

Graphical abstract

Keywords: Tricyclic penciclovir (PCV) derivatives, Tricyclic hydroxybutylguanine (HBG) derivatives, Herpes simplex virus (HSV), Herpes simplex encephalitis (HSE), Molecular modelling
Abstract
A series of tricyclic penciclovir (PCV) and hydroxybutylguanine (HBG) derivatives have been prepared with enhanced lipophilicity following an efficient synthetic route. All the novel tricyclic derivatives were evaluated for inhibitory activity against herpes simplex virus 1 and 2 (HSV-1, HSV-2) and thymidine kinase deficient (ACV resistant) HSV-1. The tricyclic HBG derivatives were devoid of inhibitory activity however several of the tricyclic PCV derivatives showed promising antiviral activity, in particular 9g (R = 4-MeO-C6H4) displayed good inhibitory activity (HSV-1 EC50 1.5 μM, HSV-2 EC50 0.8 μM) and retained inhibitory activity in HSV-1 TK− cells (EC50 0.8 μM). Computational docking experiments supported the biological data observed and this preliminary study provides useful data for further development of tricyclic acyclic nucleoside derivatives with improved lipophilicity and retention of activity in HSV-1 TK deficient strains. Also, the new tricyclic derivatives were evaluated against a broad range of other DNA and RNA viruses, but were found to be inactive at subtoxic concentrations. In addition, weak to moderate cytostatic effect was observed for the new compounds.
1. Introduction
The herpes simplex virus (HSV), like all herpes viruses, establishes lifelong latency following a primary infection. HSV-associated diseases can range from mild orolabial ulcers (cold sores) to severe encephalitis.1 Herpes simplex viruses are categorised into two types: HSV-1, which is mainly transmitted through oral-oral contact resulting mainly in cold sores;1 and HSV-2, which is mainly sexually transmitted resulting in the most common infection of genital herpes.2 Globally two thirds of the population under the age of 50 are estimated to be infected with HSV-1.3
Herpes simplex encephalitis (HSE) is the most common cause of sporadic fatal viral encephalitis, accounting for almost 20% of all cases of encephalitis.4 HSE presents as an acute focal necrotising infection of the brain, which, if untreated, has a 70% mortality rate.5 The first line treatment of HSE is aciclovir (ACV), administered intravenously for 14–21 days, however even when treated, HSE is associated with a 30% mortality rate and permanent neurological damage.6, 7, 8 Limitations of ACV therapy for HSE are the development of viral resistance9 and low CNS uptake.10
Tricyclic ACV and ganciclovir (GCV) derivatives (Fig. 1 ) were shown to be effective inhibitors of HSV-1 thymidine kinase (TK) and are of interest as they maintain the inhibitory activity against the enzyme and, as they are more lipophilic than the current medications, may be valuable agents for the treatment of CNS infections.11, 12, 13 Moreover, these tricyclic compounds were found to exhibit strong intrinsic fluorescent properties, which allows for sensitive concentration monitoring and optimal therapeutic dosing.11, 12, 13 Penciclovir (PCV) and hydroxybutylguanine (HBG) (Fig. 1) are the carbocyclic analogues of GCV and ACV, respectively. The spectrum of activity of PCV against human herpesviruses was found to be similar to that of ACV, however in certain circumstances PCV was more active than ACV owing to the higher stability of PCV-TP in infected cells relative to ACV-TP.14 Also, at concentrations up to 100 µg/mL, it was confirmed that PCV, like ACV, was highly selective and subsequently did not exhibit any toxicity in cell culture.14 Similarly, HBG is reported to have higher affinity for HSV-1 TK than ACV in addition to relatively comparable selectivity for inhibition of viral DNA synthesis and good safety profile in cell culture.15
Fig. 1.

Parent acyclic nucleosides and tricyclic derivatives.
Application of the tricyclic scaffold modification to PCV and HBG was of interest to determine whether antiviral activity was maintained and, importantly, modification of the nucleobase moiety with an additional ring may result in beneficial physicochemical properties (enhanced lipophilicity, fluorescence) and/or enhanced enzyme binding interaction. A library of tricyclic PCV and HBG derivatives (Fig. 2 ) was designed and synthesised to evaluate antiviral activity and study structure–activity relationships, supported by computational docking analysis.
Fig. 2.

Library for the designed tricyclic PCV and HBG nucleosides.
2. Results and discussion
2.1. Chemistry
PCV (6) and HBG (7) were prepared as previously described by reaction of 2-amino-6-chloropurine (1) with 5-(2-bromoethyl)-2-phenyl-1,3-dioxane (2)16 or 4-bromobutylacetate (3)17 with subsequent acid hydrolysis of the protected derivatives (4) and (5) (Scheme 1 ).
Scheme 1.

Reagents and conditions: (i) K2CO3, 18-crown-6, DMF, r.t. 72 h for synthesis of 4, 94%, 16 h for synthesis of 5, 81% (ii) (a) 2 M aqueous HCl, 90 °C, 3 h (b) 10% aqueous NaOH, 4 °C 24 h, 77% (iii) (a) 2 M aqueous HCl, THF, 75 °C overnight (b) 10% aqueous NaOH, 4 °C 24 h, 81% (iv) NaH, DMF, r.t. 6 h, 38–79%.
The new tricyclic analogues of PCV and HBG, 3-[4-hydroxy-3-(hydroxymethyl)butyl]-6-(aryl)-3,5 dihydro-9H-imidazo[1,2-a]purin-9-one (9) and 3-(4-hydroxybutyl)-6-(aryl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10) were obtained upon reaction of the sodium salt of PCV (6) and HBG (7), generated in situ on treatment with NaH, and the appropriate bromoketone (8) in 38–79% yield (Table 1 ). The new compounds were purified by flash column chromatography using a gradient eluting mixture of CH2Cl2-MeOH followed by recrystallisation (Scheme 1). The procedure used for the preparation of the new derivatives mostly followed a previously reported method for the preparation of analogously modified guanosine and other tricyclic derivatives.12
Table 1.

| No. | R | Yield (%) | m.p. (oC) | No. | R | Yield (%) | m.p. (oC) |
|---|---|---|---|---|---|---|---|
| 9a | 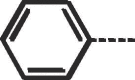 |
68 | 212–214 | 10a | 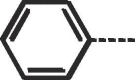 |
61 | 248–250 |
| 9b | 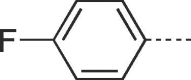 |
65 | >300 | 10b | 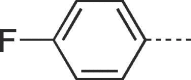 |
62 | 288–290 |
| 9c | 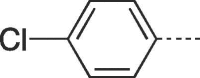 |
62 | 258–260 | 10c | 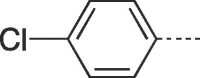 |
62 | 280–282 |
| 9d | 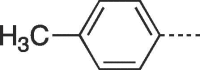 |
61 | 112–114 | 10d | 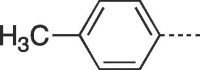 |
67 | 260–261 |
| 9e | 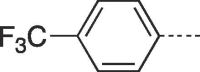 |
55 | >300 | 10e | 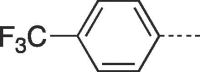 |
71 | 292–293 |
| 9f | 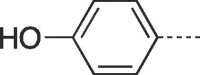 |
38 | >300 | 10f | 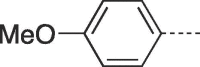 |
61 | 252–254 |
| 9g | 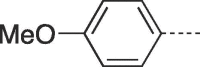 |
79 | 202–203 | 10g | 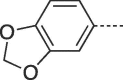 |
69 | 272–274 |
| 9h | 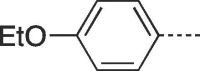 |
77 | 276–278 | 10h | 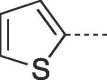 |
64 | 246–247 |
| 9i | 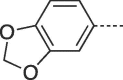 |
68 | 259–260 | ||||
| 9j | 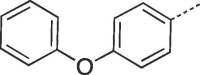 |
75 | 276–278 |
The tricyclic derivatives showed some sensitivity towards light and air, so were protected by wrapping with foil and rapid storage in amber coloured vials. The 6-thien-2-yl group was highly unstable to light and air and in the case of the PCV derivative decomposed rapidly during collection. Conversely, its HBG congener (10h) was stable enough to be collected safely.
2.2. Biological evaluation
2.2.1. Antiviral activity
The tricyclic compounds (9 and 10) were screened as inhibitors of HSV using herpes simplex virus-1 (KOS strain), herpes simplex virus-2 (G strain), thymidine kinase deficient (aciclovir resistant) herpes simplex virus-1 (HSV-1 TK–KOS ACVr strain) in human embryonic lung (HEL) cells. None of the tricyclic HBG derivatives (10) showed any inhibitory activity (EC50 > 100 μM) or cytotoxicity (>100 μM) against HSV-1 or HSV-2. However, some of the tricyclic PCV derivatives (9) exhibited anti-herpetic activity (Table 2 ).
Table 2.
Inhibitory activity of tricyclic PCV derivatives (9) against HSV and cytotoxicity data. The grey shades highlight the most interesting data.
 |
aRequired to reduce virus-induced cytopathogenicity by 50%.
bRequired to cause a microscopically detectable alteration of normal cell morphology.
Aciclovir (ACV), Brivudin (BVDU), Cidofovir (CDV), Ganciclovir (GCV).
Of the newly synthesised tricyclic PCV derivatives, compounds 9a, 9c, 9d and 9j were inactive as antiviral agents. Compounds 9h and 9i showed anti-HSV-1 activity only at a relatively high concentration (EC50 = 76.5 and 44.7 µM, respectively), compounds 9b and 9e showed moderate anti-HSV-1 activity (EC50 = 8.9 and 20.00 µM/mL, respectively), whereas 9g and 9f were the most active and were inhibitory to HSV-1 replication with EC50 = 1.5 and 7.6 µM, respectively. Similar to anti-HSV-1 results, among the synthesised tricyclic PCV compounds, compounds 9g and 9f were the most active against the HSV-2 strain and showed marked inhibition (EC50 = 0.8 and 2.3 µM, respectively). Also, compounds 9b, 9e and 9i showed moderate activity against HSV-2 strain (EC50 = 28–40.9 µM), whereas the remaining compounds did not show any activity against the HSV-2 strain. Two compounds, 9b and 9g, retained activity against the TK− ACV resistant strain of HSV-1. Compound 9g showed potent inhibitory activity against HSV-1 TK− (EC50 = 0.8 µM), whereas compound 9b was moderately active against HSV-1 TK− (EC50 = 12 µM). The data suggest that in the case of the resistant strain TK−, compounds 9f and 9g are independent of viral TK catalysed phosphorylation for antiviral activity and/or may use a different metabolic activation pathway and/or display a unique mechanism of antiviral action by the un-metabolised nucleoside analogue.
In addition to HSV all the tricyclic derivatives (9 and 10) were screening against a broad range of DNA and RNA viruses including vaccinia virus, adenovirus-2, human coronavirus (229E), human cytomegalovirus (HCMV) (AD-169 strain and Davis strain), varicella-zoster virus (TK+VZV OKA strain and TK− VZV 07–1 strain), vesicular stomatitis virus, Coxsackie virus B4, respiratory syncytial virus, parainfluenza-3 virus, reovirus-1, Sindbis virus, Punta Toro virus, yellow fever virus, influenza A virus (H1N1 and H3N2 subtypes) and influenza B virus. Of all the compounds, only a few displayed any inhibitory activity <100 μM against VZV and HCMV (Table 3 ): 9e (R = CF3-C6H4-) showed weak inhibitory activity against cytomegalovirus AD-169 strain and Davis strain (EC50 = 63.1 µM and 76.5 µM respectively) compared with GCV as the reference compound (EC50 = 6.5 µM and 4.6 µM respectively) and the TK + VZV Oka strain (EC50 = 49.5 µM); 9f (R = HO-C6H4-) showed inhibitory activity against varicella-zoster virus TK+VZV OKA strain and TK− VZV 07–1 strain (EC50 = 9.6 µM and ≥ 74.5 µM respectively) compared with ACV as the reference compound (EC50 = 5.0 µM and 59.2 µM respectively); 9d and 9j only displayed minimal activity against the VZV TK + strain Oka (EC50 = 63 µM). It should be noted that in contrast to HSV, HBG (7) proved inactive against HCMV and poorly inhibited the VZV TK + strain (EC50 = 76.5 µM). PCV was not inhibitory to HCMV but inhibited VZV with EC50 values comparable with those of ACV, i.e. 3.5 and 33.4 µM, respectively for TK + and TK- VZV strains (Table 3).
Table 3.
Inhibitory activity of tricyclic PCV derivatives (9) against VZV and HCMV and cytotoxicity data. The grey shades highlight the most interesting data.
 |
aRequired to reduce virus-induced cytopathogenicity (HCMV) or plaque formation (VZV) by 50%.
bRequired to cause a microscopically detectable alteration of normal cell morphology.
Aciclovir (ACV), Brivudin (BVDU), Cidofovir (CDV), Ganciclovir (GCV).
ND: not determined.
2.2.2. Cytostatic activity
The new compounds were screened for their potential cytostatic activity against eight human cancer cell lines. The 50% cytostatic inhibitory concentration (IC50) causing a 50% decrease in cell proliferation was determined against retina (non-cancerous) hTERT RPE-1, pancreatic adenocarcinoma Capan-1, chronic myeloid leukaemia Hap1, lung carcinoma NCI-H460, acute lymphoblastic leukaemia DND-41, acute myeloid leukaemia HL-60, chronic myeloid leukaemia K-562 and non-Hodgkin lymphoma Z-138 (Table 4 ).
Table 4.
Cytostatic activity results of compounds 9 and 10. The grey shades highlight the most interesting data.
 |
a50% Inhibitory concentration or compound concentration required to inhibit cell proliferation by 50%.
hTERT RPE-1 retina (non cancerous); Capan-1 pancreatic adenocarcinoma; Hap1 chronic myeloid leukemia; NCI-H460 lung carcinoma; DND-41 acute lymphoblastic leukemia; HL-60 acute myeloid leukemia; K-562 chronic myeloid leukemia; Z-138 non-Hodgkin lymphoma.
Among the tricyclic PCV derivatives compounds 9e (R = CF3-C6H4-) and 9j (R = C6H5-O-C6H4-) showed moderate cytostatic activity against at least six cell lines in the range of 12.5–76.1 µM and 16.9–90.4 µM respectively. The tricyclic HBG derivatives (10) were generally inactive or displayed very weak cytostatic activity. Of the tricyclic HBG derivatives the trifluoromethyl aryl derivative of HBG 10e (R = CF3-C6H4-) was the most effective against the range of cancer cell lines. Proliferation of the pancreatic adenocarcinoma Capan-1 cell line was slightly inhibited by most of the new tricyclic compounds as well as the parent compounds PCV and HBG (Table 4).
2.3. Molecular modelling
Molecular modelling of the compounds was performed using Molecular Operating Environment (MOE) software18 and the crystal structure of HSV-1 thymidine kinase co-crystallised with PCV (PDB 1KI3)19 and 9-(4-hydroxybutyl)-N2-phenylguanine (HBPG) (PDB 1QHI).20
The crystal structures of HSV-1 TK do not include an ATP molecule and a sulphate ion is observed at the supposed position of the β-phosphate of ATP.19, 20 Therefore, the distance between OH of the acyclic chain and SO4 was measured to give an idea about the proper orientation of the ligand within the active site and the possibility for the compound acyclic OH to be phosphorylated by the enzyme. Results indicated that PCV analogues 9a-j display distances in the range of 2.73–6.82 Å compared with PCV (4.61 Å). The HSV-1 TK pocket is predominately lipophilic with several regions of hydrophilicity. PCV and HBPG probe the space of the enzyme active site in a manner that maintains both the base moiety (guanine) and the acyclic sugar hydroxyl groups in a good geometry.
Overall docking results revealed some important interactions for the binding of the reference ligands PCV and HBPG within the active site (Fig. 3 ). The base moiety is lying in a location that stacks with Tyr172 and makes hydrophobic interactions with the amino acid residues; Tyr172, Met128, His58. Also, H-bond interactions are observed between the carbonyl group as well as the top ring nitrogens with Arg176 and Gln125. The acyclic sugar moiety forms two important H-bond interactions with amino acid residues Arg163 and Glu83 (Fig. 3).
Fig. 3.

2D Ligplots illustrating the amino acids in HSV-1 TK active site involved in binding interactions with (A) PCV and (B) HBPG.
The tricyclic PCV compounds lead to interactions with additional amino acids and, amongst the new compounds, the observed interactions of the most active compounds 9f and 9g (R = 4-HO-C6H4, and 4-MeO-C6H4) closely mimic the interactions observed with PCV binding. However, rather than Gln125, the 4-HO-C6H4 and 4-MeO-C6H4 form a H-bond interaction with Ala168 or water-bridged H-bonds with Ala168 and Gln125 (Fig. 4 ). The moderately active fluoro substituted analogues, 9b and 9e, showed almost similar binding modes, although the small fluoro substituent was better docked within the base pocket with the tricyclic moiety forming π–π interaction with Tyr172.
Fig. 4.

Docking of representative tricyclic PCV derivative 9f (cyan) in the HSV-1 TK active site compared with PCV (magenta).
As the tricyclic purine moiety is longer than the guanine base of PCV, the orientation of the 6-arylgroup has an impact on the geometry of the acyclic hydroxyl groups and hence the stability of the formed complex for further phosphorylation. PCV analogues 9h, 9i and 9j with relatively more bulky ether moieties (4-EtO-C6H4, 3-OCH2O-4-C6H3and 4-C6H5O-C6H4) formed weak interactions within the acyclic sugar binding pocket. In addition a steric clash was observed in the case of compound 9j. The steric effect might consequently diminish or abolish their complex stability.
Analysis of the docked HBG analogues 10a–h compared with HBPG suggests that the ligand likely competes between two different binding orientations within the active site. In one orientation the tricyclic base moiety stacks with Tyr172 with the carbonyl group forming a H-bond with Gln125, while the acyclic hydroxyl group is situated 5.73–7.13 Å from the sulphate group (HPBG 5.66 Å) and donates a H-bond to Glu83 and/or accepts water bridged H-bonds from Arg163, Glu83, Arg222 and Lys62. Also, Arg176, in some derivatives, donates a H-bond to N-1 (Fig. 5 A). In the other orientation the tricyclic structure is lying in a location that allows a stacking interaction with His58 and the carbonyl group is being upturned to accept a H-bond from Arg163 and/or water-bridged H-bonds from Arg163, Glu83, Arg222 and Lys62. In contrast, the same acyclic hydroxyl group in the second ligand orientation is 12.75 Å away from the sulphate compared with the former orientation where it accepts a H-bond from Arg176 (Fig. 5B). In both orientations no contribution in ligand binding was observed for the 6-aryl group resulting in more flexibility and weaker binding of the whole compound within the active site.
Fig. 5.

Docking of HBG analogues 10a–h showing two different orientations adopted by the compounds on binding within the HSV-1 TK active site.
3. Conclusion
Comparing antiviral activity to compound structure provides an insight into which functional groups may be beneficial towards HSV-1 inhibitory activity.
The SAR study suggested that a di-hydroxyl acyclic sugar moiety was essential for the activity. For the PCV derivatives, activity was only retained in those that have a hydrogen bond forming group on the tricyclic ring, however the size of the hydrogen bond forming group is limited with larger groups inducing a steric effect that exhibits an apparent impact on activity, which was supported by molecular docking (Fig. 6 ).
Fig. 6.

Summary for SAR of the new tricyclic PCV compounds.
The new tricyclic analogues of PCV and HBG are the carbocyclic analogues of the previously reported tricyclic analogues of GCV and ACV,11, 12 (Fig. 1). For both the tricyclic ACV and GCV derivatives, inhibitory activity against HSV-1 and HSV-2 were generally comparable, however this activity was not retained in HSV-1 TK–KOS ACVr strain. Replacing oxygen in the acyclic chain of the tricyclic ACV analogues by carbon in the new tricyclic HBG derivatives abolished their activity, highlighting the preference of O rather than CH2 in the acyclic chain of the tricyclic guanine derivative. In the case of the tricyclic PCV derivatives the activity generally decreased noticeably however activity was retained only in the tricyclic PCV derivatives with R = 6-(4-MeO-C6H4) and R = 6-(4-HO-C6H4) and R = 6-(4-F-C6H4) groups as discussed in the present study. Importantly, for two of the tricyclic PCV derivatives with R = 6-(4-MeO-C6H4) and R = 6-(4-F-C6H4) groups activity was retained in the ACV resistant HSV-1 TK–KOS strain, which was not observed for the tricyclic GCV compounds.11, 12
This preliminary study provides useful data for further development of tricyclic acyclic nucleoside derivatives with improved lipophilicity and retention of activity in TK deficient strains.
4. Experimental
4.1. General experimental
All reagents and solvents were of general purpose or analytical grade and purchased from Sigma-Aldrich Ltd, Fisher Scientific, Fluka and Acros. 1H, 13C and 19F NMR spectra were recorded with a Bruker Avance DPX500 spectrometer operating at 500, 125 and 470 MHz respectively, with Me4Si as internal standard. Elemental analysis was performed (for compounds 10e and 10g) by MEDAC Ltd (Chobham, Surrey, UK). High resolution mass spectra (HRMS) were determined (for compounds 9a–j, 10a–d, 10f, and 10h) at the EPSRC National Mass Spectrometry Facility at Swansea University and Medac Ltd (Chobham, Surrey, UK), using ESI (Electrospray Ionisation). Flash column chromatography was performed with silica gel 60 (230–400 mesh) (Merck) and TLC was carried out on precoated silica plates (kiesel gel 60 F254, BDH). Compounds were visualised by illumination under UV light (254 nm). Melting points were determined on an electrothermal instrument and are uncorrected. All solvents were dried prior to use and stored over 4 Å molecular sieves, under nitrogen. All the compounds were ≥95% pure.
The following compounds were prepared as previously described: PCV (6),16 HBG (7),17 and the bromoketones (8).21, 22, 23, 24, 25
4.2. Chemistry
4.2.1. General method for the synthesis of tricyclic PCV and HBG derivatives 9 and 10
To a suspension of penciclovir (6) or hydroxybutylguanine (7) (0.2 g, 0.8 mmol or 0.9 mmol, respectively) in dry dimethylformamide (16 mL), sodium hydride (60% suspension in mineral oil, 1.2 eq.) was added and the reaction stirred at room temperature (21 °C) for 1.5 h. Bromoketone (8) (1.3 eq.) was then added and the reaction stirred for 6 h. Aqueous ammonia (25% solution 5 mL) was added to quench the reaction, which was concentrated under reduced pressure and the residual oil purified by flash column chromatography using gradient elution (CH2Cl2-MeOH). The obtained products were recrystallised from a mixture of CH2Cl2 and MeOH.
4.2.1.1. 3-(4-Hydroxy-3-(hydroxymethyl)butyl)-6-phenyl-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9a)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(phenyl)ethan-1-one (8a) (0.21 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 90:10 v/v to give the product as a white solid. Yield: 0.19 g (68%), m.p. 212–214 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.26. 1H NMR (DMSO‑d6): δ 1.51 (septet, 1H, J = 6.0 Hz, H-3′), 1.82 (q, 2H, J = 7.0 Hz, H-2′), 3.40 and 3.47 (two quint, 4H, J = 5.4 Hz, H-4′), 4.17 (t, 2H, J = 7.5 Hz, H-1′), 4.48 (t, 2H, J = 5.3 Hz, 2 × OH), 7.39 (t, 1H, J = 7.0 Hz, H-4″), 7.49 (t, 2H, J = 7.7 Hz, H-3″ and H-5″), 7.91 (d, 2H, J = 7.4 Hz, H-2″ and H-6″), 7.95 (s, 1H, H-2), 8.21 (s, 1H, H- 7), 13.07 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 29.1 (C-2′), 41.3 (C-3′), 42.0 (C-1′), 61.8 (C-4′), 103.6 (C-7), 115.9 (C-9a), 125.4 (C-4″), 128.4 (C-6), 129.2 (C-2″ and C-6″), 129.4 (C-1″), 129.5 (C-3″ and C-5″), 139.0 (C-2), 146.9 (C-4a), 150.6 (C-3a), 151.7 (C-9). [ESI-HRMS] calculated for C18H20N5O3: 354.1561 [M+H]+. Found: 354.1562 [M+H]+.
4.2.1.2. 6-(4-Fluorophenyl)-3-(4-hydroxy-3-(hydroxymethyl)butyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9b)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(4-fluorophenyl)ethanone (8b) (0.23 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 98:2 to 91:9 v/v to give the product as a white solid. Yield: 0.19 g (65%), m.p. > 300 °C, TLC (CH2Cl2-MeOH 9:1 v/v) Rf 0.49. 1H NMR (DMSO‑d6): δ 1.50 (septet, 1H, J = 6.0 Hz, H-3′), 1.81 (q, 2H, J = 7.0 Hz, H-2′), 3.38 and 3.46 (two quint, 4H, J = 5.4 Hz, H-4′), 4.17 (t, 2H, J = 7.5 Hz, H-1′), 4.48 (t, 2H, J = 5.25 Hz, 2 × OH), 7.34 (t, 2H, JHF = 8.8 Hz, H-3″ and H-5″), 7.97 (s, 1H, H-2), 7.98 (dd, 2H, JHH,JHF = 6.0, 8.5 Hz, H-2″ and H-6″), 8.21 (s, 1H, H-7), 13.10 (br.s, 1H, H-5). 13C NMR(DMSO‑d6): δ 29.1 (C-2′), 41.3 (C-3′), 42.0 (C-1′), 61.8 (C-4′), 103.6 (C-7), 116.1 (C-9a), 116.6 (d, 2C, JCF = 22 Hz, C-3″ and C-5″), 125.1 (C-1″), 127.7 (d, 2C, JCF = 8.0 Hz, C-2″ and C-6″), 128.5 (C-6), 139.6 (C-2), 146.8 (C-4a), 150.6 (C-3a), 151.8 (C-9), 162.6 (d, 1C, JCF = 245 Hz, C-4″). 19F NMR(DMSO‑d6): δ-112.40. [ESI-HRMS] calculated for C18H19N5O3F: 372.1472 [M+H]+. Found: 372.1483 [M+H]+.
4.2.1.3. 6-(4-Chlorophenyl)-3-(4-hydroxy-3-(hydroxymethyl)butyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one, (9c)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(4-chlorophenyl)ethanone (8c) (0.24 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 90:10 v/v to give the product as a white solid. Yield: 0.19 g (62%), m.p. 258–260 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.29. 1H NMR (DMSO‑d6): δ 1.50 (septet, 1H, J = 6.0 Hz, H-3′), 1.81 (q, 2H, J = 7.0 Hz, H-2′), 3.38 and 3.46 (two quint, 4H, J = 5.5 Hz, H-4′), 4.17 (t, 2H, J = 7.5 Hz, H-1′), 4.48 (t, 2H, J = 5.2 Hz, 2 × OH), 7.56 (d, 2H, J = 8.6 Hz, H-2″ and H-6″), 7.94 (s, 1H, H-2), 7.95 (d, 2H, J = 8.6 Hz, H-3″ and H-5″), 8.29 (s, 1H, H-7), 13.13 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 29.1 (C-2′), 41.3 (C-3′), 42.0 (C-1′), 61.8 (C-4′), 104.3 (C-7), 116.0 (C-9a), 127.2 (C-2″ and C-6″), 127.5 (C-1″), 128.4 (C-6), 129.6 (C-3″ and C-5″), 133.7 (C-4″), 139.5 (C-2), 146.9 (C-4a), 150.7 (C-3a), 151.8 (C-9). [ESI-HRMS] calculated for C18H19N5O3Cl: 388.1176 [M+H]+. Found: 388.1187 [M+H]+.
4.2.1.4. 3-(4-Hydroxy-3-(hydroxymethyl)butyl)-6-(p-tolyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9d)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(p-tolyl)ethanone (8d) (0.22 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 100:0 to 85:15 v/v to give the product as a white solid. Yield: 0.18 g (61%), m.p. 112–114 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.47. 1H NMR (DMSO‑d6): δ 1.50 (septet, 1H, J = 6.0 Hz, H-3′), 1.82 (q, 2H, J = 7.0 Hz, H-2′), 2.34 (s, 3H, CH3), 3.39 and 3.46 (two quint, 4H, J = 5.4 Hz, H-4′), 4.17 (t, 2H, J = 7.5 Hz, H-1′), 4.48 (t, 2H, J = 5.3 Hz, 2 × OH), 7.28 (d, 2H, J = 8.0 Hz, H-3″ and H-5″), 7.79 (d, 2H, J = 8.0 Hz, H-2″ and H-6″), 7.95 (s, 1H, H-2), 8.13 (s, 1H, H-7), 13.01 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 21.3 (CH3), 29.1 (C-2′), 41.3 (C-3′), 44.0 (C-1′), 61.8 (C-4′), 103.0 (C-7), 116.0 (C-9a), 125.4 (C-3″ and C-5″), 125.7 (C-1″), 129.5 (C-6), 130.0 (C-2″ and C-6″), 138.8 (C-4″), 139.5 (C-2), 146.8 (C-4a), 150.6 (C-3a), 151.8 (C-9). [ESI-HRMS] calculated for C19H21N5O3Na: 390.1537[M + Na]+. Found: 390.1533[M + Na]+.
4.2.1.5. 3-(4-Hydroxy-3-(hydroxymethyl)butyl)-6-(4-(trifluoromethyl)phenyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9e)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(4-(trifluoromethyl)phenyl)ethanone (8e) (0.28 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 92:8 v/v to give the product as a white solid. Yield: 0.18 g (55%), m.p. > 300 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.43. 1H NMR (DMSO‑d6): δ 1.51 (septet, 1H, J = 6.0 Hz, H-3′), 1.82 (q, 2H, J = 7.0 Hz, H-2′), 3.39 and 3.46 (two quint, 4H, J = 5.4 Hz, H-4′), 4.18 (t, 2H, J = 7.0 Hz, H-1′), 4.45 (br s, 2H, 2 × OH), 7.85 (d, 2H, J = 7.8 Hz, H-2″ and H-6″), 7.98 (s, 1H, H-2), 8.15 (d, 2H, J = 7.7 Hz, H-3″ and H-5″), 8.42 (s, 1H, H-7), 13.24 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 29.2 (C-2′), 42.0 (C-3′), 42.3 (C-1′), 61.8 (C-4′), 105.7 (C-7), 116.1 (C-9a), 124.5 (d, 1C, JCF = 272 Hz, CF3), 126.1(C-2″ and C-6″), 126.4 (d, 2C, JCF = 3.8 Hz, C-3″ and C-5″), 128.0 (C-6), 128.5 (q, 1C, JCF = 31.8 Hz, C-4″), 132.6 (C-1″), 139.7 (C-2), 147.0 (C-4a), 150.8 (C-3a), 151.7 (C-9). 19F NMR(DMSO‑d6): δ-61.11.[ESI-HRMS] calculated for C19H19N5O3F3: 422.1440 [M+H]+, Found: 422.1430 [M+H]+.
4.2.1.6. 3-(4-Hydroxy-3-(hydroxymethyl)butyl)-6-(4-hydroxyphenyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9f)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(4-hydroxyphenyl)ethanone (8f) (0.22 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 90:10 v/v to give the product as a pale yellow solid. Yield: 0.11 g (38%), m.p. > 300 °C, TLC (CH2Cl2-MeOH 9:1 v/v) Rf 0.25. 1H NMR (DMSO‑d6): δ 1.51 (septet, 1H, J = 6.0 Hz, H-3′), 1.81 (q, 2H, J = 7.0 Hz, H-2′), 3.38 and 3.45 (two quint, 4H, J = 5.4 Hz, H-4′), 4.17 (t, 2H, J = 7.0 Hz, H-1′), 4.47 (br s, 2H, 2 × OH), 6.86 (d, 2H, J = 8.7 Hz, H-3″ and H-5″), 7.72 (d, 2H, J = 8.7 Hz, H-2″ and H-6″), 7.95 (br s, 2H, H-2 and H-7) 9.87 (br s, 1H, phenolic OH), 12.89 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 29.1 (C-2′), 41.3 (C-3′), 42.0 (C-1′), 61.8 (C-4′), 101.4 (C-7), 116.0 (C-9a), 116.3 (C-3″ and C-5″), 119.2 (C-1″), 127.1 (C-2″ and C-6″), 129.9 (C-6), 139.6 (C-2), 146.6 (C-4a), 150.5 (C-3a) 151.8 (C-9), 158.6 (C- 4″). [ESI-HRMS] calculated for C18H18N5O4: 368.1359 [M−H]+. Found: 368.1349 [M−H]+.
4.2.1.7. 3-(4-Hydroxy-3-(hydroxymethyl)butyl)-6-(4-methoxyphenyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9g)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(4-methoxyphenyl)ethanone (8g) (0.24 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 97.5:2.5 to 87:13 v/v to give the product as a white solid. Yield: 0.24 g (79%), m.p. 202–203 °C, TLC (CH2Cl2-MeOH 9:1 v/v) Rf 0.45. 1H NMR (DMSO‑d6): δ 1.51 (septet, 1H, J = 6.0 Hz, H-3′), 1.81 (q, 2H, J = 7.0 Hz, H-2′), 3.39 and 3.46 (two quint, 4H, J = 5.5 Hz, H-4′), 3.81 (s, 3H, OCH3), 4.17 (t, 2H, J = 7.5 Hz, H-1′), 4.47 (t, 2H, J = 5.2 Hz, 2 × OH), 7.04 (d, 2H, J = 8.0 Hz, H-3″ and H-5″), 7.85 (d, 2H, J = 8.0 Hz, H-2″ and H-6″), 7.95 (s, 1H, H-2), 8.07 (s, 1H, H-7), 12.97 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 29.1 (C-2′), 41.3 (C-3′), 42.0 (C-1′), 55.8 (OCH3), 61.8 (C-4′), 102.2 (C-7), 115.0 (C-3″ and C-5″), 116.1 (C-9a), 120.9 (C-1″), 127.0 (C-2″ and C-6″), 129.4 (C-6), 139.5 (C-2), 146.7 (C-4a), 150.5 (C-3a), 151.8 (C-9), 160.1 (C-4″). [ESI-HRMS] calculated for C19H22N5O4: 384.1666 [M+H]+. Found: 384.1666 [M+H]+.
4.2.1.8. 6-(4-Ethoxyphenyl)-3-(4-hydroxy-3-(hydroxymethyl)butyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9h)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(4-ethoxyphenyl)ethanone (8h) (0.25 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 91:9 v/v to give the product as a pale yellow solid. Yield: 0.24 g (77%), m.p. 276–278 °C, TLC (CH2Cl2-MeOH 9:1 v/v) Rf 0.5. 1H NMR (DMSO‑d6): δ 1.35 (t, 3H, J = 7.0 Hz, CH3), 1.51 (septet, 1H, J = 6.0 Hz, H-3′), 1.82 (q, 2H, J = 7.0 Hz, H-2′), 3.39 and 3.46 (two quint, 4H, J = 5.5 Hz, H-4′), 4.08 (q, 2H, J = 7.0 Hz, OCH2), 4.17 (t, 2H, J = 7.0 Hz, H-1′), 4.44 (br s, 2H, 2 × OH), 7.02 (d, 2H, J = 8.3 Hz, H-3″ and H-5″), 7.83 (d, 2H, J = 8.3 Hz, H-2″ and H-6″), 7.94 (s, 1H, H-2), 8.05 (s, 1H, H-7), 12.93 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 15.1 (CH3), 29.2 (C-2′), 41.4 (C-3′), 42.0 (C-1′), 61.8 (C-4′), 63.7 (OCH2), 102.1 (C-7), 115.4 (C-3″ and C-5″), 116.1 (C-9a), 120.8 (C-1″), 127.0 (C-2″ and C-6″), 129.4 (C-6), 140.0 (C-2), 146.7 (C-4a), 150.5 (C-3a), 151.8 (C-9), 159.4 (C-4″). [ESI-HRMS] calculated for C20H22N5O4: 396.1672 [M−H]+, Found: 396.1668 [M−H]+.
4.2.1.9. 6-(Benzo[d][1,3]dioxol-5-yl)-3-(4-hydroxy-3-(hydroxymethyl)butyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9i)
Prepared from PCV (0.2 g, 0.8 mmol) and 1-(benzo[d][1,3]dioxol-5-yl)-2-bromoethanone (8i) (0.25 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH-acetone 80:10:10 v/v/v to give the product as a pale yellow solid. Yield: 0.21 g (68%), m.p. 259–260 °C, TLC (CH2Cl2-MeOH 9:1 v/v) Rf 026. 1H NMR (DMSO‑d6): δ 1.51 (septet, 1H, J = 6.0 Hz, H-3′), 1.82 (q, 2H, J = 7.0 Hz, H-2′), 3.38 and 3.46 (two quint, 4H, J = 5.5 Hz, H-4′), 4.17 (t, 2H, J = 7.5 Hz, H-1′) 4.45 (t, 2H, J = 5.2 Hz, 2 × OH), 6.09 (s, 2H, H-2″), 7.03 (d, 1H, J = 8.1 Hz, H-7″), 7.41 (dd, 1H, J = 1.8, 8.1 Hz, H-6″), 7.53 (d, 1H, J = 1.7 Hz, H-4″), 7.94 (s, 1H, H-2), 8.11 (s, 1H, H-7), 12.92 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 29.2 (C-2′), 41.4 (C-3′), 42.0 (C-1′), 61.8 (C-4′), 101.9 (C-2″), 102.8 (C-7), 106.0 (C-4″), 109.2 (C-7″), 116.1 (C-9a), 119.5 (C- 6″), 122.4 (C-5″), 129.3 (C-6), 139.6 (C-2), 146.7 (C-4a), 148.2 (C-7a''), 148.5 (C-3a''), 150.6 (C-3a) 151.8 (C-9). [ESI-HRMS] calculated for C19H20N5O5: 398.1459 [M+H]+. Found: 398.1460 [M+H]+.
4.2.1.10. 3-(4-Hydroxy-3-(hydroxymethyl)butyl)-6-(4-phenoxyphenyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (9j)
Prepared from PCV (0.2 g, 0.8 mmol) and 2-bromo-1-(4-phenoxyphenyl)ethanone (8j) (0.3 g, 1.04 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 100:0 to 92:8 v/v to give the product as a white solid. Yield: 0.26 g (75%), m.p. 278–280 °C, TLC (CH2Cl2-MeOH 9:1 v/v) Rf 0.53. 1H NMR (DMSO‑d6): δ 1.52 (septet, 1H, J = 6.0 Hz, H-3′), 1.82 (q, 2H, J = 7.0 Hz, H-2′), 3.38 and 3.46 (two quint, 4H, J = 5.5 Hz, H-4′), 4.18 (t, 2H, J = 7.5 Hz, H-1′), 4.44 (br s, 2H, 2 × OH), 7.09 (d, 4H, J = 8.1 Hz, H-3″, H-5″, H-2‴ and H-6‴), 7.20 (t, 1H, J = 7.4 Hz, H-4‴), 7.44 (t, 2H, J = 8.0 Hz, H-3‴ and H-5‴), 7.93 (d, 2H, J = 8.8 Hz, H- 2″ and H-6″), 7.98 (s, 1H, H-2), 8.14 (s, 1H, H-7), 13.04 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 29.1 (C-2′), 41.4 (C-3′), 42.0 (C-1′), 61.8 (C-4′), 103.1 (C-7), 115.9 (C-9a), 119.2 and 119.6 (C-3″, C-5″, C-2‴ and C-6‴), 123.6 (C-1″), 124.5 (C-4‴), 127.4 and 130.7 (C-2″, C-6″, C-3‴ and C-5‴), 129.0 (C-6), 139.5 (C-2), 146.8 (C-4a), 150.6 (C-3a), 151.7 (C-9), 156.5 and 157.7 (C- 4″ and C-1‴). [ESI-HRMS] calculated for C24H22N5O4:444.1672 [M−H]+, Found: 444.1675 [M−H]+.
4.2.1.11. 3-(4-Hydroxybutyl)-6-phenyl-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10a)
Prepared from HBG (0.2 g, 0.9 mmol) and 2-bromo-1-(phenyl)ethan-1-one (8a) (0.23 g, 1.17 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 93:7 v/v to give the product as a white solid. Yield: 0.18 g (61%), m.p. 248–250 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.32. 1H NMR (DMSO‑d6): δ 1.43 (quint., 2H, J = 7.0 Hz, H-3′), 1.85 (quint, 2H, J = 7.0 Hz, H- 2′), 3.42 (q, 2H, J = 5.5 Hz, H-4′), 4.11 (t, 2H, J = 7.2 Hz, H-1′), 4.49 (t, 1H, J = 4.8 Hz, OH), 7.39 (t, 1H, J = 7.4 Hz, H-4″), 7.48 (t, 2H, J = 7.7 Hz, H-3″ and H-5″), 7.91 (d, 2H, J = 7.6 Hz, H-2′' and H-6″), 7.95 (s, 1H, H-2), 8.22 (s, 1H, H-7), 13.07 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 26.7 (C-2′), 30.0 (C-3′), 43.5 (C-1′), 60.6 (C-4′), 103.6 (C-7), 116.1 (C-9a), 125.4 (C-4″), 128.5 (C-6), 129.2 (C-2″ and C-6″), 129.4 (C-1″), 129.5 (C-3″ and C-5″), 139.7 (C-2), 146.9 (C-4a), 150.9 (C-3a), 151.8 (C-9). [ESI-HRMS] calculated for C17H28N5O2: 322.1309 [M+H]+. Found: 322.1302 [M+H]+.
4.2.1.12. 6-(4-Fluorophenyl)-3-(4-hydroxybutyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10b)
Prepared from HBG (0.2 g, 0.9 mmol) and 2-bromo-1-(4-fluorophenyl)ethanone (8b) (0.25 g, 1.17 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 100:0 to 94:6 v/v to give the product as a white solid. Yield: 0.19 g (62%), m.p. 288–290 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.31. 1H NMR (DMSO‑d6): δ 1.43 (quint, 2H, J = 7.5 Hz, H-3′), 1.85 (quint, 2H, J = 7.5 Hz, H-2′), 3.42 (q, 2H, J = 5.4 Hz, H-4′), 4.11 (t, 2H, J = 7.0 Hz, H-1′), 4.49 (t, 1H, J = 5.0 Hz, OH), 7.34 (t, 2H, JHF = 8.9 Hz, H-3″ and H-5″), 7.96 (dd, 2H, JHF = 5.0, 9.0 Hz, H-2″ and H-6″), 7.96 (s, 1H, H-2), 8.22 (d, 1H, J = 2.0 Hz, H-7), 13.09 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 26.7 (C-2′), 30.0 (C-3′), 43.5 (C-1′), 60.6 (C-4′), 103.6 (C-7), 116.1 (C-9a), 116.6 (d, 2C, JCF = 22.6 Hz, C-3″ and C-5″), 125.1 (d, 1C, J = 3.8 Hz, C-1″), 127.7 (d, 2C, J = 7.5 Hz, C-2″ and C-6″), 128.5 (C-6), 139.7 (C- 2), 146.8 (C-4a), 150.7 (C-3a), 151.8 (C-9), 162.6 (d, 1C, J = 246.5 Hz, C-4″). 19F NMR(DMSO‑d6): δ-112.40.[ESI-HRMS] calculated for C17H17N5O2F: 342.1366 [M+H]+. Found: 342.1373 [M+H]+.
4.2.1.13. 6-(4-Chlorophenyl)-3-(4-hydroxybutyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10c)
Prepared from HBG (0.2 g, 0.9 mmol) and 2-bromo-1-(4-chlorophenyl)ethanone (8c) (0.27 g, 1.17 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 92:8 v/v to give the product as a white solid. Yield: 0.2 g (62%), m.p. 280–282 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.36. 1H NMR (DMSO‑d6): δ 1.43 (quint, 2H, J = 7.0 Hz, H-3′), 1.85 (quint, 2H, J = 7.5 Hz; H-2′), 3.42 (q, 2H, J = 5.5 Hz, H-4′), 4.10 (t, 2H, J = 7.0 Hz, H-1′), 4.48 (t, 1H, J = 5.0 Hz, OH), 7.53 (d, 2H, J = 8.4 Hz, H-2″ and H-6″), 7.92 (d, 2H, J = 8.4 Hz, H-3″ and H-5″), 7.94 (s, 1H, H-2), 8.27 (s, 1H, H-7), 14.00 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 26.7 (C-2′), 30.0 (C-3′), 43.5 (C-1′), 60.6 (C-4′), 104.3 (C-7), 116.1 (C-9a), 127.1 (C-2″ and 6″), 127.4 (C-1″), 128.3 (C-6), 129.5 (C-3″ and C-5″), 133.6 (C-4″), 139.7 (C-2), 146.9 (C-4a), 150.7 (C-3a), 151.7 (C-9).[ESI-HRMS] calculated for C17H17N5O2Cl: 358.1071 [M+H]+. Found: 358.1075 [M+H]+.
4.2.1.14. 3-(4-Hydroxybutyl)-6-(p-tolyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10d)
Prepared from HBG (0.2 g, 0.9 mmol) and 2-bromo-1-(p-tolyl)ethanone (8d) (0.25 g, 1.17 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 100:0 to 94:6 v/v to give the product as a white solid. Yield: 0.2 g (67%), m.p. 260–261 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.63. 1H NMR (DMSO‑d6): δ 1.43 (quint, 2H, J = 7.0 Hz, H-3′), 1.85 (quint, 2H, J = 7.5 Hz, H-2′), 2.34 (s, 3H, CH3), 3.42 (q, 2H, J = 5.5 Hz, H-4′), 4.11 (t, 2H, J = 7.2 Hz, H-1′), 4.49 (t, 1H, J = 5 Hz, OH), 7.28 (d, 2H, J = 8.0 Hz, H-3″ and H-5″), 7.79 (d, 2H, J = 8.2 Hz, H-2″ and H-6″), 7.95 (s, 1H, H-2), 8.14 (s, 1H, H-7), 13.01 (br.s, 1H, H- 5). 13C NMR (DMSO‑d6): δ 21.3 (CH3), 26.7 (C-2′), 30.0 (C-3′), 43.5 (C-1′), 60.6 (C-4′), 103.0 (C-7), 116.0 (C-9a), 125.4 (C-3″ and 5″), 125.6 (C-1″), 129.5 (C- 6), 130.1 (C-2″ and C-6″), 138.8 (C-4″), 139.6 (C-2), 146.8 (C-4a), 150.6 (C- 3a), 151.8 (C-9). [ESI-HRMS] calculated for C18H20N5O2: 338.1617 [M+H]+. Found: 338.1626 [M+H]+.
4.2.1.15. 3-(4-Hydroxybutyl)-6-(4-(trifluoromethyl)phenyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10e)
Prepared from HBG (0.2 g, 0.9 mmol) and 2-bromo-1-(4-(trifluoromethyl)phenyl)ethanone (8e) (0.3 g, 1.17 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 97.5:2.5 to 92:8 v/v to give the product as a white solid. Yield: 0.25 g (71%), m.p. 292–293 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.58. 1H NMR (DMSO‑d6): δ 1.44 (quint, 2H, J = 7.0 Hz, H-3′), 1.86 (quint, 2H, J = 7.5 Hz, H-2′), 3.43 (q, 2H, J = 5.5 Hz, H-4′), 4.12 (t, 2H, J = 7.0 Hz, H-1′), 4.45 (t, 1H, J = 5.0 Hz, OH), 7.84 (d, 2H, J = 8.0 Hz, H-2″ and H-6″), 7.96 (s, 1H, H-2), 8.14 (d, 2H, J = 8.0 Hz, H-3″ and H-5″), 8.42 (s, 1H, H-7), 13.23 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 26.7 (C-2′), 30.0 (C-3′), 43.5 (C-1′), 60.6 (C-4′), 105.7 (C-7), 116.1 (C-9a), 124.5 (d, 1C, J = 272.0 Hz, CF3), 126.0 (C-2″ and C-6″), 126.4 (d, 2C, J = 3.6 Hz, C-3″ and C-5″), 128.0 (C-6), 128.5 (q, 1C, JCF = 32.0 Hz, C-4″), 132.6 (C-1″), 139.8 (C-2), 147.1 (C-4a), 150.8 (C-3a), 151.7 (C-9). 19F NMR(DMSO‑d6): δ −61.14. Anal. Calcd for C18H16F3N5O2·0.5H2O (400.1309): C, 54.00; H, 2.38; N, 17.49. Found: C, 53.56; H, 3.84; N, 17.33.
4.2.1.16. 3-(4-Hydroxybutyl)-6-(4-methoxyphenyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10f)
Prepared from HBG (0.2 g, 0.9 mmol) and 2-bromo-1-(4-methoxyphenyl)ethanone (8g) (0.27 g, 1.17 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 93:7 v/v to give the product as a white solid. Yield: 0.19 g (61%), m.p. 252–254 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.23. 1H NMR (DMSO‑d6): δ 1.43 (quint, 2H, J = 7.0 Hz, H-3′), 1.85 (quint, 2H, J = 7.5 Hz; H-2′), 3.42 (q, 2H, J = 5.4 Hz, H-4′), 3.81 (s, 3H, OCH3) 4.11 (t, 2H, J = 7.0 Hz, H-1′), 4.48 (t, 1H, J = 5.0 Hz, OH), 7.04 (d, 2H, J = 8.8 Hz, H-3″ and 5″), 7.84 (d, 2H, J = 8.8 Hz, H-2″ and H-6″), 7.94 (s, 1H, H-2), 8.08 (s, 1H, H-7), 12.97 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 26.7 (C-2′), 30.1 (C-3′), 43.5 (C-1′), 55.7 (OCH3), 60.6 (C-4′), 102.2 (C-7), 115.0 (C-3″ and C-5″), 116.0 (C-9a), 120.9 (C-1″), 126.9 (C-2″ and C-6″), 129.4 (C-6), 139.6 (C-2), 146.7 (C-4a), 150.5 (C-3a), 151.8 (C-9), 160.1 (C-4″).[ESI-HRMS] calculated for C18H20N5O3: 354.1566 [M+H]+. Found: 354.1563 [M+H]+.
4.2.1.17. 6-(Benzo[d][1,3]dioxol-5-yl)-3-(4-hydroxybutyl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10g)
Prepared from HBG (0.2 g, 0.9 mmol) and 1-(benzo[d][1,3]dioxol-5-yl)-2-bromoethanone (8i) (0.28 g, 1.17 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 97.5:2.5 to 92:8 v/v to give the product as a pale yellow solid. Yield: 0.23 g (69%), m.p. 272–274 °C, TLC (CH2Cl2-MeOH 95:5 v/v) Rf 0.38. 1H NMR (DMSO‑d6): δ 1.43 (quint, 2H, J = 7.5 Hz, H-3′), 1.85 (quint, 2H, J = 7.5 Hz; H-2′), 3.42 (q, 2H, J = 5.5 Hz, H-4′), 4.11 (t, 2H, J = 7 Hz, H-1′), 4.45 (t, 1H, J = 5 Hz, OH), 6.09 (s, 1H, H-2″), 7.03 (d, 1H, J = 8.2 Hz, H-7″), 7.41 (d, 1H, J = 8.1 Hz, H-6″), 7.53 (s, 1H, H-4″), 7.93 (s, 1H, H-2), 8.12 (s, 1H, H-7), 12.92 (br.s, 1H, H- 5). 13C NMR (DMSO‑d6): δ 26.7 (C-2′), 30.1 (C-3′), 43.5 (C-1′), 60.7 (C- 4′), 101.9 (C-2″), 102.8 (C-7), 106.0 (C-4″), 109.2 (C-7″), 116.0 (C-9a), 119.4 (C-6″), 122.5 (C-5″), 129.4 (C-6), 139.9 (C-2), 146.7 (C-4a), 148.2 (C-7a''), 148.5 (C-3a''), 150.6 (C-3a), 151.8 (C-9). Anal. Calcd for C18H17N5O4·0.3H2O (372.5312): C, 58.00; H, 4.76; N, 18.79. Found: C, 57.60; H, 4.37; N, 18.56.
4.2.1.18. 3-(4-Hydroxybutyl)-6-(thiophen-2-yl)-3,5-dihydro-9H-imidazo[1,2-a]purin-9-one (10h)
Prepared from HBG (0.2 g, 0.9 mmol) and 2-bromo-1-(thiophen-2-yl)ethanone (8 k) (0.24 g, 1.17 mmol) and purified by flash column chromatography eluting with CH2Cl2-MeOH 95:5 to 92:8 v/v to give the product as a pale yellow solid. Yield: 0.19 g (64%), m.p. 246–247 °C, TLC (CH2Cl2-MeOH 9:1 v/v) Rf 0.67. 1H NMR (DMSO‑d6): δ 1.43 (quint., 2H, J = 7.0 Hz, H-3′), 1.85 (quint, 2H, J = 7.5 Hz; H-2′), 3.42 (q, 2H, J = 5.5 Hz, H-4′), 4.11 (t, 2H, J = 7.0 Hz, H-1′), 4.45 (t, 1H, J = 5.0 Hz, OH), 7.18 (t, 1H, J = 4.2 Hz, H-4″), 7.61 (d, 1H, J = 3.3 Hz, H-3″), 7.66 (d, 1H, J = 4.9 Hz, H-5″), 7.91 (s, 1H, H-2), 7.95 (s, 1H, H-7), 13.14 (br.s, 1H, H-5). 13C NMR (DMSO‑d6): δ 26.6 (C-2′), 30.0 (C-3′), 43.5 (C-1′), 60.6 (C-4′), 102.7 (C-7), 116.1 (C-9a), 124.6 (C-6), 126.6 (C-3″), 127.5 (C-4″), 128.6 (C-5″), 139.7 (C-2), 146.5 (C-4a), 150.6 (C-3a), 151.7 (C-9). [ESI-HRMS] calculated for C15H16N5O2S: 330.1019 [M+H]+. Found: 330.1021 [M+H]+.
4.3. Antiviral and cytotoxicity assays
The antiviral assays were based on inhibition of virus-induced cytopathicity or plaque formation in HEL [herpes simplex virus 1 (HSV-1) (KOS), HSV-2 (G), vaccínia virus, vesicular stomatitis virus, cytomegalovirus (HCMV), varicella-zoster virus (VZV), adenovirus-2, and human corona virus (299E)], Vero (para-influenza-3, reovirus-1, Sindbisvirus, and Coxsackie B4), HeLa (vesicular stomatitis virus, Coxsackie virus B4, and respiratorysyncytial virus), or MDCK [influenza A (H1N1; H3N2) and influenza B] cell cultures. Confluent cell cultures (or nearly confluent for MDCK cells) in microtiter 96-well plates were inoculated with 100 CCID50of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) or with 20 plaque- forming units (PFU). After 1–2 h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations (200, 40, 8, 1.6, 0.32 μM) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 (50% effective concentration) or compound concentration required to reduce virus-induced cytopathogenicity or viral plaque formation by 50%. The MCC (minimal cytotoxic concentration) values were determined as the compound concentration required to afford a microscopically visible alteration of cell morphology. The new compounds were screened for their potential cytostatic activity against eight human cancer cell lines. The 50% cytostatic inhibitory concentration (IC50) causing a 50% decrease in cell proliferationrelative to the number of cells in the untreated controls was determined against retina (non cancerous) hTERT RPE-1, pancreatic adenocarcinomaCapan-1, chronic myeloid leukaemia Hap1, lung carcinoma NCI-H460, acute lymphoblastic leukaemia DND-41, acute myeloid leukaemia HL-60, chronic myeloid leukaemia K-562 and non-Hodgkin lymphoma Z-138.
4.4 Molecular modelling
Docking studies were performed using the MOE18 crystal structure of HSV-1 thymidine kinase co-crystallised with PCV (PDB 1KI3)19 and 9-(4-hydroxybutyl)-N2-phenylguanine (HBPG) (PDB 1QHI).20 All minimisations were performed with MOE until a RMSD gradient of 0.01 Kcal/mol/A with the MMFF94 forcefield and partial charges were automatically calculated. The Alpha Triangle placement, which derives poses by random superposition of ligand atom triplets through alpha sphere dummies in the receptor site, was chosen to determine the poses. The London ΔG scoring function estimates the free energy of binding of the ligand from a given pose. Refinement of the results was done using the MMFF94 forcefield, and rescoring of the refined results using the London ΔG scoring function was applied. The output database dock file was created with different poses for each ligand and arranged according to the final score function (S), which is the score of the last stage that was not set to zero.
Acknowledgements
We thank the Egyptian Government for a Channel research scholarship to AFM and the EPSRC Mass Spectrometry Centre, Swansea, U.K. for mass spectroscopy data. The authors wish to express their gratitude to Mrs Leentje Persoons, Mrs Ellen De Waegenaere, Mrs Bianca Stals, Mrs Kirsten Lepaige, and Mrs. Nathalie Van Winkel for excellent technical assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2019.02.005.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.World Health Organization. Herpes simplex virus. Fact sheets. http://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus 2017(accessed 5/10/2018).
- 2.Gupta R., Warren T., Wald A. Genital herpes. Lancet. 2007;370:2127–2137. doi: 10.1016/S0140-6736(07)61908-4. [DOI] [PubMed] [Google Scholar]
- 3.Looker K.J., Margaret A.S., May M.T., Turner K.M.E., Vickerman P., Gottlieb S.L., Newman L.M. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS One. 2015;10 doi: 10.1371/journal.pone.0140765. e0140765.17pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Granerod J., Ambrose H.E., Davies N.W. Health Protection Agency (HPA) Aetiology of Encephalitis Study Group. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis. 2010;10:835–844. doi: 10.1016/S1473-3099(10)70222-X. [DOI] [PubMed] [Google Scholar]
- 5.Whitley R.J. Herpes simplex encephalitis: adolescents and adults. Antiviral Res. 2006;71:141–148. doi: 10.1016/j.antiviral.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Rozenberg F., Deback C., Agut H. Herpes simplex encephalitis: from virus to therapy. Infectious Disorders – Drug Targets. 2011;11:235–250. doi: 10.2174/187152611795768088. [DOI] [PubMed] [Google Scholar]
- 7.Zuo X.-Z., Tang W.-J., Chen X.-Y., Huang W. A review with comments on herpes simplex virus encephalitis in adults. Neuroimmunol Neuroinflammation. 2017;4:24–27. [Google Scholar]
- 8.Bradshaw M.J., Venkatesan A. Herpes simplex virus-1 encephalitis in adults: pathophysiology, diagnosis, and management. Neurotherapeutics. 2016;13:493–508. doi: 10.1007/s13311-016-0433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piret J., Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011;55:459–472. doi: 10.1128/AAC.00615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawaguchi T., Kusumi M., Hasegawa T. Roles of hydrophobicity, protein binding and the probenecid-sensitive transport system in the cerebrospinal fluid delivery of nucleoside analogues with antiviral activity. Biol Pharm Bull. 2000;23:979–983. doi: 10.1248/bpb.23.979. [DOI] [PubMed] [Google Scholar]
- 11.Golankiewicz B., Ostrowski T., Goslinski T. Fluorescent tricyclic analogues of acyclovir and ganciclovir. A structure-antiviral activity study. J Med Chem. 2001;44:4284–4287. doi: 10.1021/jm010922s. [DOI] [PubMed] [Google Scholar]
- 12.Goslinski T., Golankiewicz B., De Clercq E., Balzarini J. Synthesis and biological activity of strongly fluorescent tricyclic analogues of acyclovir and ganciclovir. J Med Chem. 2002;45:5052–5057. doi: 10.1021/jm020827z. [DOI] [PubMed] [Google Scholar]
- 13.Golankiewicz B., Ostrowski T. Tricyclic nucleoside analogues as antiherpes agents. Antiviral Res. 2006;71:134–140. doi: 10.1016/j.antiviral.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Boyd M.R., Safrin S., Kern E.R. Penciclovir: a review of its spectrum of activity, selectivity, and cross-resistance pattern. Antivir Chem Chemother. 1993;4:3–11. [Google Scholar]
- 15.Larsson A., Alenius S., Johansson N.G., Oberg B. Antiherpetic activity and mechanism of action of 9-(4-hydroxybutyl)guanine. Antiviral Res. 1983;3:77–86. doi: 10.1016/0166-3542(83)90028-1. [DOI] [PubMed] [Google Scholar]
- 16.Toyokuni T., Walsh J.C., Namavari M. Selective and practical synthesis of penciclovir. Synth Commun. 2003;33:3897–3905. [Google Scholar]
- 17.Volpini R., Mishra R.C., Kachare D.D. Adenine based acyclic nucleotides as novel P2X3 receptor ligands. J Med Chem. 2009;52:4596–4603. doi: 10.1021/jm900131v. [DOI] [PubMed] [Google Scholar]
- 18.Molecular Operating Environment (MOE), 2014.0901; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2016.
- 19.Champness J.N., Bennett M.S., Wien F. Exploring the active site of herpes simplex virus type-1 thymidine kinase by X-ray crystallography of complexes with aciclovir and other ligands. Proteins Struct Funct Genet. 1998;32:350–361. doi: 10.1002/(sici)1097-0134(19980815)32:3<350::aid-prot10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 20.Bennett M.S., Wien F., Champness J.N. Structure to 1.9 Å resolution of a complex with herpes simplex virus type-1 thymidine kinase of a novel, non-substrate inhibitor: X-ray crystallographic comparison with binding of aciclovir. FEBS Lett. 1999;443:121–125. doi: 10.1016/s0014-5793(98)01619-6. [DOI] [PubMed] [Google Scholar]
- 21.Cowper R.M., Davidson L.H., Smith L.I., Kaiser E.W. Organic Syntheses. John Wiley & Sons, Inc.; 2003. Phenacyl bromide. [Google Scholar]
- 22.Bellale E., Naik M., Varun V.B. Diarylthiazole: an antimycobacterial scaffold potentially targetingPrrB-PrrA two-component Ssystem. J Med Chem. 2014;57:6572–6582. doi: 10.1021/jm500833f. [DOI] [PubMed] [Google Scholar]
- 23.Joy A, Sun S. Substituted phenacyl molecules and photoresponsive polymers. WO2013090892A1, 2013.
- 24.Abdel-Aziz H.A., Ghabbour H.A., Eldehna W.M. 2-((Benzimidazol-2-yl)thio)-1-arylethan-1-ones: synthesis, crystal study and cancer stem cells CD133 targeting potential. Eur J Med Chem. 2015;104:1–10. doi: 10.1016/j.ejmech.2015.09.023. [DOI] [PubMed] [Google Scholar]
- 25.Wei S., Feng X., Du H. A metal-free hydrogenation of 3-substituted 2H–1,4- benzoxazines. Org Biomol Chem. 2016;14:8026–8029. doi: 10.1039/c6ob01556e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.