

Graphical abstract
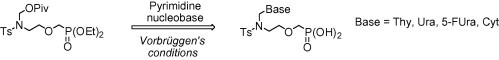
Keywords: Azanucleosides, Acyclic nucleoside phosphonates, Nucleoside analogs, Antiviral agents, Sulfonamides
Abstract
Acyclic 2′-azanucleosides with a phosphonomethoxy function in the side chain were obtained by coupling of diethyl {2-[N-(pivaloyloxymethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonate with the pyrimidine nucleobases via the Vorbrüggen-type protocol. The compounds were evaluated in vitro for activity against a broad variety of RNA and DNA viruses.
1. Introduction
Nucleoside analogues need to be in vivo phosphorylated to their triphosphates to display antiviral activity.1 The first step of this three-step phosphorylation sequence, leading to the corresponding monophosphates, is essential for their activity. Low activity of nucleoside analogues may often result from poor in vivo monophosphorylation. To circumvent this problem, acyclic nucleoside phosphonates (ANPs), that is, derivatives with a phosphonate P–C–O-linkage instead of a phosphoester P–O–C one, were developed.2 Since ANPs are stable analogues of nucleoside monophosphates, in contrast to the ‘classical’ acyclic nucleoside analogues (such as acyclovir, ganciclovir or penciclovir),1 they do not require activation by the first phosphorylation.3 Moreover, the presence of the phosphonate group makes ANPs resistant towards phosphomonoesterases and nucleotidases, enzymes involved in nucleotide catabolism. Owing to the specific mode of action, ANPs enhance a broad spectrum of antiviral activity.3 Several of them (i.e., cidofovir, adefovir, and tenofovir, Fig. 1 ) have been approved for the treatment of viral infections.3 However the therapeutic use of the ANP drugs is limited by poor oral bioavailability, potential nephrotoxicity or development of resistant viral strains.3 In attempts to overcome these problems, a variety of structural analogues were synthesized and biologically evaluated.4 Among them, compounds with the aza function at the 1′-, 3′-, or 4′-position of the acyclic moiety (Fig. 1, analogues 1, 2 and 3, respectively)5 as well as N-(phosphonomethyl)prolinol derivatives 4 6 (Fig. 1) have also been developed. Among the synthesized derivatives, compounds 2 (B = Gua or Cyt) were endowed with a weak anti-herpes virus activity.5a
Figure 1.
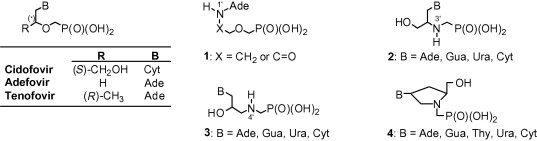
The ANP drugs and their aza analogues.
These findings inspired us to evaluate the antiviral activity of compounds 8 (Scheme 1 ) being phosphonomethyl ethers of the previously reported, antivirally inactive N-(2-hydroxyethyl) azanucleosides.7c Considering a unique mode of action of ANPs, we decided to examine the effect of the O-phosphonomethyl modification of the parent hydroxy derivatives on their antiviral activity. Compounds 8 can be regarded as aza-congeners of cidofovir. Taking into account the location of the aza function at the 2′-position of the side chain, they can be also considered as complementary to the abovementioned 1′-, 3′-, and 4′-aza analogues of the acyclic nucleoside phosphonates.
Scheme 1.
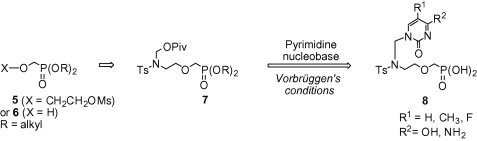
Synthetic strategy for the target 2’-azanucleosides 8.
2. Results and discussion
Generally, the synthetic approaches reported for the aforementioned acyclic aza-analogues 1–3 (Fig. 1) involved two key steps: (a) transformation of a nucleobase into the corresponding N-amino- or N-aminoalkyl derivative; and (b) alkylation (or acylation) of the resultant precursor at the exocyclic amino group with a specific synthon bearing a dialkylphosphoryl function, followed by deprotection of the intermediate phosphonate esters.5 Our strategy for the synthesis of the target 2′-azanucleosides 8 is outlined in Scheme 1. Based on our previous experience in synthesizing nucleoside aza-analogues,7 we decided to achieve compounds 8 via simultaneous introduction of the aza and dialkylphosphoryl function to the nucleobase in one reaction step, that is, by N-alkylation of the nucleobase with the phosphonylated N-(pivaloyloxymethyl)sulfonamide 7 under Vorbrüggen’s conditions.
Synthesis of N-(pivaloyloxymethyl)sulfonamide 7 from diethyl (2-mesyloxyethoxy)methylphosphonate8 5 or diethyl hydroxymethylphosphonate 6 is shown in Scheme 2 . Starting from diethyl (2-mesyloxyethoxy)methylphosphonate 5, the p-toluenesulfonylamino function of 7 was introduced by two methods [Scheme 2, conditions (i)–(iii) or (iv)]. The first one involved the following steps: (i) transformation of 5 into azide 9 using sodium azide at room temperature;9 (ii) reduction of 9 to amine 10 under Staudinger’s conditions;9 and (iii) N-sulfonation of 10 with p-toluenesulfonyl chloride; the overall yield of 11 was 59%. The second method [conditions (iv)], that is, direct reaction of 5 with p-toluenesulfonamide in the presence of anhydrous potassium carbonate, afforded 11 in 68% yield. We also tried to prepare 11 by reaction of diethyl hydroxymethylphosphonate 6 with N-(p-toluenesulfonyl)aziridine10 12 in the presence of sodium hydride [conditions (v)]. However, this approach was not satisfactory; 11 was obtained in a low yield of 23%. In the next step [conditions (vi)], N-alkylation of 11 with chloromethyl pivalate in the presence of anhydrous potassium carbonate gave N-(pivaloyloxymethyl)sulfonamide 7 in a high yield of 96%. Reaction of 7 with the pyrimidine nucleobases (thymine, 5-fluorouracil or uracil) was performed according to the one-pot base silylation/nucleoside coupling procedure using N,O-bis(trimethylsilyl)acetamide (BSA) as the silylating agent and tin(IV) chloride as the Lewis acid [Scheme 3 , conditions (i)]. The thymine derivative 13a and the 5-fluorouracil derivative 13b were obtained as the only products in a yield of 83% and 55%, respectively. The only exception was the reaction of 7 with uracil; the desired azanucleoside 13c (the major product, 73%) and 1,3-disubstituted uracil 14 (7%) were obtained. The minor product 14 was readily separated by column chromatography. Coupling of 7 with N 4-benzoylcytosine under the same conditions, followed by treatment of the crude product (not shown) with ammonium hydroxide gave the cytosine derivative 13d in 80% yield [Scheme 3, conditions (ii)].
Scheme 2.
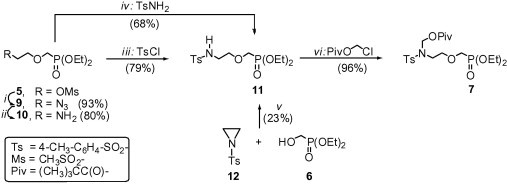
Reagents and conditions: (i) NaN3, DMF, rt, 3 d; (ii) (1) Ph3P, toluene, rt, 1 h; (2) H2O; (iii) NEt3, DCM, rt, 20 h; (iv) K2CO3, DMF, 50 °C, 3 d; (v) NaH, DMF, rt, 10 h; (vi) K2CO3, DMF, rt, 3 d.
Scheme 3.
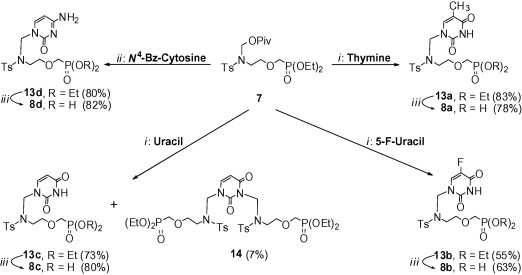
Reagents and conditions: (i) (1) nucleobase (thymine, 5-F-uracil or uracil), BSA, CH3CN, rt, 1 h; (2) 7, SnCl4/CH2Cl2, CH3CN, rt, 2 d; (ii) (1) N4-Bz-cytosine, BSA, CH3CN, rt, 1 h; (2) 7, SnCl4/CH2Cl2, CH3CN, rt, 2 d; (3) NH4OH, MeOH, rt, 20 h; (iii) (1) TMSBr, CH3CN, rt, 20 h; (2) acetone, H2O, rt, 2 h.
The N-1 substitution pattern of the aza analogues 13 was confirmed by the interactions observed in the 2D NMR spectra of 13a and 13b (Fig. 2 ). In the 1H–1H ROESY spectrum of 13a, the 1H–1H interaction between H-6 of thymine and the H-1′ protons was seen. In the 1H–13C HMBC spectrum of 13b, in turn, the 1H–13C couplings of the H-1′ protons with both C-6 and C-2 of 5-fluorouracil, as well as the coupling between H-6 and C-1′ were observed.
Figure 2.
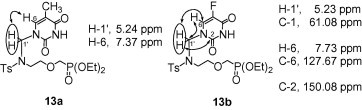
The principal 1H–1H ROESY correlation and the 1H–13C HMBC couplings observed for 13a and 13b, respectively.
The final step [Scheme 3, conditions (iii)], dealkylation of 13 with bromotrimethylsilane (TMSBr) followed by treatment of the reaction mixture with a water/acetone mixture, afforded the desired phosphonic acids 8 in 63–82% yield. Structure and purity of derivatives 8a–c was verified by standard NMR and IR techniques as well as by elemental analysis. In contrast to 8a–c, solubility of the cytosine derivative 8d in water (5 mg/ca. 100 ml of water) and in conventional organic solvents, including dimethylformamide and dimethylsulfoxide, was very low. For this reason, only IR spectrum and elemental analysis was performed for 8d. Therefore, in an attempt to transform 8d into the corresponding sodium salt, a suspension of 8d in a water/DMSO mixture was shaken with DOWEX® 88 ion-exchange resin (Na-form). Contrary to our expectation that 8d would gradually dissolve upon the treatment with the ion-exchange resin, the suspension (which was then identified by elemental analysis as the unchanged 8d) was still observed. Therefore, the time of shaking was prolonged for six months. Nevertheless, the conversion of 8d to its sodium salt was not satisfactory. Filtration of the unchanged 8d and evaporation of the solvents from the filtrate afforded a small amount of a solid (8d–s, 15 mg from 50 mg of 8d) which was identified by elemental analysis as an equimolar mixture of 8d and its monosodium salt (not shown). The 1H NMR spectrum of the mixture allowed us to confirm the structure of 8d. Unfortunately, solubility of 8d–s in DMSO or in water was too low to measure the 13C NMR spectrum.
3. Antiviral activity evaluation
Compounds 8a–d and the 8d–s mixture were evaluated in vitro for activity against a variety of RNA and DNA viruses, using the following cell-based assays: (a) HEL cells infected with herpes simplex virus type 1 (KOS), herpes simplex virus type 2 (G), acyclovir-resistant herpes simplex virus type 1 (TK− KOS ACVr), vaccinia virus, or vesicular stomatitis virus; (b) HeLa cells infected with vesicular stomatitis virus, Coxsackie B4 virus, or respiratory syncytial virus; (c) Vero cells infected with parainfluenza-3 virus, reovirus-1, Sindbis virus, Coxsackie B4 virus, or Punta Toro virus; (d) HEL cells infected with cytomegalovirus (AD-169 or Davis strain); (e) HEL cells infected with varicella-zoster virus (TK+ strain OKA or TK− strain 07/1); and (f) Crandell-Rees Feline Kidney (CRFK) cells infected with feline corona virus or feline herpes virus. The antiviral activity was expressed as (i) the effective concentration (EC50) required to reduce virus-induced cytopathic effect by 50%, as determined by visual scoring of the CPE [assays (a)–(c)] or by measuring the cell viability with the colorimetric formazan-based MTS assay [assays (f)]; or (ii) the effective concentration (EC50) required to reduce virus plaque formation by 50% [assays (d) and (e)]. None of the evaluated compounds, at the highest (subtoxic) concentrations used (i.e., 10–100 μg/mL), was active against the viruses examined.
4. Conclusion
A series of 2′-azanucleosides 8a–d containing the phosphonomethoxy function in the side chain were obtained by coupling of diethyl {2-[N-(pivaloyloxymethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonate 7 with the pyrimidine nucleobases under the Vorbrüggen’s conditions followed by dealkylation of the intermediate phosphonate esters. The biological results demonstrate that replacement of the 3-hydroxy-2-(phosphonomethoxy)-propyl moiety in cidofovir with the N-(2-phosphonomethoxyethyl)-N-(p-toluenesulfonyl)aminoethyl function causes annihilation of the antiviral activity at subtoxic concentrations. This lack of antiviral activity may be due to the poor cell membrane permeability of the free phosphonic acids 8, resulting from the polarity of the phosphono group.2 Considering the fact that masking of the phosphono group with a lipophilic function decreases the polarity of ANPs,2, 11 we have undertaken research on such modification of 8. This study as well as antiviral activity evaluation of masked derivatives of 8, including diesters 13, will be a subject of a forthcoming publication.
5. Experimental
5.1. Materials and methods
Pre-coated Merck Silica Gel 60 F-254 plates were used for thin-layer chromatography (TLC, 0.2 mm), spots were detected under UV light (254 nm). Column chromatography was performed using silica gel (200–400 mesh, Merck). High Resolution Mass Spectra (Electrospray Ionisation, ESI) were performed on a Mariner® spectrometer in positive ionization mode. The IR spectra were recorded on a Perkin–Elmer System 2000 spectrometer in KBr disc; resolution was 2 cm−1; absorption maxima (ν max) are given in cm−1 and quoted as ‘s’ strong, ‘m’ moderate, ‘w’ weak, ‘br’ broad. The 1H NMR spectra were measured on a Varian Gemini-200BB (200 MHz) or on a Varian Mercury-400BB spectrometer (400 MHz). The 13C NMR spectra were recorded on a Varian Gemini-200BB spectrometer at 50 MHz. 1H and 13C chemical shifts (δ) are reported in parts per million (ppm) relative to the solvent signals (CDCl3, δ H (residual CHCl3) 7.26 ppm, δ C 77.16 ppm; or DMSO-d 6, δ H (residual DMSO-d 5) 2.50 ppm, δ C 39.52 ppm), or relative to 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS), which was used as an internal standard for the spectra taken in D2O; signals are quoted as ‘s’ (singlet), ‘d’ (doublet), ‘t’ (triplet), ‘q’ (quartet), ‘m’ (multiplet), and ‘br s’ (broad singlet). Coupling constants (J) are reported in Hertz. The 1H–1H ROESY (Rotating frame Overhauser Effect Spectroscopy) for 13a was measured on a Varian Mercury-400BB spectrometer in CDCl3. The 1H–13C HMBC (Heteronuclear Multiple Bond Correlation) for 13b was measured on a Varian VNMRS 500 spectrometer in CDCl3. Elemental analyses were performed in a Perkin Elmer 2400 apparatus. Anhydrous MgSO4 was employed as a drying agent. Solvents were distilled off under reduced pressure in a rotating evaporator.
5.2. Preparation of diethyl [2-(p-toluenesulfonylamino)ethoxy]methylphosphonate (11)
5.2.1. Method A [Scheme 2, conditions (i)–(iii)]
5.2.1.1. Diethyl (2-azidoethoxy)methylphosphonate (9)9
A mixture of diethyl (2-mesyloxyethoxy)methylphosphonate 5 (3.0 g, 10.3 mmol), sodium azide (6.72 g, 103.0 mmol) and anhydrous DMF (60 mL) was stirred at room temperature for 3 days and then evaporated to dryness under vacuum at 80 °C (0.05 mmHg). The residue was diluted with water (50 mL) and extracted with dichloromethane (3 × 20 mL). The combined extracts were dried, filtered, and evaporated to dryness under reduced pressure to give 9 as an oil (2.28 g, 93%). An analytical sample was obtained by column chromatography (chloroform). δ H (CDCl3, 200 MHz) 1.34 (td, 3 J H–H 7.0, 4 J H–P 0.4, 6H), 3.37–3.42 (triplet-like multiplet, 2H), 3.73–3.78 (triplet-like multiplet, 2H), 3.84 (d, 2 J H–P 8.2, 2H), 4.17 (dq, 3 J H–P 8.2, 3 J H–H 7.0, 4H). δ C (CDCl3, 50 MHz) 16.61 (d, 3 J C–P 5.7), 50.77, 62.71 (d, 2 J C–P 6.5), 65.57 (d, 1 J C–P 165.0), 72.20 (d, 3 J C–P 10.2).
5.2.1.2. Diethyl (2-aminoethoxy)methylphosphonate (10)9
A mixture of triphenylphosphine (4.88 g, 18.6 mmol) and dry toluene (20 mL) was cooled on an ice-water bath and a solution of 9 (2.94 g, 12.4 mmol) in dry toluene (2.5 mL) was added dropwise within 30 min. The mixture was stirred at room temperature for 1 h. Water was then added and stirring was continued for 15 min. The aqueous layer was separated, extracted with diethyl ether (3 × 20 mL), and evaporated to dryness under vacuum at 80 °C (0.05 mmHg) to afford 10 as an oil (2.09 g, 80%). An analytical sample was obtained by column chromatography (chloroform/methanol, 95:5, v/v). δ H (CDCl3, 200 MHz) 1.34 (td, 3 J H–H 7.0, 4 J H–P 0.4, 6H), 1.55 (br s, 2H, NH 2), 2.84–2.89 (triplet-like multiplet, 2H), 3.57–3.63 (triplet-like multiplet, 2H), 3.80 (d, 2 J H–P 8.4, 2H), 4.17 (dq, 3 J H–P 8.0, 3 J H–H 7.0, 4H). δ C (CDCl3, 50 MHz) 16.63 (d, 3 J C–P 5.3), 41.77, 62.53 (d, 2 J C–P 6.8), 62.30 (d, 1 J C–P 165.8), 75.96 (d, 3 J C–P 10.7).
5.2.1.3. Diethyl [2-(p-toluenesulfonylamino)ethoxy]methylphosphonate (11)
A mixture of 10 (2.09 g, 9.86 mmol), triethylamine (2.0 g, 19.7 mmol, 2.8 mL) and dichloromethane (10 mL) was cooled in an ice-water bath and a solution of p-toluenesulfonyl chloride (2.8 g, 14.8 mmol) in dichloromethane (10 mL) was then added dropwise within 15 min. The mixture was stirred at room temperature overnight, diluted with dichloromethane (100 mL), washed with brine (2 × 20 mL), and dried. The solvent was distilled off. The residue was purified by column chromatography (chloroform/acetone, 98:2→80:20, v/v) to give 11 as an oil (2.85 g, 79%). δ H (CDCl3, 400 MHz) 1.34 (t, 3 J H–H 7.0, 6H), 2.43 (s, 3H), 3.12–3.16 (m, 2H), 3.62–3.65 (triplet-like multiplet, 2H), 3.73 (d, 2 J H–P 8.0, 2H), 4.16 (dq, 3 J H–P 8.4, 3 J H–H 7.0, 4H), 5.29 (triplet-like multiplet, 1H, NH), 7.29–7.31 (m, 2H), 7.73–7.76 (m, 2H). δ C (CDCl3, 50 MHz) 16.55 (d, 3 J C–P 5.7), 21.52, 42.87, 62.54 (d, 2 J C–P 6.5), 65.33 (d, 1 J C–P 166.2), 71.98 (d, 3 J C–P 9.9), 127.09, 129.70, 137.08, 143.40. ν max (NaCl) 1162s (S O), 1330s (S O), 3151 m (N–H). HRMS m/z C14H24NO6NaPS (M+Na)+ 388.0954, found 388.0951.
5.2.2. Method B [Scheme 2, conditions (iv)]
A mixture of 5 (1.3 g, 4.5 mmol), p-toluenesulfonamide (7.67 g, 45.0 mmol), anhydrous potassium carbonate (0.62 g, 4.5 mmol), and anhydrous DMF (15 mL) was stirred at 50 °C (an oil bath) for 3 days and then evaporated to dryness under vacuum at 80 °C (0.05 mmHg). The residue was diluted with ethyl ether (100 mL) and filtered through a Celite pad. The filtrate was washed with water (3 × 20 mL), dried, filtered, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography (chloroform/acetone, 98:2→80:20, v/v) to afford 11 as an oil (1.12 g, 68%).
5.2.3. Method C [Scheme 2, conditions (v)]
A mixture of diethyl hydroxymethylphosphonate 6 (0.57 g, 3.4 mmol), sodium hydride (60% suspension in mineral oil, 0.14 g, 3.4 mmol) and anhydrous DMF (10 mL) was stirred in an ice-water bath for 1 h under argon atmosphere. N-(p-Toluenesulfonyl)aziridine 12 (0.34 g, 1.7 mmol) was added in one portion. After 1 day of stirring at room temperature, the mixture was poured into water (75 mL) and extracted with ethyl acetate (3 × 20 mL). The combined extracts were washed with water (2 × 10 mL) and dried. The low boiling solvents were distilled off under reduced pressure and residual DMF was removed under vacuum at 80 °C (0.05 mmHg). The residue was purified by column chromatography (chloroform) to yield 11 as an oil (0.14 g, 23%).
5.3. Diethyl {2-[N-(pivaloyloxymethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonate (7)
Chloromethyl pivalate (6.39 g, 42.4 mmol, 6.1 mL) was added to a stirred mixture of 11 (3.1 g, 8.5 mmol) and anhydrous potassium carbonate (11.7 g, 85.0 mmol) in dry DMF (50 mL) at room temperature. The mixture was stirred for 5 days at room temperature and then poured into ice-cold water (250 mL). The organic phase was extracted with dichloromethane (3 × 50 mL). The extracts were combined and washed with water (2 × 20 mL), dried and filtered. The solvent was distilled off. The residue was purified by column chromatography (chloroform) to give 7 as an oil (3.92 g, 96%). δ H (CDCl3, 400 MHz) 0.99 (s, 9H), 1.35 (t, 3 J H–H 7.2, 6H), 2.42 (s, 3H), 3.36–3.42 (triplet-like multiplet, 2H), 3.78–3.81 (m, 4H), 4.17 (dq, 3 J H–P 8.0, 3 J H–H 7.2, 4H), 5.53 (s, 2H), 7.28–7.30 (m, 2H), 7.73–7.76 (m, 2H). δ C (CDCl3, 50 MHz) 16.63 (d, 3 J C–P 6.1), 21.64, 26.93, 45.91, 62.68 (d, 2 J C–P 6.5), 65.36 (d, 1 J C–P 165.8), 71.86 (d, 3 J C–P 9.4), 73.16, 127.73, 129.98, 137.08, 143.99, 177.43. ν max (NaCl) 1729s (C O). HRMS m/z C20H34NO8NaPS (M+Na)+ 502.1635, found 502.1653.
5.4. General procedure for coupling of 7 with thymine or 5-fluorouracil
A mixture of a nucleobase (thymine or 5-fluorouracil) (2.0 mmol) and N,O-bis(trimethylsilyl)acetamide (BSA, 815 mg, 4.0 mmol, 1.0 mL) in dry acetonitrile (10 mL) was stirred at room temperature for 1 h under argon atmosphere. A solution of 7 (1.0 mmol) in dry acetonitrile (1 mL), and subsequently 1 M solution of tin(IV) chloride in dichloromethane (3 mL) was added. The reaction mixture was kept at room temperature for 2 days. Ethyl acetate (50 mL) and then a saturated sodium bicarbonate solution (1 mL) was added. The mixture was stirred for 1 h and filtered through a Celite pad. The organic phase was separated, washed with brine (3 × 10 mL), dried and filtered. The volatiles were distilled off. The residue was purified by column chromatography (chloroform/methanol, 98:2, v/v) to yield azanucleoside 13a or 13b.
5.4.1. Diethyl {2-[N-(5-methyl-1H,3H-pyrimidin-2,4-dion-1-ylmethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonate (13a)
According to the general procedure, 13a was obtained from 7 (960 mg, 2.0 mmol) and thymine. Chromatographic purification gave 13a (834 mg, 83%) as an oil. δ H (CDCl3, 400 MHz) 1.32 (t, 3 J H–H 7.2, 6H), 1.90 (d, 4 J H–H 0.8, 3H), 2.41 (s, 3H), 3.55–3.58 (triplet-like multiplet, 2H), 3.68–3.72 (m, 4H), 4.14 (dq, 3 J H–P 8.2, 3 J H–H 7.2, 4H), 5.24 (s, 2H), 7.26–7.31 (m, 2H), 7.60–7.65 (m, 2H), 7.37 (q, 4 J H–H 0.8, 1H), 8.98 (br s, 1H, NH). δ C (CDCl3, 50 MHz) 12.40, 16.62 (d, 3 J C–P 5.7), 21.65, 48.26, 60.99, 62.58 (d, 2 J C–P 6.5), 65.25 (d, 1 J C–P 165.8), 72.20 (d, 3 J C–P 10.2), 111.16, 126.98, 130.03, 136.93, 139.85, 144.39, 151.37, 164.05. ν max (KBr) 1689br s (C O). HRMS m/z C20H30N3O8NaPS (M+Na)+ 526.1389, found 526.1390.
5.4.2. Diethyl {2-[N-(5-fluoro-1H,3H-pyrimidin-2,4-dion-1-ylmethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonate (13b)
According to the general procedure, 13b was obtained from 7 (487 mg, 1.0 mmol) and 5-fluorouracil. Chromatographic purification gave 13b (285 mg, 55%) as an oil. δ H (CDCl3, 400 MHz) 1.32 [t, 3 J H–H 7.2, 6H, (CH3–CH2–O)2P(O)–], 2.43 (s, 3H, CH3–C6H4–SO2–), 3.52–3.54 (triplet-like multiplet, 2H, –O–CH2–CH2–N–), 3.68–3.72 [m, 4H, –O–CH2–CH2–N–, –P(O)–CH 2–O–], 4.14 [dq, 3 J H–P 8.4, 3 J H–H 7.2, 4H, (CH3–CH 2–O)2P(O)–], 5.23 (s, 2H, –N–CH2–N–), 7.31–7.34 (m, 2H, Ar-H-ortho to CH3), 7.65–7.68 (m, 2H, Ar-H-ortho to SO2), 7.73 (d, 3 J H–F 5.2, 1H, –N–CH CF–), 9.31 (br s, 1H, NH). δ C (CDCl3, 50 MHz) 16.23 [d, 3 J C–P 5.3, (CH3–CH2–O)2P(O)–], 21.31 (CH3–C6H4–SO2–), 48.14 (–O–CH2–CH2–N–), 61.08 (–N–CH2–N–), 62.45 [d, 2 J C–P 6.5, (CH3–CH2–O)2P(O)–], 64.74 [d, 1 J C–P 165.4, –P(O)–CH2–O–], 71.74 (d, 3 J C–P 11.0, –O–CH2–CH2–N–), 126.77 (Ar-C-ortho to SO2), 127.67 (d, 2 J C–F 33.4, –N–CH CF–), 129.84 (Ar-C-ortho to CH3), 136.09 (Ar-C-ipso to SO2), 140.17 [d, 1 J C–F 236.0, –CH CF–C(O)–], 144.23 (Ar-C-ipso to CH3), 150.08 [–N–C(O)–N–], 157.39 [d, 2 J C–F 26.2, CF–C(O)–N–]. ν max (KBr) 1706br s (C O), 1672s (C O). HRMS m/z C19H27N3O8FNaPS (M+Na)+ 530.1133, found 530.1125.
5.5. Coupling of 7 with uracil
According to the procedure used for the coupling of 7 with thymine or 5-fluorouracil, a solution of the silylated uracil in dry acetonitrile (15 mL) was prepared from uracil (361 mg, 3.2 mmol) and BSA (1.3 g, 6.4 mmol, 1.6 mL) and then treated with a solution of 7 (746 mg, 1.6 mmol) in dry acetonitrile (1.5 mL) and 1 M solution of tin(IV) chloride in dichloromethane (4.8 mL). Column chromatography (chloroform/methanol, 98:2, v/v) provided azanucleoside 13c (558 mg, 73%) as an oil and 14 (49 mg, 7%) as an oil.
5.5.1. Diethyl {2-[N-(1H,3H-pyrimidin-2,4-dion-1-ylmethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonate (13c)
δH (CDCl3, 400 MHz) 1.32 (t, 3 J H–H 7.2, 6H), 2.41 (s, 3H), 3.54–3.56 (triplet-like multiplet, 2H), 3.69–3.72 (m, 4H), 4.14 (dq, 3 J H–P 8.2, 3 J H–H 7.2, 4H), 5.26 (s, 2H), 5.71 (dd, 3 J H–H 8.2, 4 J H–H 2.2, 1H), 7.29–7.31 (m, 2H), 7.61–7.64 (m, 3H), 9.14 (br s, 1H, NH). δ C (CDCl3, 50 MHz) 16.63 (d, 3 J C–P 5.3), 21.66, 48.43, 61.35, 62.58 (d, 2 J C–P 6.5), 65.27 (d, 1 J C–P 165.4), 72.33 (d, 3 J C–P 10.6), 102.64, 126.99, 130.10, 136.68, 144.25, 144.46, 151.24, 163.47. ν max (KBr) 1694br s (C O). HRMS m/z C19H28N3O8NaPS (M+Na)+ 512.1227, found 512.1240.
5.5.2. 1,3-Bis{N-(p-toluenesulfonyl)-N-[2-(diethylphosphorylmethoxy)ethyl]aminomethyl}-1H,3H-pyrimidin-2,4-dione (14)
δH (CDCl3, 200 MHz) 1.19 (m, 12H), 2.39 (s, 6H), 3.54–3.74 (m, 12H), 4.06–4.20 (m, 8H), 5.24 (s, 2H), 5.33 (s, 2H), 5.64 (d, 3 J H–H 8.0, 1H), 7.28–7.37 (m, 4H), 7.52 (d, 3 J H–H 8.0, 1H), 7.60–7.76 (m, 4H). δ C (CDCl3, 50 MHz) 16.60 (d, 3 J C–P 5.3), 21.63, 47.97, 49.48, 54.57, 61.85, 62.56 (d, 2 J C–P 6.5), 65.17 (d, 1 J C–P 165.4), 72.17 (d, 3 J C–P 10.4), 72.31 (d, 3 J C–P 10.8), 102.00, 127.02, 127.54, 129.70, 130.08, 136.67, 136.92, 142.58, 143.69, 144.39, 151.67, 162.54. ν max (KBr) 1699br s (C O). HRMS m/z C34H52N4NaO14P2S2 (M+Na)+ 889.2289, found 889.2311.
5.6. Diethyl {2-[N-(4-amino-1H-pyrimidin-2-on-1-ylmethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonate (13d)
According to the procedure used for the coupling of 7 with thymine or 5-fluorouracil, a solution of the silylated N 4-benzoylcytosine in dry acetonitrile (10 mL) was prepared from N 4-benzoylcytosine (435 mg, 2.0 mmol) and BSA (815 mg, 4.0 mmol, 1.0 mL) and then treated with a solution of 7 (480 mg, 1.0 mmol) in dry acetonitrile (1 mL) and 1 M solution of tin(IV) chloride in dichloromethane (3 mL). After workup, the reaction mixture was passed through a short chromatographic column (chloroform/methanol, 98:2, v/v) in order to separate the unreacted N 4-benzoylcytosine. Fractions with R F value of 0.25 were combined and evaporated to dryness. The residue was dissolved in methanol (10 mL) and aqueous ammonium hydroxide (25 mL) was added. The mixture was kept at room temperature overnight. The volatiles were distilled off, and the residue was purified by column chromatography (chloroform/methanol, 95:5, v/v) to afford 13d (392 mg, 80%) as a white solid; mp 135–137 °C. δ H (CDCl3, 400 MHz) 1.32 (t, 3 J H–H 7.2, 6H), 2.40 (s, 3H), 3.59–3.61 (triplet-like multiplet, 2H), 3.67–3.73 (m, 4H), 4.14 (dq, 3 J H–P 8.0, 3 J H–H 7.2, 4H), 5.29 (s, 2H), 5.86 (d, 3 J H–H 7.2, 1H), 6.38 (br s, 2H, NH 2), 7.25–7.28 (m, 2H), 7.60–7.63 (m, 3H). δ C (CDCl3, 50 MHz) 16.70, 21.66, 47.75, 61.64, 62.72, 65.12 (d, 1 J C–P 166.2), 72.14 (d, 3 J C–P 10.4), 95.55, 127.00, 130.02, 136.88, 144.11, 145.00, 156.98, 166.53. ν max (KBr) 1656s (C O), 3118 m (N–H), 3363 m (N–H). HRMS m/z C19H29N4O7NaPS (M+Na)+ 511.1387, found 511.1412.
5.7. General procedure for the dealkylation of 13
A solution of 13 (1.0 mmol) in dry acetonitrile (10 mL) was cooled in an ice-water bath under argon atmosphere and trimethylsilyl bromide (TMSBr, 10 mmol) was added dropwise. The mixture was left at room temperature overnight and evaporated to dryness under reduced pressure. The residue was diluted with an acetone/water mixture (1:9, v/v, 10 mL). The resulting solution was stirred at room temperature until a white solid precipitated out (ca. 2 h). The suspension was cooled in the refrigerator (5 °C) overnight. The solid was filtered off, washed with the acetone/water mixture (1:9, v/v), and purified by crystallization (acetone/water, 1:9, v/v) or boiling with the same solvent to yield 8a–d.
5.7.1. {2-[N-(5-Methyl-1H,3H-pyrimidin-2,4-dion-1-ylmethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonic acid (8a)
According to the general procedure, 8a was obtained from 13a (582 mg, 1.2 mmol). Crystallization gave 8a (405 mg, 78%) as a white powder; mp 119–123 °C. δ H (DMSO-d 6, 400 MHz) 1.74 (d, 4 J H–H 0.8, 3H), 2.37 (s, 3H), 3.47–3.51 (m, 4H), 3.58–3.60 (triplet-like multiplet, 2H), 3.84 (br s, 2H, OH), 5.15 (s, 2H), 7.31–7.33 (m, 2H), 7.35 (q, 4 J H–H 0.8, 1H), 7.65–7.67 (m, 2H), 11.22 (br s, 1H, NH). δ C (DMSO-d 6, 50 MHz) 12.08, 21.15, 48.09, 60.87, 66.55 (d, 1 J C–P 159.4), 71.48 (d, 3 J C–P 10.6), 108.95, 126.92, 129.96, 136.83, 140.54, 143.91, 151.26, 164.21. ν max (KBr) 2771br w (PO–H). Anal. Calcd for C16H22N3O8PS·H2O: C, 41.29; H, 5.20; N, 9.03; S, 6.89. Found: C, 41.36; H, 5.35; N, 9.08; S, 6.46.
5.7.2. {2-[N-(5-Fluoro-1H,3H-pyrimidin-2,4-dion-1-ylmethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonic acid (8b)
According to the general procedure, 8b was obtained from 13b (277 mg, 0.5 mmol). Crystallization gave 8b (156 mg, 63%) as a white powder; mp 166–168 °C. δ H (DMSO-d 6, 400 MHz) 2.38 (s, 3H), 3.46 (d, 2 J H–P 8.8, 2H), 3.51–3.59 (m, 4H), 5.14 (s, 2H), 7.38–7.40 (m, 2H), 7.71–7.73 (m, 2H), 7.91 (d, 3 J H–F 6.4, 1H), 11.83 (d, 4 J H–F 4.4, 1H, NH). δ C (DMSO-d 6, 50 MHz) 21.03, 48.15, 61.31, 66.48 (d, 1 J C–P 159.4), 71.20 (d, 3 J C–P 10.6), 126.87, 129.10 (d, 2 J C–F 33.4), 129.89, 136.54, 139.25 (d, 1 J C–F 229.2), 143.85, 149.74, 157.24 (d, 2 J C–F 25.8). ν max (KBr) 1662s, 1688s, 1723s, 2695br w (PO–H). Anal. Calcd for C15H19FN3O8PS: C, 39.91; H, 4.24; N, 9.31; S, 7.10. Found: C, 39.86; H, 4.24; N, 9.29; S, 7.21.
5.7.3. {2-[N-(1H,3H-Pyrimidin-2,4-dion-1-ylmethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonic acid (8c)
According to the general procedure, 8c was obtained from 13c (465 mg, 0.9 mmol). Crystallization gave 8c (331 mg, 80%) as a white powder; mp 184–186 °C. δ H (DMSO-d 6, 400 MHz) 2.37 (s, 3H), 3.46–3.50 (m, 4H), 3.56–3.59 (triplet-like multiplet, 2H), 4.19 (br s, 2H, OH), 5.15 (s, 2H), 5.59 (dd, 3 J H–H 8.0, 4 J H–H 1.6, 1H), 7.37–7.39 (m, 2H), 7.49 (d, 3 J H–H 8.0, 1H), 7.68–7.70 (m, 2H), 11.28 (br s, 1H, NH). δ C (DMSO-d 6, 50 MHz) 21.07, 48.13, 61.10, 66.55 (d, 1 J C–P 159.4), 71.26 (d, 3 J C–P 10.5), 101.34, 126.88, 129.95, 136.47, 143.86, 145.04, 151.18, 163.53. ν max (KBr) 2823br w (PO–H). Anal. Calcd for 2C15H20N3O8PS·H2O: C, 40.73; H, 4.78; N, 9.50; S, 7.25. Found: C, 40.76; H, 4.55; N, 9.56; S, 7.35.
5.7.4. {2-[N-(4-Amino-1H-pyrimidin-2-on-1-ylmethyl)-N-(p-toluenesulfonyl)amino]ethoxymethyl}phosphonic acid (8d)
According to the general procedure, 8d was obtained from 13d (374 mg, 0.8 mmol). Boiling of the crude product with a mixture of acetone and water (2 × 5 mL, 1:9, v/v) gave 8d (272 mg, 82%) as a white powder; mp >218 °C (dec). Its solubility in DMSO or in water (5 mg/ca. 100 ml of water) was too low to measure the 1H and 13C NMR spectra. ν max (KBr) 1688s, 1725s, 2737br w (PO–H). Anal. Calcd for C15H21N4O7PS: C, 41.67; H, 4.90; N, 12.96; S, 7.42. Found: C, 41.58; H, 4.81; N, 12.74; S, 7.40.
A suspension of 8d (50 mg) in the DMSO (50 mL)/water (50 mL) mixture was shaken with Dowex® 88 ion-exchange resin (Na-form, 500 mg) for six months. The suspension was decanted from ion-exchange resin. The unchanged 8d (identified by elemental analysis, 23 mg) was filtered off, was washed with the DMSO/water mixture and dried under high vacuum. The filtrate was evaporated to dryness under high vacuum. The residue was twice crystallized from water to give a white solid 8d–s (15 mg) which was identified by elemental analysis as an equimolar mixture of 8d and its monosodium salt. δ H (D2O, DSS, 200 MHz) 2.39 (s, 3H), 3.59 (d, 2 J H–P 8.4, 2H), 3.96–4.10 (m, 4H), 5.30 (s, 2H), 5.81 (d, 3 J H–H 7.2, 1H), 7.34–7.38 (m, 2H), 7.54 (d, 3 J H–H 7.2, 1H), 7.68–7.62 (m, 2H). The solubility of 8d–s in DMSO or in water was too low to measure the 13C NMR spectrum. Anal. Calcd for C15H21N4O7PS·C15H20N4NaO7PS: C, 40.63; H, 4.66; N, 12.64; S, 7.23. Found: C, 40.43; H, 4.96; N, 12.34; S, 6.88.
5.8. Antiviral activity assays
The compounds were evaluated against the following viruses: herpes simplex virus type 1 (HSV-1) strain KOS, thymidine kinase-deficient (TK−) HSV-1 strain KOS resistant to ACV (ACVr), herpes simplex virus type 2 (HSV-2) strain G, varicella zoster virus (VZV) strain Oka, TK- VZV strain 07-1, human cytomegalovirus (HCMV) strains AD-169 and Davis, vaccinia virus Lederle strain, respiratory syncitial virus (RSV) strain Long, vesicular stomatitis virus (VSV), Coxsackie B4, Parainfluenza 3, Reovirus-1, Sindbis, Punta Toro, feline coronavirus and feline herpes virus. The antiviral assays were based on inhibition of virus-induced cytopathicity or plaque formation in human embryonic lung (HEL) fibroblasts, African green monkey cells (Vero), human epithelial cells (HeLa) or MTT dye staining of virus-infected feline kidney (CRFK) cell cultures. Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1CCID50 being the virus dose to infect 50% of the cell cultures), or with 20 plaque forming units (PFU) (for VZV) or with 100 PFU (for HCMV). After a 1–2 h adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations of the test compounds. Viral cytopathicity or plaque formation (VZV) was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 or compound concentration required to reduce virus-induced cytopathicity or viral plaque formation by 50%.
5.9. Cytotoxicity assays
Cytostatic measurements were based on the inhibition of cell growth. HEL cells were seeded at 5 × 103 cells/well into 96-well microtiter plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37 °C, the cell number was determined with a Coulter counter. The cytostatic concentration was calculated as the CC50, or the compound concentration required to reduce cell proliferation by 50% relative to the number of cells in the untreated controls. CC50 values were estimated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds. Alternatively, cytotoxicity for cell morphology was expressed as the minimum cytotoxic concentration (MCC) or the compound concentration that caused a microscopically detectable alteration of cell (i.e., HEL, Vero, HeLa) morphology.
Acknowledgments
The synthetic part of this work was financially supported by Ministry of Science and Higher Education, Poland, Project No. 1 T09B 013 30. The virological part of the work was supported by the Concerted Research Actions (GOA), Project No. 05/19 of the K. U. Leuven. The technical assistance of Mrs. Leentje Persoons, Frieda Demeyer, Vicky Broeckx, Anita Camps, Lies Van den Heurck and Steven Carmans was highly appreciated.
References and notes
- 1.De Clercq E. J. Clin. Virol. 2004;30:115. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Holý A. Curr. Pharm. Des. 2003;9:2567. doi: 10.2174/1381612033453668. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq E., Holý A. Nat. Rev. Drug Discovery. 2005;4:928. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 4.(a) Jeffery A.L., Kim J-H., Wiemer D.F. Tetrahedron. 2000;56:5077. For the selected papers on the ANP analogues with modifications in acyclic moiety, see: [Google Scholar]; (b) Esteban-Gamboa A., Balzarini J., Esnouf R., De Clercq E., Camarasa J-M., Pérez-Pérez M-J. J. Med. Chem. 2000;43:971. doi: 10.1021/jm9911377. [DOI] [PubMed] [Google Scholar]; (c) Rejman D., Masojidkova M., De Clercq E., Rosenberg I. Nucleosides Nucleotides. 2001;20:1497. doi: 10.1081/NCN-100105244. [DOI] [PubMed] [Google Scholar]; (d) Choi J.-R., Cho D.-G., Roh K.Y., Hwang J.-T., Ahn S., Jang H.S., Cho W.-Y., Kim, K W., Cho Y.-G., Kim J., Kim Y.-Z. J. Med. Chem. 2004;47:2864. doi: 10.1021/jm0305265. [DOI] [PubMed] [Google Scholar]; (e) Kim A., Hong J.H. Arch. Pharm. (Weinheim, Ger.) 2005;338:528. doi: 10.1002/ardp.200500187. [DOI] [PubMed] [Google Scholar]; (f) Wu M., El-Kattan Y., Lin T.-H., Ghosh A., Kumar V.S., Kotian P.L., Cheng X., Bantia S., Babu Y.S., Chand P. Nucleosides Nucleotides. 2005;24:1569. doi: 10.1080/15257770500265315. [DOI] [PubMed] [Google Scholar]; (g) Ivanov A.V., Andronova V.L., Galegov G.A., Jasko M.V. Russ. J. Bioorg. Chem. 2005;31:58. (Bioorg.Khim.2005, 31, 65) [Google Scholar]; (h) Oh C.H., Hong J.H. Arch. Pharm. 2006;339:507. doi: 10.1002/ardp.200600031. [DOI] [PubMed] [Google Scholar]; (i) Pomeisl K., Votruba I., Holý A., Pohl R. Collect. Czech. Chem. Commun. 2006;71:595. [Google Scholar]; (j) Kim A., Hong J.H. Nucleosides Nucleotides. 2006;25:1399. doi: 10.1080/15257770600918920. [DOI] [PubMed] [Google Scholar]; (k) Vrobska S., Holý A., Pohl R., Masojidkova M. Collect. Czech. Chem. Commun. 2006;71:543. [Google Scholar]; (l) Kim A., Hong J.H. Nucleosides Nucleotides. 2006;25:941. doi: 10.1080/15257770600793927. [DOI] [PubMed] [Google Scholar]; (m) Pham P.-T., Vince R. Phosphorus, Sulfur Silicon Relat. Elem. 2007;182:779. [Google Scholar]; (n) Vrbkova S., Dracínsky M., Holý A. Tetrahedron: Asymmetry. 2007;18:2233. [Google Scholar]
- 5.(a) Zhou D., Lagoja I.M., Van Aerschot A., Herdewijn P. Collect. Czech. Chem. Commun. 2006;71:15. [Google Scholar]; (b) Harnden M.R., Jarvest R.L. Bioorg. Med. Chem. Lett. 1992;2:1559. [Google Scholar]
- 6.Vanĕk V., Budĕšinský M., Rinnová M., Rosenberg I. Tetrahedron. 2009;65:862. [Google Scholar]
- 7.(a) Koszytkowska-Stawińska M., Sas W. Tetrahedron Lett. 2004;45:5437. [Google Scholar]; (b) Koszytkowska-Stawińska M., Sas W., De Clercq E. Tetrahedron. 2006;62:10325. [Google Scholar]; (c) Koszytkowska-Stawińska M., Kaleta K., Sas W., De Clercq E. Nucleosides Nucleotides. 2007;26:51. doi: 10.1080/15257770601052281. [DOI] [PubMed] [Google Scholar]; (d) Koszytkowska-Stawińska M., Kołaczkowska E., Adamkiewicz E., De Clercq E. Tetrahedron. 2007;63:10587. [Google Scholar]; (e) Gawin R., De Clercq E., Naesens L., Koszytkowska-Stawińska M. Bioorg. Med. Chem. 2008;16:8379. doi: 10.1016/j.bmc.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bronson J.J., Ghazzouli I., Hitchcock M.J.M., Webb R.R., Martin J.C. J. Med. Chem. 1989;32:1457. doi: 10.1021/jm00127a010. [DOI] [PubMed] [Google Scholar]
- 9.Eger K., Kluender E., Schmidt M. J. Med. Chem. 1994;37:3057. doi: 10.1021/jm00045a010. The preparation of 9 in 68% yield form diethyl (2-chloroethoxy)methylphosphonate and sodium azide under phase transfer catalysis conditions at 90 °C was also reported: [DOI] [PubMed] [Google Scholar]
- 10.Martin A.E., Ford T.M., Bulkowski J.E. J. Org. Chem. 1982;47:412. [Google Scholar]
- 11.(a) Vrbková S., Dracínský M., Holý A. Tetrahedron. 2007;63:11391. [Google Scholar]; (b) Khandazhinskaya A., Yasko M., Shirokova E. Curr. Med. Chem. 2006;13:2953. doi: 10.2174/092986706778521896. [DOI] [PubMed] [Google Scholar]; (c) Starrett J.E., Jr., Tortolani D.R., Russell J., Hitchcock M.J.M., Whiterock V., Martin J.C., Mansuri M.M. J. Med. Chem. 1994;37:1857. doi: 10.1021/jm00038a015. [DOI] [PubMed] [Google Scholar]