

The binding analysis of SARS-CoV main proteinase with HIV, psychotic and parasite drugs (lopinavir, ritonavir, niclosamide and promazine) suggests that these existing drugs can be used as starting points for designing SARS-CoV proteinase inhibitors.
Keywords: SARS-CoV, Drugs, Binding, Proteinase
Abstract
The SARS-associated coronavirus (SARS-CoV) main proteinase is a key enzyme in viral polyprotein processing. To allow structure-based design of drugs directed at SARS-CoV main proteinase, we predicted its binding pockets and affinities with existing HIV, psychotic and parasite drugs (lopinavir, ritonavir, niclosamide and promazine), which show signs of inhibiting the replication of SARS-CoV. Our results suggest that these drugs and another two HIV inhibitors (PNU and UC2) could be used as templates for designing SARS-CoV proteinase inhibitors.
1. Introduction
Reemergence of severe acute respiratory syndrome (SARS) is a distinct possibility. Currently neither antiviral therapy nor vaccine is available. Viral replicase and protease are preferred targets for the screening and design of antiviral compounds and have been successfully targeted in several viral diseases. The SARS-associated coronavirus (SARS-CoV) main proteinase (Mpro or 3CL pro) plays a key role in proteolytic processing of the replicase polyproteins 1a and 1ab, which makes it an attractive target for developing drugs against this new disease. Recent report indicated that the proteinase inhibitor kaletra, a mixture of protease inhibitors––lopinavir and ritonavir, approved for treating HIV in 2000, shows signs of effectiveness against the SARS virus.1 In particular, researchers in Taiwan discovered that two existing medicines, which have significant effect in inhibiting the replication of SARS-CoV (http://www.etaiwannews.com/Taiwan/2003/10/31/1067562739.htm). One is an anti-parasite drug niclosamide, and another is anti-psychotic drug promazine. The purpose of this study is to analyze whether the SARS-CoV main proteinase could be the target of these existing drugs. We performed in silico binding studies of the drugs using the recently identified crystal structure of Mpro,[2], [3] to provide information for anti-SARS inhibitor design.
2. Materials and methods
The atomic coordinates of SARS-CoV main proteinase were downloaded from Protein Data Bank (PDB ID 1Q2W). Another crystal structure of SARS-CoV main proteinase is also available (PDB ID 1UJ1), the superposition of 1Q2W A chain and 1UJ1 A chain is shown in Figure 1 , they overlap very well (rmsd = 0.64), here we chose 1Q2W as docking studies, which was released early. The overall structure of a monomer of SARS-CoV main proteinase is composed of three domains: domain I (residues 1–101), domain II (residues 102–200) and III (residues 201–303), represented by green, pink and white trace in Figure 1. The cleft between domains I and II is its substrate-binding site.
Figure 1.
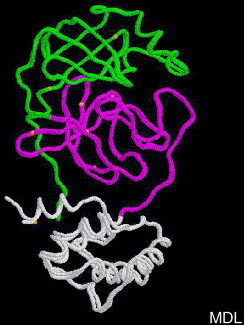
Superposition of two crystal structures from SARS-CoV main proteinase: 1Q2W A chain and 1UJ1 A chain. Domain I (residues 1–101, green trace), domain II (residues 102–200, pink trace) and III (residues 201–303, white trace).
Except four drugs (lopinavir, ritonavir, niclosamide and promazine), we also conducted the docking studies of two other molecules, PNU and UC2, for their molecular formulas are close to those of niclosamide and promazine, respectively (Fig. 2 ), and they both are the inhibitors of HIV-1 reverse transcriptase.[4], [5] The program Hex was employed to conduct the docking of the ligands to the SARS-CoV main proteinase, its basic approach to the docking problem is to model each molecule using 3D parametric functions, which encode both surface shape, electrostatic charge and potential distributions. The surface shape representation uses a novel 3D surface skin model of protein topology, and a novel soft molecular mechanics energy minimization procedure is used to refine the candidate docking solutions. Unlike conventional 3D fast Fourier transform (FFT) docking approaches, Hex uses spherical polar Fourier correlations to accelerate the docking between 10 and 100 times faster than FFT docking algorithm.6 Here we used the following parameter set: correlation type = shape + three probes, post-processing = MM minimization, steric scan = 20 (maximum), final search = 32 (maximum), the others are default set. The structural comparison was performed by LGA.7 The visualization of 3D structure was generated by PROTEINEXPLORER (http://www.proteinexplorer.org).
Figure 2.
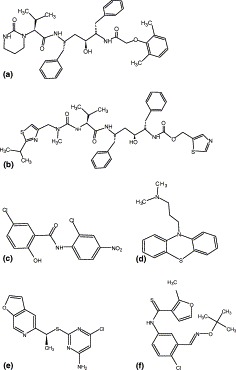
Chemical structures of drugs and inhibitors mentioned in this study: (a) lopinavir (C37H48N4O5), (b) ritonavir (C37H48N6O5S2), (c) niclosamide (C13H8Cl2N2O4), (d) promazine (C17H20N2S), (e) PNU (C13H11ClN4OS), (f) UC2 (C17H19ClN2O2S).
3. Results and discussion
Figure 3 displays the overall structures of docking for four drugs (lopinavir, ritonavir, niclosamide and promazine) and two inhibitors (PNU and UC2) to SARS-CoV main proteinase. The binding pockets of these compounds in SARS-CoV main protease are shown in Table 1 , which is defined by those residues that have at least one heavy atom (other than hydrogen) with a distance less than 5 Å from a heavy atom of inhibitors, as described by Chou et al.8 The results show that the binding pockets of six compounds can be divided into three classes: (1) residues 40–86 and 181–192 for four drugs/inhibitors (lopinavir, niclosamide, promazine and PNU); (2) residues 41–51 and 164–194 for UC2 inhibitor; (3) residues 19–57 and 117–193 for drug ritonavir. All these pockets locate in domain I (residues 8–101), domain II (residues 102–184) and a long loop region (residues 185–200) connecting domains I and II in SARS-CoV main proteinase. Thus, the four drugs and two inhibitors studied here can basically bind to the active site of SARS-CoV main proteinase, a cleft between domains I and II.
Figure 3.
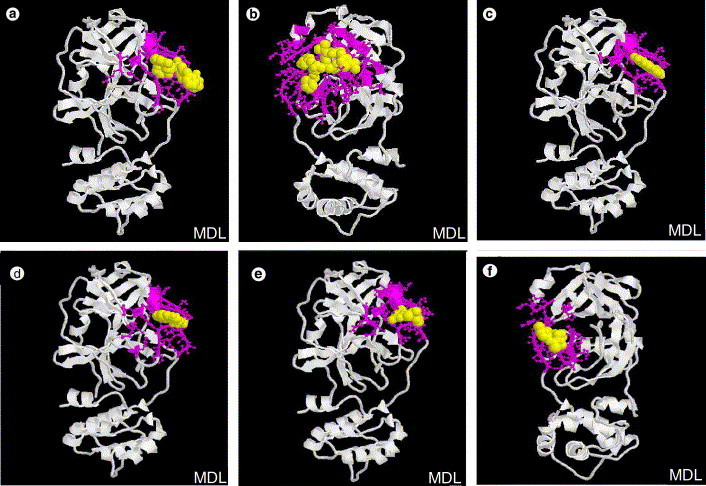
The binding pockets (pink ball-stick) of SARS-associated coronavirus main proteinase (white cartoon) with drugs and inhibitors (yellow spacefill): Anti-HIV drugs lopinavir (a) and ritonavir (b), anti-parasite drug niclosamide (c), anti-psychotic drug promazine (d), HIV inhibitors PNU (e) and UC2 (f).
Table 1.
Binding pockets for SARS-CoV main proteinase with different drugs
| Lopinavir | Ritonavir | Niclosamide | Promazine | PNU | UC2 | |
|---|---|---|---|---|---|---|
| ARG 40 | GLN 19 | SER 123 | ARG 40 | ARG 40 | ARG 40 | HIS 41 |
| CYS 44 | VAL 20 | PHE 140 | MET 49 | ILE 43 | HIS 41 | MET 49 |
| MET 49 | THR 21 | LEU 141 | LEU 50 | CYS 44 | ILE 43 | LEU 50 |
| LEU 50 | CYS 22 | ASN 142 | ASN 51 | MET 49 | CYS 44 | ASN 51 |
| ASN 51 | GLY 23 | GLY 143 | PRO 52 | LEU 50 | MET 49 | HIS 164 |
| PRO 52 | THR 24 | SER 144 | ASN 53 | ASN 51 | LEU 50 | MET 165 |
| ASN 53 | THR 25 | CYS 145 | TYR 54 | PRO 52 | ASN 51 | GLU 166 |
| TYR 54 | THR 26 | GLY 146 | GLU 55 | ASN 53 | PRO 52 | LEU 167 |
| GLU 55 | LEU 27 | SER 147 | ASP 56 | TYR 54 | ASN 53 | PRO 168 |
| ASP 56 | ASN 28 | HIS 163 | LEU 57 | GLU 55 | TYR 54 | THR 169 |
| LEU 57 | PRO 39 | HIS 164 | MET 82 | ASP 56 | GLU 55 | GLY 170 |
| MET 82 | ARG 40 | MET 165 | VAL 186 | LEU 57 | ASP 56 | VAL 171 |
| ASN 84 | HIS 41 | GLU 166 | ASP 187 | LEU 58 | LEU 57 | HIS 172 |
| CYS 85 | VAL 42 | LEU 167 | ARG 188 | MET 82 | LEU 58 | ALA 173 |
| PHE 181 | ILE 43 | PRO 168 | GLN 189 | ASN 84 | MET 82 | PHE 181 |
| PHE 185 | CYS 44 | VAL 171 | THR 190 | CYS 85 | GLN 83 | PRO 184 |
| VAL 186 | MET 49 | HIS 172 | VAL 186 | ASN 84 | PHE 185 | |
| ASP 187 | LEU 50 | ALA 173 | ASP 187 | CYS 85 | VAL 186 | |
| ARG 188 | ASN 51 | GLY 174 | ARG 188 | LEU 86 | ASP 187 | |
| GLN 189 | PRO 52 | PHE 181 | GLN 189 | VAL 186 | ARG 188 | |
| THR 190 | ASN 53 | PHE 185 | THR 190 | ASP 187 | GLN 189 | |
| ALA 191 | TYR 54 | VAL 186 | ARG 188 | THR 190 | ||
| GLN 192 | LEU 57 | ASP 187 | GLN 189 | ALA 191 | ||
| CYS 117 | ARG 188 | THR 190 | GLN 192 | |||
| TYR 118 | GLN 189 | ALA 193 | ||||
| ASN 119 | THR 190 | ALA 194 | ||||
| GLY 120 | ALA 191 | |||||
| SER 121 | GLN 192 | |||||
| ALA 193 | ||||||
To estimate the binding affinities of each compound, the inhibitory constant (K i, mole) was calculated from the equation:ΔG=−RTlnKiwhere ΔG is the free energy of binding (kJ/mol) (here refers to the final docked energy), R is the gas constant 8.31 J/K/mol and T is the absolute temperature (at 300 K), as did in Jenwitheesuk and Samudrala.9 The results indicate that the inhibitory constants of six compounds are: 8.7 × 10−20 (lopinavir), 5.6 × 10−25 (ritonavir), 4.2 × 10−22 (niclosamide), 6.2 × 10−21 (promazine), 3.5 × 10−23 (PNU), 2.1 × 10−19 (UC2). It is noted that these values are too low, for example, the inhibitory constant of lopinavir was determined as ∼10−7 by Jenwitheesuk and Samudrala.10 The reason for this difference is that the docked energy value from Hex program is a pseudo-energy, which is designed to give reasonably consistent units with conventional energy calculations, not based on experimentally derived parameters, and as a theoretical reference value only when performing the docking algorithm. Thus we do not expect these values are the genuine representations of inhibitory constants and we use them primarily for qualitative comparison among the drugs/inhibitors studied here. Because the lower the K i is, the greater the binding affinity is, hence HIV drug ritonavir is the compound that bind to the substrate binding site of SARS-CoV proteinase with the highest binding affinity, followed by HIV inhibitor PNU and anti-parasite drug niclosamide, and UC2 is the compound with the lowest binding affinity. Moreover, the inhibitory constants of ritonavir, PNU, niclosamide, promazine and UC2 are about 10−5, 10−3, 10−2, 10−1 and 10-fold inhibitory constant of lopinavir, respectively, if we assume that a value of 10−7 mol for lopinavir's inhibitory constant is correct, the inhibitory constants of ritonavir, PNU, niclosamide, promazine and UC2 could be estimated as 10−12, 10−10, 10−9, 10−8 and 10−6 mol, respectively.
The close views of the interactions between SARS-CoV main proteinase and these drugs/inhibitors are exhibited in Figure 4 . The results show that half of lopinavir is left outside the catalytic site (Fig. 4a), for ritonavir, the thiazole group (P1) and a benzene group (P2) are inserted into S1 and S2 specificity pockets, respectively, while another benzene side chain (P3) might be too long to fit the substrate binding pocket perfectly (Fig. 4b), there is similar situation in the inhibitor AG7088,11 which has been experimentally shown to not bind with high affinity to the SARS-CoV proteinase (http://www.nature.com/nsu/030512/030512-11.html). Thus the efficacy of lopinavir/ritonavir could be poor. Indeed, consistent with our predictions, experimental observation data indicated that both lopinavir and ritonavir individually have only a weak in vitro activity against SARS-CoV. However, the addition of lopinavir/ritonavir to ribavirin and corticosteroid treatment regimens appears to reduce incubation and mortality rates, especially when administered early.12 Similarly, the half of niclosamide or promazine is left outside the active site (Fig. 4c and d), obviously the propane side chain in promazine is too long. For PNU inhibitor, seems it can basically fit into the active cleft, except the dihydrofuran side chain is a little bit long (Fig. 4e). Finally, the inhibitor UC2 binds to a position that is slightly away from the active centre (Fig. 4f), its neopentane or methylfuran side chain is a little long and makes it unable to insert into the active pocket properly. Indeed UC2 is the compound with lowest binding affinity as mentioned above. Taken together, our study illustrates that existing drugs/inhibitors may be used as starting points for the discovery of rationally designed anti-SARS proteinase drugs.
Figure 4.
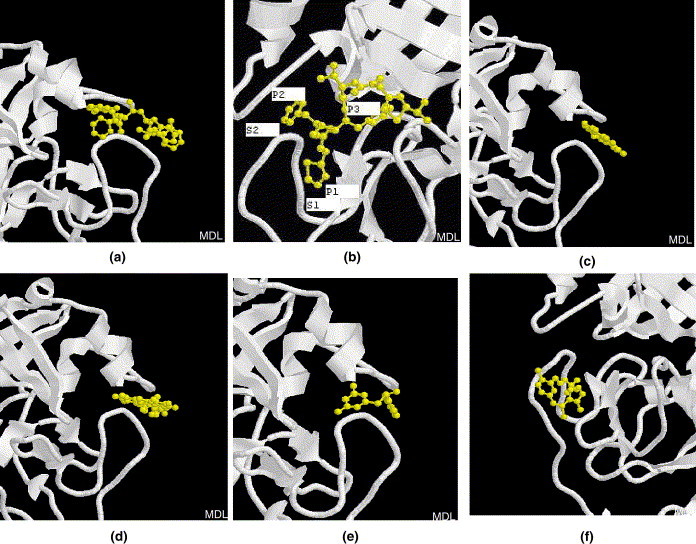
A close view of the interactions between SARS-associated coronavirus main proteinase (white cartoon) with drugs and inhibitors (yellow ball-stick): (a) lopinavir, (b) ritonavir, (c) niclosamide, (d) promazine, (e) PNU and (f) UC2.
References
- 1.Vastag B. Old drugs for a new bug. JAMA. 2003;290:1695–1696. doi: 10.1001/jama.290.13.1695. [DOI] [PubMed] [Google Scholar]
- 2.Bonanno, J. B.; Fowler, R.; Gupta, S.; Hendle, J.; Lorimer, D.; Romero, R.; Sauder, M.; Wei, C. L.; Liu, E. T.; Burley, S. K.; Harris, T. X-ray Crystal Structure of the SARS Coronavirus Main Protease, to be published (http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1Q2W), 2003
- 3.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindberg J., Sigursson S., Lowgren S., Andersson H.O., Sahlberg C., Noreen R., Fridborg K., Zhang H., Unge T. Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 2002;269:1670–1677. doi: 10.1046/j.1432-1327.2002.02811.x. [DOI] [PubMed] [Google Scholar]
- 5.Ren J., Esnouf R.M., Hopkins A.L., Warren J., Balzarini J., Stuart D.I., Stammers D.K. Crystal structures of HIV-1 reverse transcriptase in complex with carboxanilide derivatives. Biochemistry. 1998;37:14394–14403. doi: 10.1021/bi981309m. [DOI] [PubMed] [Google Scholar]
- 6.Ritchie D.W. Evaluation of protein docking predictions using Hex 3.1 in CAPRI rounds 1 and 2. Proteins: Struct. Funct. Genet. 2003;52:98–106. doi: 10.1002/prot.10379. [DOI] [PubMed] [Google Scholar]
- 7.Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucl. Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chou K., Wei D., Zhong W. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenwitheesuk E., Samudrala R. Improved prediction of HIV-1 protease-inhibitor binding energies by molecular dynamics simulations. BMC Struct. Biol. 2003;3(1):2–10. doi: 10.1186/1472-6807-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenwitheesuk E., Samudrala R. Identifying inhibitors of the SARS coronavirus proteinase. Bioorg. Med. Chem. Lett. 2003;13(22):3989–3992. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 12.Chan K.S., Lai S.T., Chu C.M., Tsui E., Tam C.Y., Wong M.M., Tse M.W., Que T.L., Peiris J.S., Sung J., Wong V.C., Yuen K.Y. Treatment of severe acute respiratory syndrome with lopinavir/ritonavir: a multicentre retrospective matched cohort study. Hong Kong Med. J. 2003;9(6):399–406. [PubMed] [Google Scholar]