eq.) and THF (4.5 ml) was then added to reaction mixture and stirred an hour at −40 to −50 °C, under inert atmosphere of argon. 6-Hydroxy-2,3-di-O-benzyl-4,5-didehydro-l-ascorbic acid (HdBAA, 0.9–1.0 eq.) was soluted in absolute dry THF (5.0 ml) and was added to the mixture and stirred at −40 to −50 °C which was gradually risen to room temperature and stirred overnight. Crude reaction product was retained by evaporation of the raw reaction mixture in vacuo and was purified by silica gel column chromatography (n-hexane:ethyl-acetate = 2:1, dichloromethane:methanol = 175:1) and rechromatography (n-hexane:ethyl-acetate = 1:1, dichloromethane:methanol = 120:1) which gave compounds 13–15.
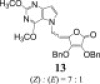
3.2.8.1. 3,4-di-O-benzyl-5-(7-(2,4-dimethoxy-5H-pyrrolo[3,2-d]pyrimidine-7-yl)ethylidene)furan-2(5H)-one (13)
According to procedure D, solution of 2,6-dimethoxy-9-deazapurine (F, 250.0 mg; 1.4 mmol), PPh3 (587.5 mg; 2.24 mmol), DEAD (0.44 ml; 2.24 mmol) and HdBAA (410.0 mg; 1.2 mmol) in THF (13.5 ml) was stirred under inert argon atmosphere at −40 to −50 °C for 3 h and then at room temperature overnight. Crude reaction product was purified by silica gel column chromatography (n-hexane:ethyl-acetate = 2:1) and rechromatoraphy (n-hexane:ethyl-acetate = 1:1) which resulted in isolation of transparent oil of compound 13 (106.0 mg; 18.18%). MS (ESI): m/z = 500.3 ([M+H]+). Anal. Calcd. for C28H25N3O6 (499.17): C, 67.33; H, 5.04; N, 8.41. Found: C, 67.18; H, 5.05; N, 8.42.
(Z)-13: 1H NMR (DMSO-d 6): δ 3.84 (s, 3H, OCH 3-2′), 3.94 (s, 3H, OCH 3-6′), 5.10 (d, 2H, 3 J = 7.4 Hz, H-6), 5.16 (s, 2H, OCH 2-2), 5.29 (s, 2H, OCH 2-3), 5.47 (t, 1H, 3 J = 7.4 Hz, H-5), 6.34 (d, 1H, 3 J = 3.0 Hz, H-9′), 7.22–7.50 (m, 10H, C6 H 5), 7.58 (d, 1H, 3 J = 3.0 Hz, H-8′) ppm.
(Z)-13: 13C NMR (DMSO-d 6): δ 43.25 (C-6), 53.45 (OCH3-6′), 53.95 (OCH3-2′), 72.95 (OCH2-3), 73.90 (OCH2-2), 100.83 (C-9′), 104.73 (C-5), 111.27 (C-5′), 123.09 (C-2), 127.5–129.3 (C 6H5), 134.09 (C-8′), 135.36, 135.69 (C-2a, C-3a), 141.94 (C-4), 148.09 (C-3), 151.62 (C-4′), 157.03 (C-6′), 159.45 (C-2′), 163.79 (C-1) ppm.
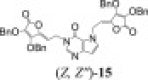
3.2.8.2. 3,4-di-O-benzyl-5-(1-(3H-pyrrolo[3,2-d]pyrimidine-4(5H)-one-1-yl)ethylidene)furan-2(5H)-one (14) and 1,7-di-(5-(3,4-di-O-benzyl-furan-2(5H)-one)etiliden)-3H-pirolo[3,2-d]pyrimidine-4(5H)-on (15)
According to procedure D, solution of 9-deazahypoxanthine (G, 200.0 mg; 1.48 mmol), PPh3 (587.5 mg; 2.24 mmol), DIAD (0.44 ml; 2.24 mmol) and HdBAA (500.0 mg; 1.48 mmol) in THF (13.5 ml) was stirred under inert argon atmosphere at −40 to −50 °C for 3 h and overnight at room temperature. Purification of crude product by silica gel column chromatography (n-hexane:ethyl-acetate = 1:1) and rechromatography with the same eluent, gave white powders of compounds 14 (47.6 mg; 28.81%; m.p. = 114–117 °C) and 15 (97.1 mg; 8.29%; m.p. = 54–57 °C). (Z)-14: MS (ESI): m/z = 457.3 ([M]+). Anal. Calcd. for C26H23N3O5 (457.16): C, 68.26; H, 5.07; N, 9.19. Found: C, 68.31; H, 5.06; N, 9.21. (Z, Z″)-15: MS (ESI): m/z = 776.4 ([M+H]+). Anal. Calcd. for C46H37N3O9 (775.25): C, 71.22; H, 4.81; N, 5.42. Found: C, 71.20; H, 4.81; N, 5.43.
(Z)-14: 1H NMR (DMSO-d 6): δ 4.82 (d, 2H, 3 J = 6.9 Hz, H-6), 5.14 (s, 2H, OCH 2-2), 5.29 (s, 2H, OCH 2-3), 5.54 (t, 1H, 3 J = 6.9 Hz, H-5), 6.37 (dd, 1H, 3 J = 2.7, 1.9 Hz, H-9′), 7.25–7.45 (m, 10H, C6 H 5), 7.38 (m, 1H, H-8′), 8.10 (s, 1H, H-2′), 12.11 (s, 1H, NH-7′) ppm.
(Z)-14: 13C NMR (DMSO-d 6): δ 40.5 (C-6), 72.93 (OCH2-3), 74.00 (OCH2-2), 102.96 (C-9′), 104.39 (C-5), 116.99 (C-5′), 122.91 (C-2), 127.8 (C-8′), 127.9–128.8 (C 6H5), 135.38, 135.73 (C-2a, C-3a), 142.23 (C-4), 144.07 (C-4′), 144.19 (C-2′), 148.12 (C-3), 152.98 (C-6′), 163.75 (C-1) ppm.
(Z, Z′)-15: 1H NMR (DMSO-d 6): δ 4.79 (d, 2H, 3 J = 6.8 Hz, H-6″), 5.13* (s, 2H, OCH 2-2, OCH 2-2″), 5.14* (s, 2H, OCH 2-2, OCH 2-2″), 5.24 (d, 2H, 3 J = 7.1 Hz, H-6), 5.29 (s, 2H, OCH 2-3), 5.29 (s, 2H, OCH 2-3″), 5.53 (t, 1H, 3 J = 6.8 Hz, H-5″), 5.56 (t, 1H, 3 J = 7.1 Hz, H-5), 6.36 (d, 1H, 3 J = 3.0 Hz, H-9′), 7.2–7.5 (m, 20H, C6 H 5, C6 H 5″), 7.46 (d, 1H, 3 J = 3.0 Hz, H-8′), 8.10 (s, 1H, H-2′) ppm. *Chemical shifts could not be unequivocally assigned.
(Z, Z″)-15: 13C NMR (DMSO-d 6): δ 40.3 (C-6), 42.71 (C-6″), 72.94 (OCH2-3), 72.94 (OCH2-3″), 73.97/73.99* (OCH2-2, OCH2-2″), 102.80 (C-9′), 104.38 (C-5), 104.85 (C-5″), 115.99 (C-5′), 122.94/123.14* (C-2, C-2″), 127.9–128.8 (C 6H5, C 6H5″), 131.60 (C-8′), 135.35, 135.73 (C-2a, C-2a″, C-3a, C-3a″), 141.91 (C-4″), 142.20 (C-4), 144.49 (C-4′), 144.78 (C-2′), 148.04/148.07* (C-3, C-3″), 153.26 (C-6′), 163.70 (C-1), 163.70 (C-1″) ppm. * Chemical shifts could not be unequivocally assigned.
3.2.9. Method E. General procedure for the preparation of 9-deazapurine derivatives of imino-l-ascorbic acid (16–19)
Ammonia was induced in stirred solution of compounds 13–15 (0.06–0.21 mmol) and anhydrous methanol (1.5–3.0 ml) and dioxane (1.0–3.0 ml) in ice-cold bath (0 °C). After saturation of ammonia, mixture was stirred overnight at room temperature. Methanol, 1,4-dioxane and surplus ammonia were removed in vacuum. Purification of crude residue by silica gel column chromatography using dichloromethane:methanol (25:1 or 15:1 ratio), obtained compounds 16–19 as solids, respectively.
3.2.9.1. 3,4-di-O-benzyl-5-hydroxy-5-(7-(2,4-dimethoxy-5H-pyrrolo[3,2-d]pyrimidine-7-yl)ethyl)-1H-pyrrolo-2(5H)-one (16)
Solution of compound 13 (92.5 mg; 0.19 mmol) in accordance with method D gave crude residue which was purified by silica gel column chromatography using dichloromethane:methanol = 25:1 and resulted in isolation of white powder of compound 16 (31.65 mg; 33.01%; m.p. = 85–88 °C). HRMS Calcd. for C28H28N4O6: m/z = 517.2081 ([M+H]+). Found MS (TOF): m/z = 517.2084 ([M+H]+).
1H NMR (DMSO-d6): δ 2.12 (m, 2H, H-5), 3.83 (s, 3H, OCH 3-2′), 3.91 (s, 3H OCH 3-6′), 4.16 (t, 2H, 3 J = 7.5 Hz, H-6), 4.94 (d, 1H, 2 J = 11.9 Hz, OCH 2-3), 4.97 (s, 2H, OCH 2-2), 5.11 (d, 1H, 2 J = 11.9 Hz, OCH 2-3), 6.17 (s, 1H, OH-4), 6.31 (d, 1H, 3 J = 3.0 Hz, H-9′), 7.20–7.40 (m, 10H, C6 H 5), 7.41 (d, 1H, 3 J = 3.0 Hz, H-8′), 8.18 (s, 1H, NHimino-l-AA) ppm.
13C NMR (DMSO-d6): δ 38.25 (C-5), 44.04 (C-6), 53.46 (OCH3-6′), 53.89 (OCH3-2′), 71.92 (OCH2-3), 73.14 (OCH2-2), 81.72 (C-4), 100.20 (C-9′), 111.22 (C-5′), 123.06 (C-2), 127.4–128.5 (C 6H5), 133.90 (C-8′), 136.32, 136.75 (C-2a, C-3a), 151.39 (C-4′), 153.11 (C-3), 156.88 (C-6′), 159.29 (C-2′), 167.93 (C-1) ppm.
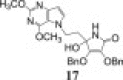
3.2.9.2. 3,4-di-O-benzyl-5-hydroxy-5-(1-(3H-pyrrolo[3,2-d]pyrimidine-4(5H)-one-1-yl)ethyl)-1H-pyrrolo-2(5H)-one (17)
In accordance with method D, ammonolysis of compound 14 (94.0 mg; 0.21 mmol) resulted in gaining crude residue which was purified by silica gel column chromatography using dichloromethane:methanol = 15:1 and white powder of compound 17 was obtained (28.5 mg; 28.73%; m.p. = 205–208 °C). HRMS Calcd. for C26H24N4O5: m/z = 495.1639 ([M+Na]+). Found MS (TOF): m/z = 495.1638 ([M+Na]+).
1H NMR (DMSO-d6): δ 2.08 (m, 2H, H-5), 3.95 (m, 2H, H-6), 5.01 (d, 1H, 2 J = 11.8 Hz, OCH 2-3), 5.02 (s, 2H, OCH 2-2), 5.13 (d, 1H, 2 J = 11.8 Hz, OCH 2-3), 6.16 (s, 1H, OH-4), 6.36 (d, 1H, 3 J = 2.3 Hz, H-9′), 7.20–7.45 (m, 10H, C6 H 5), 7.37 (m, 1H, H-8′), 7.93 (s, 1H, H-2′), 8.23 (s, 1H, NHimino-l-AA), 12.10 (s, 1H, NH-7′) ppm.
13C NMR (DMSO-d6): δ 36.24 (C-5), 41.05 (C-6), 72.04 (OCH2-3), 73.27 (OCH2-2), 81.90 (C-4), 102.80 (C-9′), 117.18 (C-5′), 123.08 (C-2), 127.5 (C-8′), 127.7–128.6 (C 6H5), 136.30, 136.77 (C-2a, C-3a), 143.94 (C-4′), 144.34 (C-2′), 153.16 (C-6′), 153.38 (C-3), 167.96 (C-1) ppm.
3.2.9.3. 1-(5-(3,4-di-O-benzyl-5-hydroxy-furan-2(5H)-one)ethyl)-7-(5-(3,4-di-O-benzyl-5-hydroxy-furan-2(5H)-one)ethyl)-3H-pyrrolo[3,2-d]pyrimidine-4(5H)-one (18 and 19)
In accordance with method D, ammonolysis of compound 15 (43.2 mg; 0.06 mmol) resulted in gaining crude residue which was purified by silica gel column chromatography using dichloromethane:methanol = 15:1 and white powders of compounds 18 (11.45 mg; 50.44%; m.p. = 103–106 °C) and 19 (11.78 mg; 51.89%; m.p. = 101–103 °C) were isolated. 18: HRMS Calcd. for C46H43N5O9: m/z = 832.2952 ([M+Na]+). Found MS (TOF): m/z = 832.2951 ([M+Na]+). 19: HRMS Calcd. for C46H43N5O9: m/z = 832.2952 ([M+Na]+). Found MS (TOF): m/z = 832.2946 ([M+Na]+).
18: 1H NMR (DMSO-d6): δ 2.07 (m, 2H, H-5″), 2.18 (m, 2H, H-5), 3.92 (t, 2H, 3 J = 7.2 Hz, H-6″), 4.31 (m, 2H, H-6), 4.95–5.15 (m, 4H, OCH 2-3, OCH 2-3″), 5.01 (s, 2H, OCH 2-2), 5.01 (s, 2H, OCH 2-2″), 6.15 (s, 1H, OH-4), 6.17 (s, 1H, OH-4″), 6.30 (d, 1H, 3 J = 2.9 Hz, H-9′), 7.20–7.40 (m, 20H, C6 H 5, C6 H 5″), 7.26 (d, 1H, 3 J = 2.9 Hz, H-8′), 7.91 (s, 1H, H-2′), 8.15 (s, 1H, NHimino-l-AA), 8.24 (s, 1H, NH″imino-l-AA) ppm.
18: 13C NMR (DMSO-d6): δ 36.19 (C-5), 38.6 (C-5″), 41.21 (C-6), 43.88 (C-6″), 71.96/72.04* (OCH2-3, OCH2-3″), 73.29 (OCH2-2), 73.29 (OCH2-2″), 81.75 (C-4″), 81.91 (C-4), 101.97 (C-9′), 116.15 (C-5′), 123.08/123.12* (C-2, C-2″), 127.6–128.5 (C 6H5, C 6H5″), 131.44 (C-8′), 136.30, 136.77 (C-2a, C-2a″, C-3a, C-3a″), 144.39 (C-4′), 144.70 (C-2′), 153.17 (C-6′), 153.34/153.40* (C-3, C-3″), 167.95 (C-1), 167.95 (C-1″) ppm. *Chemical shifts could not be unequivocally assigned.
19: 1H NMR (DMSO-d6): δ 2.07 (m, 2H, H-5″), 2.17 (m, 2H, H-5), 3.91 (t, 2H, 3 J = 7.3 Hz, H-6″), 4.31 (m, 2H, H-6), 4.95–5.20 (m, 4H, OCH 2-3, OCH 2-3″), 5.01 (s, 2H, OCH 2-2), 5.01 (s, 2H, OCH 2-2″), 6.14 (s, 1H, OH-4), 6.16 (s, 1H, OH-4″), 6.30 (d, 1H, 3 J = 2.9 Hz, H-9′), 7.20–7.50 (m, 20H, Ph, Ph″), 7.27 (d, 1H, 3 J = 2.9 Hz, H-8′), 7.91 (s, 1H, H-2′), 8.15 (s, 1H, NHimino-l-AA), 8.24 (s, 1H, NH″imino-l-AA) ppm.
19: 13C NMR (DMSO-d6): δ 36.16 (C-5), 38.6 (C-5″), 41.17 (C-6), 43.86 (C-6″), 71.95/72.03* (OCH2-3, OCH2-3″), 73.26 (OCH2-2), 73.26 (OCH2-2″), 81.75 (C-4″), 81.90 (C-4), 101.95 (C-9′), 116.14 (C-5′), 123.06/123.09* (C-2, C-2″), 127.6–128.5 (C 6H5, C 6H5″), 131.41 (C-8′), 136.29, 136.76 (C-2a, C-2a″, C-3a, C-3a″), 144.37 (C-4′), 144.68 (C-2′), 153.15 (C-6′), 153.33/153.38* (C-3, C-3″), 167.93 (C-1), 167.93 (C-1″) ppm. *Chemical shifts could not be unequivocally assigned.
3.3. Biological tests
3.3.1. Cytostatic and antiviral activity assays
The compounds were evaluated against the following viruses: herpes simplex virus type 1 (HSV-1) strain KOS, thymidine kinase-deficient (TK-) HSV-1 KOS strain resistant to ACV (ACVr), herpes simplex virus type 2 (HSV-2) strains Lyons and G, varicella-zoster virus (VZV) strain Oka, TK- VZV strain 07-1, human cytomegalovirus (HCMV) strains AD-169 and Davis, vaccinia virus Lederle strain, human immunodeficiency virus (HIV) type 1 (IIIB) and type 2 (ROD), respiratory syncytial virus (RSV) strain Long, vesicular stomatitis virus (VSV), Coxsackie B4, Parainfluenza 3, Reovirus-1, Sindbis, Punta Toro, feline coronavirus (FIPV), influenza A virus subtypes H1N1 and H3N2, and influenza B virus. The antiviral assays, other than HIV, were based on inhibition of virus-induced cytopathicity or plaque formation in human embryonic lung (HEL) fibroblasts, African green monkey kidney cells (Vero), human epithelial cervix carcinoma cells (HeLa), Crandell-Rees feline kidney cells (CRFK), or Madin Darby canine kidney cells (MDCK). Confluent cell cultures (or nearly confluent for MDCK cells) in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) in the presence of varying concentrations (5-fold compound dilutions) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. The minimal cytotoxic concentration (MCC) of the compounds was defined as the compound concentration that caused a microscopically visible alteration of cell morphology. The methodology of the anti-HIV assays was as follows: human CEM (∼3 × 105 cells/cm3) cells were infected with 100 CCID50 of HIV(IIIB) or HIV-2(ROD)/ml and seeded in 200 μL wells of a microtiter plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37 °C, HIV-induced CEM giant cell formation was examined microscopically.
For the HCMV assays, confluent human embryonic lung (HEL) fibroblasts were grown in 96-well microtiter plates and infected with the human cytomegalovirus (HCMV) strains, AD-169 and Davis at 100 PFU per well. After a 2-h incubation period, residual virus was removed and the infected cells were further incubated with medium containing different concentrations of the test compounds (in duplicate). After incubation for 7 days at 37 °C, virus-induced cytopathogenicity was monitored microscopically after ethanol fixation and staining with Giemsa. Antiviral activity was expressed as the EC50 or compound concentration required to reduce virus-induced cytopathogenicity by 50%. EC50's were calculated from graphic plots of the percentage of cytopathogenicity as a function of concentration of the compounds.
The laboratory wild-type VZV strain Oka and the thymidine kinase-deficient VZV strain 07-1 were used for VZV infections. Confluent HEL cells grown in 96-well microtiter plates were inoculated with VZV at an input of 20 PFU per well. After a 2-h incubation period, residual virus was removed and varying concentrations of the test compounds were added (in duplicate). Antiviral activity was expressed as EC50, or compound concentration required to reduce viral plaque formation by 50% after 5 days as compared with untreated controls.
The cytostatic activity measurements were based on the inhibition of cell growth. HEL cells were seeded at a rate of 5 × 103 cells/well into 96-well microtiter plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37 °C, the cell number was determined with a Coulter counter. The cytostatic concentration was calculated as the CC50, or the compound concentration required to reduce cell proliferation by 50% relative to the number of cells in the untreated controls. CC50 values were estimated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds. Alternatively, cytotoxicity of the test compounds was expressed as the minimum cytotoxic concentration (MCC) or the compound concentration that caused a microscopically detectable alteration of cell morphology.
3.3.2. Cell culturing
Cell lines HeLa (cervical carcinoma), SW620 (colorectal adenocarcinoma, metastatic), MiaPaCa-2 (pancreatic carcinoma), HepG2 (hepatocellular carcinoma), CFPAC-1 (pancreatic adenocarcinoma cells) and 3T3 (mouse embryonic fibroblast cell line), were cultured as monolayers and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere with 5% CO2 at 37 °C.
3.3.3. Antiproliferative assays
The panel cell lines were inoculated onto a series of standard 96-well microtiter plates on day 0, at 5000 cells per well. Test agents were then added in five, 10-fold dilutions (0.01–100 μM) and incubated for further 72 h. Working dilutions were freshly prepared on the day of testing in the growth medium. The solvent (DMSO) was also tested for eventual inhibitory activity by adjusting its concentration to be the same as in the working concentrations (DMSO concentration never exceeded 0.1%). After 72 h of incubation, the cell growth rate was evaluated by performing the MTT assay: experimentally determined absorbance values were transformed into a cell percentage growth (PG) using the formulas proposed by NIH and described previously [25]. This method directly relies on control cells number at the day of assay because it compares the growth of treated cells with the growth of untreated cells in control wells on the same plate – the results are therefore a percentile difference from the calculated expected value.
The IC50 values for each compound were calculated from dose–response curves using linear regression analysis by fitting the mean test concentrations that give PG values above and below the reference value. If, however, all of the tested concentrations produce PGs exceeding the respective reference level of effect (e.g. PG value of 50) for a given cell line, the highest tested concentration is assigned as the default value (in the screening data report that default value is preceded by a “>” sign). Each test point was performed in quadruplicate in three individual experiments. The results were statistically analysed (ANOVA, Tukey post-hoc test at p < 0.05). Finally, the effects of the tested substances were evaluated by plotting the mean percentage growth for each cell type in comparison to control on dose response graphs.
4. Conclusion
In this paper we describe the synthesis of new types of 3-, 7- and 9-deazapurine derivatives of l-ascorbic (1–4, 8–10 and 13–15) or imino-l-ascorbic acid (5–7, 11, 12 and 16–19) and their in vitro effects on the growth of tumour cell lines, normal fibroblasts as well as their antiviral activities. Compounds with the most pronounced antitumoural activities, the 9-deazapurine derivative of l-AA (13) and the 9-deazapurine derivatives of imino-l-AA (15 and 18) will serve as leading molecules for synthetic structure modifications with the aim to develop more selective and more active antitumoural substrates in this class of compounds. In addition to that, compound 3 will also be a model skeleton for developing even more effective anti-HCMV substrate.
Acknowledgements
Support for this study was provided by the Ministry of Science of the Republic of Croatia grants (no. 125-0982464-2922 and 335-0982464-2393), University of Rijeka research support grants (no. 511-10 and 511-21) and KU Leuven (University of Leuven) grant no. GOA 10/14. The authors would like to thank Leentje Persoons, Lies van den Heurck, Steven Carmans, Anita Camps and Lizette van Berckelaer for excellent technical assistance.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2015.08.008.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Legraverend M., Grierson D.S. Bioorg. Med. Chem. 2006;14:3987. doi: 10.1016/j.bmc.2005.12.060. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E., Field H.J. Br. J. Pharmacol. 2006;147:1. doi: 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Clercq E., Holý A. J. Med. Chem. 1979;22:510. doi: 10.1021/jm00191a010. [DOI] [PubMed] [Google Scholar]
- 4.Long M.C., Allan P.W., Luo M.-Z., Liu M.-C., Sartorelli A.C., Parker W.B. J. Antimicrob. Chemother. 2007;59:118. doi: 10.1093/jac/dkl448. [DOI] [PubMed] [Google Scholar]
- 5.Vittori S., Dal Ben D., Lambertucci C., Marucci G., Volpini R., Cristalli G. Curr. Med. Chem. 2006;13:29. doi: 10.2174/092986706779026228. [DOI] [PubMed] [Google Scholar]
- 6.Hasobe M., Mckee D.R., Borcherding D.R., Borchardt R.T. Antimicrob. Agents Chemother. 1987;31:1849. doi: 10.1128/aac.31.11.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg L.M., Chaffee S., Hershfiled S.M. J. Biol. Chem. 1989;15:795. [PubMed] [Google Scholar]
- 8.Ault-Riche D.B., Lee Y., Yuan C.S., Hasobe M., Wolfe M.S., Borcherding D.R., Borchardt R.T. Mol. Pharmacol. 1993;43:989. [PubMed] [Google Scholar]
- 9.Liu M.C., Luo M.Z., Mozdziesz D.E., Lin T.S., Dutschman G.E., Gullen E.A., Cheng Y.C., Sartorelli A.C. Nucleosides Nucleotides Nucleic Acids. 2001;20:1975. doi: 10.1081/NCN-100108327. [DOI] [PubMed] [Google Scholar]
- 10.Migliacco G., Tomassini J.E., Carroll S.S., Tomei L., Altamura S., Bhat B., Bartholomew L., Bosserman M.R., Ceccacci A., Colwell L.F., Cortese R., De Francesco R., Eldrup A.B., Getty K.L., Hou X.S., LaFemina R.L., Ludmerer S.W., MacCoss M., McMasters D.R., Stahlhut M.W., Olsen D.B., Hazuda D.J., Flores O.A. J. Biol. Chem. 2003;49:49164. doi: 10.1074/jbc.M305041200. [DOI] [PubMed] [Google Scholar]
- 11.De Clercq E. Nat. Rev. Drug Discov. 2007;6:1001. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 12.Shi J., Zhou L., Zhang H., McBrayer T.R., Detorio M.A., Johns M., Bassit L., Powdrill M.H., Whitaker T., Coats S.J., Götte M., Schinazi R.F. Bioorg. Med. Chem. Lett. 2011;21:7094. doi: 10.1016/j.bmcl.2011.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu M.-C., Luo M.-Z., Mozdziesz D.E., Sartorelli A.C. Nucleosides Nucleotides Nucleic Acids. 2005;24:45. doi: 10.1081/NCN-46784. [DOI] [PubMed] [Google Scholar]
- 14.Liu M.-C., Luo M.-Z., Mozdziesz D.E., Dutschman G.E., Gullen E.A., Cheng Y.-C., Sartorelli A.C. Nucleosides Nucleotides Nucleic Acids. 2005;24:135. doi: 10.1081/NCN-51898. [DOI] [PubMed] [Google Scholar]
- 15.Raić-Malić S., Svedružić D., Gazivoda T., Marunović A., Hergold-Brundić A., Nagl A., Balzarini J., De Clercq E., Mintas M. J. Med. Chem. 2000;43:4806. doi: 10.1021/jm0009540. [DOI] [PubMed] [Google Scholar]
- 16.Gazivoda T., Šokčević M., Kralj M., Suman L., Pavelić K., De Clercq E., Andrei G., Snoeck R., Balzarini J., Mintas M., Raić-Malić S. J. Med. Chem. 2007;50:4105. doi: 10.1021/jm070324z. [DOI] [PubMed] [Google Scholar]
- 17.Wittine K., Stipković Babić M., Košutić M., Cetina M., Rissanen K., Kraljević Pavelić S., Tomljenović Paravić A., Sedić M., Pavelić K., Mintas M. Eur. J. Med. Chem. 2011;46:2770. doi: 10.1016/j.ejmech.2011.03.066. [DOI] [PubMed] [Google Scholar]
- 18.Wittine K., Stipković Babić M., Makuc D., Plavec J., Kraljević Pavelić S., Sedić M., Pavelić K., Leyssene P., Neyts J., Balzarini J., Mintas M. Bioorg. Med. Chem. 2012;20:3675. doi: 10.1016/j.bmc.2012.01.054. [DOI] [PubMed] [Google Scholar]
- 19.Gazivoda T., Plevnik M., Plavec J., Kraljević S., Kralj M., Pavelić K., Balzarini J., De Clercq E., Mintas M., Raić-Malić S. Bioorg. Med. Chem. 2005;13:131. doi: 10.1016/j.bmc.2004.09.052. [DOI] [PubMed] [Google Scholar]
- 20.Padayatty S.J., Katz A., Wang Y., Eck P., Kwon O., Lee Y.-H., Chen S., Corpe C., Dutta A., Dutta S.K., Levine M. J. Am. Coll. Nutr. 2003;22:18. doi: 10.1080/07315724.2003.10719272. [DOI] [PubMed] [Google Scholar]
- 21.Naidu A.K., Wiranowska M., Kori S.H., Prockop L.D., Kulkarni A.P. Anticancer Res. 1993;13:1469. [PubMed] [Google Scholar]
- 22.Naidu A.K., Wiranowska M., Kori S.H., Prockop L.D., Kulkarni A.P. J. Neuro-Oncol. 1993;16:1. [Google Scholar]
- 23.Makino Y., Sakagami H., Takeda M. Anticancer Res. 1999;19:3125. [PubMed] [Google Scholar]
- 24.Khan M.A., Adams H. Carbohydr. Res. 1999;322:279. [Google Scholar]
- 25.Gazivoda T., Raić-Malić S., Krištafor V., Makuc D., Plavec J., Bratulic S., Kraljević Pavelić S., Pavelić K., Naesens L., Andrei G., Snoeck R., Balzarini J., Mintas M. Bioorg. Med. Chem. 2008;16:5624. doi: 10.1016/j.bmc.2008.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.