

Abstract
Numerous modifications of the well-known antimalarial drug primaquine, both at the quinoline ring and at the primary amino group, have been reported, mostly to obtain antimalarial agents with improved bioavailability, reduced toxicity and/or prolonged activity. Modifications of the terminal amino group were made with the main idea to prevent the metabolic pathway leading to inactive and toxic carboxyprimaquine (follow-on strategy), but also to get compounds with different activity (repurposing strategy). The modifications undertaken until 2009 were included in a review published in the same year. The present review covers various classes of primaquine N-derivatives with diverse biological profiles, prepared in the last decade by our research group as well as the others. We have summarized the synthetic procedures applied for their preparation and discussed the main biological results. Several hits for the development of novel antiplasmodial, anticancer, antimycobacterial and antibiofilm agents were identified.
Keywords: Primaquine derivatives, Antiplasmodial activity, Antiproliferative activity, Antibacterial activity, Antibiofilm, Antioxidative activity
Graphical abstract

1. Introduction
Although the development of first-in-class drugs with a novel mode of action is considered to be truly innovative, there are many other approaches used in drug research and development, like follow-on and analogue-based drug discovery, product-related research and platform drugs [1,2]. These strategies could result in lower costs, lower risk of failure, and shorter time to the market. Drug repositioning or repurposing, i.e. finding new indications for existing/approved drugs, is an increasingly utilized strategy in search of novel medicines [3,4], for example antiparasitic [5,6] and antimycobacterial use of antimalarial drugs [7,8]. Repurposing of antimalarial drugs as anticancer agents is particularly interesting, since different classes of antimalarials display either direct or adjuvant activity against cancer, act as sensitivity reversers of resistant tumour cell lines, inhibitors of drug resistance development, or synergistic agents with known anticancer drugs [[9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22]]. Hydroxychloroquine, chloroquine and other antimalarial agents (artesunate, quinacrine, atovaquone, doxycycline) were evaluated or are currently under evaluation in numerous clinical trials (approximately 150) against different cancer types, alone or in combination with conventional anticancer drugs/treatments [23]. Derivatization of the registered antimalarial drugs, particularly their hybridization with various scaffolds, is also a popular strategy in search of novel biologically active agents with diverse biological profiles [[24], [25], [26], [27], [28], [29], [30], [31], [32]].
We have focused our research on the antimalarial drug primaquine (PQ) and used it as a starting compound for further derivatization. PQ is an 8-aminoquinoline drug, developed and marketed in the 1940s. Today, after more than 70 years, it is still on the World Health Organization's List of Essential Medicines [33]. Primaquine is active against gametocytes and exoerythrocytic forms of all species of the parasite causing human malaria, including chloroquine-resistant Plasmodium falciparum [34]. Until the registration of tafenoquine in 2018, it was the only drug available worldwide against the latent liver forms of relapsing malaria caused by P. vivax and P. ovale. In general, it is considered as a safe and well-tolerated drug. However, applying primaquine at a broad scale is complicated due to the severe toxicity in a subset of patients with an inherited glucose-6-phosphate dehydrogenase (G6PD) deficiency [35]. The other pitfalls of primaquine are quick metabolism, degradation to inactive carboxyprimaquine and altering the treatment outcome depending on CYP 2D6 enzyme activity [36,37].
In order to get the antimalarial agents with improved bioavailability, reduced toxicity and/or prolonged activity, modifications of the primaquine structure, both at the quinoline heterocycle and at the primary amino group, were performed. The main goal of the terminal amino group modifications was to prevent the metabolic pathway leading to the inactive and toxic carboxyprimaquine [38]. The modifications undertaken before 2009 have been reviewed in detail by Vale et al. [35]. Until 2009, two main sets of compounds were described: primaquine imidazolidin-4-one derivatives (imidazoquines) [[39], [40], [41]] and amino acid/peptide derivatives [[42], [43], [44], [45]] or their combination – primaquine peptide derivatives bearing the imidazolidin-4-one moiety [46,47]. These compounds were evaluated primarily as antimalarial agents, but some of them were tested for anticancer [48] or antifungal activities [49,50]. However, the present review covers various classes of primaquine N-derivatives prepared in the last decade by our research group and others with two goals: to get novel antiplasmodial agents (follow-on strategy) or to get novel compounds with different biological activities (repurposing strategy).
2. N-modified primaquine derivatives prepared by our research group
Our research group has prepared various classes of primaquine derivatives: amides, carbamates, ureas, hydroxyureas, acylsemicarbazides, bis-ureas, ureidoamides and dicarboxylic acid derivatives. Their general structures are portrayed in Fig. 1 and the structures of the whole library of compounds are given in SI Tables 1–7. Most of the PQ-derivatives were prepared by common synthetic procedures or by the use of 1H-benzo[d][1,2,3]triazole (BtH) as a synthetic auxiliary. Versatile use of BtH in synthetic organic chemistry is well documented in the scientific literature [51,52]. Our research group also has a huge experience in the benzotriazole-assisted syntheses [53].
Fig. 1.

Chemical classes of the PQ-derivatives prepared by our research group.
The results have been published in 17 scientific papers. Here we have summarized the synthetic procedures applied for the preparation of various classes of primaquine derivatives and discussed the main biological results.
2.1. Syntheses of N-modified primaquine derivatives
2.1.1. Primaquine amide, carbamate, urea and semicarbazide derivatives
We shall start this review with the hybrid molecules bearing primaquine and cinnamic acid (CA) motifs. Cinnamic acid and its derivatives (CADs) are naturally occurring substances with various pharmacological activities: antimicrobial, antifungal, antiviral, antimycobacterial, antiinflammatory and antioxidative [[54], [55], [56], [57], [58], [59]]. The findings of their anticancer [[60], [61], [62]] and antimalarial activities [63] prompted us to prepare the PQ-CAD hybrids of amide and acylsemicarbazide type [64]. Cinnamic acid and the following CADs were used: α-methylcinnamic acid, methoxy, dimethoxy, trimethoxy, methylenedioxy, chloro, fluoro, trifluoromethyl and bis-trifluoromethyl cinnamic acid. The amides 1–11 were prepared by the simple acylation of PQ with CAD chlorides (CADCl) or CAD-benzotriazolide (CADBt) (Scheme 1 ). On the other hand, PQ-CAD acylsemicarbazides were obtained by acylation of precursor 61 with the corresponding CAD chlorides.
Scheme 1.

Synthesis of primaquine amides 1–18, carbamates 19, 20 and urea 42.
The amides of primaquine and cinnamic acids with alkyl, halogen or nitro substituents at different positions in the benzene moiety (N-cinnamoyl-primaquine conjugates, primacins) were prepared by Pérez et al. [66]. Primaquine was coupled with the corresponding cinnamic acids in the presence of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) and N,N-diisopropylethylamine (DIEA). Primacins have shown increased in vitro activity against the liver stage of the rodent malaria parasite P. berghei, compared to the parent drug primaquine [66] and only a modest activity against Leishmania infantum [67]. They were inactive against erythrocytic stage of P. falciparum. In 2017, the same Portuguese research group reported triple-stage antimalarial ionic liquids based on primaquine and cinnamic acids and found them superior to the analogous amide hybrids [68].
In order to obtain potential antioxidative and/or anticancer agents, we have prepared amides 12–18, conjugates of primaquine with non-steroidal anti-inflammatory drugs (NSAIDs): ibuprofen, ketoprofen, fenoprofen, hydroxy and methylene ketoprofen analogues, diclofenac and indomethacin (Scheme 1) [69]. These derivatives were designed bearing in mind several facts: i) inflammation is often accompanied by the excessive formation of reactive oxygen and nitrogen species that are potentially damaging to DNA and cell membranes; ii) oxidative stress is a major contributing factor to high mortality rates associated with several diseases, including malaria and cancer; iii) long-term use of NSAIDs is associated with a lower risk of some cancer types; iv) NSAID derivatives with pronounced cytostatic activity were described in the literature [[70], [71], [72], [73], [74]]. However, the results of biological screening of amides 12–18 were rather disappointing: our compounds exerted moderate activities in the DPPH free radical test and β-carotene-linoleic acid assay and negligible antiproliferative effects, although ketoprofen derivatives 13 and 15 demonstrated a notable Fe2+-chelating ability.
Acylation of primaquine with 1-benzotriazolecarboxylic acid chloride (BtcCl) [75,76] gave derivative 21 (BtcPQ), the common precursor for the synthesis of carbamates 19 and 20 and ureas, respectively (Scheme 1). Alcoholysis of benzotriazolide 21 was successful with methanol and ethanol, but failed with other alcohols [77]. A few years later, several O-alkyl and O-aryl primaquine carbamates were reported as potential prodrugs that prevent oxidative deamination to the inactive metabolite carboxyprimaquine [78]. The antiplasmodial screening revealed that O-alkyl carbamates have the potential to be developed as transmission-blocking antimalarial agents.
Aminolysis of benzotriazolide 21 proceeded under mild conditions, especially in the presence of triethylamine (TEA). More than twenty ureas were prepared (22–37, 40, 53–58), including twin derivative 41, in which two PQ-moieties were linked with carbonyl group (Scheme 2 ) [77,[79], [80], [81]]. Bis-derivative 41 was previously prepared by Kaur et al. [82]. The reaction of BtcPQ with O-benzylhydroxylamine gave compound 59, which afforded N-hydroxyurea 60 after catalytic hydrogenation [77]. Finally, compound 21 with hydrazine hydrate gave PQ-semicarbazide 61, a useful intermediate in the further reaction steps (Scheme 3 ) [80].
Scheme 2.

Synthesis of primaquine ureas.
Scheme 3.

Synthesis of primaquine hydroxyurea, bis-urea and acylsemicarbazide derivatives.
Another set of urea derivatives (38, 39, 43–52) was prepared in the opposite way, by the reaction of primaquine with the active ureas BtcNRR1, obtained from BtcCl and various amines [80,83]. Only urea 42 was synthesized by the classic isocyanate method, e.g. directly from PQ and 4-chloro-3-(fluoromethyl)phenyl isocyanate [80].
Syntheses of bis-urea derivatives were more complex. Two synthetic methods for the bis-ureas growing from the PQ side were applied: i) the reaction of the active ureas BtcNRR1 with the common precursor 61 (bis-ureas 75, 76) [80]; ii) the reaction of various amines with the active bis-urea intermediate obtained from 61 and BtcCl (bis-ureas 73, 74, 77, 78, 80–94, 100–104) [80,81,83]. When primaquine was used as an amine, symmetric bis-urea 78 bearing two primaquine moieties was prepared. The invert reactions were applied for the synthesis of bis-ureas 73, 74, 95–99, 105, i.e., the amine component was introduced in the first and the primaquine moiety in the final reaction step. Bis-urea 79 was obtained by the direct reaction of 61 with 4-chloro-3-(fluoromethyl)phenyl isocyanate. Finally, symmetric bis-urea 107 was prepared by the reaction of BtcPQ 21 and ethylenediamine in a molar ratio 2:1 (Scheme 3, Scheme 4 ) [77].
Scheme 4.

Synthesis of primaquine urea and bis-urea derivatives.
In order to obtain urea and bis-urea derivatives of different molecular size, lipophilicity, flexibility, metabolic stability and biological activity, various primary or secondary amines were used: alkyl and cycloalkyl amines [79], amino alcohols of different length and number of hydroxyl groups [77,80], more rigid amino alcohols bearing small cycloalkane moieties, fluoro-substituted amino alcohols [81], amines with one, two or three benzene rings [80,84], aniline derivatives with halogen (F, Cl, CF3) in m- or p-position or aminophenols [83]. These modifications were undertaken following common strategies in the drug design and discovery: conformation restrictions might increase selectivity, enhance lipophilicity and bioavailability, while the presence of fluorine atom(s) might increase bioavailability and selectivity [85].
The dimerization of the known antimalarial drugs is an old strategy to overcome drug resistance and improve antimalarial therapy outcomes [86]. One of the symmetric bisquinoline derivatives, piperaquine, was registered in the 1960s and was used in malaria prophylaxis and therapy for more than twenty years [87]. Today, it is approved only in a fixed-dose combination with other antimalarial drugs (dihydroartemisinin or arterolane) [88]. It is considered that bulky bisquinoline compounds accumulate in the cell compartment with a decreased pH gradient, inhibit parasite hem-digestive pathway and contribute to the inhibition of chloroquine efflux transporters [82,89]. The dimerization strategy became attractive in the discovery effective anticancer drugs among antimalarials as well (see for example ref. [[90], [91], [92]]). Having these facts in mind, we have prepared three bis-PQ derivatives (41, 78 and 107) with two primaquine moieties connected by linkers bearing one or two urea groups.
2.1.2. Primaquine ureidoamides
A quantitative structure-activity relationship (QSAR) study using the Support Vector Machines learning method yielded a highly accurate statistical model, which was used to prioritize novel candidate compounds, PQ-ureidoamides 108–113 [93,94]. These compounds were prepared by the reaction of PQ with N-Btc-amino acid amides, obtained from N-Btc-amino acid chlorides and the corresponding amines or hydrazone (Scheme 5 ). Three amino acids (l-leucine, d-phenylglycine, d,l-p-chlorophenylglycine), four amines (p-bromoaniline, diphenylmethanamine, (4-chlorophenyl)(phenyl)methanamine)), N-methyl-1,1-diphenylmethanamine and one hydrazone (4-methoxybenzophenone hydrazone) were used.
Scheme 5.

Synthesis of PQ-ureidoamides 108–113.
2.1.3. SAHAquines and related compounds
Our next goal was to prepare SAHAquines 129–132, hybrid molecules that combine moieties of suberoylanilide hydroxamic acid (SAHA), an anticancer agent with weak antiplasmodial activity, and primaquine, an antimalarial drug with low antiproliferative activity (Fig. 2 ). These compounds were designed as potential histone deacetylase (HDAC) inhibitors and as such, potential cytostatic and/or antiplasmodial agents. Several pan-HDAC inhibitors (SAHA, belinostat, panobinostat), marketed as anticancer drugs, exert antiplasmodial activity as well [[95], [96], [97], [98], [99]]. Their hydroxamic group binds zinc ion in Zn-dependent HDACs, leading to the accumulation of acetylated histones and other proteins [100]. In addition, three Zn-dependent HDACs prone to inhibition by SAHA and related metal-chelating drugs were identified in P. falciparum [101,102], thus represent viable targets for drug development [103].
Fig. 2.

Dicarboxylic acid derivatives 114–149.
Reaction pathway leading to SAHAquines 129–132 is presented in Scheme 6 [104,105]. In the first reaction step, dicarboxylic acid mono-esters were coupled with primaquine to give products 114–118, using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) as the coupling reagent, along with Hünig's base (DIEA). Basic hydrolysis of esters 114–118 afforded the corresponding acids 119–123, which were coupled with O-benzyl- or O-methylhydroxyl amine in the presence of HATU/DIEA and gave the corresponding O-protected hydroxamic acids 124–128 and 133–137, respectively. SAHAquines 129–132 were obtained by catalytic hydrogenation of O-benzyl hydroxamic acids. Derivatives of succinic, fumaric, glutaric, adipic and terephthalic acids were prepared.
Scheme 6.

Synthesis of dicarboxylic acid derivatives 114–149.
The synthetic procedure shown in Scheme 6 was applied for the synthesis of PQ-halogenaniline asymmetric diamides of fumaric acid (138–143) and succinic acid (144–149) [106]. Fumardiamides were designed as Michael acceptor analogues of bis-ureas 87–93, and succindiamides as the saturated counterparts of fumardiamides missing α,β-unsaturated carbonyl group (Michael acceptor moiety). Michel acceptors are often used in the drug design since they might assure the irreversible covalent binding to a cysteine residue of a specific protein [107]. Afatinib, neratinib, osimertinib, ibrutinib and exemestane are examples of registered anticancer drugs with Michael acceptors, while entacapone, dimethyl fumarate, rupintivir and ethacrynic acid are drugs used in the therapy of Parkinson's disease, psoriasis, multiple sclerosis, viral infections or hypertension, respectively. The above mentioned bis-ureas 87–93, fumardiamides and succindiamides differ only in the central part of the molecule (NHNH, CH CH or CH2CH2), while the PQ-residue and the amine component (3- or 4-fluoroaniline, 3- or 4-chloroaniline, 3- or 4-trifluoromethylaniline) remains unchanged. Structural formulas of bis-ureas, fumardiamides and succindiamides are compared in Fig. 3 .
Fig. 3.

Comparison of bis-urea derivatives 87–93, fumardiamides 138–143 and succindiamides 144–149.
2.1.4. Primaquine-polymer and glyco-conjugates
Primaquine conjugates 150–153 with glucosamine and two polymers of the polyaspartamide type, poly[α,β-(N-2-hydroxyethyl-d,l-aspartamide)] (PHEA) and poly[α,β-(N-3-hydroxypropyl-d,l-aspartamide)] (PHPA), were synthesized (Scheme 7 ) [108]. The conjugates differed in the type of covalent bonding (urea bond in 150, carbamate bond in 151–153), length of the spacer between the polymeric carrier and the drug, molecular mass and drug-loading.
Scheme 7.

Synthesis of PQ-glucosamine conjugate 150 and PQ-polymer conjugates 151–153.
2.2. Biological evaluation of primaquine derivatives
Our main goal was to find potential anticancer agents derived from primaquine, so the effectiveness of all novel compounds (approximately 150) against various cancer cell lines was evaluated. The second, but not less important goal was to find novel antiplasmodial agents. To get the complete biological profile of novel derivatives, their antimicrobial (antibacterial, antimycobacterial, antifungal, antibiofilm), antiviral, antioxidative potential and, for selected compounds, activity on the central nervous system (CNS) was evaluated as well. In general, bis-ureas and hydroxamic acids were superior in antiproliferative screening, ureidoamides and fumardiamides in antiplasmodial and antibacterial evaluation, ureas and fumardiamides in antimycobacterial, while acylsemicarbazides and fumardiamides in antiviral screening. Several hits, which might present a basis for the discovery of novel drug leads were identified.
2.2.1. Antiproliferative screening
To gain an insight into the antiproliferative profile of the synthesized primaquine derivatives, we evaluated their cytostatic activity in vitro against a panel of human cancer cell lines (SI Table 15). The results indicate that scaffold attached to primaquine has a significant impact both on activity and selectivity. Urea and bis-urea derivatives with one or two trifluoromethyl groups, chloro or methoxybenzhydryl moieties or trityl group, and SAHAquines with hydroxamic acid scaffold showed high activity to all tested cell lines, with the highest activity against human breast adenocarcinoma cell line (MCF-7) (for structures and references see Table 1 ). Urea 33 showed high selectivity towards the colorectal adenocarcinoma (SW620), whereas 29 was active against SW620 and murine lymphocytic leukaemia cell line (L120). However, the majority of the compounds displayed high selectivity towards MCF-7 cells, e.g. symmetric bis-PQ derivatives 41 and 78 and bis-ureas with hydroxy or halogenphenyl moieties 80–82, 85–91, 106. Such observation is not surprising since the sensitivity of MCF-7 cell line to primaquine and other antimalarial drugs has been documented earlier [47]. Bis-urea derivatives p-F (88) and p-OH (86) substituted benzene or hydroxypentyl (82) moieties, and twin primaquine derivative 78 were the most promising compounds in antiproliferative screening. Due to their high activity against MCF-7 cell line, extreme selectivity and favourable druglike properties, they may be considered as lead compounds in the discovery of breast carcinoma drugs.
Table 1.
The most important outcomes of antiproliferative screening in vitro.
| Compd. | Structure | IC50 (μM) | Ref. |
|---|---|---|---|
| All tested cell lines | |||
| 50, 51 | 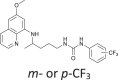 |
1.9–24 | [83] |
| 52 | 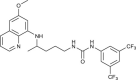 |
1.6–12 | [83] |
| 74 | 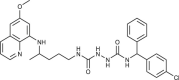 |
2–9.6 | [80] |
| 75 | 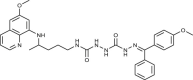 |
2–18 | [80] |
| 77 | 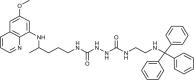 |
2–10 | [80] |
| 92, 93 | 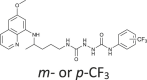 |
0.10–36 | [83] |
| 94 | 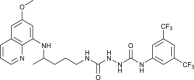 |
2.4–22 | [83] |
| 129–132 | 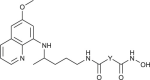 |
1.6–28 | [104] |
| MCF-7 | |||
| 11 | 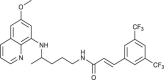 |
2.6 ± 0.5 | [64] |
| 38 | 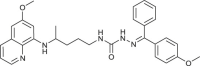 |
5 ± 3 | [80] |
| 39 | 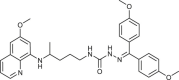 |
6 ± 2 | [80] |
| 41 | 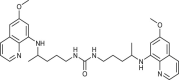 |
6 ± 3 | [80] |
| 78 | 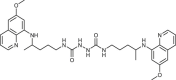 |
0.2 ± 0.1 | [80] |
| 80 | 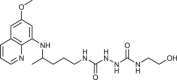 |
13 ± 4 | [80] |
| 81 | 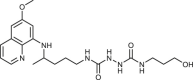 |
13 ± 0.5 | [80] |
| 82 | 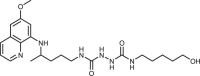 |
4 ± 0.9 | [80] |
| 85–91 | 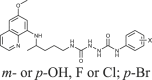 |
0.1–2.6 | [83] |
| 97 | 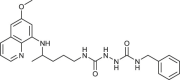 |
1 ± 0.2 | [84] |
| 98 | 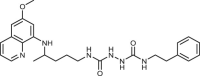 |
4 ± 0.3 | [84] |
| 106 | 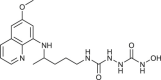 |
3 ± 0.2 | [84] |
| SW620 | |||
| 29 | 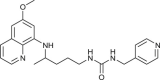 |
2 ± 1 | [79] |
| 33 | 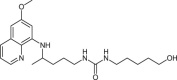 |
0.2 ± 1 | [77] |
| L120 | |||
| 29 | 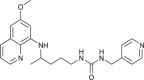 |
3.0 ± 0.4 | [79] |
In general, ureidoamides 108–113 and various dicarboxylic acid derivatives 114–128 and 133–149 were inactive [93,104], whereas the most active PQ-cinnamic acid derivatives were compounds 8, 64 and 71. Amide 8 with o-fluorine moiety significantly inhibited the growth of SW620, MCF-7 and cervical carcinoma (HeLa) cells (IC50 = 0.3–2.1 μM), acylsemicarbazides 64 (p-methoxy) against MCF-7, L1210, CEM and HeLa (IC50 = 0.4–2.4 2.4 μM), whereas 71 (p-CF3) showed activity towards MCF-7 cells in the nanomolar scale (IC50 = 0.03 ± 0.02 μM) and modest activity towards other tested cell lines [64]. To better understand the underlying mechanisms, the effects of compounds 8, 64 and 71 on MCF-7 cells were studied [65]. Morphological studies using haematoxylin and eosin staining revealed that compounds 8 and 71 induced morphological changes in cells characteristic for the apoptosis. Cell cycle analysis showed that cells treated with 8 and 71 were in the sub-G1, while cells treated with 64 were mainly in the interphase (G1 phase). The treatment of MCF-7 cells with 8 and 71 resulted in the poly ADP ribose polymerase (PARP) cleavage as well as caspase-9 activation, indicating the induction of apoptotic cell death. Compounds 8 and 71 inhibited the migration and invasion of MCF-7 cells, while compound 64 had marginal effect. Further biological experiments showed that bis-urea 88 induced growth inhibition in MCF-7 cell line through at least two different mechanisms: by induction of the cell cycle arrest in G1 phase and inhibition of the cell cycle progression through transient M phase accumulation and subsequent G2 arrest, and by inducing apoptosis [83]. On the other hand, SAHAquines 129–132 containing free hydroxamic acid proved to be HDAC inhibitors. The antibodies against acetylated histone H3K9/H3K14 in MCF-7 cells revealed a significant enhancement following treatment with 130 [104]. We have also shown that SAHAquine 130 selectively inhibits cytoplasmic HDAC6 in nanomolar concentration without markedly suppressing class I HDACs and suppresses epidermal growth factor receptor and downstream kinase activity, which are prominent therapeutic targets in glioblastoma multiforme [105]. In addition, compound 130 significantly reduces the viability and invasiveness of glioblastoma tumoroids, as well as brain tumour stem cells, which are crucial in the tumour survival and recurrence. These effects are augmented with the combination of SAHAquine 130 with temozolomide, the natural compound quercetin or buthionine sulfoximine, an inhibitor of glutathione biosynthesis.
2.2.2. Antiplasmodial evaluation
Antiplasmodial screening was performed in vitro and in vivo (only for the selected compounds). Five subsets of PQ derivatives with amide or urea moieties (57 compounds) were evaluated for antiplasmodial activity in vitro against the erythrocytic stage of P. falciparum NF54 [93,109,110]. Out of them, 33 compounds showed weak activity (IC50 = 10–20 μM) and 7 compounds strong activity in low micromolar range. QSAR analysis of the obtained data yielded a highly accurate statistical model which was used to prioritize novel candidate compounds: PQ-ureidoamides 108–113. These compounds were active against P. falciparum while exhibiting a very favourable toxicity profile towards human cells in vitro, which was retained even when the glucose-6-phosphate dehydrogenase (G6PD) was inhibited. On the other hand, bis-ureas 90, 93 and 76 with p-chlorobenzene, p-CF3-benzene and dimethoxybenzhydryl moieties displayed antiplasmodial activity in vitro against liver stage P. berghei in the nanomolar scale (IC50 = 42.0, 74.9 and 82.8 nM) [109]. Compound 93 showed antiproliferative activity in low micromolar concentrations as well. Asymmetric primaquine/halogenaniline fumardiamides 138–143 also exhibited high activity against P. berghei hepatic stages, higher than the parent drug (IC50 values ranging between 0.11 and 0.39 μM; IC50 of primaquine 8.4 ± 3.4 μM) and moderate activity against P. falciparum erythrocytic stages, PfDd2 and Pf3D7 strains (IC50 = 7.74–18.43 μM) [111]. The most active compounds 142 and 143 showed low cytotoxicity towards human cells (MCF-7 and HEK293T), also when G6PD enzyme was inhibited. On the other hand, SAHAquines 129–132 showed dual stage antiplasmodial activity which correlated with their cytostatic activity [104]. Finally, blood-schizontocidal activity of primaquine conjugates 150–153 with glucosamine and two polymers of polyaspartamide type (PHEA and PHPA) was tested against P. berghei infection in Swiss mice [108]. On the day 4, all the mice treated with polymeric conjugates were either aparasitemic or had significantly (p < 0.001) lower parasitaemia compared to negative-controls and the glucosamine conjugate. Among polymeric conjugates, compound 152 appeared to be the best with respect to the suppression of parasitaemia.
2.2.3. Antibacterial, antifungal and antibiofilm screening
Numerous quinoline-bearing compounds are well known antibacterial drugs. To mention just the most important ones: 8-hydroxyquinoline, nitroxoline and several generations of quinolone/fluoroquinolone derivatives (norfloxacin, ciprofloxacin, enoxacin, ofloxacin, moxifloxacin, etc.). Based on these facts, the in vitro antimicrobial activity of selected PQ derivatives (ureas 43–52, bis-ureas 85–94, ureidoamides 108–113, PQ-fumardiamides 138–143 and succindiamides 144–149) was carried out against a panel of Gram-positive bacteria, Gram-negative bacteria and fungi [83,94,106]. A list of microorganisms is given in Supplementary Material, SI Table 16. The tested compounds varied in activity (inactive, slightly active or active in low micromolar concentrations, but cytotoxic as well). However, some ureidoamides showed selective antimicrobial activity without haemolytic and cytotoxic effects (108 and 111 vs. Staphylococcus aureus strain, MIC = 6.5 μg/ml; 110 and 111 vs. Escherichia coli and Pseudomonas aeruginosa, MIC = 25 μg/ml). In addition, all ureidoamides had high efficiency against the biofilm formation of E. coli (the minimum biofilm eradication concentration, MBEC, from 6.25 to 50 μg/ml), and some of them against P. aeruginosa and Candida albicans [94]. On the other hand, fumardiamides 138–143 showed significant antimicrobial activity (e.g. compound 141 vs. S. aureus, Streptococcus pneumoniae, Acinetobacter baumannii, C. albicans, MIC = 6.1–12.5 μg/ml). Two microorganisms, namely E. faecalis and S. aureus showed high susceptibility to all fumardiamides, while E. coli, S. pneumoniae and P. aeruginosa were susceptible to five out of six fumardiamides. In biofilm eradication assay, majority of the bacteria, particularly S. aureus, showed susceptibility to all fumardiamides [106]. The most active compounds were fluoro-derivatives 138 and 139 (the lowest MBEC 6.3 μg/ml). A high biofilm eradication potential of fumardiamides might be explained by the reaction of Michael acceptor with cysteine thiol, which could prevent disulfide bond formation. It is a well-known fact that cysteine homeostasis impacts biofilm formation and production of extracellular matrix components, as well as folding and stability of extracytoplasmic proteins [112].
2.2.4. Antimycobacterial evaluation
Taking into account the fact that quinoline or quinolone pharmacophore is present in the antitubercular drugs bedaquiline and ciprofloxacin, antimycobacterial potential of more than seventy primaquine derivatives was evaluated against a number of Mycobacterium species (SI Table 16) [81,106,109]. After comparison of their activity and cytotoxicity several hits with MIC values from 2 to 16 μg/ml were identified: m-CF3-phenylurea 50, dimethoxybenzhydryl urea 76, urea 54 derived from (1-aminocyclobutyl)methanol, urea 58 and bis-urea 86 with phenol moieties [81]. Fumardiamides 139, 141 and 143 with p-fluorine, chlorine or CF3 substituents and bis-CF3 cinnamamide 11 also exerted significant antimycobacterial activity (MIC = 16–64 μg/ml) [106,109].
2.2.5. Antiviral evaluation
PQ-urea derivatives 22–30 [79], bis-urea derivatives 95–99, 105, 106 [84], PQ-cinnamic acid derivatives 1–11 and 62–72 [64], PQ-fumardiamides 138–143 and PQ-succindiamides 144–149 [106] were evaluated against a broad variety of viruses (SI Table 17). The ureas with phenylethyl and pyridine moieties (28 and 29) showed specific activity against human cytomegalovirus (HCMV) (EC50 = 1.2–13.4 μM), while cinnamic acylsemicarbazide derivatives 63, 67, 70–72 exerted moderate activity against human coronavirus (229E) with EC50 values between 7.9 and 15.0 μΜ. m-CF3 and m-chloroaniline fumardiamides 142 and 140 showed significant antiviral activity against reovirus-1, sindbis virus and Punta Toro virus (EC50 = 3.1–5.5 μM), while 142 was active against coxsackie virus B4 (EC50 = 3.1 μM) as well. Bis-urea derivatives, cinnamamides and PQ-succindiamides were inactive towards all tested viruses.
2.2.6. Antioxidative activity
A number of studies have shown the prooxidant effects of primaquine: treatment with primaquine causes an oxidative stress by generating reactive oxygen species within liver, kidney and erythrocytes [113,114]. Therefore, we found it worth to evaluate oxidant/antioxidant ability and lipoxygenase (LOX) inhibition of primaquine derivatives of amide and urea type [64,69,80,83,94]. Two antioxidant assays were used: (i) interaction with the stable free 1,1-diphenyl-2-picrylhydrazyl radical (DPPH test) and (ii) interaction with the water-soluble azo compound 2,2’-(2-amidinopropane) dihydrochloride (AAPH) (inhibition of lipid peroxidation, LP). Urea 44 [83], hydroxyurea 60 [77], hydroxy bis-urea 106 [84], semicarbazide 61 [80], PQ-cinnamic acid acylsemicarbazides 65–72 [64] and almost all bis-ureas 79, 85–94 [80,83] exerted potent activity, while PQ-NSAIDs 12–18 showed moderate antioxidative activity in DPPH test. Ureas 36–42 and bis-ureas 73–77 with lipophilic substituents showed stronger LP inhibition [80]. PQ-ureidoamides 109–113 significantly inhibited lipid peroxidation [94], whereas compound 110, the primaquine twin drug 107, cinnamic derivative 65, methoxy- or chlorobenzhydryl substituted ureas 37–39 and bis-ureas 74–76 presented higher LOX inhibition [77,80,82,94].
2.2.7. Activity of selected primaquine derivatives on CNS
Semicarbazide/urea PQ-derivatives are possible central nervous system effecting agents as well [[115], [116], [117]]. Selected compounds bearing these features, namely compounds 37, 74, 78 and 83 were screened for CNS activity [110]. Several behavioural tests were performed on mice: 'head-twitch' test, nociceptive reactions, influence on body temperature, locomotor activity (spontaneous and amphetamine-induced hyperactivity), motor coordination and pentylenetetrazole-induced convulsions. Compound 78 decreased the body temperature of normothermic mice, inhibited significantly 5-hydroxy-l-tryptophan-induced 'head-twitch' responses and protected mice against clonic seizures. All compounds decreased locomotor activity of mice, while derivatives 74 and 83 increased the hyperactivity caused by the amphetamine administration. All tested compounds showed antinociceptive activity in the ‘writhing’ test. Compounds 37 and 78 were active in a wide range of doses up to 0.00125 ED50 and their antinociceptive effect was reversed by naloxone. In the tail immersion test compound 37 exerted significant antinociceptive effect, which was also reversed by naloxone. None of the compounds caused coordination impairments in the rota-rod and chimney tests.
3. N-modified primaquine derivatives reported by other research groups
Various classes of primaquine derivatives were prepared by other research groups. Their general structures are presented in Fig. 4 and structural formula in SI Tables 8–14. Since these compounds were prepared as potential antiplasmodial agents, their biological outcomes are presented together with their chemical descriptions.
Fig. 4.

Chemical classes of PQ-derivatives prepared by other research groups.
3.1. Primaquine glycosides
Recently, Azad et al. prepared N-primaquine glycosides 154–161 to overcome the premature oxidative deamination and to increase the life span of the drug in the biological system [118]. The sugar part of the hybrid molecules was thought to direct the drug in the liver, where hypnozoites reside. The title conjugates were prepared by coupling of primaquine with the appropriate monosaccharide: d-glucose (154), d-galactose (155), d-mannose (156), d-arabinose (157), l-rhamnose (158), d-ribose (159), d-xylose (160) and N-acetyl-d-glucosamine (161) (Scheme 8 ). Compound 155 showed twofold the activity than that of the parent drug in experiments performed on rhesus monkeys.
Scheme 8.

Synthesis of PQ-glycosides 154–161.
3.2. Primaquine conjugates with other antimalarial drugs
3.2.1. Primaquine-artemisinin conjugates
Hybrid molecules capable of hitting more than one molecular target are an attractive alternative to currently approved antimalarial drugs. Thus, two antiplasmodial drugs with different modes of action, primaquine and artemisinin, were combined in hybrids 162–164 [119,120]. Amine and amide covalent bonds between primaquine and artemisinin were achieved according to the procedures presented in Scheme 9 .
Scheme 9.

Synthesis of PQ-artemisinin hybrids 162–164.
Hybrid 162 was prepared through several intermediates: artemisinin, dihydroartemisinin, artelinic acid, hydroxamate and corresponding aldehyde, which in reductive amination with primaquine afforded the title hybrid. Hybrid 164 was prepared from 10β-allyldeoxoartemisinin in two reaction steps, involving oxidation of the allyl to corresponding carboxylic acid and coupling of the latter with primaquine. Activities of primaquine-artemisinin conjugates against the liver and blood stages of Plasmodium were compared in vitro and in vivo with those of the parent drugs. Both hybrids displayed enhanced in vitro activities against P. berghei liver stages and similar activities as artemisinin against P. falciparum. In addition, compound 162 showed significant in vivo efficacy in controlling parasitaemia.
A small library of hybrid compounds 165–168 combining 1,2,4,5-tetraoxane (endoperoxide system similar to 1,3,4-trioxane present in artemisinin) and primaquine or other 8-aminoquinoline moieties was prepared and screened for the antimalarial activity (Scheme 10 ) [120,121]. These hybrids showed high potency against both exoerythrocytic and erythrocytic forms of malaria parasites and efficiently blocked the development of the sporogonic cycle in the mosquito vector. Compound 165 cleared P. berghei infection in mice. Similar hybrids with a trioxane motif (synthetic peroxide that causes damage in the parasites via the production of free radicals) linked to chloroquine and related aminoquinoline entities (which easily penetrate and accumulate within parasites) were also developed and extensively evaluated [[122], [123], [124], [125], [126]].
Scheme 10.

Synthesis of PQ-1,2,4,5-tetraoxane hybrids 165–168.
3.2.2. Primaquine-chloroquine conjugate
Primaquine-chloroquine conjugate 169 was obtained by heating of primaquine with 4,7-dichloroquinoline (Scheme 11 ) [127].
Scheme 11.

Synthesis of PQ-chloroquine hybrid 169.
The conjugate consisting of primaquine and chloroquine components was prepared to validate the concept of utilizing one compound to combine different modes of action that attack different Plasmodium stages. Indeed, conjugate 169 showed activity against both Plasmodium liver and blood stages, high activity against chloroquine resistant P. falciparum strain K1 and was superior to the equimolar combination of the parent drugs.
3.3. Primacenes – primaquine-ferrocene conjugates
Primaquine-ferrocene conjugates (primacenes) 170–180 appeared in 2012 as a new class of organometallic compounds. They were synthesized according to the Scheme 12 [128]. Ferrocene moiety was introduced in the molecule using ferrocene-carboxylic acid (FcCOOH) or 6-bromohexylferrocene (BHFc) and amino acid moiety with standard methods in peptide chemistry using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) hydrochloride/TEA or 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluoroborate (HBTU)/DIEA as the coupling reagents, tert-butoxycarbonyl (Boc) as N-protecting and succinimidyl (Su) as C-activating group.
Scheme 12.

Synthesis of PQ-ferrocene conjugates (primacenes) 170–180.
Primacenes were screened for their activity against blood and liver stage of Plasmodium in vitro and host-vector transmission in vivo. Both transmission-blocking and blood-schizontocidal activity of primaquine was conserved only in primacenes bearing a basic aliphatic amine group. The liver stage activity did not require this structural feature, and all tested metallocenes were comparable to the parent drug or even more active. The replacement of primaquine aliphatic chain by hexylferrocene led to a ∼45-fold-higher activity against liver stage parasitaemia in comparison to primaquine. Several primacenes displayed significant activity against Leishmania infantum with one of them being active against the clinically relevant intramacrophagic amastigote form of the parasite [129].
3.4. Primaquine derivatives with heterocyclic or carbocyclic scaffold
3.4.1. Primaquine-pyrimidine hybrids
A novel class of hybrids 181–190 combining primaquine and pyrimidine-5-carboxylate motif was prepared following Scheme 13 [130].
Scheme 13.

Synthesis of PQ-pyrimidine hybrids 181–190.
Three-component Biginelli condensation of various alkyl/aryl acetoacetates, urea and appropriate aldehyde gave 3,4-dihydropyrimidin-2(1H)-ones, which were further oxidized to pyrimidin-2(1H)-ones, chlorinated to 2-chloropyrimidin-2(1H)-ones and condensed with primaquine to give the target compounds. Effectiveness of PQ-pyrimidine hybrids 181–190 against blood and liver stages of malaria parasites was evaluated. The title compounds exhibited enhanced liver stage antiplasmodial activity in comparison to primaquine.
3.4.2. Primaquine-thiazolidinone hybrids
Primaquine-thiazolidinones 191–205 were recently prepared by multicomponent one-pot reactions from primaquine, mercaptoacetic acid and aromatic aldehyde bearing halogen, nitro, methoxy, methyl or cyano substituents at various positions (Scheme 14 ) [131].
Scheme 14.

Synthesis of PQ-thiazolidinone hybrids 191–205.
The title compounds were evaluated in several biological assays, including the ability to block malaria transmission to mosquitoes. All primaquine derivatives exhibited lower cell toxicity than primaquine and none caused haemolysis to normal or G6PD-deficient human erythrocytes in vitro. Several conjugates significantly inhibited sporogony in avian (P. gallinaceum) and rodent malaria (P. berghei). p-Methoxy primaquine-thiazolidinone hybrid was the most promising, blocking malaria transmissions and reducing the number of exoerythrocytic forms of P. berghei in hepatoma cells in vitro and in mice in vivo. The same compound also caused a 3-day delay in the malaria pre-patent period.
It is worth to note that the introduction of a similar 5-member heterocyclic ring, i.e. dihydrofuran-2-one with exocyclic C C bond conjugated with lactone C O group (Michael acceptor), to primaquine primary amine led to the discovery of antimalarial drug bulaquine (elubaquine) [132]. Since 2000, the bulaquine/chloroquine combination has been marketed in India for the treatment and prevention of P. vivax.
3.4.3. Primaquine-quinoxaline 1,4-di-N-oxide hybrids
PQ-quinoxaline hybrids 206–210 were recently prepared by a Brasilian research group [133]. The general synthetic approach for the synthesis of the title hybrids is presented in Scheme 15 . Reaction of primaquine and diketene provided the β-acetoacetamide derivative, which reacted with benzofuroxans and gave products 206–210.
Scheme 15.

Synthesis of PQ-quinoxaline 1,4-di-N-oxide hybrids 206–210.
Compound 206 and 207 were identified as the most active PQ-based hybrids against exoerythrocytic stages, displaying enhanced liver activity against P. yoelii and P. berghei. They also inhibited sporogony of P. berghei. In vivo liver efficacy assays revealed that compound 206 showed causal prophylactic activity affording parasitaemia reduction of up to 95% on day 4. On the other hand, analogous quinoxaline 1,4-di-N-oxide hybrids containing chloroquine showed high activity against the erythrocytic stage of the parasite [133].
3.4.4. Primaquine-squaramides
Squaramide is a unique scaffold widely used in medicinal chemistry. Primaquine mono-squaramide derivatives 211–213 with different alkoxy side chain were synthesized by reacting primaquine with 3,4-dialkoxy-3-cyclobutene-1,2-dione, whereas derivative 214 was prepared by the reaction of 211 with butylamine [134]. The preparative route leading to squaramides is given in Scheme 16 . Compound 213 was 7.3-fold more potent against liver stage parasites than the positive control primaquine. Antiplasmodial activity of this compound, similar 8- and 4-aminoquinoline squaramides and squaramide derivative with two primaquine moieties 215 against P. falciparum W2 strain was also reported, illustrating the importance of the squarate moiety for the activity [134,135].
Scheme 16.

Synthesis of PQ-squaramides 211–215.
3.4.5. Primaquine-atorvastatin hybrid
Atorvastatin is HMG CoA reductase inhibitor used in treatment of hypercholesterolemia. It also exerts antiplasmodial activity [136] and has synergistic effect with dihydroartemisinin [137,138]. These facts inspired Carvalho et al. to prepare PQ-atorvastatin hybrid 216 and several related aminoquinoline-atorvastatin conjugates [139]. Their preparation was achieved through Paal_Knorr reaction between 1,4-diketone derivative and corresponding aminoquinoline. Synthesis of compounds 210 is outlined in Scheme 17 .
Scheme 17.

Synthesis of PQ-atorvastatin hybrid 216.
Antiplasmodial activity in vitro against P. falciparum W2 clone (chloroquine resistant strain) and cytotoxicity assay against a monkey kidney cell line (BGM) showed that PQ-derivative was more active and less toxic than the parent drug.
3.5. Primaquine-amino acid/peptide conjugates
Numerous primaquine conjugates with amino acids, oligopeptides or pseudopeptides were reported before 2009 [34,42,45,47,140] and in the last decade [141,142]. Primaquine was conjugated to amino acids containing: i) a free terminal amino group, ii) a side-chain amino group of lysine or ornithine, iii) a carboxylic group of C-terminus or iv) a side-chain carboxylic group of aspartic or glutamic acid. Standard synthetic methods common in the peptide synthesis were applied for their preparation using adequate protecting groups and coupling reagents.
Antiplasmodial screening revealed that the conjugates containing cationic amino acids exhibit higher activities as compared to those containing anionic and lipophilic acids. For example, Lys-Lys-PQ and Arg-Arg-PQ were among the most active peptide conjugates against chloroquine-sensitive (D6) and chloroquine-resistant (W2) P. falciparum strains [141]. The same conjugates produced suppressive activity and partial cure in P. berghei mouse malaria model. On the other hand, Phe-Ala-PQ showed high activity against P. vivax and a more favourable pharmacokinetic profile compared to the parent drug [142], while Val-Leu-Lys-PQ expressed reduced toxicity and increased activity [41]. Despite the improved activity/toxicity ratio, most of these derivatives are rapidly hydrolysed to primaquine. However, introduction of imidazolidin-4-one moiety protects the N-terminal amino acid residue of di-, tri- and pentapeptides against aminopeptidase-catalysed hydrolysis [46].
Binding of primaquine to various carriers (lysosomotropic carriers, gum arabic microspheres) is an old idea [143,144]. Gomes and collaborators made this approach interesting again. They have recently reported synthesis of primaquine conjugates with cell-penetrating peptides transportan and transportan 10 [145]. Generally, the conjugates were more active against liver stage P. berghei parasites than the parent peptides and, in some cases, than the parent drug.
4. Conclusions
Derivatizations of the primaquine structure were primarily undertaken in search of novel antiplasmodial agents. Although the modification of the quinoline part of primaquine is not covered in this review, we want to stress that such efforts led to the discovery of a novel antimalarial drug tafenoquine. On the other hand, modification of the primaquine primary amino group was awarded by the registration of bulaquine (elubaquine). However, increasing resistance to the currently available antimalarial drugs, including artemisinin-based combination therapies, urges the need to develop novel and more efficient agents.
The literature survey revealed that the modification of primaquine molecule was quite explored in the last decade and successful in finding novel pharmacologically active agents. Highly reactive terminal amino group allowed modification to various derivatives (amide, carbamate, urea, semicarbazide, ureidoamide) or introduction of glycoside, peptide or heterocyclic (tetraoxane, pyrimidine, thiazolidone, quinoxaline, squaramide) moieties. Hybrids consisting of primaquine and another antiplasmodial drug (artemisinin, chloroquine) or drug from different therapeutic group (atorvastatin) As a result, high diversity of molecules with broad spectrum of activity and selectivity was achieved. Most of the prepared primaquine hybrids displayed more pronounced effect on the Plasmodium liver stages or better activity/toxicity ratio than the parent drug. On the other hand, some hybrids exerted dual activity against both stages of malaria parasite and were capable of hitting more than one molecular target. These compounds offer possibility for development of novel antiplasmodial agents active against resistant Plasmodium strains.
The molecular hybridization strategy was also successful in search of other biologically active agents outside the antiplasmodial field. Several compounds with encouraging antimycobacterial or antibiofilm activities were identified. Especially important are findings of novel antiproliferative agents displaying high potency to several cancer cell lines or high selectivity towards MCF-7 cells. A number of primaquine derivatives may be considered as lead compounds in the discovery of breast carcinoma drugs. In our opinion, this research area is still not exhausted and has a bright perspective with a lot of work that could be done in the future.
Acknowledgments
The study was supported by the Croatian Science Foundation through the research project IP-09-2014-1501. The work of doctoral student M. Beus has been fully supported by the Young researcher’s career development project – training of doctoral students of the Croatian Science Foundation founded by the European Union from the European Social Fund.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2019.111640.
Abbreviations
- AAPH
2,2’-(2-amidinopropane) dihydrochloride
- BHFc
6-bromohexylferrocene
- Bn
benzyl
- Boc
tert-butoxycarbonyl
- Bt
benzotriazolyl
- Btc
1-benzotriazolecarbonyl
- BtH
1H-benzo[d][1,2,3]triazole
- CA
cinnamic acid
- CAD
cinnamic acid derivative
- DIEA
N,N-diisopropylethylamine
- DPPH
2,2-diphenyl-1-picrylhydrazyl
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- Fc
ferrocene
- G6PD
glucose-6-phosphate dehydrogenase
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluoroborate
- HDAC
histone deacetylase
- IC50
the concentration that causes 50% growth inhibition
- LP
lipid peroxidation
- LOX
soybean lipoxygenase
- MBEC
minimum biofilm eradication concentration
- MIC
minimum inhibitory concentration
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NSAID
non-steroidal anti-inflammatory drug
- PARP
poly ADP ribose polymerase
- PHEA
poly[α,β-(N-2-hydroxyethyl-dl-aspartamide)]
- PHPA
poly[α,β-(N-3-hydroxypropyl-dl-aspartamide)]
- PQ
primaquine
- QSAR
quantitative structure-activity relationship
- SAHA
suberoylanilide hydroxamic acid
- Su
succinmidyl
- TBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate
- TEA
trimethylamine.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Giordanetto F., Bostrom J., Tyrchan C. Follow-on drugs: how far should chemists look? Drug Discov. Today. 2011;16:722–732. doi: 10.1016/j.drudis.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 2.Zhao H., Guo Z. Medicinal chemistry strategies in follow-on drug discovery. Drug Discov. Today. 2009;14:516–522. doi: 10.1016/j.drudis.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Oprea T.I., Mestres J. Drug repurposing: far beyond new targets for old drugs. AAPS. J. 2012;14:759–763. doi: 10.1208/s12248-012-9390-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashburn T.T., Thor K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 5.Rufener R., Ritler D., Zielinski J., Dick L., da Silva E.T., da Silva Araujo A., Joekel D.E., Czock D., Goepfert C., Moraes A.M., de Souza M.V.N., Müller J., Mevissen M., Hemphill A., Lundström-Stadelmann B. Activity of mefloquine and mefloquine derivatives against Echinococcus multilocularis. Int. J. Parasitol. Drugs Drug Resist. 2018;8:331–340. doi: 10.1016/j.ijpddr.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rufener R., Ritler D., Zielinski J., Dick L., da Silva E.T., da Silva Araujo A., Joekel D.E., Czock D., Goepfert C., Moraes A.M., de Souza M.V.N., Müller J., Mevissen M., Hemphill A., Lundström-Stadelmann B. Activity of mefloquine and mefloquine derivatives against Echinococcus multilocularis. Int. J. Parasitol. Drugs Drug Resist. 2018;8:331–340. doi: 10.1016/j.ijpddr.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krieger D., Vesenbeckh S., Schönfeld N., Bettermann G., Bauer T.T., Russmann H., Mauch H. Mefloquine as a potential drug against multidrug-resistant tuberculosis. Eur. Respir. J. 2015;46:1503–1505. doi: 10.1183/13993003.00321-2015. [DOI] [PubMed] [Google Scholar]
- 8.Bermudez L.E., Meek L. Mefloquine and its enantiomers are active against Mycobacterium tuberculosis in vitro and in macrophages. Tuberc. Res. Treat. 2014;2014(5):530815. doi: 10.1155/2014/530815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duffy R., Wade C., Chang R. Discovery of anticancer drugs from antimalarial natural products: a MEDLINE literature review. Drug Discov. Today. 2012;17:942–953. doi: 10.1016/j.drudis.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Solomon V.R., Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009;625:220–233. doi: 10.1016/j.ejphar.2009.06.063. [DOI] [PubMed] [Google Scholar]
- 11.Kimura T., Takabatake Y., Takahashi A., Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 12.Abdel-Aziz A.K., Shouman S., El-Demerdash E., Elgendy M., Abdel-Naim A.B. Chloroquine as a promising adjuvant chemotherapy together with sunitinib. Sci. Proc. 2014;1 Article ID e384. [Google Scholar]
- 13.Liu F., Shang Y., Chen S.-Z. Chloroquine potentiates the anti-cancer effect of lidamycin on non-small cell lung cancer cells in vitro. Acta Pharmacol. Sin. 2014;35:645–652. doi: 10.1038/aps.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ganguli A., Choudhury D., Datta S., Bhattacharya S., Chakrabarti G. Inhibition of autophagy by chloroquine potentiates synergistically anti-cancer property of artemisinin by promoting ROS dependent apoptosis. Biochimie. 2014;107:338–349. doi: 10.1016/j.biochi.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Choi A.R., Kim J.H., Woo Y.H., Kim H.S., Yoon S. Anti-malarial drugs primaquine and chloroquine have different sensitization effects with anti-mitotic drugs in resistant cancer cells. Anticancer Res. 2016;36:1641–1648. [PubMed] [Google Scholar]
- 16.Verbaanderd C., Maes H., Schaaf M.B., Sukhatme V.P., Pantziarka P., Sukhatme V., Agostinis P., Bouche G. Repurposing drugs in oncology (ReDO) – chloroquine and hydroxychloroquine as anti-cancer agents. eCancer. 2017;11 doi: 10.3332/ecancer.2017.781. Article ID 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L., Han C., Yu H., Zhu W., Cui H., Zheng L., Zhang C., Yue L. Chloroquine inhibits cell growth in human A549 lung cancer cells by blocking autophagy and inducing mitochondrial-mediated apoptosis. Oncol. Rep. 2018;39:2807–2816. doi: 10.3892/or.2018.6363. [DOI] [PubMed] [Google Scholar]
- 18.Wang F., Tang J., Li P., Si S., Yu H., Yang X., Tao J., Lv Q., Gu M., Yang H., Wang Z. Chloroquine enhances the radiosensitivity of bladder cancer cells by inhibiting autophagy and activating apoptosis. Cell. Physiol. Biochem. 2018;45:54–66. doi: 10.1159/000486222. [DOI] [PubMed] [Google Scholar]
- 19.van Huijsduijnen R.H., Kiplin Guy R., Chibale K., Haynes R.K., Peitz I., Kelter G., Phillips M.A., Vennerstrom J.L., Yuthavong Y., Wells T.N.C. Anticancer properties of distinct antimalarial drug classes. PLoS One. 2013;8:e82962. doi: 10.1371/journal.pone.0082962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamal A., Aziz A., Shouman S., El-Demerdash E., Elgendy M., Abdel-Naim A.B. Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries. Chem. Biol. Interact. 2014;217:28–40. doi: 10.1016/j.cbi.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Soo G.W., Law J.H., Kan E., Tan S.Y., Lim W.Y., Chay G., Bukhari N.I., Segarra I. Differential effects of ketoconazole and primaquine on the pharmacokinetics and tissue distribution of imatinib in mice. Anti Canccer Drugs. 2010;21:695–703. [PubMed] [Google Scholar]
- 22.Wong Y.K., Xu C., Kalesh K.A., He Y., Lin Q., Wong W.S.F., Shen H.M., Wang J. Artemisinin as an anticancer drug: recent advances in target profiling and mechanisms of action. Med. Res. Rev. 2017;37:1492–1517. doi: 10.1002/med.21446. [DOI] [PubMed] [Google Scholar]
- 23.https://clinicaltrials.gov/ct2/home
- 24.Gonçalves R.S., Kaiser C.R., Lourenço M.C., Bezerra F.A., de Souza M.V., Wardell J.L., Wardell S.M., Henriques M.D., Costa T. Mefloquine-oxazolidine derivatives, derived from mefloquine and arenecarbaldehydes: in vitro activity including against the multidrug-resistant tuberculosis strain T113. Bioorg. Med. Chem. 2012;20:243–248. doi: 10.1016/j.bmc.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Mao J., Yuan H., Wang Y., Wan B., Pak D., He R., Franzblau S.G. Synthesis and antituberculosis activity of novel mefloquine-isoxazole carboxylic esters as prodrugs. Bioorg. Med. Chem. Lett. 2010;20:1263–1268. doi: 10.1016/j.bmcl.2009.11.105. [DOI] [PubMed] [Google Scholar]
- 26.Eswaran S., Adhikari A.V., Chowdhury I.H., Pal N.K., Thomas K.D. New quinoline derivatives: synthesis and investigation of antibacterial and antituberculosis properties. Eur. J. Med. Chem. 2010;45:3374–3383. doi: 10.1016/j.ejmech.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 27.Pérez B.C., Fernandes I., Mateus N., Teixeira C., Gomes P. Recycling antimalarial leads for cancer: antiproliferative properties of N-cinnamoyl chloroquine analogues. Bioorg. Med. Chem. Lett. 2013;23:6769–6772. doi: 10.1016/j.bmcl.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 28.Slezakova S., Ruda-Kucerova J. Anticancer activity of artemisinin and its derivatives. Anticancer Res. 2017;37:5995–6003. doi: 10.21873/anticanres.12046. [DOI] [PubMed] [Google Scholar]
- 29.Sun Q., Wang J., Li Y., Zhuang J., Zhang Q., Sun X., Sun D. Synthesis and evaluation of cytotoxic activities of artemisinin derivatives. Chem. Biol. Drug Des. 2017;90:1019–1028. doi: 10.1111/cbdd.13016. [DOI] [PubMed] [Google Scholar]
- 30.Gomes A., Fernandes I., Teixeira C., Mateus N., Sottomayor M.J., Gomes P. A quinacrine analogue selective against gastric cancer cells: insight from biochemical and biophysical studies. ChemMedChem. 2016;11:2703–2712. doi: 10.1002/cmdc.201600477. [DOI] [PubMed] [Google Scholar]
- 31.Zhang F.F., Li J. Inhibitory effect of chloroquine derivatives on presenilin 1 and ubiquilin 1 expression in Alzheimer's disease. Int. J. Clin. Exp. Pathol. 2015;8:7640–7643. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Zhang Y., Xu G., Zhang S., Wang D., Prabha P.S., Zuo Z. Antitumor research on artemisinin and its bioactive derivatives. Nat. Prod. Bioprospect. 2018;8:303–319. doi: 10.1007/s13659-018-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.https://en.wikipedia.org/wiki/WHO_Model_List_of_Essential_Medicines
- 34.Kaur K., Jain M., Khan S.I., Jacob M.R., Tekwani B.L., Singh S., Singh P.P., Jain R. Amino acid, dipeptide and pseudodipeptide conjugates of ring-substituted 8-aminoquinolines: synthesis and evaluation of anti-infective, β-haematin inhibition and cytotoxic activities. Eur. J. Med. Chem. 2012;52:230–241. doi: 10.1016/j.ejmech.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 35.Vale N., Moreira R., Gomes P. Primaquine revisited six decades after its discovery. Eur. J. Med. Chem. 2009;44:937–953. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Pybus B.S., Marcsisin S.R., Jin X., Deye G., Sousa J.C., Li Q., Caridha D., Zeng Q., Reichard G.A., Ockenhouse C., Bennett J., Walker L.A., Ohrt C., Melendez V. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar. J. 2013;12:e212. doi: 10.1186/1475-2875-12-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcsisin S.R., Reichard G., Pybus B.S. Primaquine pharmacology in the context of CYP 2D6 pharmacogenomics: current state of the art. Pharmacol. Ther. 2016;161:1–10. doi: 10.1016/j.pharmthera.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 38.Kaur K., Jain M., Khan S.I., Jacob M.R., Tekwani B.L., Singh S., Pal Singh P., Jain R. Extended side chain analogues of 8-aminoquinolines: synthesis and evaluation of antiprotozoal, antimicrobial, β-hematin inhibition, and cytotoxic activities. MedChemComm. 2011;2:300–307. [Google Scholar]
- 39.Gomes P., Araújo M.R., Rodrigues M., Vale N., Azevedo Z., Iley J., Chambel P., Morais J., Moreira R. Synthesis of imidazolidin-4-one and 1H-imidazo[2,1-a]isoindole-2,5(3H,9bH)-dione derivatives of primaquine: scope and limitations. Tetrahedron. 2004;60:5551–5562. [Google Scholar]
- 40.Araújo M.J., Bom J., Capela R., Casimiro C., Chambel P., Gomes P., Iley J., Lopes F., Morais J., Moreira R., de Oliveira E., do Rosário V., Vale N. Imidazolidin-4-one derivatives of primaquine as novel transmission-blocking antimalarials. J. Med. Chem. 2005;48:888–892. doi: 10.1021/jm0494624. [DOI] [PubMed] [Google Scholar]
- 41.Vale N., Matos J., Gut J., Nogueira F., do Rosário V., Rosenthal P.J., Moreira R., Gomes P. Imidazolidin-4-one peptidomimetic derivatives of primaquine: synthesis and antimalarial activity. Bioorg. Med. Chem. Lett. 2008;18:4150–4153. doi: 10.1016/j.bmcl.2008.05.076. [DOI] [PubMed] [Google Scholar]
- 42.Philip A., Kepler J.A., Johnson B.H., Carroll F.I. Peptide derivates of primaquine as potential antimalarial agents. J. Med. Chem. 1988;31:870–874. doi: 10.1021/jm00399a032. [DOI] [PubMed] [Google Scholar]
- 43.Jain R., Jain S., Gupta R.C., Anand N., Dutta G.P., Puri S.K. Synthesis of amino acid derivatives of 8-[(4-amino-1-methylbutyl)amino]-6-methoxy-4-substituted/4,5-disubstituted-quinolines as potential antimalarial agents. Indian J. Chem. 1994;33B:251–254. [Google Scholar]
- 44.Portela M.J., Moreira R., Valente E., Constantino L., Iley J., Pinto J., Rosa R., Cravo P., do Rosário V.E. Dipeptide derivatives of primaquine as transmission-blocking antimalarials: effect of aliphatic side-chain acylation on the gametocytocidal and on the formation of carboxyprimaquine in rat liver homogenates. Pharm. Res. 1999;16:949–955. doi: 10.1023/a:1018922425551. [DOI] [PubMed] [Google Scholar]
- 45.Vangapandu S., Sachdeva S., Jain M., Singh S., Singh P.P., Kaul C.L., Jain R. 8-Quinolinamines conjugated with amino acids are exhibiting potent blood-schizontocidal antimalarial activities. Bioorg. Med. Chem. 2004;12:239–247. doi: 10.1016/j.bmc.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 46.Vale N., Matos J., Moreira R., Gomes P. Amino acids as selective acylating agents: regioselective N1-acylation of imidazolidin-4-one derivatives of the antimalarial drug primaquine. Tetrahedron. 2008;64:11144–11149. [Google Scholar]
- 47.Vale N., Nogueira F., do Rosário V.E., Gomes P., Moreira R. Primaquine dipeptide derivatives bearing an imidazolidin-4-one moiety at the N-terminus as potential antimalarial prodrugs. Eur. J. Med. Chem. 2009;44:2506–2516. doi: 10.1016/j.ejmech.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 48.Fernandes I., Vale N., de Freitas V., Moreira R., Mateus N., Gomes P. Anti-tumoral activity of imidazoquines, a new class of antimalarials derived from primaquine. Bioorg. Med. Chem. Lett. 2009;19:6914–6917. doi: 10.1016/j.bmcl.2009.10.081. [DOI] [PubMed] [Google Scholar]
- 49.Vale N., Collins M.S., Gut J., Ferraz R., Rosenthal P.J., Cushion M.T., Moreira R., Gomes P. Anti-Pneumocystis carinii and antiplasmodial activities of primaquine-derived imidazolidin-4-ones. Bioorg. Med. Chem. Lett. 2008;18:485–488. doi: 10.1016/j.bmcl.2007.11.105. [DOI] [PubMed] [Google Scholar]
- 50.Vale N., Prudêncio M., Marques C.A., Collins M.S., Gut J., Nogueira F., Matos J., Rosenthal P.J., Cushion M.T., do Rosário V.E., Mota M.M., Moreira R., Gomes P. Imidazoquines as antimalarial and anti-pneumocystis agents. J. Med. Chem. 2009;52:7800–7807. doi: 10.1021/jm900738c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katritzky A.R., Lan X., Yang J.Z., Denisko O.V. Properties and synthetic utility of N-substituted benzotriazoles. Chem. Rev. 1998;98:409–548. doi: 10.1021/cr941170v. [DOI] [PubMed] [Google Scholar]
- 52.Monbaliu J.-C.M., editor. The Chemistry of Benzotriazole Derivatives: A Tribute to Alan Roy Katritzky (Topics in Heterocyclic Chemistry) Springer; New York: 2016. [Google Scholar]
- 53.Zorc B., Rajić Džolić Z., Butula I. Benzotriazole as a synthetic auxiliary. Croat. Chem. Acta. 2012;85:595–602. [Google Scholar]
- 54.Carvalho S.A., da Silva E.F., de Souza M.V., Lourenco M.C., Vicente F.R. Synthesis and antimycobacterial evaluation of new trans-cinnamic acid hydrazide derivatives. Bioorg. Med. Chem. Lett. 2008;18:538–541. doi: 10.1016/j.bmcl.2007.11.091. [DOI] [PubMed] [Google Scholar]
- 55.Bairwa R., Kakwani M., Tawari N.R., Lalchandani J., Ray M.K., Rajan M.G.R., Degani M.S. Novel molecular hybrids of cinnamic acids and guanylhydrazones as potential antitubercular agents. Bioorg. Med. Chem. 2010;20:1623–1625. doi: 10.1016/j.bmcl.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 56.Kakwani M.D., Suryavanshi P., Ray M., Rajan M.G.R., Majee S., Samad A., Devarajan P., Degani M.S. Design, synthesis and antimycobacterial activity of cinnamide derivatives: a molecular hybridization approach. Bioorg. Med. Chem. Lett. 2011;21:1997–1999. doi: 10.1016/j.bmcl.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 57.Lone R., Shuab R., Koul K.K. Role of cinnamate and cinnamate derivatives in pharmacology. Glob. J. Pharmacol. 2014;8:328–335. [Google Scholar]
- 58.Yoya G.K., Bedos-Belval F., Constant P., Duran H., Daffé M., Baltas M. Synthesis and evaluation of a novel series of pseudo-cinnamic derivatives as antituberculosis agents. Bioorg. Med. Chem. Lett. 2009;19:341–343. doi: 10.1016/j.bmcl.2008.11.082. [DOI] [PubMed] [Google Scholar]
- 59.Sharma P.J. Cinnamic acid derivatives: a new chapter of various pharmacological activities. Chem. Pharm. Res. 2011;3:403–423. [Google Scholar]
- 60.Pontiki E., Hadjipavlou-Litina D., Litinas K., Geromichalos G. Novel cinnamic acid derivatives as antioxidant and anticancer agents: design, synthesis and modeling studies. Molecules. 2014;19:9655–9674. doi: 10.3390/molecules19079655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De M., Baltas P., Bedos-Belval F. Cinnamic acid derivatives as anticancer agents – a review. Curr. Med. Chem. 2011;18:1672–1703. doi: 10.2174/092986711795471347. [DOI] [PubMed] [Google Scholar]
- 62.Pérez B.C., Fernandes I., Mateus N., Teixeira C., Gomes P. Recycling antimalarial leads for cancer: antiproliferative properties of N-cinnamoyl chloroquine analogues. Bioorg. Med. Chem. Lett. 2013;23:6769–6772. doi: 10.1016/j.bmcl.2013.10.025. [DOI] [PubMed] [Google Scholar]
- 63.Kanaani J., Ginsburg H. Effects of cinnamic acid derivatives on in vitro growth of Plasmodium falciparum and on the permeability of the membrane of malaria-infected erythrocytes. Antimicrob. Agents Chemother. 1992;36:1102–1108. doi: 10.1128/aac.36.5.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pavić K., Perković I., Gilja P., Kozlina F., Ester K., Kralj M., Schols D., Hadjipavlou-Litina D., Pontiki E., Zorc B. Design, synthesis and biological evaluation of novel primaquine-cinnamic acid conjugates of amide and acylsemicarbazide type. Molecules. 2016;21:1629–1653. doi: 10.3390/molecules21121629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mabeta P., Pavić K., Zorc B. Insights into mechanism of antiproliferative effect of primaquine-cinnamic acid conjugates on MCF-7. Acta Pharm. 2018;68:337–348. doi: 10.2478/acph-2018-0021. [DOI] [PubMed] [Google Scholar]
- 66.Pérez B., Teixeira C., Albuquerque I.S., Gut J., Rosenthal P.J., Prudencio M., Gomes P. PRIMACINS, N-cinnamoyl-primaquine conjugates, with improved liver-stage antimalarial activity. MedChemComm. 2012;3:1170–1172. [Google Scholar]
- 67.Vale-Costa S., Costa-Gouveia J., Pérez B., Silva T., Teixeira C., Gomes P., Gomes M.S. N-cinnamoylated aminoquinolines as promising antileishmanial agents. Antimicrob. Agents Chemother. 2013;57:5112–5115. doi: 10.1128/AAC.00557-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferraz R., Pinheiro M., Gomes A., Teixeira C., Prudêncio C., Reis S., Gomes P. Effects of novel triple-stage antimalarial ionic liquids on lipid membrane models. Bioorg. Med. Chem. Lett. 2017;27:4190–4193. doi: 10.1016/j.bmcl.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 69.Rajić Z., Zovko Končić M., Miloloža K., Perković I., Butula I., Bucar F., Zorc B. Primaquine-NSAID twin drugs: synthesis, radical scavenging, antioxidant and Fe2+ chelating activity. Acta Pharm. 2010;60:325–337. doi: 10.2478/v10007-010-0024-9. [DOI] [PubMed] [Google Scholar]
- 70.Andrews P., Zhao X., Allen J., Li F., Chang M. A comparison of the effectiveness of selected non-steroidal anti-inflammatory drugs and their derivatives against cancer cells in vitro, Cancer Chemother. Pharmacol. 2008;61:203–214. doi: 10.1007/s00280-007-0462-3. [DOI] [PubMed] [Google Scholar]
- 71.Rayburn E.R., Ezell S.J., Zhang R. Anti-inflammatory agents for cancer therapy. Mol. Cell. Pharmacol. 2009;1:29–43. doi: 10.4255/mcpharmacol.09.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang D., DuBois R.N. Eicosanoids and cancer. Nat. Rev. Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zawidlak-Węgrzyńska B., Kawalec M., Bosek I., Łuczyk-Juzwa M., Adamus G., Rusin A., Filipczak P., Głowala-Kosińska M., Wolańska K., Krawczyk Z., Kurcok P. Synthesis and antiproliferative properties of ibuprofen-oligo(3-hydroxybutyrate) conjugates. Eur. J. Med. Chem. 2010;45:1833–1842. doi: 10.1016/j.ejmech.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 74.Fogli S., Banti I., Stefanelli F., Picchianti L., Digiacomo M., Macchia M., Breschi M.C., Lapucci A. Therapeutic potential of sulindac hydroxamic acid against human pancreatic and colonic cancer cells. Eur. J. Med. Chem. 2010;45:5100–5107. doi: 10.1016/j.ejmech.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 75.Butula I., Proštenik M.V., Vela V. Reactions with 1-benzotriazole carboxylic acid chloride. I. Synthesis of the 2,6-bis(hydroxymethyl)pyridine dicarbamates. Croat. Chem. Acta. 1977;49:837–842. [Google Scholar]
- 76.Kalčić I., Zovko M., Jadrijević-Mladar Takač M., Zorc B., Butula I. Synthesis and reactions of some azolecarboxylic acid derivatives. Croat. Chem. Acta. 2003;76:217–228. [Google Scholar]
- 77.Šimunović M., Perković I., Zorc B., Ester K., Kralj M., Hadjipavlou-Litina D., Pontiki E. Urea and carbamate derivatives of primaquine: synthesis, cytostatic and antioxidant activities. Bioorg. Med. Chem. 2009;17:5605–5613. doi: 10.1016/j.bmc.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 78.Mata G., do Rosário V.E., Iley J., Constantino L., Moreira R. A carbamate-based approach to primaquine prodrugs: antimalarial activity, chemical stability and activation. Bioorg. Med. Chem. 2012;20:886–892. doi: 10.1016/j.bmc.2011.11.059. [DOI] [PubMed] [Google Scholar]
- 79.Džimbeg G., Zorc B., Kralj M., Ester K., Pavelić K., Balzarini J., De Clercq E., Mintas M. The novel primaquine derivatives of N-alkyl, cycloalkyl or aryl urea: synthesis, cytostatic and antiviral activity evaluations. Eur. J. Med. Chem. 2008;43:1180–1187. doi: 10.1016/j.ejmech.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 80.Pavić K., Perković I., Cindrić M., Pranjić M., Martin-Kleiner I., Kralj M., Schols D., Hadjipavlou-Litina D., Katsori A.-M., Zorc B. Novel semicarbazides and ureas of primaquine with bulky aryl or hydroxyalkyl substituents: synthesis, cytostatic and antioxidative activity. Eur. J. Med. Chem. 2014;86:502–514. doi: 10.1016/j.ejmech.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 81.Pavić K., Rajić Z., Michnová H., Jampílek J., Perković I., Zorc B. Second generation of primaquine ureas and bis-ureas as potential antimycobacterial agents. Mol. Divers. 2019;23:657–667. doi: 10.1007/s11030-018-9899-z. [DOI] [PubMed] [Google Scholar]
- 82.Kaur K., Jain M., Khan S.I., Jacob M.R., Tekwani B.L., Singh S., Singh P.P., Jain R. Synthesis, antiprotozoal, antimicrobial, β-hematin inhibition, cytotoxicity and methemoglobin (MetHb) formation activities of bis(8-aminoquinolines) Bioorg. Med. Chem. 2011;19:197–210. doi: 10.1016/j.bmc.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perković I., Antunović M., Marijanović I., Pavić K., Ester K., Kralj M., Vlainić J., Kosalec I., Schols D., Hadjipavlou-Litina D., Pontiki E., Zorc B. Novel urea and bis-urea primaquine derivatives with hydroxyphenyl and halogenphenyl substituents: synthesis and biological evaluation. Eur. J. Med. Chem. 2016;124:622–636. doi: 10.1016/j.ejmech.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 84.Perković I., Tršinar S., Žanetić J., Kralj M., Martin-Kleiner I., Balzarini J., Hadjipavlou-Litina D., Katsori A.M. Novel 1-acyl-4-substituted semicarbazide derivatives of primaquine − synthesis, cytostatic, antiviral and antioxidative studies. J. Enzym. Inhib. Med. Chem. 2013;28:601–610. doi: 10.3109/14756366.2012.663366. [DOI] [PubMed] [Google Scholar]
- 85.Reddy V. first ed. Elsevier; Amsterdam: 2015. Organofluorine Compounds in Biology and Medicine. [Google Scholar]
- 86.Raynes K. Bisquinoline antimalarials: their role in malaria chemotherapy. Int. J. Parasitol. 1999;29:367–379. doi: 10.1016/s0020-7519(98)00217-3. [DOI] [PubMed] [Google Scholar]
- 87.Davis T.M., Hung T.Y., Sim I.K., Karunajeewa H.A., Ilett K.F. Piperaquine: a resurgent antimalarial drug. Drugs. 2005;65:75–87. doi: 10.2165/00003495-200565010-00004. [DOI] [PubMed] [Google Scholar]
- 88.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001199/WC500118113.pdf
- 89.Patil C.Y., Katare S.S., Baig M.S., Doifode S.M. Fixed dose combination of arterolane and piperaquine: a newer prospect in antimalarial therapy. Ann. Med. Health Sci. Res. 2014;4:466–471. doi: 10.4103/2141-9248.139270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Buragohain P., Saikia B., Surineni N., Barua N.C., Saxena A.K., Suri N. Synthesis of a novel series of artemisinin dimers with potent anticancer activity involving Sonogashira cross-coupling reaction. Bioorg. Med. Chem. Lett. 2014;24:237–239. doi: 10.1016/j.bmcl.2013.11.032. [DOI] [PubMed] [Google Scholar]
- 91.Fröhlich T., Karagöz A.Ç., Reiter C., Tsogoeva S.B. Artemisinin-derived dimers: potent antimalarial and anticancer agents. J. Med. Chem. 2016;59:7360–7388. doi: 10.1021/acs.jmedchem.5b01380. [DOI] [PubMed] [Google Scholar]
- 92.Lai H.C., Singh N.P., Sasaki T. Development of artemisinin compounds for cancer treatment. Investig. New Drugs. 2013;31:230–246. doi: 10.1007/s10637-012-9873-z. [DOI] [PubMed] [Google Scholar]
- 93.Levatić J., Pavić K., Perković I., Uzelac L., Ester K., Kralj M., Kaiser M., Rottmann M., Supek F., Zorc B. Machine learning prioritizes synthesis of primaquine ureidoamides with high antimalarial activity and attenuated cytotoxicity. Eur. J. Med. Chem. 2018;146:651–667. doi: 10.1016/j.ejmech.2018.01.062. [DOI] [PubMed] [Google Scholar]
- 94.Vlainić J., Kosalec I., Pavić K., Hadjipavlou-Litina D., Pontiki E., Zorc B. Insights into biological activity of ureidoamides with primaquine and amino acid moieties. J. Enzym. Inhib. Med. Chem. 2018;33:376–382. doi: 10.1080/14756366.2017.1423067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shahinas D., Folefoc A., Pillai D.R. Targeting Plasmodium falciparum Hsp90: towards reversing antimalarial resistance. Pathogens. 2013;2:33–54. doi: 10.3390/pathogens2010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tcherniuk S.O., Chesnokova O., Oleinikov I.V., Potopalsky A.I., Oleinikov A.V. Anti-malarial effect of semi-synthetic drug amitozyn. Malar. J. 2015;14(10):425. doi: 10.1186/s12936-015-0952-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kreidenweiss A., Kremsner P.G., Mordmüller B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar. J. 2008;7(8):187. doi: 10.1186/1475-2875-7-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Engel J.A., Jones A.J., Avery V.M., Sumanadasa S.D., Ng S.S., Fairlie D.P., Skinner-Adams T., Andrews K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015;5:117–126. doi: 10.1016/j.ijpddr.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trenholme K., Marek L., Duffy S., Pradel G., Fisher G., Hansen F.K., Skinner-Adams T.S., Butterworth A., Ngwa C.J., Moecking J., Goodman C.D., McFadden G.I., Sumanadasa S.D.M., Fairlie D.P., Avery V.M., Kurz T., Andrews K.T. Lysine acetylation in sexual stage malaria parasites is a target for antimalarial small molecules. Antimicrob. Agents Chemother. 2014;58:3666–3678. doi: 10.1128/AAC.02721-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andrews K.T., Haque A., Jones M.K. HDAC inhibitors in parasitic diseases, Immunol. Cell Biol. 2012;90:66–77. doi: 10.1038/icb.2011.97. [DOI] [PubMed] [Google Scholar]
- 101.Chaal B.K., Gupta A.P., Wastuwidyaningtyas B.D., Luah Y.H., Bozdech Z. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog. 2010;6(13):e1000737. doi: 10.1371/journal.ppat.1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hu H., Cabrera A., Kono M., Mok S., Chaal B.K., Haase S., Engelberg K., Cheemadan S., Spielmann T., Preiser P.R., Gillberger T.-W., Bozdech Z. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 2010;28:91–98. doi: 10.1038/nbt.1597. [DOI] [PubMed] [Google Scholar]
- 103.Andrews K.T., Tran T.N., Wheatley N.C., Fairlie D.P. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr. Top. Med. Chem. 2009;9:292–308. doi: 10.2174/156802609788085313. [DOI] [PubMed] [Google Scholar]
- 104.Beus M., Rajić Z., Maysinger D., Mlinarić Z., Antunović M., Marijanović I., Fontinha D., Prudêncio M., Held J., Olgen S., Zorc B. SAHAquines, novel hybrids based on SAHA and primaquine motifs, as potential anticancer and antiplasmodial agents. ChemistryOpen. 2018;7:624–638. doi: 10.1002/open.201800117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang I., Beus M., Stochaj U., Le P.U., Zorc B., Rajić Z., Petrecca K., Maysinger D. Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule. Cell Death Dis. 2019;5(14):41. doi: 10.1038/s41420-018-0103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rajić Z., Beus M., Michnova H., Vlainić J., Persoons L., Kosalec I., Jampilek J., Schols D., Keser T., Zorc B. Asymmetric primaquine and halogenaniline fumardiamides as novel biologically active Michael acceptors. Molecules. 2018;23(18):1724. doi: 10.3390/molecules23071724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jackson P.A., Widen J.C., Harki D.A., Brummond K.M. Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J. Med. Chem. 2017;60:839–885. doi: 10.1021/acs.jmedchem.6b00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rajić Z., Kos G., Zorc B., Singh P.P., Singh S. Macromolecular prodrugs. XII. Primaquine conjugates: synthesis and antimalarial evaluation. Acta Pharm. 2009;59:107–115. doi: 10.2478/v10007-009-0007-x. [DOI] [PubMed] [Google Scholar]
- 109.Pavić K., Perković I., Pospíšilová Š., Machado M., Fontinha D., Prudêncio M., Jampilek J., Coffey A., Endersen L., Rimac H., Zorc B. Primaquine hybrids as promising antimycobacterial and antimalarial agents. Eur. J. Med. Chem. 2018;143:769–779. doi: 10.1016/j.ejmech.2017.11.083. [DOI] [PubMed] [Google Scholar]
- 110.Kedzierska E., Orzelska J., Perković I., Knežević D., Fidecka S., Kaiser M., Zorc B. Pharmacological activity of primaquine ureas and semicarbazides on central nervous system in mice and antimalarial activity in vitro. Fundam. Clin. Pharmacol. 2016;30:58–69. doi: 10.1111/fcp.12161. [DOI] [PubMed] [Google Scholar]
- 111.Beus M., Fontinha D., Held J., Rajić Z., Uzelac L., Kralj M., Prudêncio M., Zorc B. Primaquine and chloroquine fumardiamides as promising antiplasmodial agents. Molecules. 2019 doi: 10.3390/molecules24152812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hufnagel D.A., Price J.E., Stephenson R.E., Kelley J., Benoit M.F., Chapman M.R. Thiol starvation induces redox-mediated dysregulation of Escherichia coli biofilm components. J. Bacteriol. 2018;200 doi: 10.1128/JB.00389-17. e00389-17 (15 pages) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vásquez-Vivar J., Augusto O. Oxidative activity of primaquine metabolites on rat erythrocytes in vitro and in vivo. Biochem. Pharmacol. 1994;47:309–316. doi: 10.1016/0006-2952(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 114.Grinberg L.N., Samuni A. Nitroxide stable radical prevents primaquine-induced lysis of red blood cells. Biochim. Biophys. Acta. 1994;1201:284–288. doi: 10.1016/0304-4165(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 115.Kashaw S.K., Kashaw V., Mishra P., Jain N.K., Stables J.P. Synthesis, anticonvulsant and CNS depressant activity of some new bioactive 1-(4-substituted-phenyl)-3-(4-oxo-2-phenyl/ethyl-4H-quinazolin-3-yl)-urea. Eur. J. Med. Chem. 2009;44:4335–4343. doi: 10.1016/j.ejmech.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 116.Jain A., Patil S., Gajbhiye A. Synthesis and anticonvulsant activity of some 1-cyclohexylidine/cycloheptylidine-4-substituted semicarbazide derivatives. Int. J. Pharmacol. Pharm. Technol. 2012;1:51–56. [Google Scholar]
- 117.Saravanan G., Alagarsamy V., Prakash C.R. Design, synthesis and anticonvulsant activities of novel 1-(substituted/unsubstituted benzylidene)-4-(4-(6,8-dibromo-2-(methyl/phenyl)-4-oxoquinazolin-3(4H)-yl)phenyl) semicarbazide derivatives. Bioorg. Med. Chem. Lett. 2012;22:3072–3078. doi: 10.1016/j.bmcl.2012.03.068. [DOI] [PubMed] [Google Scholar]
- 118.Azad C.S., Saxena M., Siddiqui A.J., Bhardwai J., Puri S.K., Dutta G.P., Anand N., Saxena A.K. Synthesis of primaquine glyco-conjugates as potential tissue schizontocidal antimalarial agents. Chem. Biol. Drug Des. 2017;90:254–261. doi: 10.1111/cbdd.12944. [DOI] [PubMed] [Google Scholar]
- 119.Capela R., Cabal G.G., Rosenthal P.J., Gut J., Mota M.M., Moreira R., Lopes F., Prudêncio M. Design and evaluation of primaquine-artemisinin hybrids as a multistage antimalarial strategy. Antimicrob. Agents Chemother. 2011;55:4698–4706. doi: 10.1128/AAC.05133-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miranda D., Capela R., Albuquerque I.S., Meireles P., Paiva I., Nogueira F., Amewu R., Gut J., Rosenthal P.J., Oliveira R., Mota M.M., Moreira R., Marti F., Prudêncio M., O'Neill P.M., Lopes F. Novel endoperoxide-based transmission-blocking antimalarials with liver- and blood-schizontocidal activities. ACS Med. Chem. Lett. 2014;5:108–112. doi: 10.1021/ml4002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Capela R., Magalhães J., Miranda D., Machado M., Sanches-Vaz M., Albuquerque I.S., Sharma M., Gut J., Rosenthal P.J., Frade R., Perry M.J., Moreira R., Prudêncio M. Endoperoxide-8-aminoquinoline hybrids as dual-stage antimalarial agents with enhanced metabolic stability. Eur. J. Med. Chem. 2018;149:69–78. doi: 10.1016/j.ejmech.2018.02.048. [DOI] [PubMed] [Google Scholar]
- 122.Dechy-Cabaret O., Benoit-Vical F., Loup C., Robert A., Gornitzka H., Bonhoure A., Vial H., Maagnaval J.F., Séguéla J.P., Meunier B. Synthesis and antimalarial activity of trioxaquine derivatives. Chemistry. 2004;10:1625–1636. doi: 10.1002/chem.200305576. [DOI] [PubMed] [Google Scholar]
- 123.Benoit-Vical F., Lelièvre J., Berry A., Deymier C., Dechy-Cabaret O., Cazelles J., Loup C., Robert A., Magnaval J.F., Meunier B. Trioxaquines are new antimalarial agents active on all erythrocytic forms, including gametocytes. Antimicrob. Agents Chemother. 2007;51:1463–1472. doi: 10.1128/AAC.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chauhan S.S., Sharma M., Chauhan P.M. Trioxaquines: hybrid molecules for the treatment of malaria. Drug News Perspect. 2010;23:632–646. doi: 10.1358/dnp.2010.23.10.1468390. [DOI] [PubMed] [Google Scholar]
- 125.Kiszewski A.E. Blocking Plasmodium falciparum malaria transmission with drugs: the gametocytocidal and sporontocidal properties of current and prospective antimalarials. Pharmaceuticals (Basel) 2011;4:44–68. [Google Scholar]
- 126.Odhiambo O.C., Wamakima H.N., Magoma G.N., Kirira P.G., Malala B.J., Kimani F.T., Muregi F.W. Efficacy and safety evaluation of a novel trioxaquine in the management of cerebral malaria in a mouse model. Malar. J. 2017;16(9):268. doi: 10.1186/s12936-017-1917-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lödige M., Lewis M.D., Paulsen E.S., Esch H.L., Pradel G., Lehmann L., Brun R., Bringmann G., Mueller A.K. A primaquine-chloroquine hybrid with dual activity against Plasmodium liver and blood stages. Int. J. Med. Microbiol. 2013;303:539–547. doi: 10.1016/j.ijmm.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 128.Matos J., da Cruz F.P., Cabrita É., Gut J., Nogueira F., do Rosário V.E., Moreira R., Rosenthal P.J., Prudêncio M., Gomes P. Novel potent metallocenes against liver stage malaria. Antimicrob. Agents Chemother. 2012;56:1564–1570. doi: 10.1128/AAC.05345-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vale-Costa S., Vale N., Matos J., Tomás A., Moreira R., Gomes P., Salomé Gomes M. Peptidomimetic and organometallic derivatives of primaquine active against Leishmania infantum. Antimicrob. Agents Chemother. 2012;56:5774–5781. doi: 10.1128/AAC.00873-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kaur H., Machado M., de Kock C., Smith P., Chibale K., Prudêncio M., Singh K. Primaquine-pyrimidine hybrids: synthesis and dual-stage antiplasmodial activity. Eur. J. Med. Chem. 2015;101:266–273. doi: 10.1016/j.ejmech.2015.06.045. [DOI] [PubMed] [Google Scholar]
- 131.Aguiar A.C.C., Figueiredo B., Jr., Neuenfeldt P.D., Katsuragawa T.H., Drawanz B.B., Cunico W., Sinnis P., Zavala F., Krettli A.U. Primaquine-thiazolidinones block malaria transmission and development of the liver exoerythrocytic forms. Malar. J. 2017;16(11):110. doi: 10.1186/s12936-017-1755-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.https://newdrugapprovals.org/2014/05/01/bulaquine/
- 133.Bonilla-Ramírez L., Rios A., Quiliano M., Ramírez-Calderon G., Beltrán-Hortelano I., Franetich J.F., Corcuera L., Bordessoulles M., Vettorazzi A., López de Cerain A., Aldana I., Mazier D., Pablón A., Galiano S. Novel antimalarial chloroquine- and primaquine-quinoxaline 1,4-di-N-oxide hybrids: design, synthesis, Plasmodium life cycle stage profile, and preliminary toxicity studies. Eur. J. Med. Chem. 2018;158:68–81. doi: 10.1016/j.ejmech.2018.08.063. [DOI] [PubMed] [Google Scholar]
- 134.Ribeiro C.J.A., Espadinha M., Machado M., Gut J., Gonçalves L.M., Rosenthal P.J., Prudêncio M., Moreira R., Santos M.M. Novel squaramides with in vitro liver stage antiplasmodial activity. Bioorg. Med. Chem. 2016;24:1786–1792. doi: 10.1016/j.bmc.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 135.Ribeiro C.J.A., Kumar S.P., Gut J., Goncalves L.M., Rosenthal P.J., Moreira R., Santos M.M.M. Squaric acid/4-aminoquinoline conjugates: novel potent antiplasmodial agents. Eur. J. Med. Chem. 2013;69:365–372. doi: 10.1016/j.ejmech.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 136.Pradines B., Torrentino-Madamet M., Fontaine A., Henry M., Baret E., Mosnier J., Briolant S., Fusai T., Rogier C. Atorvastatin is 10-fold more active in vitro than other statins against Plasmodium falciparum. Antimicrob. Agents Chemother. 2007;51:2654–2655. doi: 10.1128/AAC.01330-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Savini H., Souraud J.B., Briolant S., Baret E., Amalvict R., Rogier C., Pradines B. Atorvastatin as a potential antimalarial drug: in vitro synergy in combinational therapy with dihydroartemisinin. Antimicrob. Agents Chemother. 2010;54:966–967. doi: 10.1128/AAC.01006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dormoi J., Briolant S., Pascual A., Desgrouas C., Travaillé C., Pradines B. Improvement of the efficacy of dihydroartemisinin with atorvastatin in an experimental cerebral malaria murine model. Malar. J. 2013;12(9):302. doi: 10.1186/1475-2875-12-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Carvalho R.C.C., Martins W.A., Silva T.P., Kaiser C.R., Bastos M.M., Pinheiro L.C.S., Krettli A.U., Boechat N. New pentasubstituted pyrrole hybrid atorvastatin-quinoline derivatives with antiplasmodial activity. Bioorg. Med. Chem. Lett. 2016;26:1881–1884. doi: 10.1016/j.bmcl.2016.03.027. [DOI] [PubMed] [Google Scholar]
- 140.Jain M., Vangapandu S., Sachdeva S., Jain R. Synthesis and blood-schozontocidal antimalarial activities of 2-substituted/2,5-disubstituted-8-quinolinamines and some of their amino acid conjugates. Bioorg. Med. Chem. 2004;12:1003–1010. doi: 10.1016/j.bmc.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 141.Kaur K., Jain M., Khan S.I., Jacob M.R., Tekwani B.L., Singh S., Singh P.P., Jain R. Amino acid, dipeptide and pseudodipeptide conjugates of ring-substituted 8-aminoquinolines: synthesis and evaluation of anti-infective, β-haematin inhibition and cytotoxic activities. Eur. J. Med. Chem. 2012;52:230–241. doi: 10.1016/j.ejmech.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 142.Davanço M.G., Campos Aguiar A.C., dos Santos L.A., Padilha E.C., Campos M.L., de Andrade C.R., da Fonseca L.M., Dos Santos J.L., Chin C.M., Krettli A.U., Peccinini R.G. Evaluation of antimalarial activity and toxicity of a new primaquine prodrug. PLoS One. 2014;9:e105217. doi: 10.1371/journal.pone.0105217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nishi K.K., Jayakrishnan A. Preparation and in vitro evaluation of primaquine-conjugated gum Arabic microspheres. Trends Biomater. Artif. Organs. 2005;18(2):191–197. doi: 10.1021/bm0499435. [DOI] [PubMed] [Google Scholar]
- 144.Trouet A., Pirson P., Steiger R., Masquelier M., Baurain R., Gillet J. Development of new derivatives of primaquine by association with lysosomotropic carriers. Bull. World Health Organ. 1981;59:449–458. [PMC free article] [PubMed] [Google Scholar]
- 145.Aguiar L., Machado M., Sanches-Vaz M., Prudêncio M., Vale N., Gomes P. Coupling the cell-penetrating peptides transportan and transportan 10 to primaquine enhanced its activity against liver-stage malaria parasites. MedChemComm. 2019;10:221–226. doi: 10.1039/c8md00447a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.