

Abstract
In numerous animal clades, the evolutionary history of host species drives patterns of gut microbial community structure, resulting in more divergent microbiota with increasing phylogenetic distance between hosts. This phenomenon, termed phylosymbiosis, has been observed in diverse evolutionary lineages, but has been difficult to detect in birds. Previous tests of phylosymbiosis among birds have been conducted using wild individuals, and thus interspecific differences in diet and environment may have masked a phylogenetic signal. Therefore, we tested for phylosymbiosis among all 15 species of cranes (family Gruidae) housed in the same captive environment and maintained on identical diets. 16S rRNA sequencing revealed that crane species harbour distinct gut microbiota. Overall, we detected marginally significant patterns of phylosymbiosis, the strength of which was increased when including the estimates of absolute microbial abundance (rather than relative abundance) derived from microbial densities determined by flow cytometry. Using this approach, we detected the statistically significant signatures of phylosymbiosis only after removing male cranes from our analysis, suggesting that using mixed-sex animal cohorts may prevent the detection of phylosymbiosis. Though weak compared with mammals (and especially insects), these results provide evidence of phylosymbiosis in birds. We discuss the potential differences between birds and mammals, such as transmission routes and host filtering, that may underlie the differences in the strength of phylosymbiosis.
Keywords: gut microbiome, phylosymbiosis, birds
1. Introduction
There is mounting evidence that gut microbial community structure is strongly influenced by the evolutionary history of the host, resulting in more divergent microbiota with increased interspecific phylogenetic distance. To date, interspecific differences in gut microbial communities have been correlated with host phylogenetic relationships among natural populations of sponges [1], corals [2], arthropods [3], mammals [4] and primates [5,6]. Importantly, this phenomenon, known as phylosymbiosis, occurs even when host species are maintained in a common laboratory environment [7–9], suggesting that gut microbial communities assemble deterministically in accordance with evolutionarily divergent host phenotypes independent of extrinsic environmental variables. Therefore, a thorough investigation of phylosymbiosis across a variety of host lineages, especially those in which phylosymbiotic signatures may be subtle, is necessary to fully understand mechanistic underpinnings driving the correlation between gut microbial community composition and host evolutionary history.
Despite widespread evidence of phylosymbiosis across most major taxonomic groups, this phenomenon has yet to be observed in birds (class Aves). Birds are a diverse group of organisms that provide numerous ecological services [10], yet studies into their gut microbial ecology are extremely limited [11]. To date, there have been several attempts to link gut microbial community composition to avian phylogenetic relationships [12–18], the most extensive of which was conducted using individuals from 491 species (representing 34 orders) of birds [18]. While most of these studies detected a statistically significant effect of taxonomy (e.g. species identity) [12–17], none reported congruence between the gut microbial community composition and host phylogeny. Notably, one study found a statistically significant correlation between interspecific differences in gut microbiota and passerine phylogenetic distances, but did not explicitly test for patterns of phylosymbiosis [16]. While this lack of support for avian phylosymbiosis could be a product of avian evolutionary history (e.g. weight-saving digestive adaptations) [18], it may also be a product of their sensitivity to extrinsic environmental factors or species-specific differences in natural feeding strategies across several avian orders [11,19]. Therefore, studies that reduce the influence of these variables using captive birds on identical diets may increase the chances of detecting subtle phylosymbiotic signatures [7], thereby enabling the identification of the mechanisms and host factors driving this phenomenon [20,21].
The aim of this study was to characterize differences in gut microbiota across several closely related avian taxa in a controlled environment to determine whether some birds exhibit phylosymbiosis. As noted above, previous attempts to detect phylosymbiosis in birds may have been unsuccessful due to pronounced differences in habitat and diet across a taxonomically broad cohort spanning many avian orders. It is for these reasons that the use of computational or laboratory methods has been identified as a major goal for future phylosymbiosis studies [22]. Therefore, we sought to control for these potentially confounding factors by characterizing the gut microbiota of all 15 species of cranes (all within the family Gruidae) maintained on identical diets in a shared captive environment. We further improved upon previous investigations of phylosymbiosis by applying two recent methodological advances: (i) the correction of relative abundance values using microbial density estimates derived from flow cytometry analysis [23,24] and (ii) eco-evolutionary analysis of sequence variants via cladal taxonomic units [25]. Using these approaches, we tested whether evolutionary relationships between crane species were associated with changes in the gut microbiota, with the explicit prediction that dendrograms representing interspecific differences in gut microbial beta diversity would be more congruent with crane phylogeny than trees drawn at random.
2. Material and methods
(a). Animal husbandry and sample collection
We focused our study on a captive population of all 15 species of cranes (family Gruidae; electronic supplementary material, S1) at the International Crane Foundation (Baraboo, Wisconsin, USA). Cranes in this population are intended for education, conservation and captive breeding purposes, but offer a unique opportunity to test for patterns of phylosymbiosis in the absence of confounding environmental factors. With the exception of two individuals, all cranes were born in captivity (electronic supplementary material, S1). Some cranes in this population have a history of antibiotic treatment, but not within two months of faecal sample collection (electronic supplementary material, S1). Captive cranes were housed individually (or in breeding pairs) in 15 × 18 m outdoor pens with chain-link fencing along each side and grass covered soil as a substrate. Each pen was covered by flight netting and included a 4.2 × 4.2 m indoor enclosure. Cranes are naturally omnivorous and received identical diets in the form of pelleted food (Zeigler Crane Breeder or Maintenance Diet, Gardners, PA, USA; electronic supplementary material, S2), and fresh water was provided ad libitum in buckets within the enclosures. The birds were exposed to ambient temperature and natural photoperiod. None of the cranes had a history of chronic infectious disease, malnutrition or husbandry-related problems, and each bird was visually normal according to experienced keepers in the weeks preceding sample collection.
Because previous research has shown that faeces provides the best non-invasive approximation of the avian microbiome [26], we collected faecal samples from a total of 44 individuals across 15 crane species over a one-week period in October 2017 (range of 2–4 individuals per species; electronic supplementary material, S1). Cranes in this population varied in age (ranging from 1 to 55 years old) and sex (electronic supplementary material, S1). All samples consisted of a single, fresh-appearing voided faecal mass collected into sterile Whirl-Pak collection bags (Nasco, Fort Atkinson, WI) with a sterile cotton swab or sterilized tongue depressor. Faecal samples were immediately frozen at −80°C. All faecal samples were collected over the same 10-day period (electronic supplementary material, S1).
(b). Microbial density via flow cytometry
For flow cytometry, 200–500 mg of faecal sample was fixed in 4% paraformaldehyde in DMSO at a final dilution of 1 : 100 000. Samples were then syringe filtered (5 µm) to remove large particulates and eukaryotic host cells. Then, 949 µl of the filtered sample was stained with 1 µl SYBR green and spiked with 50 µl of CountBright counting beads (ThermoFisher product number C36950) as an internal reference, bringing the total volume of each sample to 1 ml. The flow cytometry analysis of the microbial cells present in each faecal sample was performed using a BD Biosciences LSR II Flow Cytometer with FACSDiva software (version 8). We used three gating parameters to exclude non-fluorescent, eukaryotic and clustered cell complexes: forward-angle light scatter (FSC), linear side scatter (SSC) and log fluorescein isothiocyanate (FITC). FITC versus event counting data plots were gated to distinguish SYBR-positive fluorescent events from non-fluorescent events. FSC-area versus SSC-area data plots were gated to distinguish CountBright internal reference beads from all other recorded events. FSC-height versus FSC-width data plots were gated to identify single prokaryotic cell events. These settings allowed the software to count 10 000 SYBR-positive (at emission wavelength of 530 nm) counting events that were distinguishable as single-cell bacteria and internal reference beads. Instrument, gating and event recording settings were identical for all samples. Raw flow cytometry data were used to estimate microbial density of faecal samples using the number of internal reference bead counts according to the manufacturer's instructions.
(c). Molecular analyses and bioinformatics
We isolated DNA from approximately 250 mg of crane faeces using the Qiagen PowerFecal DNA Kit with an overnight incubation in lysis buffer at 65°C to increase extraction yields [27]. Additionally, we conducted four ‘blank' extractions alongside the faecal extractions to account for possible microbial DNA contaminants within commercial kits [28]. We used polymerase chain reaction (PCR) to amplify a portion of the bacterial 16S rRNA gene for Illumina sequencing using the primers 515F and 806R targeting the V4 region of microbial small subunit ribosomal RNA gene [29]. Amplified products were pooled in equimolar concentrations and loaded onto an Illumina MiniSeq mid-output flow cell (2 × 153 base paired-end reads). All library preparation, pooling and sequencing was performed at the DNA Services facility at the University of Illinois at Chicago. Sequence reads have been deposited in the NCBI SRA database under PRJNA553772.
Illumina sequencing reads were filtered and processed using the DADA2 pipeline [30] in QIIME2 version 2018.8 [31]. Bacterial 16S rRNA sequence variants (hereafter ASVs) were identified using the Greengenes reference database version 13.8 [32]. Illumina sequencing generated a total of 1.75 million reads (mean of 39 860 per sample) and 1339 ASVs after DADA2 processing. These sequences were further processed by removing non-bacterial ASVs (archaea, chloroplasts and mitochondria) in QIIME2, reducing our total number of reads to 1.6 million (mean of 36 104 per sample ±2393 SE) and 1300 ASVs. We detected a total of 29 ASVs in four negative controls. Overall, the bacterial communities of these negative controls exhibited little overlap with crane faecal samples (electronic supplementary material, S3). Based on previous evidence suggesting that computationally removing contaminant ASVs may strongly influence our results and interpretation [33,34], we simply summarized the occurrence of these reads among kit controls and crane faecal samples (electronic supplementary material, S3).
Next, we generated two types of ASV tables for downstream comparisons of diversity metrics and tests of phylosymbiosis. First, we generated an evenly sampled ASV table using the standard approach of rarefying to the lowest sequence return among all samples (in our case, 8838) in QIIME2. This table was used to calculate metrics of alpha diversity (ASV richness and Faith's phylogenetic diversity) in QIIME2 and beta diversity (Bray–Curtis, unweighted UniFrac, weighted UniFrac) in the R package Phyloseq (v. 1.22.3) [35]. As a final step, we averaged ASV abundances by host species of origin for downstream analysis in ClaaTU [25] and tests of phylosymbiosis. Second, we generated an unrarefied ASV table that accounted for differences in absolute microbial abundances across samples by correcting the number of reads assigned to each ASV using flow cytometry derived microbial densities [23,24]. This was accomplished by first calculating the relative abundance of each ASV within a given sample, then multiplying the relative abundance value by the microbial density as estimated by flow cytometry. Again, we averaged ASV abundances by host species of origin for later tests of phylosymbiosis.
(i). Phylogenetic and statistical analyses
Using the molecular phylogeny of cranes [36], we investigated whether gut microbial ASV richness, Faith's phylogenetic diversity and microbial density were associated with interspecific crane phylogenetic distances using Moran's index of autocorrelation (I) [37,38] in the R package Phylosignal (v. 1.2) [39]. We then tested whether crane species deviated from expected (based on 999 random permutations) ASV richness, Faith's phylogenetic diversity and microbial density values using the local indicator of phylogenetic association (Ii) [40] in Phylosignal [39]. For both I and Ii, values range from −1 (perfect clustering of dissimilar samples) to 1 (perfect clustering of similar samples), with 0 indicating a perfect random association between microbial communities and host phylogeny.
For the metrics of beta diversity, we visualized differences in gut microbial community structure via principal coordinate analysis (PCoA) of Bray–Curtis distances using both rarefied and microbial density-corrected ASV relative abundances in the R package Phyloseq [35]. Additionally, we implemented this same approach using unweighted and weighted UniFrac distances, which incorporate microbial phylogeny into analyses [41]. We then used permutational analysis of variance (PERMANOVA) with random effects for sex in PRIMER with PERMANOVA+ v. 7.0.13 to determine whether gut microbial communities differed across (i) crane species and (ii) the five currently recognized clades within the crane molecular phylogeny (as defined in [36,42]): Balearica, Leucogeranus, Antigone/Canadensis, Anthropoides and Americana.
We used two approaches to investigate the distribution of microbial taxa across the crane phylogeny. First, we used linear discriminant analysis effect size (LEfSe version 1.0) [43] on rarefied ASV tables to identify microbial taxa with statistically significant differences in relative abundances across crane species. LEfSe was conducted using default settings (LDA ≥ 2.0, p ≤ 0.05) at the bacterial phylum and genus levels in Galaxy (http://huttenhower.sph.harvard.edu/galaxy/). However, this approach is limited by Linnaean taxonomic classifications and thus may not fully capture differences in gut microbial communities that occur at intermediate levels of taxonomy [25]. To address this limitation, we investigated the distribution of microbial taxa across the crane phylogeny using rarefied ASV tables and cladal taxonomic units with ClaaTU (v. 0.1) [25], which groups microbial lineages into ecologically relevant taxonomic units. In our case, we used ClaaTU to identify microbial taxa that were more prevalent among crane clades (as defined above) than expected by chance. We corrected p-values for multiple comparisons in R using a relatively permissive false-discovery rate [44] threshold of p ≤ 0.2 to identify microbial taxa that may explain the patterns of phylosymbiosis among cranes in this dataset.
Finally, we constructed microbial dendrograms from both rarefied and microbial density-corrected ASV tables to test whether host microbial community dendrograms exhibited the patterns of phylosymbiosis with the crane phylogeny. Using averaged ASV tables (see details above), we calculated Bray–Curtis, unweighted UniFrac and weighted UniFrac distances in the R package Phyloseq [35]. Beta diversity distance matrices were then clustered using the UPGMA method in R (function: hclust(), method = ‘average') [45]. We tested for patterns of phylosymbiosis by comparing gut microbiota dendrograms with the crane phylogeny via the Robinson–Foulds and matching cluster metrics with 100 000 random trees using a previously published Python script [7]. While both of these methods are acceptable for assessing topological congruency between trees, the matching cluster method accounts for incongruences between closely related branches and is therefore considered a more refined approach for detecting phylosymbiosis [7]. Both of these approaches produce a normalized score between 0 (complete congruence) and 1 (complete incongruence). p-values were determined by the probability of 100 000 randomized dendrogram topologies yielding equivalent or more congruent phylosymbiotic patterns than the actual microbiota dendrogram.
3. Results
First, we investigated whether interspecific variation in metrics of crane gut alpha diversity richness was correlated with host phylogenetic distances using Moran's index [37]. We observed a non-significant correlation between host interspecific phylogenetic distances and gut bacterial richness (I = −0.069, p = 0.423; figure 1a), Faith's phylogenetic diversity (I = −0.055, p = 0.354; figure 1b) and microbial density (I = −0.165, p = 0.934; figure 1c). We then tested whether individual crane species exhibited gut microbial richness, diversity and density that differed from random permutations using the local indicator of phylogenetic association (Ii). Using this approach, G. americana exhibited higher gut microbial richness than expected at random (Ii = −0.521, p = 0.013; figure 1a). For Faith's phylogenetic diversity, we observed a trend (0.05 < p < 0.1) in local phylogenetic signal for all three members of the Anthropoides clade (figure 1b): G. carunculatus (Ii = −0.297, p = 0.059), A. virgo (Ii = 0.133, p = 0.072) and A. paradiseus (Ii = −0.298, p = 0.083). Only one species, G. vipio, exhibited higher microbial density than expected at random (Ii = −0.696, p = 0.017; figure 1c). Within a crane species, males and females harboured similar gut microbiota (electronic supplementary material, figure S1).
Figure 1.

Phylogenetic patterns of crane gut microbial alpha diversity. (a) Observed bacterial ASV richness. (b) Faith's bacterial phylogenetic diversity. (c) Microbial density in cells/gram of faeces. Points represent means and error bars represent standard deviation. Red points indicate statistically significant (p < 0.05) local Moran's Index (Ii) values and orange points indicate trending Ii values (0.05 ≤ p < 0.1). Sample size ranged from 2 to 4 individuals per species. (Online version in colour.)
Next, we investigated whether gut bacterial communities were distinct among crane species and phylogenetic clades. Using the standard rarefaction-based analysis and Bray–Curtis distances, we found a statistically significant effect of species (PERMANOVA pseudo-F44,15 = 1.76, p = 0.001) and clade (PERMANOVA pseudo-F44,15 = 1.75, p = 0.006; figure 2a). When correcting the relative abundance of sequencing reads by microbial density estimates (derived from flow cytometry analysis), we were able to detect both species- (PERMANOVA pseudo-F44,5 = 1.52, p = 0.001) and clade-level (PERMANOVA pseudo-F44,5 = 1.60, p = 0.001) differences in gut microbial community structure (figure 2b). Additionally, we performed this same analysis using phylogenetically aware metrics of beta diversity (i.e. unweighted and weighted UniFrac). For unweighted UniFrac, we found a statistically significant effect of species on crane gut microbiota using both standard rarefaction-based (PERMANOVA pseudo-F44,5 = 1.37, p = 0.010) and microbial density analyses (PERMANOVA pseudo-F44,5 = 1.35, p = 0.015; electronic supplementary material, figure S2). By contrast, there was no effect of crane clade using either approach (p > 0.05; electronic supplementary material, figure S2). For weighted UniFrac, there was no effect of species or clade in any case (p > 0.05; electronic supplementary material, figure S2).
Figure 2.

Taxonomic- and clade-level patterns of crane gut microbial beta diversity using Bray–Curtis distances. (a) PCoA plot based on the relative abundance of ASVs between crane species and clades. (b) PCoA ordination-based microbial density-corrected ASV tables. (Online version in colour.)
From a total of 1300 ASVs, we identified a total of 19 bacterial phyla and 215 genera across all crane species. Regardless of host species, crane gut microbiota was almost entirely composed of the phyla Firmicutes (mean ± standard error: 76 ± 3%) and Proteobacteria (15 ± 2%; figure 3a), which translated to an average of 17 × 107 (±2.55 SE) and 3.6 × 107 (±0.55 SE) microbial cells/gram of faecal material, respectively (figure 3b). Despite these broad-scale similarities based on Linnaean taxonomy, clades within the crane phylogeny exhibited statistically significant differences in the presence of eco-phylogenetic groups of bacteria using the ClaaTU workflow (figure 4). Among the 484 eco-phylogenetic groups that were conserved in at least one crane clade, we found 87 instances where a group occurred within a specific crane clade more than expected at random (FDR adjusted P ≤ 0.2; figure 4). Most of these groups were classified as Firmicutes (70%), specifically those in the orders Clostridiales (33%) and Lactobacillales (17%). At the genus level, LEfSe analysis revealed that G. canadensis exhibited significantly higher relative abundances of the bacterial genus Lactobacillus compared with other crane species (LDA = 5.50, p = 0.040). By contrast, the A. paradiseus exhibited a significantly lower relative abundance of the bacterial genera Corynebacterium (LDA = 5.14, p = 0.020) and Streptococcus (LDA = 4.90, p = 0.017). There were no differentially abundant phyla across crane species (LDA > 2.0, p > 0.05).
Figure 3.
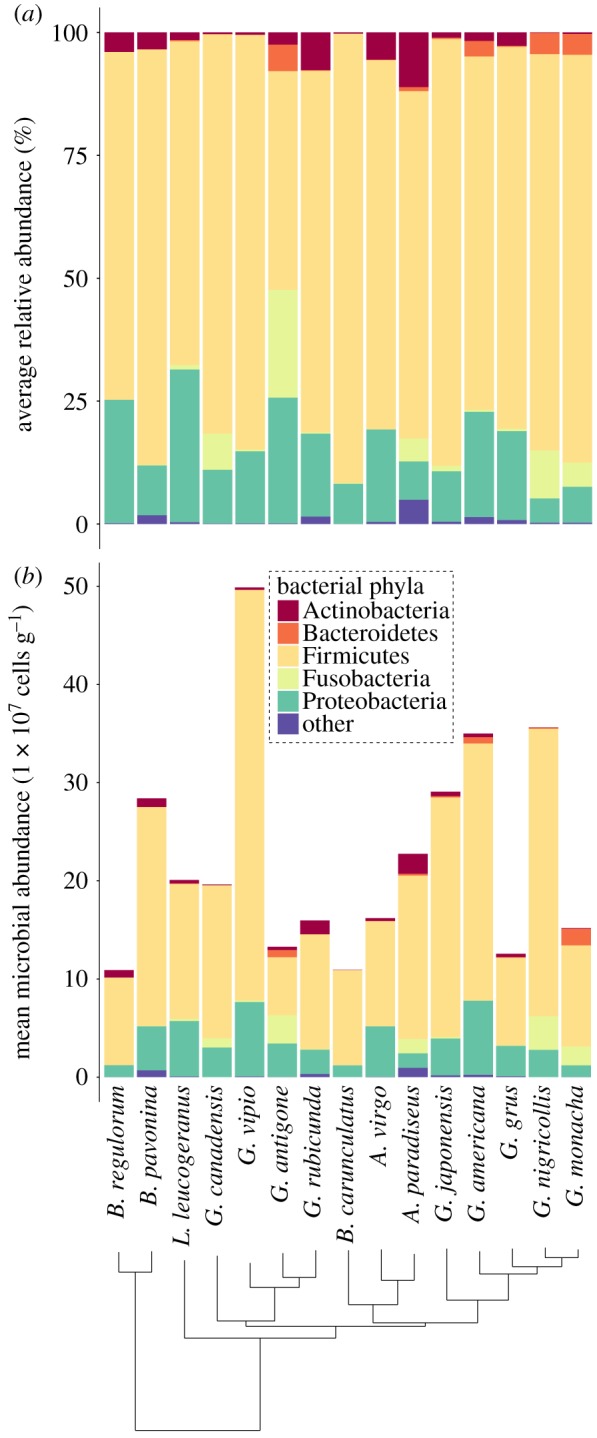
Relative versus absolute abundances of microbial phyla in the crane gut microbiota. Phylum-level gut microbial community composition among all 15 species of cranes (n = 2 to 4 individuals per species). (a) Standard approach using rarefied ASV tables to estimate the relative abundance of bacterial phyla. (b) Mean microbial abundance estimates based on un-rarefied, microbial density-corrected ASV tables. Only the five most abundant microbial phyla are presented, with less abundant phyla pooled into ‘other'. (Online version in colour.)
Figure 4.

Patterns of conserved bacterial eco-phylogenetic groups across the crane phylogeny. The 484 eco-phylogenetic groups that were conserved in at least one crane clade are plotted as columns in a heatmap that illustrates their occurrence across the crane phylogeny. Black ticks indicate an eco-phylogenetic group was detected in a given crane species. Red ticks indicate an eco-phylogenetic group with a statistically significant (FDR adjusted p ≤ 0.2) association with a crane clade. (Online version in colour.)
Finally, we investigated for the patterns of phylosymbiosis by comparing microbial community dendrograms and the crane phylogenetic tree. Because phylosymbiosis predicts that phylogenetic relatedness will correlate with beta diversity relationships of microbial communities [7], we expected that microbiota and crane dendrograms would exhibit more congruence than expected at random. Using the UPGMA trees constructed using the standard rarefaction pipeline and Bray–Curtis distances, we found no statistically significant congruence between crane phylogeny and microbiota dendrograms using normalized Robinson–Foulds (nRF = 1.0, p = 1.0) and a non-significant trend when using the matching cluster method (nMC = 0.518, p = 0.086; table 1). UPGMA trees based on weighted and unweighted and UniFrac distances produced similar non-significant results (table 1). When using ASV relative abundances that were corrected for microbial density, we detected a slightly stronger signal of phylosymbiosis using both normalized Robinson–Foulds (nRF = 0.917, p = 0.193) and matching cluster methods (nMC = 0.506, p = 0.063; table 1, figure 5). Again, weighted and unweighted and UniFrac distances produced similar non-significant results (table 1). As a final test, we also conducted an analysis using only female cranes to determine if sex affected our ability to detect phylosymbiosis. Using this approach, we detected a statistically significant topological congruence using normalized Robinson–Foulds with uncorrected Bray–Curtis distances (nRF = 0.470, p = 0.011) as well as matching cluster method using weighted UniFrac distances (nMC = 0.494, p = 0.017; table 1).
Table 1.
Summary of phylosymbiosis test statistics using Robinson–Foulds and matching cluster methods on rarefaction-based and microbial density-corrected distance matrices. Both of these approaches produce a normalized score between 0 (complete congruence) and 1 (complete incongruence). p-values were determined by the probability of 100 000 randomized dendrogram topologies yielding equivalent or more congruent phylosymbiotic patterns than the actual microbiota dendrogram. *p < 0.05; **0.05 ≤ p < 0.1.
| distance metric | host-microbe score | max congruence | normalized score | p | ||
|---|---|---|---|---|---|---|
| Robinson–Foulds | all cranes | Bray–Curtis | 12 | 12 | 1.000 | 1.000 |
| unweighted UniFrac | 12 | 12 | 1.000 | 1.000 | ||
| weighted UniFrac | 12 | 12 | 1.000 | 1.000 | ||
| corrected Bray–Curtis | 11 | 12 | 0.917 | 0.193 | ||
| corrected unweighted UniFrac | 12 | 12 | 1.000 | 1.000 | ||
| corrected weighted UniFrac | 11 | 12 | 0.916 | 0.194 | ||
| females only | Bray–Curtis | 12 | 12 | 1.000 | 1.000 | |
| unweighted UniFrac | 12 | 12 | 1.000 | 1.000 | ||
| weighted UniFrac | 12 | 12 | 1.000 | 1.000 | ||
| corrected Bray–Curtis | 12 | 12 | 1.000 | 1.000 | ||
| corrected unweighted UniFrac | 11 | 12 | 0.916 | 0.194 | ||
| corrected weighted UniFrac | 12 | 12 | 1.000 | 1.000 | ||
| matching cluster | all cranes | Bray–Curtis | 44 | 85 | 0.518 | 0.086** |
| unweighted UniFrac | 44 | 85 | 0.518 | 0.086** | ||
| weighted UniFrac | 46 | 79 | 0.582 | 0.171 | ||
| corrected Bray–Curtis | 43 | 85 | 0.506 | 0.063** | ||
| corrected unweighted UniFrac | 50 | 81 | 0.617 | 0.485 | ||
| corrected weighted UniFrac | 51 | 85 | 0.600 | 0.592 | ||
| females only | Bray–Curtis | 39 | 83 | 0.470 | 0.011* | |
| unweighted UniFrac | 51 | 83 | 0.615 | 0.591 | ||
| weighted UniFrac | 48 | 81 | 0.593 | 0.306 | ||
| corrected Bray–Curtis | 44 | 83 | 0.530 | 0.086** | ||
| corrected unweighted UniFrac | 46 | 85 | 0.541 | 0.170 | ||
| corrected weighted UniFrac | 40 | 81 | 0.494 | 0.017* |
Figure 5.

Comparison of host and gut microbiome dendrograms. Host phylogenetic tree is from Krajewski et al. [36] and microbiome tree was constucted by UPGMA clustering of microbial density-corrected ASV tables and Bray–Curtis distances.
4. Discussion
The aim of this study was to characterize differences in gut microbiota across crane species to determine whether the gut communities of birds exhibit phylosymbiosis. Indeed, we detected the effects of crane species and clade on gut microbial community structure using standard rarefaction-based analysis and when correcting relative abundances with estimates of microbial density. These interspecific differences in gut microbial community structure yielded a statistically significant pattern of phylosymbiosis in this dataset containing all 15 species of cranes, but only once all male cranes were removed from our analysis. Below, we contextualize these findings with respect to previous research on the avian gut microbiome and phylosymbiosis in general, as well as discuss the potential differences between birds and mammals that may underlie the differences in the strength of phylosymbiosis.
When comparing across host species, we found that the crane gut microbiota exhibited major similarities at the phylum level while harbouring distinct gut microbial communities at finer taxonomic scales. In terms of similarities, we found that the crane gut microbiota, regardless of host species, was primarily composed of taxa in the phylum Firmicutes (76%). The dominance of Firmicutes is consistent with previous descriptions of crane gut microbiota [46,47] and the avian gut microbiome in general [11]. However, cranes in our study harboured a higher relative abundance of Firmicutes compared with the previous studies of crane gut microbiota [46,47] and was similar to that reported in mammals [48,49]. While the potential effects of Firmicutes on crane physiology are currently unknown, studies in mammals [50] and chickens [51] have demonstrated that Firmicutes are important to host metabolism and digestive health, most likely through their ability to produce short-chain fatty acids through the breakdown of dietary carbohydrates and polysaccharides [52]. In terms of differences in gut microbial communities, ClaaTU analysis revealed that taxa within the phylum Firmicutes and the order Lactobacillales were differentially conserved among crane clades. Further investigation using LEfSe revealed that Lactobacillus (phylum Firmicutes) was differentially abundant across crane species, which is notable since bacteria in this genus are associated with weight gain and increased concentrations of short-chain fatty acids in chickens [51,53]. Further, the bacterial genus Corynebacterium (phylum Actinobacteria) was less abundant in the short-range migrant A. paradiseus. This is notable since previous research has demonstrated that non-migratory resident individuals exhibit a reduced relative abundance of Corynebacterium compared with migratory conspecifics [54]. While crane migratory behaviours vary across species and populations, the reduced relative abundance of this bacterial genus in a short-range migrant suggests that the microbiome could contribute to some aspect of migratory physiology, though these functional roles are currently unknown and warrant further investigation.
Another major finding of our study is that correcting ASV relative abundances using microbial densities improved the distinguishability of gut microbial communities across host species and enabled the detection of a statistically significant signature of phylosymbiosis. To date, the qualitative and quantitative investigations of phylosymbiosis have focused solely on the relative abundances of microbial taxa [7,21]. However, there have been numerous calls to incorporate cell quantification techniques into gut microbiome studies [23,24,55], as differences in absolute abundances could confound results based on relative abundances. Consistent with previous studies in mammals [56], we observed statistically significant interspecific differences in microbial density across crane species with substantial variation among conspecifics (figure 1). Analysing microbial densities separately for males and females greatly reduced intraspecific variation (electronic supplementary material, figure S1), suggesting that host sex may affect microbial carrying capacity. The incorporation of absolute abundance data into our analyses increased the distinguishability and revealed a statistically significant signal of phylosymbiosis among female cranes. This result is noteworthy since previous research has been unable to demonstrate that avian lineages exhibit patterns of phylosymbiosis [12–16]. One caveat to these findings is that a given bacterial species can possess anywhere between 1 and 15 copies of the 16S rRNA gene [57], and thus 16S rRNA relative abundance values should be interpreted with caution [58]. While correcting 16S rRNA relative abundance values by absolute microbial abundance cannot address this fundamental limitation, the substantial variation in microbial densities across crane species provide sufficient justification for this approach. Therefore, we argue that the future studies of phylosymbiosis should incorporate the measurements of microbial density and possibly corrections for 16S rRNA gene copy number.
Overall, the phylosymbiotic pattern of cranes was weak compared with those observed in mammals (and especially insects) [7]. These differences may be due to the unique life-history traits exhibited by avian host species [11,19]. For example, mammals are thought to inherit their gut microbiota through contact with vaginal and faecal microbes during the birth process [59]. Vertical transmission of the microbiota in birds may occur through parental care, though this has not been explicitly demonstrated, especially in birds like cranes that do not regurgitate food to their young. Additionally, some evidence exists for potential maternal microbial transmission through eggs [27]. Theoretical simulations suggest that ecological filtering or selection of microbes by the host underlies the emergent property of phylosymbiosis [21]. Thus, physiological differences between mammals and birds may explain the differences in the strength of phylosymbiosis across these groups. For example, the aspects of ‘oral tolerance', or the immune non-responsiveness to ingested antigens varies between mammals and birds, with birds having a limited developmental window where oral tolerance occurs [60]. Additionally, birds (especially those in the order Gruiformes) exhibit a much more acidic stomach than mammals [61], thereby acting as a microbial filter that may limit the degree to which host species can differ in gut microbial communities. Understanding the mechanisms by which mammals and birds differentially structure their gut microbiome according to host evolutionary history is an open question in the field.
In this study, we provide the most explicit and well-controlled test of phylosymbiosis in birds and demonstrate statistically significant (but overall weak) patterns of phylosymbiosis among a distinct avian clade. Because phylosymbiosis has been demonstrated to have a functional role in other host groups [7], these patterns likely influence the health and performance of crane species, and thus may have implications for crane conservation. More broadly, this study demonstrates that even weak patterns of phylosymbiosis may be detected with controlled experimental design and inclusion of microbial densities into analyses. These findings represent a substantial contribution to the microbiome research community, as the detection of even weak patterns of phylosymbiosis may be necessary to understand mechanisms driving the correlation between gut microbial community composition and host evolutionary history.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Data accessibility
Raw sequence reads have been deposited in the NCBI SRA database under PRJNA553772. R code and associated data files are available as part of the electornic supplementary material.
Authors' contributions
B.K.T. conducted DNA extractions, analysed data and wrote the paper. J.S. conducted flow cytometry experiments. B.K.H. and K.D.K. conceived study design and supervised the project. All authors edited the manuscript and approved the final version.
Competing interests
We declare we have no competing interests.
Funding
We received no funding for this study.
References
- 1.Easson CG, Thacker RW. 2014. Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Front. Microbiol. 5, 532 ( 10.3389/fmicb.2014.00532) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schöttner S, Hoffmann F, Cárdenas P, Rapp HT, Boetius A, Ramette A. 2013. Relationships between host phylogeny, host type and bacterial community diversity in cold-water coral reef sponges. PLoS ONE 8, e55505 ( 10.1371/journal.pone.0055505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanders JG, Powell S, Kronauer DJ, Vasconcelos HL, Frederickson ME, Pierce NE. 2014. Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Mol. Ecol. 23, 1268–1283. ( 10.1111/mec.12611) [DOI] [PubMed] [Google Scholar]
- 4.Phillips CD, et al. 2012. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol. Ecol. 21, 2617–2627. ( 10.1111/j.1365-294X.2012.05568.x) [DOI] [PubMed] [Google Scholar]
- 5.Moeller AH, Li Y, Ngole EM, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, Peeters M, Hahn BH, Ochman H. 2014. Rapid changes in the gut microbiome during human evolution. Proc. Natl Acad. Sci. USA 111,16 431–16 435. ( 10.1073/pnas.1419136111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ochman H, Worobey M, Kuo C-H, Ndjango J-BN, Peeters M, Hahn BH, Hugenholtz P. 2010. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 8, e1000546 ( 10.1371/journal.pbio.1000546) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR. 2016. Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 14, e2000225 ( 10.1371/journal.pbio.2000225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohl KD, Dearing MD, Bordenstein SR. 2018. Microbial communities exhibit host species distinguishability and phylosymbiosis along the length of the gastrointestinal tract. Mol. Ecol. 27, 1874–1883. ( 10.1111/mec.14460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franzenburg S, Walter J, Künzel S, Wang J, Baines JF, Bosch TC, Fraune S. 2013. Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc. Natl Acad. Sci. USA 110, E3730–E3738. ( 10.1073/pnas.1304960110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sekercioglu CH. 2006. Increasing awareness of avian ecological function. Trends Ecol. Evol. 21, 464–471. ( 10.1016/j.tree.2006.05.007) [DOI] [PubMed] [Google Scholar]
- 11.Grond K, Sandercock BK, Jumpponen A, Zeglin LH. 2018. The avian gut microbiota: community, physiology and function in wild birds. J. Avian Biol. 49, e01788 ( 10.1111/jav.01788) [DOI] [Google Scholar]
- 12.Hird SM, Sánchez C, Carstens BC, Brumfield RT. 2015. Comparative gut microbiota of 59 neotropical bird species. Front. Microbiol. 6, 1403 ( 10.3389/fmicb.2015.01403) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bodawatta KH, Sam K, Jønsson KA, Poulsen M. 2018. Comparative analyses of the digestive tract microbiota of New Guinean passerine birds. Front. Microbiol. 9, 1830 ( 10.3389/fmicb.2018.01830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.García-Amado MA, Shin H, Sanz V, Lentino M, Martínez LM, Contreras M, Michelangeli F, Domínguez-Bello MG. 2018. Comparison of gizzard and intestinal microbiota of wild neotropical birds. PLoS ONE 13, e0194857 ( 10.1371/journal.pone.0194857) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Chen Z, Gao G, Sun C, Li Y, Zhu Y. 2019. Characterization and comparison of gut microbiomes in nine species of parrots in captivity. Symbiosis 78, 241–250. ( 10.1007/s13199-019-00613-7) [DOI] [Google Scholar]
- 16.Kropáčková L, et al. 2017. Codiversification of gastrointestinal microbiota and phylogeny in passerines is not explained by ecological divergence. Mol. Ecol. 26, 5292–5304. ( 10.1111/mec.14144) [DOI] [PubMed] [Google Scholar]
- 17.Capunitan DC, Johnson O, Terrill RS, Hird SM. Evolutionary signal in the gut microbiomes of 74 bird species from Equatorial Guinea. Mol. Ecol. 29, 829–847. [DOI] [PubMed] [Google Scholar]
- 18.Song SJ, et al. 2020. Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. mBio 11, e02901-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohl KD. 2012. Diversity and function of the avian gut microbiota. J. Comp. Physiol. B 182, 591–602. ( 10.1007/s00360-012-0645-z) [DOI] [PubMed] [Google Scholar]
- 20.Youngblut ND, Reischer GH, Walters W, Schuster N, Walzer C, Stalder G, Ley RE, Farnleitner AH. 2019. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 10, 1–15. ( 10.1038/s41467-019-10191-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazel F, Davis KM, Loudon A, Kwong WK, Groussin M, Parfrey LW. 2018. Is host filtering the main driver of phylosymbiosis across the tree of life? mSystems 3, e00097-18 ( 10.1128/mSystems.00097-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim SJ, Bordenstein SR. 2019. An introduction to phylosymbiosis. PeerJ Preprints 7, e27879v2. [Google Scholar]
- 23.Vandeputte D, et al. 2017. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511. ( 10.1038/nature24460) [DOI] [PubMed] [Google Scholar]
- 24.Props R, Kerckhof F-M, Rubbens P, De Vrieze J, Sanabria EH, Waegeman W, Monsieurs P, Hammes F, Boon N. 2017. Absolute quantification of microbial taxon abundances. ISME J. 11, 584–587. ( 10.1038/ismej.2016.117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaulke CA, Arnold HK, Humphreys IR, Kembel SW, O'Dwyer JP, Sharpton TJ. 2018. Ecophylogenetics clarifies the evolutionary association between mammals and their gut microbiota. mBio 9, e01348-18 ( 10.1128/mBio.01348-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Videvall E, Strandh M, Engelbrecht A, Cloete S, Cornwallis CK. 2018. Measuring the gut microbiome in birds: comparison of faecal and cloacal sampling. Mol. Ecol. Resour. 18, 424–434. ( 10.1111/1755-0998.12744) [DOI] [PubMed] [Google Scholar]
- 27.Trevelline BK, MacLeod KJ, Knutie SA, Langkilde T, Kohl KD. 2018. In ovo microbial communities: a potential mechanism for the initial acquisition of gut microbiota among oviparous birds and lizards. Biol. Lett. 14, 20180225 ( 10.1098/rsbl.2018.0225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salter SJ, et al. 2014. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 ( 10.1186/s12915-014-0087-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl Acad. Sci. USA 108(Suppl. 1), 4516–4522. ( 10.1073/pnas.1000080107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. ( 10.1038/nmeth.3869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolyen E, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. ( 10.1038/s41587-019-0209-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeSantis TZ, et al. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. ( 10.1128/AEM.03006-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. 2018. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. ( 10.1016/j.tim.2018.11.003) [DOI] [PubMed] [Google Scholar]
- 34.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, Knight R, Ley RE. 2014. Conducting a microbiome study. Cell 158, 250–262. ( 10.1016/j.cell.2014.06.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 ( 10.1371/journal.pone.0061217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krajewski C, Sipiorski JT, Anderson FE. 2010. Complete mitochondrial genome sequences and the phylogeny of cranes (Gruiformes: Gruidae). The Auk 127, 440–452. ( 10.1525/auk.2009.09045) [DOI] [Google Scholar]
- 37.Gittleman JL, Kot M. 1990. Adaptation: statistics and a null model for estimating phylogenetic effects. Syst. Zool. 39, 227–241. ( 10.2307/2992183) [DOI] [Google Scholar]
- 38.Moran PA. 1948. The interpretation of statistical maps. J. R. Stat. Soc. B (Methodol.) 10, 243–251. [Google Scholar]
- 39.Keck F, Rimet F, Bouchez A, Franc A. 2016. phylosignal: an R package to measure, test, and explore the phylogenetic signal. Ecol. Evol. 6, 2774–2780. ( 10.1002/ece3.2051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anselin L. 1995. Local indicators of spatial association—LISA. Geogr. Anal. 27, 93–115. ( 10.1111/j.1538-4632.1995.tb00338.x) [DOI] [Google Scholar]
- 41.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. ( 10.1128/AEM.71.12.8228-8235.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krajewski C. 1989. Phylogenetic relationships among cranes (Gruiformes: Gruidae) based on DNA hybridization. The Auk 106, 603–618. [Google Scholar]
- 43.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 ( 10.1186/gb-2011-12-6-r60) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B (Methodol.) 57, 289–300. [Google Scholar]
- 45.R Core Team. 2013. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See https://www.r-project.org/. [Google Scholar]
- 46.Dong Y, Xiang X, Zhao G, Song Y, Zhou L. 2019. Variations in gut bacterial communities of hooded crane (Grus monacha) over spatial-temporal scales. PeerJ 7, e7045 ( 10.7717/peerj.7045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie Y, Xia P, Wang H, Yu H, Giesy JP, Zhang Y, Mora MA, Zhang X. 2016. Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Sci. Rep. 6, 33350 ( 10.1038/srep33350) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ley RE, et al. 2008. Evolution of mammals and their gut microbes. Science 320, 1647–1651. ( 10.1126/science.1155725) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maurice CF, Knowles SC, Ladau J, Pollard KS, Fenton A, Pedersen AB, Turnbaugh PJ. 2015. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 9, 2423–2434. ( 10.1038/ismej.2015.53) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clemente JC, Ursell LK, Parfrey LW, Knight R. 2012. The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. ( 10.1016/j.cell.2012.01.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Angelakis E, Raoult D. 2010. The increase of Lactobacillus species in the gut flora of newborn broiler chicks and ducks is associated with weight gain. PLoS ONE 5, e10463 ( 10.1371/journal.pone.0010463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. 2013. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340. ( 10.1194/jlr.R036012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin L, Ho Y, Abdullah N, Ali M, Jalaludin S. 1998. Effects of adherent Lactobacillus cultures on growth, weight of organs and intestinal microflora and volatile fatty acids in broilers. Anim. Feed Sci. Technol. 70, 197–209. ( 10.1016/S0377-8401(97)00080-1) [DOI] [Google Scholar]
- 54.Risely A, Waite DW, Ujvari B, Hoye BJ, Klaassen M. 2018. Active migration is associated with specific and consistent changes to gut microbiota in Calidris shorebirds. J. Anim. Ecol. 87, 428–437. ( 10.1111/1365-2656.12784) [DOI] [PubMed] [Google Scholar]
- 55.Stämmler F, Gläsner J, Hiergeist A, Holler E, Weber D, Oefner PJ, Gessner A, Spang R. 2016. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome 4, 28 ( 10.1186/s40168-016-0175-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Contijoch EJ, et al. 2019. Gut microbiota density influences host physiology and is shaped by host and microbial factors. eLife 8, e40553 ( 10.7554/eLife.40553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee ZM-P, Bussema C III, Schmidt TM. 2009. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res. 37(suppl. 1), D489–D493. ( 10.1093/nar/gkn689) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kembel SW, Wu M, Eisen JA, Green JL. 2012. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput. Biol. 8, e1002743 ( 10.1371/journal.pcbi.1002743) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Funkhouser LJ, Bordenstein SR. 2013. Mom knows best: the universality of maternal microbial transmission. PLoS Biol. 11, e1001631 ( 10.1371/journal.pbio.1001631) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friedman A. 2008. Oral tolerance in birds and mammals: digestive tract development determines the strategy. J. Appl. Poultry Res. 17, 168–173. ( 10.3382/japr.2007-00099) [DOI] [Google Scholar]
- 61.Beasley DE, Koltz AM, Lambert JE, Fierer N, Dunn RR. 2015. The evolution of stomach acidity and its relevance to the human microbiome. PLoS ONE 10, e0134116 ( 10.1371/journal.pone.0134116) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence reads have been deposited in the NCBI SRA database under PRJNA553772. R code and associated data files are available as part of the electornic supplementary material.