

Abstract
Intestinal lymphatic transport has been shown to be an absorptive pathway following oral administration of lipids and an increasing number of lipophilic drugs, which once absorbed, diffuse across the intestinal enterocyte and while in transit associate with secretable enterocyte lipoproteins. The chylomicron-associated drug is then secreted from the enterocyte into the lymphatic circulation, rather than the portal circulation, thus avoiding the metabolically-active liver, but still ultimately returning to the systemic circulation. Because of this parallel and potentially alternative absorptive pathway, first-pass metabolism can be reduced while increasing lymphatic drug exposure, which opens the potential for novel therapeutic modalities and allows the implementation of lipid-based drug delivery systems. This review discusses the physiological features of the lymphatics, enterocyte uptake and metabolism, links between drug transport and lipid digestion/re-acylation, experimental model (in vivo, in vitro, and in silico) of lymphatic transport, and the design of lipid- or prodrug-based drug delivery systems for enhancing lymphatic drug transport.
Keywords: Drugs, Lymph, Absorption, Transport, Intestine, Formulation, Lipid, Oral, Delivery, Chylomicron
1. Introduction
Drug discovery programs in the pharmaceutical industry have continued to generate increasingly potent drugs that then enter the development pipeline. However, with increasing drug potency has often come decreasing ease of design of the drug delivery systems for convenient oral administration due to the generally undesirable increases in molecular weight, lipophilicity, log octanol/water partition coefficients (log P), and decreases in water solubility. Orally administered drugs generally enter the systemic circulation by having sequentially transited intestinal epithelial cells called enterocytes, entered the portal circulation, and then been exposed to the metabolically-active liver. However, for some highly lipophilic drugs having a log P > 5 and a long-chain triglyceride (TG) solubility > 50 mg/g [1], entry into the portal circulation is reduced relative to the entry into the lymphatic circulation. During transit across the enterocyte, highly lipophilic drugs will associate with secretable enterocyte lipoproteins, specifically chylomicrons. A chylomicron-associated drug is then secreted into the mesenteric lymphatic circulation, rather than the portal circulation that leads directly to the metabolically-active liver, to ultimately join and mix with blood in the systemic circulation [2]. Therefore, having drugs that are lymphatically- rather than portally-transported avoids first-pass metabolism by the liver which in turn increases drug concentration in lymph ducts and nodes, which can be the site of therapeutic drug action [2]. With those therapeutic benefits in mind and the fact that drugs increasingly have physicochemical characteristics suitable for lymphatic transport, it is important to understand the factors that affect lymphatic drug transport. This review will discuss structural and physiological features of the lymphatics, enterocyte uptake and metabolism, links between drug transport and lipid digestion/re-acylation pathway, experimental models of lymphatic transport, and the design of lipid- or prodrug-based drug delivery systems for enhancing lymphatic drug transport.
2. Overview of intestinal lymphatic drug transport
The lymphatic system exists in all parts of the body except the central nervous system. The major parts of the system are the bone marrow, spleen, thymus gland, lymph nodes/nodules, and the tonsils. Other organs, including the heart, lungs, intestines, liver, and skin also contain lymphatic tissue. This system ultimately is responsible for the transport of lymph, which is a watery clear fluid. Throughout the body, lymph distributes lymphocyte immune cells and various other immune-related factors. The lymphocyte immune cells are functionally important as they protect the body against antigens that invade/attack the body such as viruses and/or bacteria. While the lymphatic system can be thought of as an important drainage system that actually collects drainage fluid from cells and tissues to later return the fluid back to the circulating blood pool. Therefore, an acceptable definition of lymph is the fluid and protein that has been filtered and extracted/squeezed out of the blood (i.e. blood plasma). The role of the lymphatic system in fat absorption and transport is also a major functional component of the lymphatic system. In order to appreciate the importance of this function, a generalized understanding of the lymphatic system anatomy is needed.
The ducts of the lymphatic systems interconnect the lymph organs that include the bone marrow, lymph nodes, spleen, as well as the thymus gland. Lymph within the ducts to transport lymphocyte immune cells such as B-lymphocytes (B-cells) and T-lymphocytes (T-cells) that originate from precursor cells in the bone marrow. From there, B-cells mature in the bone marrow while T-cells mature in the thymus gland after having moved through lymphatic ducts. With respect to the intestinal absorption and transport of lipids, the ducts of the lymphatic system that interconnected lymph organs, are also fairly important in providing transportation for proteins, fats, and other substances in lymph. Transport typically begins with blind-ended vessels found in tissues (termed lymph capillaries) create a capillary system in which lymph is drained. These lymph capillaries are highly permeable and are not pressurized allowing the lymph fluid to drain easily from the tissue into the lymph capillaries. Therefore, lymph vessels form a network throughout the body and overall this lymphatic system functions in a unidirectional manner in which lymph is drained from the tissue and is returned to the systemic blood where the subclavian blood vessels and the thoracic lymph duct join and their contents mix just prior to entry into the heart.
2.1. Role and relevance of the gastrointestinal lymphatic transport system
The intestinal lymphatic system is a pathway through which fat-soluble vitamins,food-derived lipids (a typical Western diet has 90 to 100 g per day [3]), and water-insoluble peptide-like molecules can be transported into systemic circulation. Drug transported via the intestinal lymphatic system can bypass the liver and thus avoid hepatic first-pass metabolism. To stay within the scope of this paper, the mechanisms by which drugs enter the intestinal lymph following oral administration are complex, but are simplified as follows. Firstly, the typical intestinal tract is richly supplied with both blood and lymph because the lamina propia that surrounds the enterocytes is in close proximity blood and lymph vessels. After being absorbed and transiting across the enterocytes, two potential scenarios can occur. One situation is where drugs prefer entry into blood capillaries and the other where entry into the lymph capillaries is favored. The former pathway, in which absorbed drugs are transported into portal blood, is the more common mechanism because of the high rate of fluid flow in the portal blood as compared to that of the intestinal lymph (500-fold higher for the portal blood) [2]. Large/high molecular weight drugs have limited or are unable to diffuse across the blood capillaries; thus, will utilize the more permeable lymphatic capillaries for absorption. Upon entry into the enterocyte, lipoproteins are critical as they will bind to macromolecules to aid in the entry process and, more importantly, to facilitate movement across the enterocyte. Since the physical size of the lipoproteins varies, this subsequently limits whether there is diffusion across the vascular endothelium. Furthermore, the lipoprotein size limits that diffusion and the remaining unobstructed diffusion across the lymphatic capillary results in the preferential access of lipoproteins along with the drug to the lymphatics (Fig. 1 ).
Fig. 1.

Drug absorption via the intestinal lymphatic system and portal vein. FA = fatty acid, MG = monoglyceride, TG = triglyceride, LP = lipoprotein.
Reproduced from Trevaskis et al. [2] with permission.
The intestinal lymphatic drug transport exhibits a number of advantages over the oral absorption via the portal blood. For instance, drugs that are transported via the intestinal lymphatic system enter the systemic circulation without first passing through the liver. This is very advantageous for drugs that are highly metabolized on first pass through the liver, since it will increase oral bioavailability [2]. It has also been observed that because intestinal lymphatic transport will affect the local exposure to the lymphatics and ultimately the systemic exposure, it is possible to obtain different toxicological profiles [2]. Intestinal lymphatic transport has also been reported to be more effective for immunomodulatory and chemotherapeutic drugs since the lymphatic system is the primary route for metastasis of solid tumors and the transport pathway for T and B lymphocytes [4], [5], [6], [7]. Furthermore, recently it has been suggested that the development, sequestration, and spread of the human immunodeficiency virus (HIV), hepatitis B and C virus, morbillivirus, canine distemper virus, and severe acute respiratory syndrome (SARS) associated coronavirus in association with lymphocytes present in the lymph and lymphoid tissue [8], [9], [10], [11], [12], [13], [14]. Therefore, lipidic prodrugs of didanosine, an antiviral that target AIDS, have been developed to enhance its lymphatic delivery [15].
2.2. Digestion and absorption of lipids
Because the digestion and absorption of lipids is linked to intestinal drug transport, the process of digestion and absorption or lipids will be reviewed. However, we recommend the reader consider other excellent reports that cover these processes in detail [16], [17], [18], [19], [20], [21], [22]. The process of digesting food-derived lipids (predominantly in the form of triglycerides) starts in the stomach where pre-duodenal lingual (mouth cavity) and gastric acid lipase (gastric mucosa) that hydrolyze triglycerides to diglycerides and fatty acids [23], [24], [25]. After the initial hydrolysis action of the acid lipase, the resulting by-products and remaining solid material migrate down the stomach where it passes to the pyloric antrum. At the pyloric antrum, gastric chyme is released and in combination with t peristaltic movements results in the emulsification of food-derived triglycerides as they empty into the duodenum [8], [18]. The presence of the lipid in the duodenum stimulates the secretion of pancreatic fluids and bile (bile salts and bile lipids). The biliary lipids (phospholipid and cholesterol) adhere to the surface of the emulsion forming a colloidal-like stable emulsion with smaller droplet size (higher surface area) that allows a better action of pancreatic lipase that allows the hydrolysis of triglycerides to 2-monoacylglycerol and two fatty acids molecules from each triglyceride [26], [27], [28], [29], [30].
Phospholipids and cholesterol also get digested in the intestine. Phospholipid digestion occurs in the small intestine because gastric lipase is incapable of digesting phospholipids. The phospholipid that is mainly found in bile is phosphatidylcholine and is in mixed micelles with cholesterol and bile salts. Once bile is released in the small intestine, phosphatidylcholine is hydrolyzed by phospholipase A2 to fatty acids and lysophosphatidylcholine [31], [32]. Cholesteryl esters get hydrolyzed by cholesterol esterase in the small intestine to free cholesterol [3], [33]. Cholesterol esterase activity is enhanced by the presence of trihydroxy bile salts such as sodium cholate, but also allows for the self-aggregation of the enzyme into a polymeric form [34].
The digestion products released from the triglyceride droplets in the colloidal emulsion form liquid crystalline structures that when combined with sufficient concentrations of bile salts produce unilamellar and miltilamellar vesicles [35], [36]. Accordingly, the post-prandial intestinal lumen (having higher bile salts due to higher bile release) has a greater presence of unilamellar and miltilamellar vesicles. The lipid digestion products contained in a mixed bile salt-phospholipid micellar phase first need to be dissociated from this phase in order to be absorbed and pass into the enterocyte [37], [38], [39]. It has been reported that the enterocyte surface might be in proximity to a low pH region that allows for a change in the colloidal structure leading to release and apical absorption of lipid digestion products [37], [40], [41], [42]. The transport of lipid digestion products across the apical membrane of the enterocyte has been reported to occur via passive transport and via active transport using specific membrane-bound carrier proteins. The passive transport predominates when the luminal lipid concentrations are high as will be the case post-prandially [43], [44], [45]. The membrane-bound carrier proteins involved in fatty acid uptake involve the microvillus membrane fatty acid binding protein and the fatty acid transporter [44], [46], [47], [48].
Once the lipid has been absorbed into the enterocyte, its chain length determines its subsequent intracellular processing. Short- and medium-chain lipids (C< 12) generally diffuse across the enterocyte, while long-chain lipids (C≥ 12) generally migrate to the endoplasmic reticulum where they get re-acylated and assembled into lipoproteins before secretion into the mesenteric lymph [49], [50], [51], [52]. The re-acylation process of fatty acids and monoglycerides to triglycerides in the endoplasmic reticulum has been reported to be involved with two cytosolic fatty acid binding proteins (I-FABP and L-FABP) [20], [43], [53], [54], [55], [56], [57], [58], [59]. Furthermore, the re-acylation process appears to occur via two possible pathways. One pathway involves a two-step sequential direct acylation of 2-monoacylglycerol to triglyceride [18], [60] and accounts for the main pathway of production for triglycerides destined for chylomicrons [18], [59]. The second and minor pathway is the phospatidic acid pathway or glycerol-3-phosphate that involves the sequential acylation of endogenous glycerol-3-phosphate with three molecules of activated fatty acid [18], [59], [61], which accounts for the production of triglycerides mainly destined for very low density lipoprotein (VLDL) [62], [63], [64], [65]. The sequential steps in digestion of lipids and their absorption via the portal blood or the mesenteric lymph are represented in Fig. 2 .
Fig. 2.

Schematic diagram describing the sequential steps in the digestion of lipids and absorption via de portal blood and intestinal lymphatics.
Reproduced from Porter and Charman [241] with permission.
2.3. Lipoprotein assembly
Because of the somewhat narrow focus of this review, we will only briefly review lipoprotein assembly. However, we direct the interested reader to a number of recent reviews that contain detailed information on this topic [16], [61], [63], [66], [67]. Chylomicrons and VLDL are the primary lipoproteins secreted by the intestine. The chylomicrons appear to be the predominant lipoprotein under post-prandial conditions (higher lipid loads), while VLDL appears to be the predominant lipoprotein under pre-prandial conditions (low lipid loads) [64]. Furthermore, it has been reported that the assembly of intestinal chylomicrons and VLDL occur by two separate independent pathways [16], [18], [68], [69], [70], [71], [72], [73], [74], [75], [76].
The first step in lipoprotein assembly involves the synthesis of apo-B in the rough endoplasmic reticulum to associate with phospholipid derived from the endoplasmic reticulum membrane. Neutral triglyceride lipids are then added to the apo-B phospholipid complex forming a primordial lipoprotein that is released into the lumen of the endoplasmic reticulum [63], [66]. The size of this primordial lipoprotein depends on the length of the apo-B polypeptide [77], [78]. The next step is the formation of large triglyceride-rich lipid droplets in the smooth endoplasmic reticulum, which is independent of apo-B and is most prevalent during luminal lipid challenge (post-prandially). The last step involves the fusion of primordial lipoprotein assembled in the rough endoplasmic reticulum with the triglyceride-rich lipid droplets formed in the smooth endoplasmic reticulum, a process called core expansion, which most likely occurs at the junction between the rough and smooth endoplasmic reticulum [63]. If apo-B were not to be present, lipid droplets would accumulate in the enterocyte [79], [80]. The factor that will determine if a chylomicron or a VLDL is produced resides in the amount of lipid added to the primordial lipoprotein during core expansion [63].
2.4. Pre- and post-prandial lymphatic transport
The amount of available food-derived lipids for formation of lipoproteins and intestinal lymphatic drug transport significantly increases, as expected, after food consumption. However, substantial amounts of endogenous lipid (as free fatty acid attached to phospholipid and triglyceride) are available in the enterocytes under fasting conditions [81], [82], [83], [84]. It has been described that endogenous fatty acids can access the enterocytes from both the apical (luminal) and basolateral sides [81], [85]. Endogenous fatty acids accessing the apical side are derived from either desquamated enterocytes or from bile that deposits fatty acids necessary for phospholipid transport [81], [86]. Endogenous fatty acids accessing the basolateral side are derived from the uptake of chylomicrons and fatty acid present in any remaining intestinal blood supply [81], [87]. However, recently a de novo synthesis route has been presented as a plausible source of endogenous fatty acids in the enterocyte [65], [81], [82], [85].
Under fasting conditions, the main contribution of endogenous fatty acids is from bile [71], [81], [88], [89], followed by a minor contribution from enterocyte desquamation [81], [82], and a minimal contribution from de novo synthesis within the enterocyte [81], [82], [85]. Bile plays a critical role in solubilization and intestinal absorption of lipophilic drugs and food-derived lipids by the formation of transient mixed micelles of bile salts and phospholipids [3], [81], [90]. Once in the enterocyte, the endogenous fatty acids re-esterify to triglycerides and then self-assemble into lipoproteins.
The effect of food on systemic exposure of co-administered drug has been reported for a variety of drugs. For instance, the transport of testosterone undecanoate (a highly lipophilic prodrug of testosterone) to systemic exposure of testosterone was studied in the greyhound dog after post-prandial administration. It was determined that testosterone undecanoate prodrug was transported lymphatically intact, but only hydrolyzed to testosterone once it is in systemic circulation [91]. In humans, testosterone undecanoate exhibits a higher systemic exposure to testosterone than testosterone given to fed subjects and that exposure to testosterone and prodrug were higher in fed subjects compared to fasted subjects [92], [93]. This increase in post-prandial systemic exposure was correlated with enhanced lymphatic transport and increased luminal solubilization of testosterone undecanoate [2]. Another instance of the effect of food was shown in the lymphatic transport of halofantrine (free base) in the pre- and post-prandial state that was assessed in the dog. The results showed that the cumulative post-prandial lymphatic transport of halofantrine is 54% of the administered dose, while pre-prandially the cumulative lymphatic transport equals to only 1.3% of the administered dose [94] as show in Fig. 3 . When the hydrochloride salt of halofantrine was administered to the dogs, a similar 47.3% cumulative post-prandial lymphatic transport was achieved [95]. This would indicate that halofantrine converted from the hydrochloride salt (poorly lipid soluble) to the free base (highly lipid soluble) before absorption into the enterocyte [59]. This conversion was reported to be favored by gastrointestinal pH that would allow the free base to associate with lipid digestion products that would be incorporated into chylomicrons during lipoprotein assembly [59].
Fig. 3.

Cumulative lymphatic transport of halofantrine (Hf) (percentage of administered dose, mean ± SE) in thoracic lymph duct-cannulated dogs after fasted administration (open symbols, n = 3) or post-prandial administration (closed symbols, n = 4) of 100 mg halofantrine (free base).
Reproduced from Khoo et al. [94] with permission.
The VLDLs are the only lipoproteins produced during fasting by the small intestine [65], [69], [96], while after a meal the small intestine mainly produces chylomicrons [69], [97]. The difference between VLDL and chylomicrons reside in their size (30–80 nm diameter for VLDL, and 75–1200 nm for chylomicrons) and their Svedberg flotation (Sf) rate (20–400 for VLDL, and more than 400 for chylomicrons) [98]. The type of fatty acid infused intraduodenally also determines the output and transport of VLDL and chylomicrons indicating that both are produced via different pathways. For instance, when palmitate is infused there are increases in VLDL transport, but no changes in its output, while chylomicron output increases when linoleate and oleate are infused [65]. These differences have been well documented to be due to the presence of different pathways for assembly of VLDL and chylomicrons [68], [70], [72], [73], [74], [75], [76].
2.5. Clinical disorders of intestinal lymphatic transport
Because both apo B-100 (secreted by the liver) and apo B-48 (secreted by the small intestine) are encoded by the same gene [99], [100], [101], [102], [103], it is possible that genetic disorders affecting apo B synthesis or chylomicrons and VLDL formation can alter intestinal lymphatic transport.
For instance, abetalipoproteinemia or Bassen–Kornzweig syndrome is a rare genetic disorder in which there is a complete failure of the liver and gut to produce triglyceride-rich lipoproteins, chylomicrons and VLDL [99], [104]. Although it was originally believed that it was caused by an apo B synthesis deficiency, it was later confirmed that it is caused by a mutation of the microsomal triglyceride transfer protein gene [101], [105], [106], [107].
Anderson's disease or chylomicron retention disorder is another rare, hereditary hypocholesterolemic syndrome characterized by the absence of chylomicrons in the intercellular space although there are observable chylomicrons in the enterocytes [108], [109]. The underlying mechanism for Anderson's disease is not fully understood yet as patients don't have a defect in the genes involved with apoprotein synthesis or microsomal triglyceride transfer protein [110]. However, it has been described that Anderson's disease might be caused by a post-Golgi cargo-specific secretion defect [106], and recently it has been reported that it might be linked to a SARA2 gene mutation [111].
Because lymph nodes undergo hyperplasia as a response to most of disease processes [112], histological changes can occur leading to an increase in lymphatic flow rate, local perfusion, and capillary permeability [113]. Furthermore, because the lymphatic system is the primary route for metastasis of solid tumors [4], [5], [6], [7] it is possible that complete obstruction of lymphatic pathways may occur, which would lead to a slower lymph flow that can ultimately cause lymphedema or lymphatic insufficiency [114], [115]. However, metastasis usually leads to the appearance of lymphomas of the gut, which constitute a large proportion of human tumors that are usually inoperable [116], [117], [118]. Furthermore, a decreased lymph return has been described to occur due to lymphatic hypoplasia or aplasia or also called primary lymphedema, while the obliteration of lymph nodes and trunks has been described as secondary lymphedema [119]. Other disturbances in lymph angiogenesis have been reported more recently as lymphedema-angiodysplasia syndromes [120].
2.6. Animal models
Animal models have been employed to study lymphatic drug transport by cannulation of the intestinal lymphatic duct for direct measurement of drug concentrations [2], [121]. Because this type of cannulation requires an irreversible surgery, intestinal lymphatic drug transport is not studied directly in this way in humans. The most commonly used model is the rat [121], but other large species such as dogs [94], [122], [123], pigs [121], [124], sheep [125], [126] have also been used.
As mentioned above, an approach to studying lymphatic drug transport is to use lymph duct-cannulated animal models, which allow the complete collection of lymph flowing through the cannula. Having collected all of the lymph allows for an accurate estimate of the total extent of lymphatic drug transport [2], [127]. Another approach is to use a lymphatic-venous shunt that allows collection of lymph through a longer period of time and at fixed time points to assess concentration-time profiles. However, the latter approach might have limited applicability due to the low lymph flow rate the limited sample volumes obtained [127]. Recently, an indirect pharmacological approach has been described utilizing intestinal chylomicron flow inhibitors Pluronic-L81 and colchicine [128]. For this approach, the systemic drug exposure is compared in the presence and absence of co-administered Pluronic-L81 or colchicine to indirectly assess the impact of intestinal lymphatic drug transport in the oral bioavailability [128]. This approach is advantageous because it doesn't require a lymph duct-cannulation. However, the impact of blocking chylomicron flow in lipid processing and synthesis has to be fully characterized. For instance, Pluronic-L81 lowers plasma VLDL and low-density lipoprotein cholesterol and reduces lipid and apoprotein secretion [129]. Nonetheless, employing cannulae is still the most common approach in the small and large animal models.
2.6.1. Small animal models
The rat is the most common small animal species utilized due to ease of handling and low space requirements to house them. Most of the studies have been performed in anesthetized rats, but conscious restrained and unrestrained models have been also described [121].
The unconscious (anesthetized) rat model has been used in various studies [130], [131], [132], [133], [134], [135] with various degrees of modification in the site of cannulation, lymph fistulation, feeding and rehydration pre- and post-operative procedures, and dosing [121]. The model is well described elsewhere [131]. Briefly, a triple-cannulation of the carotid artery (systemic blood collection), the mesenteric lymph duct (intestinal lymph collection), and the duodenum (administration of rehydration solution) is performed [121]. However, it can also include a fourth cannula in the jugular vein if intravenous administration is required [121]. The mesenteric lymph cannula allows the collection of all the lymph draining to the duodenum and, consequently, the total amount of drug transported lymphatically can be calculated. The systemic blood collected from the carotid artery allows the calculation of systemic plasma exposure that can be interpreted as the absorption of drug via the portal blood. This is advantageous because it allows the assessment of portal blood absorption contribution to bioavailability by comparing the plasma exposure of orally administered drug to rats that are lymph duct-cannulated rats with the plasma exposure of intravenously administered drug rats that are not lymph duct-cannulated rats [121]. As expected with any animal model that employs cannulation, the most common complication can be cannula occlusion that is typically greater with animals allowed to move freely. This occlusion can also be attributed to the differences in gastric motility and emptying [132] and lymph flow rate between an unconscious anesthetized (0.1–0.6 mL/h) and a conscious un-anesthetized rat (1–3 mL/h) [136]. Depending on the lymph volume or the sampling robustness, the experiment may require long periods of anesthesia (up to or longer than 24 h). This can produce significant logistical concerns in maintaining and monitoring animals under anesthesia for that long, but also can produce significant increases in mortality [121].
In the conscious restrained rat model, animals are allowed to recover from surgical anesthesia before being placed in a harness or restraining cage [133], [135], [137]. This model only requires of 2 cannulae (in the jugular vein for systemic blood collection, and in the mesenteric lymph duct for intestinal lymph collection). The intraduodenal cannula is not required as a rehydrating saline solution is administered intravenously through the jugular vein cannula. Compounds can be administered intravenously because the rat is restrained. The lymph and jugular vein cannulae are advanced under the skin and externalized out from the dorsal (back of the neck) skin so they can be connected to a swivel for continuous sampling [121], [137]. Even though this model offers advantages (normal lymph flow rate, gastric emptying, and lipid digestion because in a conscious rat) over the unconscious rat model, physical restraint can affect the lymph flow rate, gastrointestinal functioning, and produce venous return [137]. Because of these possible complications, a conscious un-restrained model has also been developed [121], [138] that allows intravenous and oral administration of compounds with continuous lymph collection over 12 h and systemic blood collection over 30 h [121]. The surgical procedures include a triple cannula in the mesenteric lymph duct (for lymph collection), in the carotid artery (for systemic blood collection), and in the duodenum (for overnight rehydration). In order to assess oral bioavailability, non- cannulated rats (no mesenteric lymph duct-cannula) can be included as well as another orally administered group of rats that maintain the other two cannulae (in the carotid artery and in the duodenum). A third group of intravenously administered rats (via a jugular vein cannula) can be included to assess absolute bioavailability, having systemic blood collected via a carotid artery cannula and administration of a rehydrating solution via an intraduodenal cannula [121].
These three rat models (unconscious, conscious restrained and conscious un-restrained) employ surgical procedures that are performed with non-fasted animals because it is easier to visualize the mesenteric lymph duct. However, the animals are fasted during the post-operative recovery period to allow lymph and triglyceride levels to return to fasting basal levels before dosing [121]. The surgical methodology for the cannulations and post-operative recovery care in rats has been well described elsewhere [121].
Despite being widely used, the rat has some considerable limitations. For instance, the rat has a continuous rather than intermittent flow of bile into the intestine like humans, rendering the rat less clinically-relevant than the dog and pig, which have a more similar biliary secretion, transit profile, and gastrointestinal tract to humans [2]. Additionally, the rat is not a suitable model to assess post-prandial lymphatic transport since it is not possible to train the rat to eat on command, the rat diet has significantly lower amounts of lipid than human (about 5% in rat and up to 20% in human) [59], and the rat has no gallbladder and thus has no pulsatile post-prandial release of bile. Furthermore, unlike the rat, a large species such as the pig, sheep, or dog, allows the administration of more clinically-relevant doses and formulations and are amenable to studies with monitored/controlled fasted and fed conditions [2], [59], [121].
2.6.2. Large animal models
Because of the reported limitations of the rat models, larges species such as dogs [94], [122], [123], [139], [140], pigs [124], [141], [142], [143], and sheep [125], [126], [144], [145] have been described to improve the assessment of lymphatic drug transport.
2.6.2.1. Dog models
The dog is widely used as the large animal model to study lymphatic drug transport because of the similarities in gastrointestinal physiology with humans and because it is feasible to assess post-prandial lymphatic transport [94], [121], [122], [123], [139], [140], [146]. The dog model was originally implemented by cannulating the thoracic duct either using a T-shaped silicone rubber cannula [147], [148] or a double-lumen cannula [149]. However, these cannulae require continuous infusion of heparanized saline to prevent clotting and a surgical thoracotomy to access the thoracic duct, but this surgical procedure usually produces longer post-operative recovery periods [121].
To prevent the need for surgical thoracotomy, another model was employed by inserting a cannula into a ligated and isolated segment of the external jugular vein above and below the entry of the thoracic duct into the vein [137], [140], [150], [151], which allows chronic collection of lymph. Because this surgical procedure is difficult, there is a high incidence of pneurothorax and obstruction of the venous return at the caval bifurcation [121].
A triple-cannulated dog model was implemented to collect lymph via a cannula in the thoracic duct, to collect blood via a cannula in the hepatic portal vein and venous blood via the jugular vein [94], [121]. This model has also been implemented to allow collection of venous blood via a cannula in the cephalic vein, and assessment of lymphatic drug transport and first past metabolism via collection of lymph and blood from cannulae in the thoracic lymph duct and the hepatic portal vein, respectively [59], [94], [132], [133], [152]. The surgical methodology for the cannulations and post-operative recovery care in dogs has been well described elsewhere [121]. Even though the dog is the most commonly employed large animal model, the pig and the sheep have also been reported to be suitable models, but also have their limitations.
2.6.2.2. Pig models
The pig exhibits many similarities in its gastrointestinal tract compared to humans; for this reason an anesthetized [124], [141] and conscious pig model [142], [143] have been developed to assess the absorption and lymphatic transport of drugs. The anesthetized pig model involves the oral administration of the lipophilic dye Sudan black to improve the visualization of the mesenteric lymph duct before cannulation. It also permits simultaneous sampling of the hepatic portal blood and systemic blood [124], [141]. One disadvantage of this model is that it doesn't allow for the cumulative assessment of lymphatic transport as it only allows for periodic sampling instead of a continuous collection. This limitation most likely accounts for why this model has not been widely employed [121].
The conscious pig model involves the use of an external thoracic duct-venous shunt that allows the return of the thoracic lymph to the systemic circulation [142], [143]. This model has been employed to the assessment of lymphatic transport of proteins, but similar to the anesthetized model, it doesn't permit for the cumulative assessment of lymphatic transport as it only allows for periodic sampling instead of continuous sampling [121], [143].
2.6.2.3. Sheep models
A conscious model has been described for the assessment of lymphatic transport of proteins administered subcutaneously in sheep [144], [145]. Lymph is collected directly from a cannula located in the thoracic lymph duct. However, because the differences in physiology and complexity between sheep (ruminant) and human (monogastric) are significant, application of this model is limited [121].
2.7. In vitro models
The widely utilized intestinal permeability model using Caco-2 cells has been applied to assess intracellular lipoprotein assembly [61], [63], [153], [154], [155], [156] as well as to determine the effect of lipids and lipidic excipients on drug association with lipoproteins during lymphatic transport [157], [158], [159], [160]. It has also applied to assess the genetic expression and post-translational modification of apolipoproteins [161], and as a model to assess the assembly of chylomicrons and VLDLs [153], [162]. However, it has to be acknowledged that Caco-2 cells differ from enterocytes in multiple ways and that the data generated using this model should be interpreted with caution. The main difference is Caco-2 cells are derived from cancerous cells of the human colon rather than from healthy enteric cells from the small intestine. That difference surely results in different activated biochemical pathways. In addition, Caco-2 cells synthesize apo B-48 and apo B-100, while enterocytes only synthesize apo B-48 [3]. Furthermore, the pathway for formation of triglycerides and chylomicron synthesis differ between cells, while enterocytes use the monoglyceride pathway and mainly produce chylomicrons during fat absorption, the Caco-2 cells employ the glycerol-3-phosphate pathway [163] and produce various lipoproteins, but chylomicrons not being the major lipoprotein produced [3].
Another in vitro model that has been described is based on the good correlation between the degree of intestinal lymphatic drug transport and the degree of ex vivo association of the drug with chylomicrons obtained from plasma. This correlation was significantly better than the correlation of intestinal lymphatic drug transport with the log P or triglyceride solubility of the drug [164]. However, this model has not been widely adapted. More recently, biorelevant test media such as Fasted State Simulated Intestinal Fluid (FaSSIF) and Fed State Simulated Intestinal Fluid (FeSSIF) have been employed to assess drug release from lipid based delivery systems [165], [166], [167], [168]. However, as described in Section 2.2 (Digestion and absorption of lipids), lipid-based delivery systems will follow an emulsification process in the gastrointestinal tract caused by different lipolytic enzymes to release the drug and be able to pack it into chylomicrons for entry into the lymphatic system. Therefore, an in vitro model should take in account lipolysis.
A lipolysis model has been described to predict in vivo absorption by assessing the release of drug from lipid-based drug delivery systems and by assessing whether drug precipitates following lipolyis [169]. The main purpose of a lipid-based drug delivery system is to increase oral bioavailability by preventing precipitation of a poorly water-soluble drug in the gastrointestinal fluids. However, in order for the drug to be absorbed by the enterocyte, the lipid-based delivery system needs to keep drug solubilized following a lipolysis process. Thus, design of a lipid-based delivery system needs to account for its performance in vivo after lipolysis to ensure that the drug remains solubilized and does not precipitate. Failure to take this in account leads to the success or failure of a delivery system to improve drug absorption [169]. It is here where the in vitro lipolysis model becomes useful to assess the suitability of a lipid-based delivery system and to predict how it would behave in vivo.
This in vitro lipolysis model basically accounts for five main variables that occur in vivo: 1) lipolysis is dynamic model that releases a hydroxyl anion (OH−) for each liberated free fatty acid [169], 2) lipolysis requires the presence of counterions such as calcium (Ca2+) to control the process rate [168], [170], 3) lipolysis is regulated by the presence of hydrolytic enzymes, 4) lipolysis occurs at regular body temperature, and 5) upon lipolysis completion, digested fractions will either be in or not be in solution, which determines whether fractional components would be absorbed. The experimental set-up of the model accounts for each of the in vivo variables by, respectively: 1) having the pH continuously measured, to determine hydroxyl anion release as a measure of the extent of lipolysis, and held constant by an autoburette that titrates with base [169], 2) ensuring calcium is present in a constantly-stirred lipolysis medium also composed of digestion buffer, bile salts and phosphatidylcholine [169], 3) adding bio-relevant levels of pancreatic lipase and co-lipase to initiate lipolysis [171], [172], and adding 4-bromobenzene-4-boronic acid (BBBA) to terminate lipolysis [62], [65], 4) maintaining temperature at 37 °C by water bath [169], and 5) ultracentrifuging the medium to obtain a 3-phase separation: an aqueous phase containing bile salts, fatty acids and monoglycerol; a lipid phase containing undigested triglycerides and diglycerides; and a precipitate (sediment) containing un-dissolved fatty acids [64]. Each of these phases can then be analyzed for drug content. Because the dissolution of drug in the intestinal fluid is required for drug absorption, the drug solubilized in the aqueous phase of the experimental medium will be generally available for absorption while the drug in the sediment would not be available for absorption in vivo [169]. Furthermore, this model allows the simulation of fasted and fed state conditions by using different concentrations of bile salts and lecithin in the experimental medium. For instance, fasted state conditions will be simulated by 5 mM cholates and 1.25 mM lecithin, while the fed state conditions will be simulated with 20 mM cholates and 5 mM lecithin [173], [174]. More theoretical background and technical details can be found elsewhere [169], [175].
2.8. In silico models
As described in Section 2.6 on animal models, some models employ surgery and have a post-surgery recovery period that can be stressful for the animal, which could ultimately affect the physiology of the gastrointestinal tract [176]. Thus, in silico models would be desirable to provide a simple computational approach to predict intestinal lymphatic transport. The need for these models came from inadequate correlations between in vivo studies and empirically-determined in vitro measurements of solubility. Based on in vivo studies it was observed that lipophilicity [177] and triglyceride solubility [1] correlated well with the extent of lymphatic transport. For instance, it was proposed that drugs with log P values > 5 and triglyceride solubility > 50 mg/mL would have a significant lymphatic transport [1]. However, compounds like penclomedine (log P = 5.48 and triglyceride solubility = 175 mg/mL) [178] and the lipophilic lipid regulator CI-976 (log P = 5.83 and triglyceride solubility > 100 mg/mL) [179] were reported to have low lymphatic transport [176]. Therefore, it can be observed that the lipophilicity and triglyceride solubility correlation with lymphatic transport was not completely adequate and that a more adequate predictive model needed to be implemented.
The initial work to develop an in silico model for intestinal lymphatic transport was based on successful models for passive absorption [180], [181], [182]. More recently a quantitative relationship between molecular structure and the degree of intestinal lymphatic drug of lipophilic compounds co-administered with a long-chain triglyceride vehicle was examined using the computer program VolSurf [176]. For this, various drugs were selected using literature values of intestinal lymphatic drug transport. Molecular descriptors were calculated using VolSurf software [183], [184] based on the 3D molecular structures rendered using Sybyl molecular modeling system (Tripos Inc.), and their log P values were estimated using the online version of logKow [185]. To assess the similarity between the experimental and estimated lymphatic transport, partial least squares and projection to latent structures (PLS) were determined using the software Simca-P [186]. Further statistical analysis included the variable influence on projection (VIP) values and the cross-validated correlation coefficient (Q2) [176]. The PLS analysis reported that intestinal lymphatic transport of a drug can be estimated based on nine descriptors: globularity, hydrophilic surface size, local interaction minima, fraction of hydrophilic surface area, hydrophilic-lipophilic ratio of the molecule, unbalance between the center of mass and the center of the hydrophilic regions of the molecule, and unbalance between the center of mass and the center of the lipophilic regions of the molecule [176], [183], [184]. It was observed that this computational model was significantly better than the correlation of intestinal lymphatic drug transport with the empirically-determined log P and/or triglyceride solubility of a drug [176], [177].
Even though this model was successful in predicting lymphatic drug transport, it has not been validated, and it has been described by the authors as an initial model [176]. Therefore, it can be appreciated that in terms of in silico models for intestinal lymphatic drug transport this area is at a very early stage and more studies are warranted. However, it has to be acknowledged that different in vitro and in silico models have been widely applied to assess intestinal drug absorption, drug dissolution, and drug solubility [167], [187], [188].
3. Metabolic enzymes and transporters involved in intestinal lymphatic drug transport
3.1. First-pass metabolism involvement
The lymphatics absorption pathway offer a unique advantage over drug absorbed via portal blood following oral administration in that hepatic first-pass metabolism can be evaded to some extent. Drug metabolism usually involves a variety of enzymes, which by definition (from a biochemical perspective) improves the ability (acting as a catalyst) to biotransform a substrate drug to a metabolite by lowering the required activation energy for this reaction. Two categories of drug metabolism are well recognized today; phase I and phase II metabolism.
3.1.1. Phases I and II metabolism
Phase I enzymes can catalyze a wide array of chemical reactions. However, a common theme apparent for all Phase I enzyme reactions is that final products are usually modified to contain a functional group like a hydroxyl, an amine or a carboxylic acid [189]. Phase II metabolism, which does not necessarily require phase I metabolism to precede beforehand, can further modify functional groups by glucuronidation, glycosidation, sulfation, methylation, acetylation, glutathione conjugation, amino acid conjugation, fatty acid conjugation and condensation. The majority of research in the area of Phase I and II metabolism has focused on cytochrome P450 (CYPs) and UDP-glucuronosyltransferases (UGTs) proteins. UGT-catalyzed glucuronidation reactions are responsible for 35% of all drugs metabolized by phase II enzymes [190]. The human UGTs are a superfamily of enzymes that metabolize via conjugation a variety of endogenous substrates that include bile acids, fat-soluble vitamins, and drugs [191], [192]. UGTs are found bound to the internal membrane and face the luminal side of the endoplasmic reticulum [192]. This specific configuration allows these enzymes to have direct access to metabolites formed by phase I reactions.
As previously described, drugs transported through the intestinal lymphatic system are protected from first-pass hepatic metabolism because the mesenteric lymph, unlike the portal blood, empties into the systemic circulation without first passing through the liver [91], [94]. This is significant for drugs with high pre-systemic clearance like testosterone that has an extremely limited oral bioavailability [193], [194]. However, the highly lipophilic prodrug of testosterone, testosterone undecanoate, is orally bioavailable because it enters (as well as its active metabolite 5α-dihydrotestosterone) the systemic compartment via the intestinal lymph [91], [92], [195]. However, an enterocyte-based first-pass metabolism has been described.
3.1.2. Enterocyte-based first pass metabolism
The impact of lymphatic drug transport on enterocyte-based first-pass metabolism has been well described in the literature [2]. Briefly, when benzo(α)pyrene was orally co-administered with lipid to kilfish, the lipid was transported into the lymph as lipoporotein but benzo(α)pyrene only dispersed through the enterocyte without association with lipid or lipoproteins. This was attributed to the formation of a more hydrophilic metabolite of benzo(α)pyrene in the smooth endoplasmic reticulum, causing its ready absorption into the systemic compartment via the portal circulation instead of via the lymphatic circulation [196], [197]. Similarly, when halofantrine was orally administered to lymph duct-cannulated rats a lower transport rate into the lymph was observed for the drug when compared to fatty acids administered to control groups [2]. Apparently halofantrine was removed from the lymph precursor pool in the enterocyte through CYP3A4 metabolism of halofantrine to desbutylhalofantrine, which caused the differences in transport rate [2]. Furthermore, co-administration of an appreciable amount of lipid either as food-derived lipid by being fed a fatty meal or as a direct lipid dose diminishes enterocyte metabolism because of the sequestration of drug into larger lipid droplets that reduce the accessibility of drugs such as benzo(α)pyrene [196] and halofantrine [2] to metabolizing enzymes located in the surface of the smooth endoplasmic reticulum [198], [199], [200].
3.2. Transporters of lipids and drugs
Although only a few reported drugs are transported via the intestinal lymphatic system, the intestinal lymphatic system could be utilized as a transport pathway for lipophilic drugs. In order to fully benefit from lymphatic drug transport, much work is required to gain further knowledge about how enterocytes contribute the separation of lipophilic drug transport into lymph versus portal circulation.
3.2.1. Lipid transporters
While lipid transport-proteins have been identified on enterocyte membranes (both apical and basolateral membranes), families of intracellular lipid binding proteins also exist and together facilitate the entry (absorption) and movement across (intracellular transport) of endogenous lipids and lipids from diet (exogenous) [201], [202].Typically lipids can enter the enterocytes via passive diffusion or active transport. In addition to this, apical transporters have also been identified to facilitate this absorption process [47], [203]. For example, NPC1L1 has been identified as a lipid transporter protein [204], [205]. Furthermore, it is of interest to mention that proteins have also been shown to be transferred across the apical membrane of enterocytes by transporters that are less well defined.
Several ATP-binding cassette (ABC) transporters have been implicated in lipid uptake across plasma membranes and intracellular lipid trafficking [206], [207], [208], [209]. ABC transporters may therefore be involved in intestinal lipid absorption, although the role of only a few of these transporters has been demonstrated. For example, P-glycoprotein (P-gp) is believed to influence intestinal lipoprotein formation [157], [208] and it has been suggested that P-gp facilitates the absorption and intracellular trafficking of cholesterol, although the evidence for this is still circumstantial [210]. Additionally, ABCA1 appears to facilitate absorption of cholesterol across the basolateral membrane of enterocytes to plasma ApoA-1, which enhances the formation of nascent high density lipoprotein [209], [210], [211], ABCG5 and ABCG8 are thought to reduce excess intestinal cholesterol and sterol absorption by facilitating efflux from enterocytes [209], [210], [212]. Table 1 summarizes the different predominant ATP-binding cassette (ABC) transporters and metabolic enzyme families, modulating lipid excipients/surfactants and associated known drugs that have been reported to be involved in intestinal lymphatic transport.
Table 1.
Predominant ATP-binding cassette (ABC) transporters and metabolic enzyme families reported to be involved in intestinal lymphatic transport and known modulating lipid excipients/surfactants and associated known drugs.
| Transporter | Lipid excipients/surfactants | Metabolism | Examples of drugs associated with CYP3A and P-gp transport |
|---|---|---|---|
| P-gp | Polyoxyl 35 castor oil (e.g., Cremophor) | CYP3A | Cyclosporine Ketoconazole Lovastatin Ritonavir Tamoxifen Amiodarone Saquinavir Carbamezapine |
| P-gp | PEG-15-hydroxystearate (e.g., Solutol HS-15) | CYP3A | |
| P-gp | Polysorbates (Tween 80, Tween 20) | CYP3A | |
| P-gp | Polymers (Pluronic block copolymers) | CYP3A | |
| P-gp | Sucrose esters (Sucrose monolaurate) | No metabolism reported | |
| P-gp | Medium chain glycerol and PEG esters (e.g., Labrasol) | No metabolism reported | |
| MRP2 | 1-monopalmitin, 1-monoolein and 1-monostearin | No metabolism reported |
3.2.2. Drug transporters
Upon crossing the apical side of the intestinal lumen, a drug can be metabolized (phase I or phase II; described in detail in the above sections of this review). These metabolites, as well as the parent drug, can be actively transported across the basal membrane of the intestine into the blood or efflux back out into the intestinal lumen. Therefore, these processes of active transport are facilitated by many types of major active transport proteins such as P-gp (which belongs to the multidrug resistance-type), multidrug resistance-associated proteins (MRPs), and other organic anion transporters which together play a major role in governing the overall bioavailability of foods, drugs and other xenobiotics.
3.2.2.1. P-glycoprotein (P-gp)
Upon discovery of cytotoxic drugs that can destroy cancer cells, researchers also discovered that drug resistance (for multiple drugs) can impede the effects of these drugs. P-gp was the first active transporter protein to have been discovered. Today, we know that P-gp, and other more recently identified protein(s) act as efflux pumps with a broad specificity for a variety of substrates. P-gp is a member of a large diverse family (over 50 members) of ATP-binding cassette (ABC) efflux proteins. Substrates for P-gp are typically lipophilic and cationic. As previously mentioned, a large number of compounds are substrates/inhibitors for P-gp and include anticancer agents, antibiotics, antivirals, calcium channel blockers, immunosuppressive agents and plant chemicals usually found in normal diet [213]. The nature of P-gp is to limit the absorption of certain compounds by effluxing compounds from the enterocytes back into the lumen, thus limiting their bioavailabilities. Data from both in vitro and in vivo studies using human intestinal epithelial cell lines [214] and P-gp-knockout mice [215], respectively, show that the disruption of P-gp activity would lead to some potentially hazardous problems with regard to drug disposition. Because the typical substrates of P-gp are lipophilic, it is expected that P-gp plays a role in the lymphatic transport of drugs; however, direct evidence has not been reported yet.
3.2.2.2. Multidrug-resistance associated proteins (MRPs)
MRPs were discovered much later than P-gp. In general, MRP1 through MRP9 are the only currently known MRPs; however, MRP7-9 has recently been discovered, but relatively little information exist currently with regard to both function and expression patterns. The substrates for MRPs are different with regard to each individual isoform as well as their respective localization. However, the general mechanistic function of MRPs is currently believed to be the elimination of compounds from the cell via efflux. Studies have shown that this class of transporters can confer resistance to cytotoxic drugs such as vincristine [216] and peptides [217], heavy metal anions [218] as well as endogenous metabolites such as bilirubin glucuronides [219]. Because of the diversity of substrates that MRPs handle, it would be expected that MRPs play a role in the lymphatic transport of drugs; however, direct evidence has not been reported yet.
3.3. Coupling efflux transporters with metabolic enzymes
In early research focusing on cytochrome P450s, it was thought that the rate and the overall extent of metabolism were the predominant factors in the overall disposition of drugs that were subject to biotransformation [220]. As research progressed, we begin to understand that an intricate relationship exists between Phase I metabolic enzymes with efflux transporters such as P-gp [221]. This interplay, in which not only a single metabolic enzyme and/or transporter but a relationship/communication between at least 2 proteins (i.e., one metabolic enzyme and one transporter), is an area of active research [221].
To help understand the mechanics of coupling, one could imagine a traditional one-two punch scenario in which a compound enters the enterocyte, quickly gets metabolized (Phase I or Phase II or both), and then is immediately taken up by efflux transporters and efflux back across the intestinal lumen. However, this is rather a simplified one-two mechanism that was observed and reported. Current findings and literature data suggest that the mechanism(s) by which metabolic enzymes interact with efflux transporters, the way in which metabolic enzymes interact with each other as well as efflux transporters (or transporters in general), and their connection to each other are highly complex and is not as simple as a one-two process. As an example, the “double jeopardy” theory was proposed by Benet and coworkers [222], [223], where the substrate is assumed to be absorbed at the apical membrane and is met by CYPs. This substrate is subjected to metabolism and thus is termed as ‘prosecuted’ for the first time by CYPs. With respect to efflux, P-gp is assumed to take the parent compound and transport it via efflux back into the intestinal lumen and unmetabolized parent drug can pass through the other side (basolateral) of the membrane and thus has reach either portal- or lymphatic-capillaries. But if the intact parent drug returns to the intestinal lumen via efflux, it can be absorbed one more time at the apical side of the membrane (further down the intestinal tract) and thus is subjected to metabolism again. This repeated ‘prosecution’ thus gives the term “Double Jeopardy” to this theory. Consequentially, a disruption of P-gp activity would result in less repeated chance of exposure time for the substrate in question at the apical membrane. This would lead to more substrate inside the intestine (i.e., increase in absorption) and thus could potentially increase the bioavailability of both the substrate and/or its metabolite in the systemic circulation. On the other hand, if CYPs activity was disrupted, the amount of metabolite would decrease severely and thus perhaps allowing more substrate to reach systemic circulation intact. A schematic representation of the double jeopardy theory is presented in Fig. 4 .
Fig. 4.

Schematic representation of the double jeopardy theory. Substrates are represented by triangles whereas products of enzymes or metabolites are represented by pentagons.
4. Lipid-based formulation and lipidic prodrug approaches to enhance intestinal lymphatic transport
4.1. Rationale of lipid-based formulations
Lipophilic drugs have poor water-solubility and consequently tend to have low and variable absorption following oral administration. The absorption of lipophilic drugs into intestinal lymphatic and portal circulation following oral administration is often increased by post-prandial effects that result from co-administration with food. The increased drug absorption with food has been attributed to multiple effects of food-derived lipids, particularly the long-chain triglyceride lipids, by decreasing the rate of gastric emptying, stimulating the secretion of biliary lipids, increasing the rate of lipolysis, increasing the drug-to-intestinal membrane contact, increasing the drug dissolution rate and solubilization by having altered the composition of intestinal fluids, increasing the formation of triglyceride-rich chylomicrons through the lipid digestion/re-acylation pathway [152], [169], [224], [225], [226], [227], [228], [229], [230]. The glyceride-rich chylomicrons are believed to allow lymphatically-transported drugs partition into and be transported along with chylomicrons into the intestinal lymphatics [164], [231]. It is that preferential association of the administered lipophilic drug with lipid digestion/re-acylation pathway that results in the secretion of drug-containing lipoproteins into the intestinal lymphatics [232]. Thus, a common formulation approach to achieving either post- or para-prandial effects seen with food-derived lipids is to replace food- for formulation-derived lipids. Using combinations of lipid(s), surfactant(s), and/or co-solvent(s), the expanded ability to improve oral absorption by additional mechanisms than those already mentioned for food-derived lipid include protection from luminal drug degradation, enhanced membrane permeability and decreased hepatic first-pass metabolism [225]. To optimize lymphatic drug transport, the candidate drug and its lipid-based formulation need to be rationally paired.
4.2. Candidate drugs for lipid-based formulations
Because the quantity of luminal and cellular lipid is generally not limiting when drug is co-administered with either food- or formulation-derived lipids, it is often the solubility of the administered drug or its dissolution in triglyceride that is limiting to the lymphatic transport of lipophilic drugs. Knowing that drug needs to partition into triglyceride-rich chylomicrons, having a candidate drug with a high solubility in triglyceride should take primacy in selecting candidate drugs. Charman et al. have suggested that the amount of a candidate drug that may be transported by intestinal lymphatics is the product of the quantity of lipid transported in the lymph in the form of chylomicrons and the amount of drug per chylomicron [130]. Recognizing the concentration of drug per chylomicron is influenced by the partition coefficient and triglyceride solubility of the drug [233], Charman et al. proposed that drug candidate for lymphatic transport have a log P > 5 and a triglyceride solubility > 50 mg/mL [1]. However, a combined high log P and high triglyceride solubility does not necessarily result in lymphatic transport [233]. As was mentioned earlier, penclomedine, an experimental cytotoxic agent with log P of 5.48 and a triglyceride solubility of 175 mg/mL, was poorly transported in the intestinal lymph, ~ 3% of the dose [178]. Similarly, Hauss et al. [179] reported very low levels, < 1% dose, of lymphatic transport using CI-976, a lipophilic lipid regulator with a log P of 5.83 and a triglyceride solubility of > 100 mg/mL [233].
4.3. Lipid-based formulations
4.3.1. Classification of lipid-based formulations
The co-administration of drug with formulation-derived lipids is perhaps the most studied and successful approach to increasing lymphatic drug transport. Lipid-based formulations can be solutions, suspensions and self-emulsifying formulations containing liquid or semi-solid triglycerides, mixed mono- and diglycerides, surfactants, either alone or as mixtures, all in possible combinations with hydrophilic co-solvents [175]. With that variety and in an effort to standardize the description of lipid-based formulations, Pouton et al. devised the Lipid Formulation Classification System (LFCS) which has recently been published in its most updated version [234], [235], [236] (Table 2 ). Although this system is not solely directed to formulating for lymphatically-transported drugs, it does encompass those formulations suitable for those types of drugs. The LFCS conveys the characteristics and the potential limitation of the various types of formulation in achieving small particle sizes and how prone the drug is to precipitating with intestinal fluid dilution and with formulation-derived lipid digestion. The precipitation of lymphatically-transported drugs, which already require lipid-based formulations to dissolve, will not easily or at all be re-dissolved upon precipitation.
Table 2.
The lipid formulation classification system: characteristic features, advantages and disadvantages of the four essential types of lipid formulations. Reproduced from Pouton and Porter [236] with permission.
| Formulation type | Materials | Characteristics | Advantages | Disadvantages |
|---|---|---|---|---|
| Type I | Oils without surfactants (e.g. tri-, di- and monoglycerides) | Non-dispersing, requires digestion | GRAS status; simple; excellent capsule compatibility | Formulation has poor solvent capacity unless drug is highly lipophilic |
| Type II | Oils and water-insoluble surfactants | SEDDS formed without water-soluble components | Unlike to lose solvent capacity on dispersion | Turbid o/w dispersion (particle size 0.25–2 μm) |
| Type III | Oils, surfactants, cosolvents (both water-insoluble and water-soluble excipients) | SEDDS/SMEDDS formed with water-soluble components | Clear or almost clear dispersion; drug absorption without digestion | Possible loss of solvent capacity on dispersion; less easily digested |
| Type IV | Water-soluble surfactants and cosolvents (no oils) | Formulation disperses typically to form a micellar solution | Formulation has good solvent capacity for many drugs | Likely loss of solvent capacity on dispersion; may not be digestible |
Briefly, Type 1 systems contain solely lipid components, typically mixtures of glycerides, having little to no solubility in water. These systems require lipid digestion to facilitate the formation of the colloidal dispersion of the lipids, lipid digestion products, and lipophilic drug in the intestines by bile salt-phospholipid mixed micelles [234]. These systems are ideally suited for lymphatically-transported drugs that have high triglyceride solubility. Type 2 systems contain lipids, polar lipids and water insoluble surfactants. These systems will vary in their digestibility, but still can enhance lipophilic drug bioavailability by achieving a smaller dispersion than can be achieved with Type 1 systems [234]. Type 3 systems can stably self-emulsify and disperse quickly to form fine dispersions owing to appreciable amounts of surfactant. However, if that dispersion proceeds too quickly, these systems can as quickly lose solvent capacity upon dilution in intestinal fluids resulting in drug precipitation. Type 4 systems are pure surfactants or mixtures of surfactants and co-solvents. These systems are not reported as having been used for lymphatically-transported drugs because of the likelihood of lipophilic drug precipitation [235]. Early work on using these systems were recently reviewed [237].
As examples of the Type 3 system, also knows a self-emulsifying drug delivery systems (SEDDS), which form fine oil-in-water emulsions or even microemulsions (SMEDDS) when exposed to aqueous media under conditions of gentle agitation [238], [239]. The commercially available formulation of cyclosporine (Neoral™), is a microemulsion preconcentrate with improved oral bioavailability and reduced inter- and intra-subject variability compared to the original crude emulsion product, Sandimmune™ [233]. Similar lipid-based formulations of the HIV protease inhibitors, saquinavir, ritonavir and amprenavir, have also reached the market [233]. The advantages of these formulations include their ease of production, enhanced solvent capacity, increased stability, and the potential to administer the final product as oral soft gelatin capsules [233], [234]. In addition, SEDDS typically produce emulsions with a particle size between 1000 and 300 nm, while SMEDDS form transparent microemulsions with a particle size of less than 100 nm [238]. These properties have been suggested to make SEDDS and SMEDDS a good formulation alternative for oral delivery of lipophilic drugs [238].
As an example, the impact of lipid-based formulation type on in vitro dispersion and digestion properties and the relationship to oral bioavailability, using danazol as a model lipophilic poorly water-soluble drug has been studied [226]. Three lipid-based danazol formulations were assessed: solution of a long-chain triglyceride (LCT-solution), SMEDDS based on long-chain (C18) lipids (LC-SMEDDS) and medium-chain (C8–C10) lipids (MC-SMEDDS) were administered to fasted dogs and compared with a micronized danazol formulation either co-administered with food or fasted. The LCT-solution and LC-SMEDDS formulations significantly enhanced the oral bioavailability of danazol when compared to fasted administration of the powder formulation. The MC-SMEDDS resulted in little enhancement in danazol bioavailability. In support of the in vivo findings, in vitro digestion of the MC-SMEDDS formulation resulted in significant drug precipitation when compared with the LC-SMEDDS formulation.
4.4. Altering lipids in formulation to increase lymphatic transport
The potential utility of lipids to enhance bioavailability and lymphatic transport has generally been assessed by considering structural features of the lipid (chain length, lipid class, and degrees of saturation). However, consideration of the amount of formulated lipid administered, digestibility of the lipid and the extent of dispersion should also be considered. These points have been briefly reviewed here, but further details appear in previous reviews [1], [233], [240], [241].
4.4.1. Structural features of lipids
The performance of a lipid-based formulation to increase absorption of lymphatically-transported drug is based on a particular combination of formulation and drug. However, from a review of the literature some general comments can be made structural features of lipids favoring lymphatic drug transport. Lymphatically-transported drugs will tend to have higher and achieve peak concentrations quicker when formulated in lipids that require minimal digestion prior to entering the lipid digestion/re-acylation pathway. Accordingly, fatty acids can be used in place of triglycerides and phospholipids because they have no need for pre-absorptive hydrolysis [1], [240], [242], [243]. In addition, lymphatic drug transport is increased using monounsaturated and polyunsaturated fatty acids, in part because of larger lipoproteins than when using saturated fatty acids [175], [233], [244]. With each increase in the degree of fatty acid unsaturation, the lag-time to a significant increase in the chylomicron concentration was reduced compared to control [244]. Cheema et al. hypothesized that the difference on the onset of chylomicron synthesis probably reflects a more rapid rate of absorption with increasing fatty acid unsaturation. Additionally, the greater the degree of unsaturation of the fatty acid, the lower the melting point, the greater the fluidity at 37 °C and the less hydrophobic the molecule, all factors facilitating absorption [244]. Finally, lymphatic drug transport is generally enhanced by lipid formulations based on long (C18) chain lipids as opposed to medium-chain (C8–10) and short-chain (C4) lipids [152], [175], [233], [242]. Fatty acids with chain lengths of 14 and above are primarily transported into intestinal lymph and shorter-chain fatty acids are primarily transported into portal blood [175].
4.4.2. Formulated-lipid digestibility
The process of digestion, especially the rate of digestion relative to transit time of the formulated drug at absorption site, can increase or decrease the capacity of formulation-derived lipid to solubilize lipophilic drug. In understanding how digesting formulation-derived lipids can maintain drug in solution in intestinal fluids, one needs to understand the interactions between the drug, digesting formulated-lipid, and the end product of formulated-lipid digestion: the lipid-rich, bile salt-phospholipid mixed micelle. Lipolytic products of lipids in lipid-based formulations are generated by being exposed to intestinal fluids containing lipases and bile salts secreted from the pancreas and the gall bladder, respectively. During lipolysis, multi-lamellar, liquid crystalline intermediate phases, having greater polarity than initial formulated-lipid, build up on the surface of degrading lipid droplets [245]. Despite differences in polarity, a hydrophobic continuum exists going from the surface of dispersed and degrading lipid droplets to the interior that contains the intermediate product phases [245]. That continuum allows lipophilic drug to remain solubilized by allowing drug migration in the dispersed and degrading lipid droplets. The liquid crystalline phases are continuously being broken down by the action of bile salt micelles, leading to the formation of the end product of formulated-lipid digestion, the lipid-rich, bile salt-phospholipid mixed micelles. In these mixed micelles, the drug solubility is proportional to the bile salt concentration and there is an increase in lipophilic drug solubility with increase in log P of drugs and a minimum of log P of ≥ 4 to remain in solution [245]. Consequently, lymphatically-transported lipophilic drug, which has already been defined as having a logP > 5, is expected to remain in solution whether in the administered and digesting formulation-derived lipid droplets or in the end product, mixed bile salt-phospholipid micelles, which provide a strong solubilizing environment for lipophilic drugs [245]. The mixed micelles provide the source of freely diffusible drug for enterocyte uptake at the intestinal wall. Solubilization of drug in mixed micellar phase of bile salts and lipolytic digestion products increases their luminal solubility by orders-of-magnitude as well as facilitating their passage through the unstirred layer thereby enhancing the quantitative aspects of absorption [231]. Hydrophilic surfactants can inhibit significantly the lipolysis of the triglyceride component. During a screening exercise, it has been identified that Cremophor RH40, a commonly used hydrophilic ethoxylated triglyceride surfactant, completely inhibited lipolyis of MCT oil for a 90 min period in vitro, when the oil and surfactant were present in equal mass [245]. The addition of a third component, a lipophilic surfactant, in some cases caused recovery of lipolyis [245].
4.4.3. Dispersed state of the vehicle
It may not be concluded that low particle size inevitable leads to better bioavailability because the performance of lipid-based delivery systems is governed by their fate in the gastrointestinal tract rather than by the particle size on initial dispersion [246]. In general, dispersed formulations are typically preferred, whether dispersible, non-dispersible or self-emulsifying [175], [233]. It is reasonable to expect that the rate at which a lipophilic drug diffuses from the dispersed oil phase into aqueous intestinal fluids is governed by its solution in mixed micelles and the rate at which these structures are formed by lipolysis [245]. Since the latter is an interfacial process, the rate of lipolysis is itself dependent on the size of the emulsion particles [245]. Therefore, if it is necessary to maximize the rate of drug partitioning into aqueous intestinal fluids, and hence the absorption rate, the formulation should be highly dispersible [245]. Accordingly, to maximize lymphatically-absorbed lipophilic drug the lipid-based formulation should be prepared as mixed micellar system, but if not possible as crude emulsion, and if still not possible, a lipid solution.
4.4.4. Administered lipid amount
Drug transport via the intestinal lymphatics is generally enhanced by an increase in lipid amount [122], [175], [240]. It was shown in the rat that following administration of cholesterol undecanoate in oleic acid resulted in a significant lag time that was seen with administering 500 μL but not 200 μL of lipid vehicle. This was likely the result of prolonged gastric emptying that resulted from the large lipid load provided at the higher dose volume [242]. However, the amount of co-administered lipid has to be limited to ensure their proper digestion in the intestinal lumen prior to enterocyte uptake [164].
It has been reported that the dose volume, and thus dose amount, of particular lipids had no significant effect on the intestinal lymphatic transport of benzo(a)pyrene (50 μmol and 500 μmol of olive oil; [1] and DDT 50 μL and 200 μL of various lipids); [247]. Although there was no effect of dose volume on cumulative lymphatic transport of dichlorodiphenyltrichloroethane (DDT), the concentration of DDT per chylomicron was proportionally higher when administered with 50 μL compared to 200 μL of lipid vehicle. This suggests that chylomicron-mediated transport had became saturated by the finite solubility of the drug in the triglyceride core of the chylomicron [247]. Like the last study, the in vivo study of lipid effects on lymphatically-transported drugs has generally been done in the rat and using widely varying and sometimes excessive lipid volumes (50 μL to 1 mL; [240]). Using the lower volume of 50 μL, the equivalent dose in human based on body weight-adjustments, is 10 mL lipid. However, lipid-based formulations given to humans typically have dose volumes of only a few milliliters.
Although the administered lipids are excessive to rats, fortunately, the effects of lipids are often relevant to human because effects in human can be seen at low lipid amounts. The influence of oral administration of three lipid-based formulations and a negative control formulation on gastric emptying and biliary secretion was evaluated in human subjects using gamma scintigraphy, ultrasonography, and duodenal aspiration [229]. It was shown that as little as 2 g of long-chain lipid lowered gastric emptying, stimulated gall bladder contraction and elevated intestinal bile salts, phospholipid and cholesterol levels. Similar changes were not observed when a similar quantity of medium-chain lipid was administered. Therefore, the quantities of long-chain lipid that might be administered in a pharmaceutical formulation could be expected to stimulate gall bladder contraction and elevated intestinal levels of bile salt and phospholipid [229].
4.5. Lipidic prodrugs
4.5.1. Rationale
Lipidic prodrugs are comprised of drugs covalently bound to lipids such as a fatty acid, monoglyceride, diglyceride, or phosphoglyceride. The rationale to use lipidic prodrugs to increase lymphatic transport has been previously reviewed [232], [241], [248], [249]. In an attempt to ensure increased log P and triglyceride solubility of the parent drug into glyceride-rich chylomicrons, albeit in covalent-association as a prodrug, chemists have synthesized prodrugs that increase the lipophilicity of the prodrug compared to parent drug using simple esters with long-chain fatty acids. However, this approach has had limited success due to ubiquitous esterases and some peptidases that are present in most organs of the body, but are particularly active in the intestinal tract, intestinal cells, and the liver, and are capable of cleaving ester prodrugs [248].
Consequently, there is high probability that an ester prodrug would lose its enzyme-labile chemical functionality well in advance other transiting from intestinal fluids to associating with enterocyte-derived chylomicrons or still further in transit to the lymphatics. By having failed to remain intact, the net effect is no better than had the parent drug been administered in the first place provided the parent drug is metabolically stable and not a substrate to an efflux transporter. Nevertheless, ester/ether linkages have been used to increase lipophilicity of testosterone, fat soluble vitamins A and C, and non-steroidal anti-inflammatory drugs (NSAIDS) [1], [135], [241], [248], [250], [251], [252].
4.5.2. Triglyceride digestion/re-acylation pathway
In an effort to get a prodrug to participate in the lipid digestion/biochemistry pathway, three different lipid carriers are generally used: fatty acids, glycerides and phospholipids (Fig. 5 ) [228], [253], [254]. Since triglycerides themselves are not absorbed intact, pancreatic lipase driven drug delivery by metabolism at the 1- and 3-positions initially produces the symmetric 2-monoglyceride and two fatty acids. When fatty acid substituents are placed at the 1- and 3-positions and drug placed at the 2-position, it is then possible to generate a drug-attached pseudo-glyceride. Lipases rapidly hydrolyze esters at positions-1 and -3 of the glycerol backbone, but only very slow at position-2. Therefore, most lipidic prodrugs have the drug attached at position-2 by either an ester bond if the drug bears a carboxylic group or by a spacer if other functions are present [249]. After being released by lipases from the glycerol backbone, monoglycerides and fatty acids enter enterocytes. Only the long-chain fatty acid (C> 12) are re-esterified into triglycerides by the addition of the activated fatty acids in the 1 and 3 positions [241], [249].
Fig. 5.
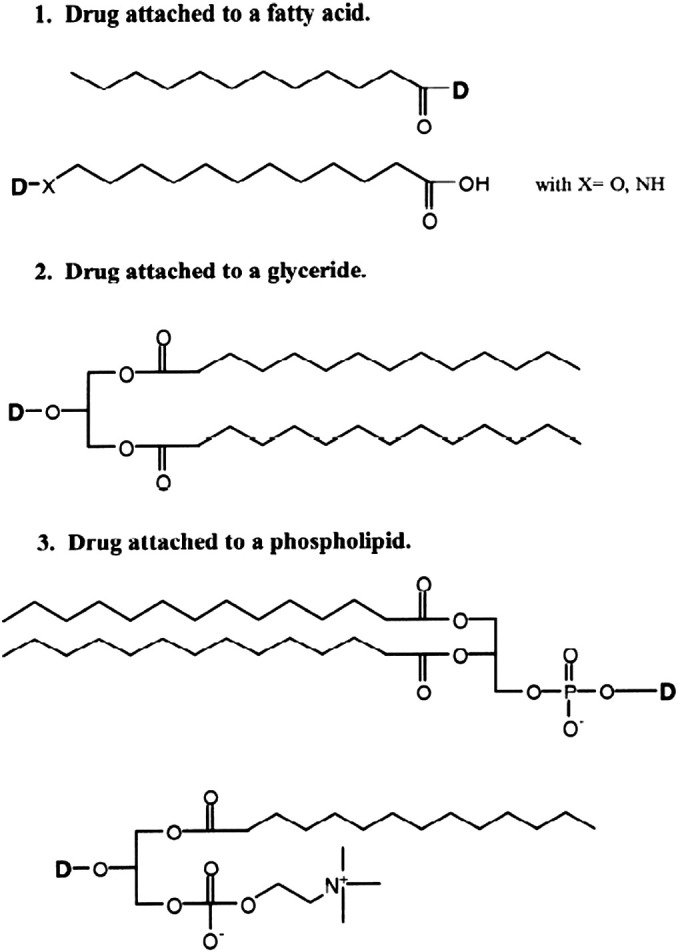
Types of lipidic carriers: fatty acids, glycerides and phospholipids. In the case of fatty acids, the drug is attached directly to the carboxylate or to a modified ω-atom. Drug-glycerides conjugates are represented here by a 1,3-diglyceride where the drug is in position-2. Phospholipid prodrugs consist either in drugs linked to the phosphate group or to the glycerol backbone, in this case, the drug replaces a fatty acid.
Reproduced from Lambert DM [249] with permission.
As examples, the monoglyceride prodrugs of the NSAIDS naproxen and acetaminophen were synthesized to both reduce adverse side effects that include stomach ulceration, bleeding and perforation, and to increase oral bioavailability [255], [256]. The oral administration of a 1-monoglyceride ester of naproxen did not result in increased recovery of total prodrug and drug recovered in the lymph relative to administration of just the parent drug. However, the intestinal lymphatic transport rate of naproxen was increased by approximately 10-fold (2.7 to 28.3% dose) when dosed as the n-alkyl chain introduced between the 1- or 2-position of glycerol backbone and drug. Acetaminophen, was similarly increased (0.8 to 13.8% dose [255]). Unexpectedly, the intestinal lymphatic transport of triglyceride derivatives with the drugs combined directly with the 2-position of glycerol were low in comparison with the mono-glyceride prodrugs. Although the authors did not give a reason for the differences, it is possible that the selection of palmitic acid, which is a saturated fatty acid, is a less favorable for increasing lymphatic transport of triglycerides than a poly-unsaturated fatty acid.
Khan et al. have taken a diglyceride prodrug approach in a set of prodrugs of diclofenac, ibuprofen, and mefenamic acid [250], [251], [252]. Prodrugs were synthesized having dipalmitate and stearate glyceride derivative at the 1- and 3-positions on the glycerol backbone, with parent drug at the 2-position of the backbone. All of the prodrugs were resistant or showed less hydrolysis under pH conditions that might be considered relevant to gastric conditions (pH ≤ 5). However, facile release of the parent drug was observed under pH conditions that might be considered relevant to intestinal conditions (pH 7.4). Peak plasma concentrations for parent drug were higher and delayed in prodrug-dosed than parent drug-dosed rats. All prodrugs showed less frequent and severe gastric ulcers compared to parent drug. The prodrugs showed better anti-inflammatory activity compared to the parent drug and analgesic activity comparable to the parent drug. Taken together, these studies show that prodrugs are a way to both increase bioavailability and decrease the gastrointestinal side effects of NSAIDS.
4.5.3. Phospholipid digestion/re-acylation product pathway
The concept of phospholipid prodrugs to help circumvent pre-systemic clearance follows from the biochemistry of normal phospholipids in the body. Phospholipids are hydrolyzed in the small intestine to lyso-phospholipid, which after absorption into the enterocyte can enter a re-acylation pathway similar to the re-acylation pathway of glycerides. Rather than be incorporated into the glyceride-rich chylomicrons, the reacylated phospholipid is associated with and stabilizes at the surface of the chylomicron. There have been a few reported examples where phospholipid prodrug and the released parent drug have been directly shown to be lymphatically-transported.
In an early example of the phospholipid prodrug approach, Sakai et al. synthesized dipalmitoyl phospholipid prodrug of the antitumor agent fluorouridine [241], [254]. A prodrug approach was needed because fluorouridine, as a small polar drug, would not be expected to be lymphatically-transported to both expose cancer cell-rich lymph nodes and minimize systemic exposure and resulting toxicity. The prodrug, dipalmitoylphosphatidylfluorouridine (DPPF), was orally dosed to rats and the extent that DPPF it and its two metabolites, fluorouridine (FUR) and fluorourcacil (5-FU), were lymphatically-transported was assessed. Lymph and blood concentrations of DPPF, FUR, and 5-FU were all low, but DPPF lymph concentrations were 30-fold higher than in blood, demonstrating the prodrug lymphotropism of DPPF. In contrast to the low blood and lymph concentrations of DPPF, high levels of DPPF congeners, different in only with respect to the position 2-acyl groups were measured, but were higher in the lymph than blood. The congeners were shown to be 1-palmitoyl-2-arachidonyl-phosphatidylfluorouridine (PAPF) and 1-palmitoyl-2-lineoyl-phosphatidyl-fluorouridine (PLPF). Because the only difference between DPPF and the congeners were the 2-acyl groups, the data suggests that DPPF had been digested and re-acylated prior to being incorporated onto chylomicrons and secreted in the lymph. Given sufficient time, the authors expected the congeners to be further hydrolyzed in the lymphatics resulting in release of phosphate-derivatized drug, which is the active form [254].
Dahan et al. have recently studied the use of a phosphatidylcholine esteric prodrug of the antiepileptic drug valproic acid [227]. As the prodrug (DP-VPA), valproic acid was directly conjugated to the phospholipid at the sn-2 position and was thus susceptible to enzymatic cleavage by phospholipase A2 (PLA2). However, administration of the prodrug to mice either homozygously-defective for PLA2 gene (C57BL/6) or having the normal gene (BALB/c) [257] showed no differences in DP-VPA plasma exposure. The stability was further confirmed by the absence of any in vitro DP-VPA degradation in serum and low (4.8%) degradation by bee venom PLA2. When DP-VPA was administered to fasted rats dissolved in a long-chain triglyceride (LCT; C12) versus a medium-chain triglyceride (MCT; C6–10) the plasma exposure was increased 3-fold and time to achieve peak concentrations in plasma was 2-fold delayed. The authors suggested the significant food effect could have resulted from improved dissolution and solubilization of the drug in intestinal fluids, as well as intestinal lymphatic transport. The combined observations of a food effect and increased absorption following administration of LCT, known to simulate chylomicron production, support the notion of intestinal lymphatic transport. When the lymphatic transport was evaluated in mesenteric lymph duct cannulated freely moving rats it was shown that 60% of the absorbed DP-VPA was associated with lymphatic transport. These results confirm the lymphatic transport of DP-VPA at least in dogs [258].
4.6. Excipient effects on metabolism and transporters
In the developing lipid-based formulations, ideally the excipient would enhance drug solubility and stability, but would be pharmacologically inert. However, increasing evidence suggests that excipients can have effects on drug metabolism and interactions with cellular transporters. Research has shown the inhibitory abilities of surfactants on the regulation, expression, and functional capacity of various CYP isoforms. More specifically, polyoxyethylated/pegylated surfactants such as Cremophor [259], [260], [261], PEG-15-hydroxystearate [260], polysorbates (e.g., Tween 80, Tween 20) [262] as well as polymers [263] have all been identified as inhibitors of CYP3A isozymes. Inhibition, in this case, is apparent in expression (molecular level) and/or intrinsic metabolic activity (functional capacity). Accordingly, surfactants should not necessarily be considered inert in that they may significantly alter drug metabolism.
Excipients used in lipid-based formulations have been found to interrupt/modulate efflux transporter protein activities. While CYPs can be inhibited/modulated via certain lipid-based excipients, these and other excipients can simultaneously affect both CYP isoform (typically CYP3As) as well as efflux transporters (i.e., P-gp). Excipients have been found to modulate efflux transporters such as P-gp at all levels including enzyme/protein alteration (e.g., disruption in drug binding) regulation/expression (i.e., amount of protein expressed/present) and functionality/functional capacity (i.e., intrinsic activity rate).
An example of a lipid excipient that modulates with specific effects on the expression of efflux transporters (i.e., P-gp) is seen with Peceol® and Gelucrie® [264]. Using Caco-2 cell monolayers, it was demonstrated that a significant decrease in P-gp efflux activity likely due to a decrease in expression. However, it is not clear that the observed decrease in P-gp efflux can be solely attributed to a decrease in expression. In another example, a non-ionic detergents (e.g., Triton X-100) have been shown to prevent drug binding and thus modulate the functional capacity of P-gp [265]. However, while this is true for the mechanism of modulation, expression is also impacted due to the sequential down regulation of P-gp. Another example of the differences in modulation caused by different excipients can be seen in fatty acid ester surfactants (e.g., Cremophor EL) [265]. For this excipient, P-gp activity/expression was modulated due to an alteration in drug binding likely caused by mutations in transmembrane domains (which are critical for proper docking of the substrate for efflux). In addition to this, polymers such as Pluronic P85 block copolymer have also been shown to inhibit P-gp [266]. However, an additional aspect of modulation was also observed in Caco-2 monolayers experiments as permeability (apical to basolateral) was increased.
As mentioned earlier, efflux transporter proteins such as P-gp and MRP2 share many similarities with respect to function (broad substrate overlaps), localization (at the apical side of enterocytes) and tissue distribution (expression patterns) [267]. Therefore, it is not surprising that lipid excipients can also have an effect on MRP2 transporters. Jia et al. recently demonstrated that 1-monoolein, 1-monopalmitin or 1-monostearin treated caco-2 cells inhibited efflux by MRP2 [268]. Furthermore, treatment using 1-monoolein was observed to cause a non-specific down-regulation of MRP2 expression [268].
4.6.1. Excipient effect on coupling
As was previously mentioned, enzymes (i.e., CYP3As) and efflux transporters (i.e., P-gp) are intricately related (i.e., coupling). Therefore, it is very plausible that when we effect/modulate CYP3As, for example, that we will be able to directly and/or indirectly measure a decrease in efflux. In several published studies, lipid excipients found in SEDDS/SMEDDS have been able to inhibit both metabolism and efflux [269], [270]. A recent study in which an aspect of coupling (e.g., presystemic metabolism) are investigated can be found in which compounds such as halofantrine are co-administered with lipids, lipid excipients and/or metabolism inhibitors (e.g., ketoconazole for CYP3A inhibition) [271]. However, it is worthwhile to note by inhibiting metabolism, it is plausible that an increase in functional capacity (i.e., increase in intrinsic rate of activity) followed with an increase in expression can compensate for the elimination of the compound in question. Therefore, inhibition of metabolism only might not translate into an observable (i.e., in vivo effect) effect. It is critical to design in vitro studies (or any other studies) that aim to investigate coupling to target many components of the coupling machinery.
5. Pharmacokinetics
Lymphatic drug transport has been reported to be a contributor in the oral bioavailability in various lipophilic drugs and xenobiotics after oral administration such as: dexabinol [272], moxidectin [123], halofantrine [94], [122], mepitiostane [243], [273], testosterone derivatives [91], penclomedine [178], naftifine [274], probucol [275], cyclosporine [276], ontazolast [277], retinoids and fat soluble vitamins [278], lycopene, DDT and analogs [247], [279], benzopyrene, polychlorinated biphenyls (PCBs) [280], and various lipophilic drugs [232], [248], [258]. Also a very small number of hydrophilic drugs such as salicylic acid, isozianid and caffeine are recovered in small quantities in lymph after oral administration [279].
For instance, in the case of DDT [281], halofantrine [152] and the lipophilic lipid regulator CI-976 [179] there was a discrepancy between the oral bioavailability of cannulated rats (sum of lymph concentrations and systemic blood concentrations) and non-cannulated rats (systemic blood concentration). Cannulated rats exhibited higher bioavailability, indicating a lack of mass balance in total absorption. This discrepancy could be the result of changes in clearance due to either drug accumulation into the adipose tissue [179], pre-systemic clearance of the drug in the lymph or lymphoid tissues, or the clearance of chylomicron-bound drug being increased upon entry into the systemic circulation [152]. However, the exact mechanism by which these changes in oral bioavailability occurred still needs to be elucidated.
In the case of human growth hormone following subcutaneous administration in sheep, a discrepancy was observed between the fraction of administered dose collected from the peripheral lymph (61.7%) via the efferent duct of the popliteal lymph node, and the 8.6% collected from the central lymph collected via the thoracic duct [144]. This discrepancy in cumulative lymphatic transport could not be explained by enzymatic degradation of human growth hormone between the efferent popliteal node and the thoracic lymph duct [233]. Thus, the exact mechanism is to still to be elucidated.
It has to be acknowledged that for a drug with very low bioavailability, the occurrence of lymphatic transport even to the extent of less than 1% of the administered dose can significantly affect the cumulative plasma exposure [179]. Furthermore, it has been recommended that if a drug is to be pharmacologically targeted to the lymphatic system, the degree of lymphatic transport should be expressed as concentration of drug in lymph rather than using the percentage of the administered dose, especially when there are differences in oral bioavailability between cannulated and non-cannulated animals [94], [282]. Table 3 summarizes the drugs that have been reported to present different degrees of intestinal lymphatic transport.
Table 3.
Summary of drugs that have been reported to present different degrees of intestinal lymphatic transport.
| Compound | Log P | Formulation and feeding status | Species | Percentage (%) of absorbed dose in lymph | Reference |
|---|---|---|---|---|---|
| Δ4-androstan-17-β-methoxycyclopentyl ether (Δ4-AE) | 6.6 | Long chain triglyceride | Rat | 96.77 | Ichihashi et al. [177] |
| 2α,3α-epithio-5α-androstan-17-one (KEP) | 3 | Long chain triglyceride | Rat | 7.98 | Ichihashi et al. [177] |
| 5α-androst-2-en-17-one (KO) | 4.6 | Long chain triglyceride | Rat | 6.49 | Ichihashi et al. [177] |
| CI-976 | 5.8 | Long chain triglyceride | Rat | 43 | Hauss et al. [179] |
| CI-976 | 5.83 | Triglyceride emulsion | Rat | < 1 | Hauss et al. [179] |
| Cyclosporin A | 2.99 | Micellar solution | Rat | ~ 2 | Takada et al. [283] |
| Cyclosporin A | 2.99 | Long chain triglyceride | Rat | 2.16 | Ueda et al. [276] |
| Dehydrotestosterone-17β-methoxycyclopentyl ether (DHTE) | 5.4 | Long chain triglyceride | Rat | 28.35 | Ichihashi et al. [177] |
| Dichlorodiphenyltrichloroethane (DDT) | 6.19 | Triglyceride emulsion | Rat | 15 | Myers and Stella [178] |
| Epitiostanol | 4.4 | Long chain triglyceride | Rat | 4.53 | Ichihashi et al. [284] |
| Etretinate | 7.8 | Long chain triglyceride | Rat | 25.71 | Nankervis et al. [278] |
| Halofantrine | 8.5 | Emulsion | Rat | 12 | Porter et al. [133] |
| Halofantrine | 8.5 | Micellar | Rat | 18 | Porter et al. [133] |
| Halofantrine | 8.5 | Triglyceride lipid | Rat | 17 | Porter et al. [132] |
| Halofantrine | 8.5 | Micellar | Rat | 20 | Porter et al. [132] |
| Halofantrine | 8.5 | Long chain triglyceride | Rat | 16 | Caliph et al. [152] |
| Halofantrine | 8.5 | Fasted | Dog | 1.3 | Khoo et al. [282] |
| Halofantrine | 8.5 | Fed | Dog | 54 | Khoo et al. [94] |
| Halofantrine (hydrochloride salt) | 8.5 | Fatty acid/monoglycerol solution | Rat | ~ 5 | Porter et al. [132] |
| Halofantrine (hydrochloride salt) | 8.5 | Fed | Dog | 44 | Khoo et al. [282] |
| Hexachlorobenzene (HCB) | 6.53 | Triglyceride emulsion | Rat | ~ 3 | Myers and Stella [178] |
| Isotretinoin | 6.8 | Long chain triglyceride | Rat | 1.99 | Nankervis et al. [278] |
| Lu28-179 | 8 | Triglyceride emulsion | Rat | 4.5 | Neilson et al. [285] |
| Mepitiostane (prodrug of Epitiostanol) | 6.06 | Triglyceride | Rat | 41 | Ichihashi et al. [243] |
| Mepitiostane (prodrug of Epitiostanol) | 6 | Long chain triglyceride | Rat | 92.59 | Ichihashi et al. [284] |
| Mepitiostaneolefin | 5.1 | Long chain triglyceride | Rat | 97.67 | Ichihashi et al. [177] |
| MK-386 | 8.6 | Triglyceride | Rat | 0.1 | Kwei et al. [286] |
| MK-386 | 8 | Long chain triglyceride | Rat | 1.67 | Kwei et al. [286] |
| Ontazolast | 4 | Triglyceride emulsion | Rat | 1.25 | Hauss et al. [277] |
| Ontazolast | 4 | SEEDS | Rat | 0.7 | Hauss et al. [277] |
| Ontazolast | 4 | Long chain triglyceride | Rat | 69.6 | Hauss et al. [179] |
| p,p-DDT | 6.2 | Long chain triglyceride | Rat | 51.6 | O'Driscoll et al. [281] |
| Penclomedine | 5.48 | Triglyceride emulsion | Rat | ~ 3 | Myers and Stella [178] |
| Progesterone | 3.9 | Long chain triglyceride | Rat | 0.82 | Ichihashi et al. [177] |
| Temarotene | 8.7 | Long chain triglyceride | Rat | 45.67 | Nankervis et al. [278] |
| Testosterone | 3.3 | Long chain triglyceride | Rat | 0.11 | Ichihashi et al. [177] |
| Testosterone-17-β-methoxycyclopentyl ether | 6.2 | Long chain triglyceride | Rat | 12.48 | Ichihashi et al. [177] |
| Vitamin D3 | 7.9 | Triglyceride emulsion | Rat | 19.2 | Liu et al. [287] |
6. Concluding remarks and future perspectives
The increasing development of highly lipophilic drugs coupled with the increasing interest in understanding the mechanisms by which drugs access the lymph is cause for a newly found interest in intestinal lymphatic drug transport. It is been recognized as a suitable alternative to enhance bioavailability and systemic exposure of high liver first-pass metabolized drugs as it initially gains access the systemic circulation without passing through the liver. Multiple formulation approaches are been developed to enhance the lymphatic transport of drugs, but also to have a better understanding at the cellular level of the digestion, uptake, intracellular metabolism, and packaging of food-derived lipids and drugs into chylomicrons, their intestinal lymphatic drug transport process and enterocyte metabolism, and how they are impacted by the co-administration of various formulation-derived lipids and excipients. Further studies are warranted to address these issues but also to identify novel applications for this process.
Footnotes
This review is part of the Advanced Drug Delivery Reviews theme issue on “Lymphatic Drug Delivery: Therapy, Imaging and Nanotechnology”.
References
- 1.Charman W.N., Stella V.J. Estimating the maximum potential for intestinal lymphatic transport of lipophillic drug molecules. Int. J. Pharm. 1986;34:175–178. [Google Scholar]
- 2.Trevaskis N.L., Charman W.N., Porter C.J. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv. Drug Deliv. Rev. 2008;60:702–716. doi: 10.1016/j.addr.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nordskog B.K., Phan C.T., Nutting D.F., Tso P. An examination of the factors affecting intestinal lymphatic transport of dietary lipids. Adv. Drug Deliv. Rev. 2001;50:21–44. doi: 10.1016/s0169-409x(01)00147-8. [DOI] [PubMed] [Google Scholar]
- 4.Cense H.A., van Eijck C.H., Tilanus H.W. New insights in the lymphatic spread of oesophageal cancer and its implications for the extent of surgical resection. Best Pract. Res. Clin. Gastroenterol. 2006;20:893–906. doi: 10.1016/j.bpg.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Arya M., Bott S.R., Shergill I.S., Ahmed H.U., Williamson M., Patel H.R. The metastatic cascade in prostate cancer. Surg. Oncol. 2006;15:117–128. doi: 10.1016/j.suronc.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Muranishi S. Lymphatic delivery of drugs and its application to cancer chemotherapy (author's transl) Yakugaku Zasshi. 1980;100:687–698. [PubMed] [Google Scholar]
- 7.Garzon-Aburbeh A., Poupaert J.H., Claesen M., Dumont P., Atassi G. 1,3-dipalmitoylglycerol ester of chlorambucil as a lymphotropic, orally administrable antineoplastic agent. J. Med. Chem. 1983;26:1200–1203. doi: 10.1021/jm00362a021. [DOI] [PubMed] [Google Scholar]
- 8.Pantaleo G., Graziosi C., Demarest J.F., Cohen O.J., Vaccarezza M., Gantt K., Muro-Cacho C., Fauci A.S. Role of lymphoid organs in the pathogenesis of human immunodeficiency virus (HIV) infection. Immunol. Rev. 1994;140:105–130. doi: 10.1111/j.1600-065x.1994.tb00867.x. [DOI] [PubMed] [Google Scholar]
- 9.Pantaleo G., Graziosi C., Fauci A.S. The role of lymphoid organs in the immunopathogenesis of HIV infection. AIDS. 1993;7(Suppl. 1):S19–S23. [PubMed] [Google Scholar]
- 10.Umeda M., Marusawa H., Seno H., Katsurada A., Nabeshima M., Egawa H., Uemoto S., Inomata Y., Tanaka K., Chiba T. Hepatitis B virus infection in lymphatic tissues in inactive hepatitis B carriers. J. Hepatol. 2005;42:806–812. doi: 10.1016/j.jhep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 11.von Messling V., Svitek N., Cattaneo R. Receptor (SLAM [CD150]) recognition and the V protein sustain swift lymphocyte-based invasion of mucosal tissue and lymphatic organs by a morbillivirus. J. Virol. 2006;80:6084–6092. doi: 10.1128/JVI.00357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lan N.T., Yamaguchi R., Inomata A., Furuya Y., Uchida K., Sugano S., Tateyama S. Comparative analyses of canine distemper viral isolates from clinical cases of canine distemper in vaccinated dogs. Vet. Microbiol. 2006;115:32–42. doi: 10.1016/j.vetmic.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 13.Spiegel M., Schneider K., Weber F., Weidmann M., Hufert F.T. Interaction of severe acute respiratory syndrome-associated coronavirus with dendritic cells. J. Gen. Virol. 2006;87:1953–1960. doi: 10.1099/vir.0.81624-0. [DOI] [PubMed] [Google Scholar]
- 14.Kessel A., Toubi E. Chronic HCV-related autoimmunity: a consequence of viral persistence and lymphotropism. Curr. Med. Chem. 2007;14:547–554. doi: 10.2174/092986707780059652. [DOI] [PubMed] [Google Scholar]
- 15.Lalanne M., Paci A., Andrieux K., Dereuddre-Bosquet N., Clayette P., Deroussent A., Re M., Vassal G., Couvreur P., Desmaele D. Synthesis and biological evaluation of two glycerolipidic prodrugs of didanosine for direct lymphatic delivery against HIV. Bioorg. Med. Chem. Lett. 2007;17:2237–2240. doi: 10.1016/j.bmcl.2007.01.062. [DOI] [PubMed] [Google Scholar]
- 16.Tso P. Intestinal lymphatic transport of lipids. Adv. Drug Deliv. Rev. 2001;50:21–44. doi: 10.1016/s0169-409x(01)00147-8. [DOI] [PubMed] [Google Scholar]
- 17.Phan C.T., Tso P. Intestinal lipid absorption and transport. Front. Biosci. 2001;6:D299–D319. doi: 10.2741/phan. [DOI] [PubMed] [Google Scholar]
- 18.Tso P. Intestinal lipid absorption. In: Johnson L.R., editor. Physiology of the Gastrointestinal Tract. Raven Press; New York: 1994. pp. 1867–1907. [Google Scholar]
- 19.Thomson A.B., Keelan M., Garg M.L., Clandinin M.T. Intestinal aspects of lipid absorption: in review. Can. J. Physiol. Pharmacol. 1989;67:179–191. doi: 10.1139/y89-031. [DOI] [PubMed] [Google Scholar]
- 20.Davidson N.O. Cellular and molecular mechanisms of small intestinal lipid transport. In: Johnson L.R., editor. Physiology of the Gastrointestinal Tract. Raven Press; New York: 1994. pp. 1909–1934. [Google Scholar]
- 21.Carey M.C., Small D.M., Bliss C.M. Lipid digestion and absorption. Annu. Rev. Physiol. 1983;45:651–677. doi: 10.1146/annurev.ph.45.030183.003251. [DOI] [PubMed] [Google Scholar]
- 22.Nutting D.F., Kumar N.S., St Hilaire R.J., Mansbach C.M., II Nutrient absorption. Curr. Opin. Clin. Nutr. Metab. Care. 1999;2:413–419. doi: 10.1097/00075197-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Abrams C.K., Hamosh M., Lee T.C., Ansher A.F., Collen M.J., Lewis J.H., Benjamin S.B., Hamosh P. Gastric lipase: localization in the human stomach. Gastroenterology. 1988;95:1460–1464. doi: 10.1016/s0016-5085(88)80063-5. [DOI] [PubMed] [Google Scholar]
- 24.Hamosh M., Scanlon J.W., Ganot D., Likel M., Scanlon K.B., Hamosh P. Fat digestion in the newborn. Characterization of lipase in gastric aspirates of premature and term infants. J. Clin. Invest. 1981;67:838–846. doi: 10.1172/JCI110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen M., Morgan R.G., Hofmann A.F. Lipolytic activity of human gastric and duodenal juice against medium and long chain triglycerides. Gastroenterology. 1971;60:1–15. [PubMed] [Google Scholar]
- 26.Mattson F.H., Volpenhein R.A. Hydrolysis of primary and secondary esters of glycerol by pancreatic juice. J. Lipid Res. 1968;9:79–84. [PubMed] [Google Scholar]
- 27.Shiau Y.F. Mechanism of intestinal fat absorption. Am. J. Physiol. 1981;240:G1–G9. doi: 10.1152/ajpgi.1981.240.1.G1. [DOI] [PubMed] [Google Scholar]
- 28.Mattson F.H., Beck L.W. The specificity of pancreatic lipase for the primary hydroxyl groups of glycerides. J. Biol. Chem. 1956;219:735–740. [PubMed] [Google Scholar]
- 29.Mattson F.H., Volpenhein R.A. The digestion and absorption of triglycerides. J. Biol. Chem. 1964;239:2772–2777. [PubMed] [Google Scholar]
- 30.Hofmann A.F., Borgstrom B. Hydrolysis of long-chain monoglycerides in micellar solution by pancreatic lipase. Biochim. Biophys. Acta. 1963;70:317–331. doi: 10.1016/0006-3002(63)90755-8. [DOI] [PubMed] [Google Scholar]
- 31.Borgstrom B., Dahlqvist A., Lundh G., Sjovall J. Studies of intestinal digestion and absorption in the human. J. Clin. Invest. 1957;36:1521–1536. doi: 10.1172/JCI103549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van den Bosch H., Postema N.M., de Haas G.H., van Deenen L.L. On the positional specificity of phospholipase A from pancreas. Biochim. Biophys. Acta. 1965;98:657–659. doi: 10.1016/0005-2760(65)90168-2. [DOI] [PubMed] [Google Scholar]
- 33.McIntyre N. Cholesterol absoprtion. In: Rommel K., Goebell H., Bhomer R., editors. Lipid Absorption: Biochemical and Clinical Aspects. MTP Press Ltd.; London: 1976. pp. 73–84. [Google Scholar]
- 34.Erlanson C. Purification, properties, and substrate specificity of a carboxylesterase in pancreatic juice. Scand. J. Gastroenterol. 1975;10:401–408. [PubMed] [Google Scholar]
- 35.Staggers J.E., Hernell O., Stafford R.J., Carey M.C. Physical-chemical behavior of dietary and biliary lipids during intestinal digestion and absorption. 1. Phase behavior and aggregation states of model lipid systems patterned after aqueous duodenal contents of healthy adult human beings. Biochemistry. 1990;29:2028–2040. doi: 10.1021/bi00460a011. [DOI] [PubMed] [Google Scholar]
- 36.Hernell O., Staggers J.E., Carey M.C. Physical–chemical behavior of dietary and biliary lipids during intestinal digestion and absorption. 2. Phase analysis and aggregation states of luminal lipids during duodenal fat digestion in healthy adult human beings. Biochemistry. 1990;29:2041–2056. doi: 10.1021/bi00460a012. [DOI] [PubMed] [Google Scholar]
- 37.Shiau Y.F. Mechanism of intestinal fatty acid uptake in the rat: the role of an acidic microclimate. J. Physiol. 1990;421:463–474. doi: 10.1113/jphysiol.1990.sp017955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffman N.E. The relationship between uptake in vitro of oleic acid and micellar solubilization. Biochim. Biophys. Acta. 1970;196:193–203. doi: 10.1016/0005-2736(70)90006-4. [DOI] [PubMed] [Google Scholar]
- 39.Simmonds W.J. The role of micellar solubilization in lipid absorption. Aust. J. Exp. Biol. Med. Sci. 1972;50:403–421. doi: 10.1038/icb.1972.35. [DOI] [PubMed] [Google Scholar]
- 40.Chijiiwa K., Linscheer W.G. Effect of intraluminal pH on cholesterol and oleic acid absorption from micellar solutions in the rat. Am. J. Physiol. 1984;246:G492–G499. doi: 10.1152/ajpgi.1984.246.5.G492. [DOI] [PubMed] [Google Scholar]
- 41.Shiau Y.F., Kelemen R.J., Reed M.A. Acidic mucin layer facilitates micelle dissociation and fatty acid diffusion. Am. J. Physiol. 1990;259:G671–G675. doi: 10.1152/ajpgi.1990.259.4.G671. [DOI] [PubMed] [Google Scholar]
- 42.Chijiiwa K., Linscheer W.G. Mechanism of pH effect on oleic acid and cholesterol absorption in the rat. Am. J. Physiol. 1987;252:G506–G510. doi: 10.1152/ajpgi.1987.252.4.G506. [DOI] [PubMed] [Google Scholar]
- 43.Thomson A.B., Schoeller C., Keelan M., Smith L., Clandinin M.T. Lipid absorption: passing through the unstirred layers, brush-border membrane, and beyond. Can. J. Physiol. Pharmacol. 1993;71:531–555. doi: 10.1139/y93-078. [DOI] [PubMed] [Google Scholar]
- 44.Schoeller C., Keelan M., Mulvey G., Stremmel W., Thomson A.B. Role of a brush border membrane fatty acid binding protein in oleic acid uptake into rat and rabbit jejunal brush border membrane. Clin. Invest. Med. 1995;18:380–388. [PubMed] [Google Scholar]
- 45.Schoeller C., Keelan M., Mulvey G., Stremmel W., Thomson A.B. Oleic acid uptake into rat and rabbit jejunal brush border membrane. Biochim. Biophys. Acta. 1995;1236:51–64. doi: 10.1016/0005-2736(95)00035-2. [DOI] [PubMed] [Google Scholar]
- 46.Stremmel W., Lotz G., Strohmeyer G., Berk P.D. Identification, isolation, and partial characterization of a fatty acid binding protein from rat jejunal microvillous membranes. J. Clin. Invest. 1985;75:1068–1076. doi: 10.1172/JCI111769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stremmel W. Uptake of fatty acids by jejunal mucosal cells is mediated by a fatty acid binding membrane protein. J. Clin. Invest. 1988;82:2001–2010. doi: 10.1172/JCI113820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poirier H., Degrace P., Niot I., Bernard A., Besnard P. Localization and regulation of the putative membrane fatty-acid transporter (FAT) in the small intestine. Comparison with fatty acid-binding proteins (FABP) Eur. J. Biochem. 1996;238:368–373. doi: 10.1111/j.1432-1033.1996.0368z.x. [DOI] [PubMed] [Google Scholar]
- 49.Bloom B., Chaikoff I.L., Reinhardt Intestinal lymph as pathway for transport of absorbed fatty acids of different chain lengths. Am. J. Physiol. 1951;166:451–455. doi: 10.1152/ajplegacy.1951.166.2.451. [DOI] [PubMed] [Google Scholar]
- 50.Chaikoff I.L., Bloom B., Stevens B.P., Reinhardt W.O., Dauben W.G. Pentadecanoic acid-5-C14; its absorption and lymphatic transport. J. Biol. Chem. 1951;190:431–435. [PubMed] [Google Scholar]
- 51.Bloom B., Chaikoff I.L., Reinhardt W.O., Dauben W.G. Participation of phospholipides in lymphatic transport of absorbed fatty acids. J. Biol. Chem. 1951;189:261–267. [PubMed] [Google Scholar]
- 52.Kiyasu J.Y., Bloom B., Chaikoff I.L. The portal transport of absorbed fatty acids. J. Biol. Chem. 1952;199:415–419. [PubMed] [Google Scholar]
- 53.Storch J., Thumser A.E. The fatty acid transport function of fatty acid-binding proteins. Biochim. Biophys. Acta. 2000;1486:28–44. doi: 10.1016/s1388-1981(00)00046-9. [DOI] [PubMed] [Google Scholar]
- 54.Luxon B.A., Milliano M.T. Cytoplasmic transport of fatty acids in rat enterocytes: role of binding to fatty acid-binding protein. Am. J. Physiol. 1999;277:G361–G366. doi: 10.1152/ajpgi.1999.277.2.G361. [DOI] [PubMed] [Google Scholar]
- 55.Ockner R.K., Kaikaus R.M., Bass N.M. Fatty acid binding proteins: recent concepts of regulation and function. Prog. Clin. Biol. Res. 1992;375:189–204. [PubMed] [Google Scholar]
- 56.Thumser A.E., Storch J. Liver and intestinal fatty acid-binding proteins obtain fatty acids from phospholipid membranes by different mechanisms. J. Lipid Res. 2000;41:647–656. [PubMed] [Google Scholar]
- 57.Alpers D.H., Bass N.M., Engle M.J., DeSchryver-Kecskemeti K. Intestinal fatty acid binding protein may favor differential apical fatty acid binding in the intestine. Biochim. Biophys. Acta. 2000;1483:352–362. doi: 10.1016/s1388-1981(99)00200-0. [DOI] [PubMed] [Google Scholar]
- 58.Levin M.S., Talkad V.D., Gordon J.I., Stenson W.F. Trafficking of exogenous fatty acids within Caco-2 cells. J. Lipid Res. 1992;33:9–19. [PubMed] [Google Scholar]
- 59.Porter C.J., Charman W.N. Intestinal lymphatic drug transport: an update. Adv. Drug Deliv. Rev. 2001;50:61–80. doi: 10.1016/s0169-409x(01)00151-x. [DOI] [PubMed] [Google Scholar]
- 60.Senior J.R., Isselbacher K.J. Direct esterification of monoglycerides with palmityl coenzyme A by intestinal epithelial subcellular fractions. J. Biol. Chem. 1962;237:1454–1459. [PubMed] [Google Scholar]
- 61.Levy E., Mehran M., Seidman E. Caco-2 cells as a model for intestinal lipoprotein synthesis and secretion. FASEB J. 1995;9:626–635. doi: 10.1096/fasebj.9.8.7768354. [DOI] [PubMed] [Google Scholar]
- 62.Ockner R.K., Hughes F.B., Isselbacher K.J. Very low density lipoproteins in intestinal lymph: origin, composition, and role in lipid transport in the fasting state. J. Clin. Invest. 1969;48:2079–2088. doi: 10.1172/JCI106174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hussain M.M. A proposed model for the assembly of chylomicrons. Atherosclerosis. 2000;148:1–15. doi: 10.1016/s0021-9150(99)00397-4. [DOI] [PubMed] [Google Scholar]
- 64.Tso P., Lindstrom M.B., Borgstrom B. Factors regulating the formation of chylomicrons and very-low-density lipoproteins by the rat small intestine. Biochim. Biophys. Acta. 1987;922:304–313. doi: 10.1016/0005-2760(87)90053-1. [DOI] [PubMed] [Google Scholar]
- 65.Ockner R.K., Hughes F.B., Isselbacher K.J. Very low density lipoproteins in intestinal lymph: role in triglyceride and cholesterol transport during fat absorption. J. Clin. Invest. 1969;48:2367–2373. doi: 10.1172/JCI106203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Greevenbroek M.M., de Bruin T.W. Chylomicron synthesis by intestinal cells in vitro and in vivo. Atherosclerosis. 1998;141(Suppl. 1):S9–S16. doi: 10.1016/s0021-9150(98)00212-3. [DOI] [PubMed] [Google Scholar]
- 67.Gordon D.A., Jamil H. Progress towards understanding the role of microsomal triglyceride transfer protein in apolipoprotein-B lipoprotein assembly. Biochim. Biophys. Acta. 2000;1486:72–83. doi: 10.1016/s1388-1981(00)00049-4. [DOI] [PubMed] [Google Scholar]
- 68.Tso P., Balint J.A., Rodgers J.B. Effect of hydrophobic surfactant (Pluronic L-81) on lymphatic lipid transport in the rat. Am. J. Physiol. 1980;239:G348–G353. doi: 10.1152/ajpgi.1980.239.5.G348. [DOI] [PubMed] [Google Scholar]
- 69.Tso P., Drake D.S., Black D.D., Sabesin S.M. Evidence for separate pathways of chylomicron and very low-density lipoprotein assembly and transport by rat small intestine. Am. J. Physiol. 1984;247:G599–G610. doi: 10.1152/ajpgi.1984.247.6.G599. [DOI] [PubMed] [Google Scholar]
- 70.Tso P., Balint J.A., Bishop M.B., Rodgers J.B. Acute inhibition of intestinal lipid transport by Pluronic L-81 in the rat. Am. J. Physiol. 1981;241:G487–G497. doi: 10.1152/ajpgi.1981.241.6.G487. [DOI] [PubMed] [Google Scholar]
- 71.Tso P., Balint J.A. Formation and transport of chylomicrons by enterocytes to the lymphatics. Am. J. Physiol. 1986;250:G715–G726. doi: 10.1152/ajpgi.1986.250.6.G715. [DOI] [PubMed] [Google Scholar]
- 72.Mahley R.W., Bennett B.D., Morre D.J., Gray M.E., Thistlethwaite W., LeQuire V.S. Lipoproteins associated with the Golgi apparatus isolated from epithelial cells of rat small intestine. Lab. Investig. 1971;25:435–444. [PubMed] [Google Scholar]
- 73.Vahouny G.V., Blendermann E.M., Gallo L.L., Treadwell C.R. Differential transport of cholesterol and oleic acid in lymph lipoproteins: sex differences in puromycin sensitivity. J. Lipid Res. 1980;21:415–424. [PubMed] [Google Scholar]
- 74.Tso P., Kendrick H., Balint J.A., Simmonds W.J. Role of biliary phosphatidylcholine in the absorption and transport of dietary triolein in the rat. Gastroenterology. 1981;80:60–65. [PubMed] [Google Scholar]
- 75.Nutting D., Hall J., Barrowman J.A., Tso P. Further studies on the mechanism of inhibition of intestinal chylomicron transport by Pluronic L-81. Biochim. Biophys. Acta. 1989;1004:357–362. doi: 10.1016/0005-2760(89)90084-2. [DOI] [PubMed] [Google Scholar]
- 76.Luchoomun J., Hussain M.M. Assembly and secretion of chylomicrons by differentiated Caco-2 cells. Nascent triglycerides and preformed phospholipids are preferentially used for lipoprotein assembly. J. Biol. Chem. 1999;274:19565–19572. doi: 10.1074/jbc.274.28.19565. [DOI] [PubMed] [Google Scholar]
- 77.Graham D.L., Knott T.J., Jones T.C., Pease R.J., Pullinger C.R., Scott J. Carboxyl-terminal truncation of apolipoprotein B results in gradual loss of the ability to form buoyant lipoproteins in cultured human and rat liver cell lines. Biochemistry. 1991;30:5616–5621. doi: 10.1021/bi00236a040. [DOI] [PubMed] [Google Scholar]
- 78.McLeod R.S., Zhao Y., Selby S.L., Westerlund J., Yao Z. Carboxyl-terminal truncation impairs lipid recruitment by apolipoprotein B100 but does not affect secretion of the truncated apolipoprotein B-containing lipoproteins. J. Biol. Chem. 1994;269:2852–2862. [PubMed] [Google Scholar]
- 79.Young S.G., Cham C.M., Pitas R.E., Burri B.J., Connolly A., Flynn L., Pappu A.S., Wong J.S., Hamilton R.L., Farese R.V., Jr. A genetic model for absent chylomicron formation: mice producing apolipoprotein B in the liver, but not in the intestine. J. Clin. Invest. 1995;96:2932–2946. doi: 10.1172/JCI118365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hamilton R.L., Wong J.S., Cham C.M., Nielsen L.B., Young S.G. Chylomicron-sized lipid particles are formed in the setting of apolipoprotein B deficiency. J. Lipid Res. 1998;39:1543–1557. [PubMed] [Google Scholar]
- 81.Trevaskis N.L., Porter C.J., Charman W.N. Bile increases intestinal lymphatic drug transport in the fasted rat. Pharm. Res. 2005;22:1863–1870. doi: 10.1007/s11095-005-6808-9. [DOI] [PubMed] [Google Scholar]
- 82.Baxter J. Origin and characteristics of endogenous lipid in thoracic duct lymph in rat. J. Lipid Res. 1966;7:158–166. [PubMed] [Google Scholar]
- 83.Byers S.O., Friedman M. Site of origin of plasma triglyceride. Am. J. Physiol. 1960;198:629–631. doi: 10.1152/ajplegacy.1960.198.3.629. [DOI] [PubMed] [Google Scholar]
- 84.Risser T.R., Reaven G.M., Reaven E.P. Intestinal contribution to secretion of very low density lipoproteins into plasma. Am. J. Physiol. 1978;234:E277–E281. doi: 10.1152/ajpendo.1978.234.3.E277. [DOI] [PubMed] [Google Scholar]
- 85.Shiau Y.F., Popper D.A., Reed M., Umstetter C., Capuzzi D., Levine G.M. Intestinal triglycerides are derived from both endogenous and exogenous sources. Am. J. Physiol. 1985;248:G164–G169. doi: 10.1152/ajpgi.1985.248.2.G164. [DOI] [PubMed] [Google Scholar]
- 86.Mansbach C.M., II, Dowell R.F. Uptake and metabolism of circulating fatty acids by rat intestine. Am. J. Physiol. 1992;263:G927–G933. doi: 10.1152/ajpgi.1992.263.6.G927. [DOI] [PubMed] [Google Scholar]
- 87.Mansbach C.M., II, Dowell R.F. Role of the intestine in chylomicron remnant clearance. Am. J. Physiol. 1995;269:G144–G152. doi: 10.1152/ajpgi.1995.269.1.G144. [DOI] [PubMed] [Google Scholar]
- 88.Mansbach C.M., II, Arnold A., Cox M.A. Factors influencing triacylglycerol delivery into mesenteric lymph. Am. J. Physiol. 1985;249:G642–G648. doi: 10.1152/ajpgi.1985.249.5.G642. [DOI] [PubMed] [Google Scholar]
- 89.Tso P., Balint J.A., Simmonds W.J. Role of biliary lecithin in lymphatic transport of fat. Gastroenterology. 1977;73:1362–1367. [PubMed] [Google Scholar]
- 90.Hofmann A.F., Borgstrom B. Physico-chemical state of lipids in intestinal content during their digestion and absorption. Fed. Proc. 1962;21:43–50. [PubMed] [Google Scholar]
- 91.Shackleford D.M., Faassen W.A., Houwing N., Lass H., Edwards G.A., Porter C.J., Charman W.N. Contribution of lymphatically transported testosterone undecanoate to the systemic exposure of testosterone after oral administration of two andriol formulations in conscious lymph duct-cannulated dogs. J. Pharmacol. Exp. Ther. 2003;306:925–933. doi: 10.1124/jpet.103.052522. [DOI] [PubMed] [Google Scholar]
- 92.Frey H., Aakvaag A., Saanum D., Falch J. Bioavailability of oral testosterone in males. Eur. J. Clin. Pharmacol. 1979;16:345–349. doi: 10.1007/BF00605634. [DOI] [PubMed] [Google Scholar]
- 93.Bagchus W.M., Hust R., Maris F., Schnabel P.G., Houwing N.S. Important effect of food on the bioavailability of oral testosterone undecanoate. Pharmacotherapy. 2003;23:319–325. doi: 10.1592/phco.23.3.319.32104. [DOI] [PubMed] [Google Scholar]
- 94.Khoo S.M., Edwards G.A., Porter C.J., Charman W.N. A conscious dog model for assessing the absorption, enterocyte-based metabolism, and intestinal lymphatic transport of halofantrine. J. Pharm. Sci. 2001;90:1599–1607. doi: 10.1002/jps.1110. [DOI] [PubMed] [Google Scholar]
- 95.Khoo S.M., Prankerd R.J., Edwards G.A., Porter C.J., Charman W.N. A physicochemical basis for the extensive intestinal lymphatic transport of a poorly lipid soluble antimalarial, halofantrine hydrochloride, after postprandial administration to dogs. J. Pharm. Sci. 2002;91:647–659. doi: 10.1002/jps.10045. [DOI] [PubMed] [Google Scholar]
- 96.Ockner R.K., Manning J.A. Fatty acid-binding protein in small intestine. Identification, isolation, and evidence for its role in cellular fatty acid transport. J. Clin. Invest. 1974;54:326–338. doi: 10.1172/JCI107768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zilversmit D.B. The composition and structure of lymph chylomicrons in dog, rat, and man. J. Clin. Invest. 1965;44:1610–1622. doi: 10.1172/JCI105267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lindgren F.T., Jensen L.C., Hatch F.T. The isolation and quantitative analysis of serum lipoproteins. In: Nelson G.J., editor. Blood lipids and Lipoproteins: Quantitation, Composition and Metabolism. Wiley; New York: 1972. pp. 181–274. [Google Scholar]
- 99.Carey M.C., Small D.M. The characteristics of mixed micellar solutions with particular reference to bile. Am. J. Med. 1970;49:590–608. doi: 10.1016/s0002-9343(70)80127-9. [DOI] [PubMed] [Google Scholar]
- 100.Edge S.B., Hoeg J.M., Schneider P.D., Brewer H.B., Jr. Apolipoprotein B synthesis in humans: liver synthesizes only apolipoprotein B-100. Metabolism. 1985;34:726–730. doi: 10.1016/0026-0495(85)90022-8. [DOI] [PubMed] [Google Scholar]
- 101.Glickman R.M., Rogers M., Glickman J.N. Apolipoprotein B synthesis by human liver and intestine in vitro. Proc. Natl. Acad. Sci. U. S. A. 1986;83:5296–5300. doi: 10.1073/pnas.83.14.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Glickman R.M., Glickman J.N., Magun A., Brin M. Apolipoprotein synthesis in normal and abetalipoproteinemic intestinal mucosa. Gastroenterology. 1991;101:749–755. doi: 10.1016/0016-5085(91)90535-s. [DOI] [PubMed] [Google Scholar]
- 103.Berriot-Varoqueaux N., Dannoura A.H., Moreau A., Verthier N., Sassolas A., Cadiot G., Lachaux A., Munck A., Schmitz J., Aggerbeck L.P., Samson-Bouma M.E. Apolipoprotein B48 glycosylation in abetalipoproteinemia and Anderson's disease. Gastroenterology. 2001;121:1101–1108. doi: 10.1053/gast.2001.29331. [DOI] [PubMed] [Google Scholar]
- 104.Levy R.I., Fredrickson D.S., Laster L. The lipoproteins and lipid transport in abetalipoproteinemia. J. Clin. Invest. 1966;45:531–541. doi: 10.1172/JCI105367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ohashi K., Ishibashi S., Osuga J., Tozawa R., Harada K., Yahagi N., Shionoiri F., Iizuka Y., Tamura Y., Nagai R., Illingworth D.R., Gotoda T., Yamada N. Novel mutations in the microsomal triglyceride transfer protein gene causing abetalipoproteinemia. J. Lipid Res. 2000;41:1199–1204. [PubMed] [Google Scholar]
- 106.Berriot-Varoqueaux N., Aggerbeck L.P., Samson-Bouma M., Wetterau J.R. The role of the microsomal triglygeride transfer protein in abetalipoproteinemia. Annu. Rev. Nutr. 2000;20:663–697. doi: 10.1146/annurev.nutr.20.1.663. [DOI] [PubMed] [Google Scholar]
- 107.Hooper A.J., van Bockxmeer F.M., Burnett J.R. Monogenic hypocholesterolaemic lipid disorders and apolipoprotein B metabolism. Crit. Rev. Clin. Lab. Sci. 2005;42:515–545. doi: 10.1080/10408360500295113. [DOI] [PubMed] [Google Scholar]
- 108.Anderson C.M., Townley R.R., Freemanm, Johansen P. Unusual causes of steatorrhoea in infancy and childhood. Med. J. Aust. 1961;48(2):617–622. doi: 10.5694/j.1326-5377.1961.tb69860.x. [DOI] [PubMed] [Google Scholar]
- 109.Roy C.C., Levy E., Green P.H., Sniderman A., Letarte J., Buts J.P., Orquin J., Brochu P., Weber A.M., Morin C.L. Malabsorption, hypocholesterolemia, and fat-filled enterocytes with increased intestinal apoprotein B, chylomicron retention disease. Gastroenterology. 1987;92:390–399. doi: 10.1016/0016-5085(87)90133-8. [DOI] [PubMed] [Google Scholar]
- 110.Dannoura A.H., Berriot-Varoqueaux N., Amati P., Abadie V., Verthier N., Schmitz J., Wetterau J.R., Samson-Bouma M.E., Aggerbeck L.P. Anderson's disease: exclusion of apolipoprotein and intracellular lipid transport genes. Arterioscler. Thromb. Vasc. Biol. 1999;19:2494–2508. doi: 10.1161/01.atv.19.10.2494. [DOI] [PubMed] [Google Scholar]
- 111.Silvain M., Bligny D., Aparicio T., Laforet P., Grodet A., Peretti N., Menard D., Djouadi F., Jardel C., Begue J.M., Walker F., Schmitz J., Lachaux A., Aggerbeck L.P., Samson-Bouma M.E. Anderson's disease (chylomicron retention disease): a new mutation in the SARA2 gene associated with muscular and cardiac abnormalities. Clin. Genet. 2008;74:546–552. doi: 10.1111/j.1399-0004.2008.01069.x. [DOI] [PubMed] [Google Scholar]
- 112.Papisov M., Weissleder R. Drug delivery to lymphatic tissue. Crit. Rev. Ther. Drug Carrier Syst. 1996;13:57–84. [PubMed] [Google Scholar]
- 113.Weiss L. The pathophysiology of metastasis within the lymphatic system. In: Weiss L., Gilbert H., Ballon S., editors. Lymphatic System Metastases. Hall; Boston: 1988. p. 2. [Google Scholar]
- 114.Ohtake E., Matsui K. Lymphoscintigraphy in patients with lymphedema. A new approach using intradermal injections of technetium-99 m human serum albumin. Clin. Nucl. Med. 1986;11:474–478. [PubMed] [Google Scholar]
- 115.Mariani G., Erba P., Manca G., Villa G., Gipponi M., Boni G., Buffoni F., Suriano S., Castagnola F., Bartolomei M., Strauss H.W. Radioguided sentinel lymph node biopsy in patients with malignant cutaneous melanoma: the nuclear medicine contribution. J. Surg. Oncol. 2004;85:141–151. doi: 10.1002/jso.20027. [DOI] [PubMed] [Google Scholar]
- 116.Hussain N., Jaitley V., Florence A.T. Recent advances in the understanding of uptake of microparticulates across the gastrointestinal lymphatics. Adv. Drug Deliv. Rev. 2001;50:107–142. doi: 10.1016/s0169-409x(01)00152-1. [DOI] [PubMed] [Google Scholar]
- 117.Tanigawa N., Satomura K., Hikasa Y., Hashida M., Muranishi S., Sezaki H. Surgical chemotherapy against lymph node metastases: an experimental study. Surgery. 1980;87:147–152. [PubMed] [Google Scholar]
- 118.Gipponi M., Bassetti C., Canavese G., Catturich A., Di Somma C., Vecchio C., Nicolo G., Schenone F., Tomei D., Cafiero F. Sentinel lymph node as a new marker for therapeutic planning in breast cancer patients. J. Surg. Oncol. 2004;85:102–111. doi: 10.1002/jso.20022. [DOI] [PubMed] [Google Scholar]
- 119.Witte C.L., Witte M.H. Disorders of lymph flow. Acad. Radiol. 1995;2:324–334. doi: 10.1016/s1076-6332(05)80193-x. [DOI] [PubMed] [Google Scholar]
- 120.Witte M.H., Jones K., Wilting J., Dictor M., Selg M., McHale N., Gershenwald J.E., Jackson D.G. Structure function relationships in the lymphatic system and implications for cancer biology. Cancer Metastasis Rev. 2006;25:159–184. doi: 10.1007/s10555-006-8496-2. [DOI] [PubMed] [Google Scholar]
- 121.Edwards G.A., Porter C.J., Caliph S.M., Khoo S.M., Charman W.N. Animal models for the study of intestinal lymphatic drug transport. Adv. Drug Deliv. Rev. 2001;50:45–60. doi: 10.1016/s0169-409x(01)00148-x. [DOI] [PubMed] [Google Scholar]
- 122.Khoo S.M., Shackleford D.M., Porter C.J., Edwards G.A., Charman W.N. Intestinal lymphatic transport of halofantrine occurs after oral administration of a unit-dose lipid-based formulation to fasted dogs. Pharm. Res. 2003;20:1460–1465. doi: 10.1023/a:1025718513246. [DOI] [PubMed] [Google Scholar]
- 123.Lespine A., Chanoit G., Bousquet-Melou A., Lallemand E., Bassissi F.M., Alvinerie M., Toutain P.L. Contribution of lymphatic transport to the systemic exposure of orally administered moxidectin in conscious lymph duct-cannulated dogs. Eur. J. Pharm. Sci. 2006;27:37–43. doi: 10.1016/j.ejps.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 124.White D.G., Story M.J., Barnwell S.G. An experimental animal model for studying the effects of a novel lymphatic drug delivery system for propranolol. Int. J. Pharm. 1991;69:169–174. [Google Scholar]
- 125.Onizuka M., Flatebo T., Nicolaysen G. Lymph flow pattern in the intact thoracic duct in sheep. J. Physiol. 1997;503(Pt 1):223–234. doi: 10.1111/j.1469-7793.1997.223bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Segrave A.M., Mager D.E., Charman S.A., Edwards G.A., Porter C.J. Pharmacokinetics of recombinant human leukemia inhibitory factor in sheep. J. Pharmacol. Exp. Ther. 2004;309:1085–1092. doi: 10.1124/jpet.103.063289. [DOI] [PubMed] [Google Scholar]
- 127.Kagan L., Gershkovich P., Mendelman A., Amsili S., Ezov N., Hoffman A. The role of the lymphatic system in subcutaneous absorption of macromolecules in the rat model. Eur. J. Pharm. Biopharm. 2007;67:759–765. doi: 10.1016/j.ejpb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 128.Dahan A., Hoffman A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur. J. Pharm. Sci. 2005;24:381–388. doi: 10.1016/j.ejps.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 129.Pidlich J., Renner F., Ellinger A., Huttinger M., Pavelka M., Gangl A. Effect of pluronic L-81 on intestinal lipoprotein secretion in the rat. Dig. Dis. Sci. 1996;41:1445–1451. doi: 10.1007/BF02088571. [DOI] [PubMed] [Google Scholar]
- 130.Charman W.N., Stella V.J. Estimating the maximal potentital for intestinal lymphatic transport of lipophilic drug molecules. Int. J. Pharm. 1986;33:175–178. [Google Scholar]
- 131.Porter C.J.H., Charman W.N. Model systems for intestinal lymphatic transport studies. In: Borchardt R.T., Smith P.L., Wilson G., editors. Models for Assessing Drug Absorption and Metabolism. Plenum Press; New York: 1996. pp. 85–102. [Google Scholar]
- 132.Porter C.J., Charman S.A., Humberstone A.J., Charman W.N. Lymphatic transport of halofantrine in the conscious rat when administered as either the free base or the hydrochloride salt: effect of lipid class and lipid vehicle dispersion. J. Pharm. Sci. 1996;85:357–361. doi: 10.1021/js9502229. [DOI] [PubMed] [Google Scholar]
- 133.Porter C.J., Charman S.A., Charman W.N. Lymphatic transport of halofantrine in the triple-cannulated anesthetized rat model: effect of lipid vehicle dispersion. J. Pharm. Sci. 1996;85:351–356. doi: 10.1021/js950221g. [DOI] [PubMed] [Google Scholar]
- 134.Jandacek R.J., Rider T., Yang Q., Woollett L.A., Tso P. Lymphatic and portal vein absorption of organochlorine compounds in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G226–G234. doi: 10.1152/ajpgi.90517.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Noguchi T., Charman W.N., Stella V.J. Lymphatic appearance of DDT in thoracic mesenteric lymph duct cannulated rats. Int. J. Pharm. 1985;24:185–192. [Google Scholar]
- 136.Turner S.G., Barrowman J.A. Intestinal lymph flow and lymphatic transport of protein during fat absorption. Q. J. Exp. Physiol. Cogn. Med. Sci. 1977;62:175–180. doi: 10.1113/expphysiol.1977.sp002387. [DOI] [PubMed] [Google Scholar]
- 137.Raub T.J., Douglas S.L., Melchior G.W., Charman W.N. Methodology for assessing intestinal lymphatic transport. In: Charman W.N., Stella V.J., editors. Lymphatic Transport of Drugs. CRC Press; Boca Raton, FL: 1992. pp. 63–111. [Google Scholar]
- 138.Hauss D., Fogal S., Ficorilli J. Chronic collection of mesenteric lymph from conscious, tethered rats. Contemp. Top. Lab. Anim. Sci. 1998;37:56–58. [PubMed] [Google Scholar]
- 139.Khoo S.M., Porter C.J.H., Edwards G.A., Charman W.N. A novel triple-canulaed conscious dog model for assessment of the absorption, enterocyte-based metablism and lymphocyte transport of candidate lipophilic drugs. Pharm. Sci. 1999;1:S647. [Google Scholar]
- 140.Rajpal S.G., Kirkpatrick J.R. Creation of a thoracic duct fistula: an improved technique. J. Surg. Res. 1972;13:260–261. doi: 10.1016/0022-4804(72)90074-1. [DOI] [PubMed] [Google Scholar]
- 141.Kararli T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995;16:351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 142.Jensen L.T., Olesen H.P., Risteli J., Lorenzen I. External thoracic duct-venous shunt in conscious pigs for long term studies of connective tissue metabolites in lymph. Lab. Anim. Sci. 1990;40:620–624. [PubMed] [Google Scholar]
- 143.Chen S.A., Sawchuk R.J., Brundage R.C., Horvath C., Mendenhall H.V., Gunther R.A., Braeckman R.A. Plasma and lymph pharmacokinetics of recombinant human interleukin-2 and polyethylene glycol-modified interleukin-2 in pigs. J. Pharmacol. Exp. Ther. 2000;293:248–259. [PubMed] [Google Scholar]
- 144.Charman S.A., Segrave A.M., Edwards G.A., Porter C.J. Systemic availability and lymphatic transport of human growth hormone administered by subcutaneous injection. J. Pharm. Sci. 2000;89:168–177. doi: 10.1002/(SICI)1520-6017(200002)89:2<168::AID-JPS4>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 145.Porter C.J., Edwards G.A., Charman S.A. Lymphatic transport of proteins after s.c. injection: implications of animal model selection. Adv. Drug Deliv. Rev. 2001;50:157–171. doi: 10.1016/s0169-409x(01)00153-3. [DOI] [PubMed] [Google Scholar]
- 146.Girardet R.E., Benninghoff D.L. Surgical techniques for long-term study of thoracic duct lymph circulation in dogs. J. Surg. Res. 1973;15:168–175. doi: 10.1016/0022-4804(73)90073-5. [DOI] [PubMed] [Google Scholar]
- 147.Fish J.C., Sarles H.E., Mattingly A.T., Ross M.V., Remmers A.R., Jr. Preparation of chronic thoracic duct lymph fistulas in man and laboratory animals. J. Surg. Res. 1969;9:101–106. doi: 10.1016/0022-4804(69)90038-9. [DOI] [PubMed] [Google Scholar]
- 148.Gralla E.J., Cappiello F.M., Jonas A.M. Technique for chronic cannulation of the canine thoracic duct. Am. J. Vet. Res. 1973;34:285–287. [PubMed] [Google Scholar]
- 149.Dedo D.D., Ogura J.H. Exteriorization of thoracic duct lymph. Theoretical considerations and an experimental model. Arch. Otolaryngol. 1975;101:671–674. doi: 10.1001/archotol.1975.00780400029008. [DOI] [PubMed] [Google Scholar]
- 150.Witte C.L., Witte M.H., Cole W.R. A simplified method for cannulation of the normal canine cervical thoracic duct. Lymphology. 1970;3:159–161. [PubMed] [Google Scholar]
- 151.Briscoe D.E. Canine thoracic duct cannulation revisited. Lymphology. 1981;14:32–34. [PubMed] [Google Scholar]
- 152.Caliph S.M., Charman W.N., Porter C.J. Effect of short-, medium-, and long-chain fatty acid-based vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymph-cannulated and non-cannulated rats. J. Pharm. Sci. 2000;89:1073–1084. doi: 10.1002/1520-6017(200008)89:8<1073::aid-jps12>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 153.Murthy S., Albright E., Mathur S.N., Field F.J. Effect of eicosapentaenoic acid on triacylglycerol transport in CaCo-2 cells. Biochim. Biophys. Acta. 1990;1045:147–155. doi: 10.1016/0005-2760(90)90144-m. [DOI] [PubMed] [Google Scholar]
- 154.Kam N.T., Albright E., Mathur S.N., Field F.J. Inhibition of acylcoenzyme A: cholesterol acyltransferase activity in CaCo-2 cells results in intracellular triglyceride accumulation. J. Lipid Res. 1989;30:371–377. [PubMed] [Google Scholar]
- 155.Kam N.T., Albright E., Mathur S., Field F.J. Effect of lovastatin on acyl-CoA: cholesterol O-acyltransferase (ACAT) activity and the basolateral-membrane secretion of newly synthesized lipids by CaCo-2 cells. Biochem. J. 1990;272:427–433. doi: 10.1042/bj2720427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Traber M.G., Kayden H.J., Rindler M.J. Polarized secretion of newly synthesized lipoproteins by the Caco-2 human intestinal cell line. J. Lipid Res. 1987;28:1350–1363. [PubMed] [Google Scholar]
- 157.Seeballuck F., Ashford M.B., O'Driscoll C.M. The effects of pluronics block copolymers and Cremophor EL on intestinal lipoprotein processing and the potential link with P-glycoprotein in Caco-2 cells. Pharm. Res. 2003;20:1085–1092. doi: 10.1023/a:1024422625596. [DOI] [PubMed] [Google Scholar]
- 158.Seeballuck F., Lawless E., Ashford M.B., O'Driscoll C.M. Stimulation of triglyceride-rich lipoprotein secretion by polysorbate 80: in vitro and in vivo correlation using Caco-2 cells and a cannulated rat intestinal lymphatic model. Pharm. Res. 2004;21:2320–2326. doi: 10.1007/s11095-004-7684-4. [DOI] [PubMed] [Google Scholar]
- 159.Karpf D.M., Holm R., Garafalo C., Levy E., Jacobsen J., Mullertz A. Effect of different surfactants in biorelevant medium on the secretion of a lipophilic compound in lipoproteins using Caco-2 cell culture. J. Pharm. Sci. 2006;95:45–55. doi: 10.1002/jps.20431. [DOI] [PubMed] [Google Scholar]
- 160.O'Driscoll C.M., Griffin B.T. Biopharmaceutical challenges associated with drugs with low aqueous solubility — the potential impact of lipid-based formulations. Adv. Drug Deliv. Rev. 2008;60:617–624. doi: 10.1016/j.addr.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 161.Murthy S., Albright E., Mathur S.N., Davidson N.O., Field F.J. Apolipoprotein B mRNA abundance is decreased by eicosapentaenoic acid in CaCo-2 cells. Effect on the synthesis and secretion of apolipoprotein B. Arterioscler. Thromb. 1992;12:691–700. doi: 10.1161/01.atv.12.6.691. [DOI] [PubMed] [Google Scholar]
- 162.Hussain M.M., Kancha R.K., Zhou Z., Luchoomun J., Zu H., Bakillah A. Chylomicron assembly and catabolism: role of apolipoproteins and receptors. Biochim. Biophys. Acta. 1996;1300:151–170. doi: 10.1016/0005-2760(96)00041-0. [DOI] [PubMed] [Google Scholar]
- 163.Trotter P.J., Storch J. Fatty acid esterification during differentiation of the human intestinal cell line Caco-2. J. Biol. Chem. 1993;268:10017–10023. [PubMed] [Google Scholar]
- 164.Gershkovich P., Hoffman A. Uptake of lipophilic drugs by plasma derived isolated chylomicrons: linear correlation with intestinal lymphatic bioavailability. Eur. J. Pharm. Sci. 2005;26:394–404. doi: 10.1016/j.ejps.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 165.Crison J.R., Shah V.P., Skelly J.P., Amidon G.L. Drug dissolution into micellar solutions: development of a convective diffusion model and comparison to the film equilibrium model with application to surfactant-facilitated dissolution of carbamazepine. J. Pharm. Sci. 1996;85:1005–1011. doi: 10.1021/js930336p. [DOI] [PubMed] [Google Scholar]
- 166.Crison J.R., Weiner N.D., Amidon G.L. Dissolution media for in vitro testing of water-insoluble drugs: effect of surfactant purity and electrolyte on in vitro dissolution of carbamazepine in aqueous solutions of sodium lauryl sulfate. J. Pharm. Sci. 1997;86:384–388. doi: 10.1021/js960105t. [DOI] [PubMed] [Google Scholar]
- 167.Dressman J.B., Vertzoni M., Goumas K., Reppas C. Estimating drug solubility in the gastrointestinal tract. Adv. Drug Deliv. Rev. 2007;59:591–602. doi: 10.1016/j.addr.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 168.Vogt M., Kunath K., Dressman J.B. Dissolution improvement of four poorly water soluble drugs by cogrinding with commonly used excipients. Eur. J. Pharm. Biopharm. 2008;68:330–337. doi: 10.1016/j.ejpb.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 169.Dahan A., Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J. Control Release. 2008;129:1–10. doi: 10.1016/j.jconrel.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 170.Alvarez F.J., Stella V.J. The role of calcium ions and bile salts on the pancreatic lipase-catalyzed hydrolysis of triglyceride emulsions stabilized with lecithin. Pharm. Res. 1989;6:449–457. doi: 10.1023/a:1015956104500. [DOI] [PubMed] [Google Scholar]
- 171.Porter C.J., Charman W.N. In vitro assessment of oral lipid based formulations. Adv. Drug Deliv. Rev. 2001;50(Suppl. 1):S127–S147. doi: 10.1016/s0169-409x(01)00182-x. [DOI] [PubMed] [Google Scholar]
- 172.Armand M., Borel P., Pasquier B., Dubois C., Senft M., Andre M., Peyrot J., Salducci J., Lairon D. Physicochemical characteristics of emulsions during fat digestion in human stomach and duodenum. Am. J. Physiol. 1996;271:G172–G183. doi: 10.1152/ajpgi.1996.271.1.G172. [DOI] [PubMed] [Google Scholar]
- 173.Galia E., Nicolaides E., Horter D., Lobenberg R., Reppas C., Dressman J.B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998;15:698–705. doi: 10.1023/a:1011910801212. [DOI] [PubMed] [Google Scholar]
- 174.Zangenberg N.H., Mullertz A., Kristensen H.G., Hovgaard L. A dynamic in vitro lipolysis model. II: Evaluation of the model. Eur. J. Pharm. Sci. 2001;14:237–244. doi: 10.1016/s0928-0987(01)00182-8. [DOI] [PubMed] [Google Scholar]
- 175.Porter C.J., Trevaskis N.L., Charman W.N. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007;6:231–248. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 176.Holm R., Hoest J. Successful in silico predicting of intestinal lymphatic transfer. Int. J. Pharm. 2004;272:189–193. doi: 10.1016/j.ijpharm.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 177.Ichihashi T., Nagasaki T., Takagishi Y., Yamada H. A quantitative concept of the mechanism of intestinal lymphatic transfer of lipophilic molecules. Pharm. Res. 1994;11:508–512. doi: 10.1023/a:1018954213469. [DOI] [PubMed] [Google Scholar]
- 178.Myers R.A., Stella V.J. Factors affecting the lymphaic transport of penclomedine (NSC-338720), a lipophilic cytotoxic drug: comparison to DDT and hexachlorobenzene. Int. J. Pharm. 1992;80:511–562. [Google Scholar]
- 179.Hauss D.J., Mehta S.C., Radebaugh G.W. Targeted lymphatic transport and modified systemic distribution of CI-976, a lipophilic lipid-regulator drug, via a formulation approach. Int. J. Pharm. 1994;108:85–93. [Google Scholar]
- 180.Palm K., Luthman K., Ungell A.L., Strandlund G., Artursson P. Correlation of drug absorption with molecular surface properties. J. Pharm. Sci. 1996;85:32–39. doi: 10.1021/js950285r. [DOI] [PubMed] [Google Scholar]
- 181.Krarup L.H., Christensen I.T., Hovgaard L., Frokjaer S. Predicting drug absorption from molecular surface properties based on molecular dynamics simulations. Pharm. Res. 1998;15:972–978. doi: 10.1023/a:1011905522110. [DOI] [PubMed] [Google Scholar]
- 182.Norinder U., Osterberg T., Artursson P. Theoretical calculation and prediction of intestinal absorption of drugs in humans using MolSurf parametrization and PLS statistics. Eur. J. Pharm. Sci. 1999;8:49–56. doi: 10.1016/s0928-0987(98)00059-1. [DOI] [PubMed] [Google Scholar]
- 183.Cruciani G., Crivori P., Carrupt P.A., Testa B. Molecular fields in quantitative structure-permeation relationships: the VolSurf approach. J. Mol. Struct. Thoechem. 2000;503:17–30. [Google Scholar]
- 184.Cruciani G., Pastor M., Guba W. VolSurf: a new tool for the pharmacokinetics optimization of lead compounds. Eur. J. Pharm. Sci. 2000;11:S29–S39. doi: 10.1016/s0928-0987(00)00162-7. [DOI] [PubMed] [Google Scholar]
- 185.Meylan W.M., Howard P.H. Atom/fragment contribution method for estimating octanol-water partition coefficients. J. Pharm. Sci. 1995;84:83–92. doi: 10.1002/jps.2600840120. [DOI] [PubMed] [Google Scholar]
- 186.Wold S., Johansson E., Cocchi M. PLS-partial least-squares projections to latent structures. In: Kubinyi H., editor. 3D QSAR in Drug Design. Escom; Leiden: 1993. pp. 523–550. [Google Scholar]
- 187.McConnell E.L., Fadda H.M., Basit A.W. Gut instincts: explorations in intestinal physiology and drug delivery. Int. J. Pharm. 2008;364:213–226. doi: 10.1016/j.ijpharm.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 188.Yu L.X., Lipka E., Crison J.R., Amidon G.L. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv. Drug Deliv. Rev. 1996;19:359–376. doi: 10.1016/0169-409x(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 189.Sue Masters B., Marohnic C.C. Cytochromes P450 — a family of proteins and scientists-understanding their relationships. Drug Metab. Rev. 2006;38:209–225. doi: 10.1080/03602530600570065. [DOI] [PubMed] [Google Scholar]
- 190.Evans W.E., Relling M.V. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 191.Ritter J.K. Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chem. Biol. Interact. 2000;129:171–193. doi: 10.1016/s0009-2797(00)00198-8. [DOI] [PubMed] [Google Scholar]
- 192.Tukey R.H., Strassburg C.P. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 193.Murad F., Haynes R.C. Androgens and anabolic steroids. In: Gilman A.G., Goodman L.S., Gilman A., editors. The Pharmacological Basis of Therapeutics. MacMillan; New York: 1980. p. 1448. [Google Scholar]
- 194.Daggett P.R., Wheeler M.J., Nabarro J.D. Oral testosterone, a reappraisal. Horm. Res. 1978;9:121–129. doi: 10.1159/000178904. [DOI] [PubMed] [Google Scholar]
- 195.Coert A., Geelen J., de Visser J., van der Vies J. The pharmacology and metabolism of testosterone undecanoate (TU), a new orally active androgen. Acta Endocrinol. (Copenh) 1975;79:789–800. doi: 10.1530/acta.0.0790789. [DOI] [PubMed] [Google Scholar]
- 196.Van Veld P.A., Vetter R.D., Lee R.F., Patton J.S. Dietary fat inhibits the intestinal metabolism of the carcinogen benzo[a]pyrene in fish. J. Lipid Res. 1987;28:810–817. [PubMed] [Google Scholar]
- 197.Vetter R.D., Carey M.C., Patton J.S. Coassimilation of dietary fat and benzo(a)pyrene in the small intestine: an absorption model using the killifish. J. Lipid Res. 1985;26:428–434. [PubMed] [Google Scholar]
- 198.Reubsaet F.A., Veerkamp J.H., Bukkens S.G., Trijbels J.M., Monnens L.A. Acyl-CoA oxidase activity and peroxisomal fatty acid oxidation in rat tissues. Biochim. Biophys. Acta. 1988;958:434–442. doi: 10.1016/0005-2760(88)90229-9. [DOI] [PubMed] [Google Scholar]
- 199.Haunerland N.H. Fatty acid binding protein in locust and mammalian muscle. Comparison of structure, function and regulation. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1994;109:199–208. doi: 10.1016/0305-0491(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 200.Veerkamp J.H., van Moerkerk H.T. Fatty acid-binding protein and its relation to fatty acid oxidation. Mol. Cell. Biochem. 1993;123:101–106. doi: 10.1007/BF01076480. [DOI] [PubMed] [Google Scholar]
- 201.Stremmel W., Pohl L., Ring A., Herrmann T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids. 2001;36:981–989. doi: 10.1007/s11745-001-0809-2. [DOI] [PubMed] [Google Scholar]
- 202.Agellon L.B., Toth M.J., Thomson A.B. Intracellular lipid binding proteins of the small intestine. Mol. Cell. Biochem. 2002;239:79–82. [PubMed] [Google Scholar]
- 203.Chow S.L., Hollander D. A dual, concentration-dependent absorption mechanism of linoleic acid by rat jejunum in vitro. J. Lipid Res. 1979;20:349–356. [PubMed] [Google Scholar]
- 204.David H.R., Jr., Zhu L.J., Hoos L.M., Tetzloff G., Maguire M., Liu J., Yao X., Iyier S.P., Lam M.H., Lund E.G., Detmers P.A., Graziano M.P., Altmann S.W. Niemann-Pick C1 Like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J. Biol. Chem. 2004;279:33586–33592. doi: 10.1074/jbc.M405817200. [DOI] [PubMed] [Google Scholar]
- 205.Altmann S.W., Davis H.R., Jr., Zhu L.J., Yao X., Hoos L.M., Tetzloff G., Iyier S.P., Maguire M., Golovko A., Zeng M., Wang L., Murgolo N., Graziano M.P. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–1204. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- 206.Sarkadi B., Homolya L., Szakacs G., Varadi A. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol. Rev. 2006;86:1179–1236. doi: 10.1152/physrev.00037.2005. [DOI] [PubMed] [Google Scholar]
- 207.Wakabayashi K., Tamura A., Saito H., Onishi Y., Ishikawa T. Human ABC transporter ABCG2 in xenobiotic protection and redox biology. Drug Metab. Rev. 2006;38:371–391. doi: 10.1080/03602530600727947. [DOI] [PubMed] [Google Scholar]
- 208.Borst P., Zelcer N., van Helvoort A. ABC transporters in lipid transport. Biochim. Biophys. Acta. 2000;1486:128–144. doi: 10.1016/s1388-1981(00)00053-6. [DOI] [PubMed] [Google Scholar]
- 209.Schmitz G., Langmann T., Heimerl S. Role of ABCG1 and other ABCG famly members in lipid metabolism. J. Lipid Res. 2001;42:1513–1520. [PubMed] [Google Scholar]
- 210.Levy E., Spahis S., Sinnett D., Peretti N., Maupas-Schwalm F., Delvin E., Lambert M., Lavoie M.A. Intestinal cholesterol transport proteins: an update and beyond. Curr. Opin. Lipidol. 2007;18:310–318. doi: 10.1097/MOL.0b013e32813fa2e2. [DOI] [PubMed] [Google Scholar]
- 211.Repa J.J., Turley S.D., Lobaccaro J.A., Medina J., Li L., Lustig K., Shan B., Heyman R.A., Dietschy J.M., Mangelsdorf D.J. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- 212.Berge K.E., Tian H., Graf G.A., Yu L., Grishin N.V., Schultz J., Kwiterovich P., Shan B., Barnes R., Hobbs H.H. Accumulation of dietary cholesterol in sitosterolemia cuased by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 213.Evans A.M. Influence of dietary components on the gastrointestinal metabolism and transport of drugs. Ther. Drug Monit. 2000;22:131–136. doi: 10.1097/00007691-200002000-00028. [DOI] [PubMed] [Google Scholar]
- 214.Hunter J., Hirst B.H., Simmons N.L. Epithelial secretion of vinblastine by human intestinal adenocarcinoma cell (HCT-8 and T84) layers expressing P-glycoprotein. Br. J. Cancer. 1991;64:437–444. doi: 10.1038/bjc.1991.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Schinkel A.H., Mayer U., Wagenaar E., Mol C.A., van Deemter L., Smit J.J., van der Valk M.A., Voordouw A.C., Spits H., van Tellingen O., Zijlmans J.M., Fibbe W.E., Borst P. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Stride B.D., Grant C.E., Loe D.W., Hipfner D.R., Cole S.P., Deeley R.G. Pharmacological characterization of the murine and human orthologs of multidrug-resistance protein in transfected human embryonic kidney cells. Mol. Pharmacol. 1997;52:344–353. doi: 10.1124/mol.52.3.344. [DOI] [PubMed] [Google Scholar]
- 217.de Jong M.C., Slootstra J.W., Scheffer G.L., Schroeijers A.B., Puijk W.C., Dinkelberg R., Kool M., Broxterman H.J., Meloen R.H., Scheper R.J. Peptide transport by the multidrug resistance protein MRP1. Cancer Res. 2001;61:2552–2557. [PubMed] [Google Scholar]
- 218.Cole S.P., Sparks K.E., Fraser K., Loe D.W., Grant C.E., Wilson G.M., Deeley R.G. Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 1994;54:5902–5910. [PubMed] [Google Scholar]
- 219.Kawabe T., Chen Z.S., Wada M., Uchiumi T., Ono M., Akiyama S., Kuwano M. Enhanced transport of anticancer agents and leukotriene C4 by the human canalicular multispecific organic anion transporter (cMOAT/MRP2) FEBS Lett. 1999;456:327–331. doi: 10.1016/s0014-5793(99)00979-5. [DOI] [PubMed] [Google Scholar]
- 220.Deguchi T., Kusuhara H., Takadate A., Endou H., Otagiri M., Sugiyama Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004;65:162–174. doi: 10.1111/j.1523-1755.2004.00354.x. [DOI] [PubMed] [Google Scholar]
- 221.Shugarts S., Benet L.Z. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm. Res. 2009;26:2039–2054. doi: 10.1007/s11095-009-9924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Benet L.Z., Cummins C.L., Wu C.Y. Unmasking the dynamic interplay between efflux transporters and metabolic enzymes. Int. J. Pharm. 2004;277:3–9. doi: 10.1016/j.ijpharm.2002.12.002. [DOI] [PubMed] [Google Scholar]
- 223.Cummins C.L., Jacobsen W., Benet L.Z. Unmasking the dynamic interplay between intestinal P-glycoprotein and CYP3A4. J. Pharmacol. Exp. Ther. 2002;300:1036–1045. doi: 10.1124/jpet.300.3.1036. [DOI] [PubMed] [Google Scholar]
- 224.Charman W.N., Porter C.J., Mithani S., Dressman J.B. Physiochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J. Pharm. Sci. 1997;86:269–282. doi: 10.1021/js960085v. [DOI] [PubMed] [Google Scholar]
- 225.Aungst B.J. Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism. J. Pharm. Sci. 1993;82:979–987. [PubMed] [Google Scholar]
- 226.Porter C.J., Kaukonen A.M., Boyd B.J., Edwards G.A., Charman W.N. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm. Res. 2004;21:1405–1412. doi: 10.1023/b:pham.0000036914.22132.cc. [DOI] [PubMed] [Google Scholar]
- 227.Dahan A., Duvdevani R., Dvir E., Elmann A., Hoffman A. A novel mechanism for oral controlled release of drugs by continuous degradation of a phospholipid prodrug along the intestine: in-vivo and in-vitro evaluation of an indomethacin-lecithin conjugate. J. Control Release. 2007;119:86–93. doi: 10.1016/j.jconrel.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 228.Dahan A., Duvdevani R., Shapiro I., Elmann A., Finkelstein E., Hoffman A. The oral absorption of phospholipid prodrugs: in vivo and in vitro mechanistic investigation of trafficking of a lecithin-valproic acid conjugate following oral administration. J. Control Release. 2008;126:1–9. doi: 10.1016/j.jconrel.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 229.Kossena G.A., Charman W.N., Wilson C.G., O'Mahony B., Lindsay B., Hempenstall J.M., Davison C.L., Crowley P.J., Porter C.J. Low dose lipid formulations: effects on gastric emptying and biliary secretion. Pharm. Res. 2007;24:2084–2096. doi: 10.1007/s11095-007-9363-8. [DOI] [PubMed] [Google Scholar]
- 230.Caliph S.M., Faassen W.A., Vogel G.M., Porter C.J. Oral bioavailability assessment and intestinal lymphatic transport of Org 45697 and Org 46035, two highly lipophilic novel immunomodulator analogues. Curr. Drug Deliv. 2009;6:359–366. doi: 10.2174/156720109789000500. [DOI] [PubMed] [Google Scholar]
- 231.Humberstone A.J., Charman W.N. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv. Drug Deliv. Rev. 1997;25:103–128. [Google Scholar]
- 232.Charman W.N., Porter C.J.H. Lipophilic prodrugs designed for intestinal lymphatic transport. Adv. Drug Deliv. Rev. 1996;19:149–169. [Google Scholar]
- 233.O'Driscoll C.M. Lipid-based formulations for intestinal lymphatic delivery. Eur. J. Pharm. Sci. 2002;15:405–415. doi: 10.1016/s0928-0987(02)00051-9. [DOI] [PubMed] [Google Scholar]
- 234.Pouton C.W. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000;11(Suppl. 2):S93–S98. doi: 10.1016/s0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 235.Pouton C.W. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006;29:278–287. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 236.Pouton C.W., Porter C.J. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv. Drug Deliv. Rev. 2008;60:625–637. doi: 10.1016/j.addr.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 237.Porter C.J., Pouton C.W., Cuine J.F., Charman W.N. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv. Drug Deliv. Rev. 2008;60:673–691. doi: 10.1016/j.addr.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 238.Holm R., Porter C.J., Edwards G.A., Mullertz A., Kristensen H.G., Charman W.N. Examination of oral absorption and lymphatic transport of halofantrine in a triple-cannulated canine model after administration in self-microemulsifying drug delivery systems (SMEDDS) containing structured triglycerides. Eur. J. Pharm. Sci. 2003;20:91–97. doi: 10.1016/s0928-0987(03)00174-x. [DOI] [PubMed] [Google Scholar]
- 239.Constantinides P.P. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm. Res. 1995;12:1561–1572. doi: 10.1023/a:1016268311867. [DOI] [PubMed] [Google Scholar]
- 240.Porter C.J., Charman W.N. Lipid-based formulations for oral administration: opportunities for bioavailability enhancement and lipoprotein targeting of lipophilic drugs. J. Recept. Signal Transduct. Res. 2001;21:215–257. doi: 10.1081/rrs-100107429. [DOI] [PubMed] [Google Scholar]
- 241.Porter C.J.H., Charman W.N. Uptake of drugs into the intestinal lymphatics after oral administration. Adv. Drug Deliv. Rev. 1997:71–89. [Google Scholar]
- 242.Noguchi T., Charman W.N., Stella V.J. The effect of drug lipophilicity and lipid vehicles on the lymphatic absorption of various testosterone esters. Int. J. Pharm. 1985;24:173–184. [Google Scholar]
- 243.Ichihashi T., Kinoshita H., Takagishi Y., Yamada H. Effect of oily vehicles on absorption of mepitiostane by the lymphatic system in rats. J. Pharm. Pharmacol. 1992;44:560–564. doi: 10.1111/j.2042-7158.1992.tb05464.x. [DOI] [PubMed] [Google Scholar]
- 244.Cheema M., Palin K.J., Davis S.S. Lipid vehicles for intestinal lymphatic drug absorption. J. Pharm. Pharmacol. 1987;39:55–56. doi: 10.1111/j.2042-7158.1987.tb07164.x. [DOI] [PubMed] [Google Scholar]
- 245.MacGregor K.J., Embleton J.K., Lacy J.E., Perry E.A., Solomon L.J., Seager H., Pouton C.W. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv. Drug Deliv. Rev. 1997:33–46. [Google Scholar]
- 246.Cuine J.F., McEvoy C.L., Charman W.N., Pouton C.W., Edwards G.A., Benameur H., Porter C.J. Evaluation of the impact of surfactant digestion on the bioavailability of danazol after oral administration of lipidic self-emulsifying formulations to dogs. J. Pharm. Sci. 2008;97:995–1012. doi: 10.1002/jps.21246. [DOI] [PubMed] [Google Scholar]
- 247.Charman W.N., Stella V.J. Effects of lipid class and lipid vehicle volume on the intestinal lymphatic transport of DDT. Int. J. Pharm. 1986;33:165–172. [Google Scholar]
- 248.Stella V.J., Pochopin N.L. Lipophilic prodrugs and the promotion of the intestinal lymphatic drug transport. In: Charman W.N., Stella V.J., editors. Lymphatic Transport of Drugs. CRC Press; Boca Raton, FL: 1992. pp. 181–210. [Google Scholar]
- 249.Lambert D.M. Rationale and applications of lipids as prodrug carriers. Eur. J. Pharm. Sci. 2000;11(Suppl. 2):S15–S27. doi: 10.1016/s0928-0987(00)00161-5. [DOI] [PubMed] [Google Scholar]
- 250.Khan M.S., Akhter M. Synthesis, pharmacological activity and hydrolytic behavior of glyceride prodrugs of ibuprofen. Eur. J. Med. Chem. 2005;40:371–376. doi: 10.1016/j.ejmech.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 251.Khan M.S., Akhter M. Glyceride derivatives as potential prodrugs: synthesis, biological activity and kinetic studies of glyceride derivatives of mefenamic acid. Pharmazie. 2005;60:110–114. [PubMed] [Google Scholar]
- 252.Khan M.S., Akhter M. Synthesis, biological evaluation and kinetic studies of glyceride prodrugs of diclofenac. Indian J. Exp. Biol. 2004;42:1066–1072. [PubMed] [Google Scholar]
- 253.Lambert D.M., Scriba G.K.E., Gallez B., Poupaert J.H., Dumont P. Glyceride conjugates as drug carriers. Curr. Med. Chem. 1995;1:376–391. [Google Scholar]
- 254.Sakai A., Mori N., Shuto S., Suzuki T. Deacylation-reacylation cycle: a possible absorption mechanism for the novel lymphotropic antitumor agent dipalmitoylphosphatidylfluorouridine in rats. J. Pharm. Sci. 1993;82:575–578. doi: 10.1002/jps.2600820606. [DOI] [PubMed] [Google Scholar]
- 255.Sugihara J., Furuuchi S., Nakano K., Harigaya S. Studies on intestinal lymphatic absorption of drugs. I. Lymphatic absorption of alkyl ester derivatives and alpha-monoglyceride derivatives of drugs. J. Pharmacobiodyn. 1988;11:369–376. doi: 10.1248/bpb1978.11.369. [DOI] [PubMed] [Google Scholar]
- 256.Sugihara J., Furuuchi S., Ando H., Takashima K., Harigaya S. Studies on intestinal lymphatic absorption of drugs. II. Glyceride prodrugs for improving lymphatic absorption of naproxen and nicotinic acid. J. Pharmacobiodyn. 1988;11:555–562. doi: 10.1248/bpb1978.11.555. [DOI] [PubMed] [Google Scholar]
- 257.Kennedy B.P., Payette P., Mudgett J., Vadas P., Pruzanski W., Kwan M., Tang C., Rancourt D.E., Cromlish W.A. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J. Biol. Chem. 1995;270:22378–22385. doi: 10.1074/jbc.270.38.22378. [DOI] [PubMed] [Google Scholar]
- 258.Shackleford D.M., Porter C.J.H., Charman W.N. Lymphatic absorption of orally administered drugs. In: Stella V.J., Borchardt R.T., Hageman M.J., Oliyai R., Maag H., Tilley J., editors. Prodrugs: Challenges and Rewards. AAPS Press; New York: 2007. pp. 653–682. [Google Scholar]
- 259.Martin-Facklam M., Burhenne J., Ding R., Fricker R., Mikus G., Walter-Sack I., Haefeli W.E. Dose-dependent increase of saquinavir bioavailability by the pharmaceutic aid cremophor EL. Br. J. Clin. Pharmacol. 2002;53:576–581. doi: 10.1046/j.1365-2125.2002.01595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Bravo Gonzalez R.C., Huwyler J., Boess F., Walter I., Bittner B. In vitro investigation on the impact of the surface-active excipients Cremophor EL, Tween 80 and Solutol HS 15 on the metabolism of midazolam. Biopharm. Drug Dispos. 2004;25:37–49. doi: 10.1002/bdd.383. [DOI] [PubMed] [Google Scholar]
- 261.Rege B.D., Kao J.P., Polli J.E. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur. J. Pharm. Sci. 2002;16:237–246. doi: 10.1016/s0928-0987(02)00055-6. [DOI] [PubMed] [Google Scholar]
- 262.Mountfield R.J., Senepin S., Schleimer M., Walter I., Bittner B. Potential inhibitory effects of formulation ingredients on intestinal cytochrome P450. Int. J. Pharm. 2000;211:89–92. doi: 10.1016/s0378-5173(00)00586-x. [DOI] [PubMed] [Google Scholar]
- 263.Johnson B.M., Charman W.N., Porter C.J. An in vitro examination of the impact of polyethylene glycol 400, Pluronic P85, and vitamin E d-alpha-tocopheryl polyethylene glycol 1000 succinate on P-glycoprotein efflux and enterocyte-based metabolism in excised rat intestine. AAPS PharmSci. 2002;4:E40. doi: 10.1208/ps040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Sachs-Barrable K., Thamboo A., Lee S.D., Wasan K.M. Lipid excipients Peceol and Gelucire 44/14 decrease P-glycoprotein mediated efflux of rhodamine 123 partially due to modifying P-glycoprotein protein expression within Caco-2 cells. J. Pharm. Pharm. Sci. 2007;10:319–331. [PubMed] [Google Scholar]
- 265.Zordan-Nudo T., Ling V., Liu Z., Georges E. Effects of nonionic detergents on P-glycoprotein drug binding and reversal of multidrug resistance. Cancer Res. 1993;53:5994–6000. [PubMed] [Google Scholar]
- 266.Batrakova E., Lee S., Li S., Venne A., Alakhov V., Kabanov A. Fundamental relationships between the composition of pluronic block copolymers and their hypersensitization effect in MDR cancer cells. Pharm. Res. 1999;16:1373–1379. doi: 10.1023/a:1018942823676. [DOI] [PubMed] [Google Scholar]
- 267.Dean M., Hamon Y., Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- 268.Jia J.X., Wasan K.M. Effects of monoglycerides on rhodamine 123 accumulation, estradiol 17 beta-d-glucuronide bidirectional transport and MRP2 protein expression within Caco-2 cells. J. Pharm. Pharm. Sci. 2008;11:45–62. doi: 10.18433/j33s3z. [DOI] [PubMed] [Google Scholar]
- 269.Dintaman J.M., Silverman J.A. Inhibition of P-glycoprotein by d-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) Pharm. Res. 1999;16:1550–1556. doi: 10.1023/a:1015000503629. [DOI] [PubMed] [Google Scholar]
- 270.Chervinsky D.S., Brecher M.L., Hoelcle M.J. Cremophor-EL enhances taxol efficacy in a multi-drug resistant C1300 neuroblastoma cell line. Anticancer. Res. 1993;13:93–96. [PubMed] [Google Scholar]
- 271.Trevaskis N.L., Shackleford D.M., Charman W.N., Edwards G.A., Gardin A., Appel-Dingemanse S., Kretz O., Galli B., Porter C.J. Intestinal lymphatic transport enhances the post-prandial oral bioavailability of a novel cannabinoid receptor agonist via avoidance of first-pass metabolism. Pharm. Res. 2009;26:1486–1495. doi: 10.1007/s11095-009-9860-z. [DOI] [PubMed] [Google Scholar]
- 272.Gershkovich P., Qadri B., Yacovan A., Amselem S., Hoffman A. Different impacts of intestinal lymphatic transport on the oral bioavailability of structurally similar synthetic lipophilic cannabinoids: dexanabinol and PRS-211,220. Eur. J. Pharm. Sci. 2007;31:298–305. doi: 10.1016/j.ejps.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 273.Ichihashi T., Kinoshita H., Takagishi Y., Yamada H. Effect of bile on absorption of mepitiostane by the lymphatic system in rats. J. Pharm. Pharmacol. 1992;44:565–569. doi: 10.1111/j.2042-7158.1992.tb05465.x. [DOI] [PubMed] [Google Scholar]
- 274.Grimus R.C., Schuster I. The role of the lymphatic transport in the enteral absorption of naftifine by the rat. Xenobiotica; the fate of foreign compounds in biological systems. 1984;14:287–294. doi: 10.3109/00498258409151414. [DOI] [PubMed] [Google Scholar]
- 275.Palin K.J., Wilson C.G. The effect of different oils on the absorption of probucol in the rat. J. Pharm. Pharmacol. 1984;36:641–643. doi: 10.1111/j.2042-7158.1984.tb04919.x. [DOI] [PubMed] [Google Scholar]
- 276.Ueda C.T., Lemaire M., Gsell G., Nussbaumer K. Intestinal lymphatic absorption of cyclosporin A following oral administration in an olive oil solution in rats. Biopharm. Drug Dispos. 1983;4:113–124. doi: 10.1002/bdd.2510040203. [DOI] [PubMed] [Google Scholar]
- 277.Hauss D.J., Fogal S.E., Ficorilli J.V., Price C.A., Roy T., Jayaraj A.A., Keirns J.J. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor. J. Pharm. Sci. 1998;87:164–169. doi: 10.1021/js970300n. [DOI] [PubMed] [Google Scholar]
- 278.Nankervis R., Davis S.S., Day N.H., Shaw P.N. Intestinal lymphatic transport of three retinoids in the rat after oral administration: effect of lipophilicity and lipid vehicle. Int. J. Pharm. 1996;130:57–64. [Google Scholar]
- 279.Sieber S.M., Cohn V.H., Wynn W.T. The entry of foreign compounds into the thoracic duct lymph of the rat. Xenobiotica; the fate of foreign compounds in biological systems. 1974;4:265–284. doi: 10.3109/00498257409052055. [DOI] [PubMed] [Google Scholar]
- 280.Laher J.M., Rigler M.W., Vetter R.D., Barrowman J.A., Patton J.S. Similar bioavailability and lymphatic transport of benzo(a)pyrene when administered to rats in different amounts of dietary fat. J. Lipid Res. 1984;25:1337–1342. [PubMed] [Google Scholar]
- 281.O'Driscoll C.M., Myers R.A., Stella V.J. Blood and lymph transport of DDT after oral and parenteral administration to anaesthetised rats. Int. J. Pharm. 1991;73:177–183. [Google Scholar]
- 282.Khoo S.M., Porter C.J.H., Edwards G.A., Charman W.N. Intestinal lymphatic transport is the primary route for halofantrine after oral postprandial administration. Pharm. Sci. 1999;1:S-624. [Google Scholar]
- 283.Takada K., Yoshimura H., Shibata N., Masuda Y., Yoshikawa H., Muranishi S., Yasumura T., Oka T. Effect of administration route on the selective lymphatic delivery of cyclosporin A by lipid-surfactant mixed micelles. J. Pharmacobiodyn. 1986;9:156–160. doi: 10.1248/bpb1978.9.156. [DOI] [PubMed] [Google Scholar]
- 284.Ichihashi T., Kinoshita H., Takagishi Y., Yamada H. Intrinsic lymphatic partition rate of mepitiostane, epitiostanol, and oleic acid absorbed from rat intestine. Pharm. Res. 1991;8:1302–1306. doi: 10.1023/a:1015864131681. [DOI] [PubMed] [Google Scholar]
- 285.Neilson P.B., Mullertz A., Norling T., Kristensen H.G. Comparison of the lymphatic transport of a lipophilic drug from vehicles containing a-tocopherol and/or triglycerides in rats. J. Pharm. Pharmacol. 2001;53:1439–1445. doi: 10.1211/0022357011777972. [DOI] [PubMed] [Google Scholar]
- 286.Kwei G.Y., Novak L.B., Hettrick L.H., Reiss E.R., Fong E.K., Olah T.V., Loper A.E. Lymphatic uptake of MK-386, a sterol 5a-reductase inhibitor, from aqueous and lipid formulations. Int. J. Pharm. 1998;164:37–44. [Google Scholar]
- 287.Liu H.X., Adachi I., Horikoshi I., Ueno M. Mechanism of promotion of lymphatic drug absorption by milk fat globule membrane. Int. J. Pharm. 1995;118:55–64. [Google Scholar]