

Abstract
The spike protein of SARS-associated coronavirus (SARS-CoV) is an important target for anti-SARS drug discovery. Its S1 domain is responsible for receptor binding and SARS-CoV entry into cells. In this study, we constructed a rational 3D model for S1 domain of SARS-CoV spike protein by fold recognition and molecular modeling techniques. We found that there is a structure similarity between S1 protein and influenza virus neuraminidase. Our analyses suggest that the existing anti-influenza virus inhibitors and anti-neuraminidase antibody could be used as a starting point for designing anti-SARS drugs, vaccines and antibodies. Interestingly, our prediction for antibody is consistent with a recently experimental discovery of anti-SARS antibody.
Keywords: SARS-CoV, S1 protein, Structure, Influenza virus, Inhibitor, Antibody
1. Introduction
There is an urgent need for effective antiviral therapy when the reemergence of the highly contagious SARS-CoV infection happens. Chou et al. [1] studied the interaction of SARS-associated coronavirus (SARS-CoV) main proteinase with two ligands. Jenwitheesuk and Samudrala [2] identified some inhibitors of the SARS-CoV proteinase. Xiong et al. [3] is screening possible 73 inhibitors of SARS-CoV 3CL proteinase. Zhang and Yap [4] explored the binding mechanism of SARS-CoV main proteinase.
It is known that SARS-CoV has the same structure proteins as three previously known groups of coronaviruses: spike glycoprotein (S), membrane protein (M), envelope protein (E) and nucleocapsid protein (N). All these proteins can be used as targets for anti-SARS drug development in principle. Among these structure proteins, S protein is a type I transmembrane glycoprotein including two functional domains S1 and S2, which are conserved among coronaviruses. S1 is responsible for the binding with its receptor angiotensin-converting enzyme 2 (ACE2) on host cells and defines the host range of the virus [5]. The goal of this study is to construct a rational 3D model of S1, to identify noncanonical interactions in the structure of S1, possible inhibitors and antibodies, hence to provide important information for anti-SARS drug, vaccine and antibody discovery.
2. Materials and methods
The sequence of spike protein was downloaded from GenBank (NP_828851). Liu et al. [6] found that the region 75-609 of SARS-CoV S protein matches to the conserved coronavirus S1 domain PF01600 in HMM database and the region 641-1247 matches to conserved coronavirus S2 domain PF01601 in HMM database. In previous study, we have predicted the structure of SARS-CoV S2 protein [7]. Here, we used the same method 3D Jury system [8] to predict the 3D structure of SARS-CoV S1 protein based on the domain (residues 75-609) mentioned above. The proteins with sufficiently high 3D score were used as templates to construct 3D models of S1 by modeller program [9]. The quality of 3D model was evaluated by ProQ program [10] and finally validated with the procheck program [11]. The best model was used for further analyses. Specifically, NCI program [12] was used to identify non-canonical interactions in protein structures. VAST (http://www.ncbi.nlm.nih.gov/Structure/VAST/vastsearch.html), DALI (http://www.ebi.ac.uk/dali/) and CE [13] programs were employed to search the structure neighbors of S1 protein. The structural comparison was performed by LGA [14]. The visualization of 3D structure was generated by PROTEINEXPLORER (http://www.proteinexplorer.org).
3. Results and discussion
Fold prediction by meta-server (3D Jury) revealed that top three significant hits (3D score>50) for S1 protein are as below: 1loq_A (Orotidine monophosphate decarboxylase (lyase), 3D score 154, threading server PCONS2), 1ijq (low-density lipoprotein receptor (lipid transport), 3D score 125, threading server PCONS2) and 2bbk_H (Methylamine dehydrogenase (electron transport), 3D score 118, threading server PCONS2). Using them as templates the corresponding 3D models of S1 were generated and the quality of protein model was evaluated by ProQ program. The results are as follows: 1loq (ProQ-LG=0.969, ProQ-MX=0.055), 1ijq (ProQ-LG=1.955, ProQ-MX=0.101), 2bbk (ProQ-LG=0.877, ProQ-MX=0.057). So the ‘correct’ model (the cutoffs for ‘correct model’ are ProQ-LG>1.5 or ProQ-MX>0.1) for S1 protein is the model built on template1ijq. The sequence alignment between template 1ijq and S1 with ClustalW [15] and the secondary structures of S1 predicted by PsiPred v2.3 [16] are displayed in Fig. 1 . Thus, the 3D model of S1 is basically composed of one long α helix and six three-stranded β-sheets arranged in a propeller fashion (among them are a couple of small helices with 3–5 residues only) (Fig. 2), comparable to the structure of its template (low-density lipoprotein receptor): consisting of six four-stranded β-sheets arranged in a propeller fashion. It is not difficult to understand such a structure similarity in terms of their functions, the low-density lipoprotein receptor is responsible for the binding and entry into cells of lipoprotein ligands [17], while SARS-CoV S1 is also responsible for receptor binding and SARS-CoV entry into cells [5].
Fig. 1.

The sequence alignment between SARS-CoV S1 and template 1ijq with ClustalW. The secondary structure of S1 predicted by PsiPred is also included.
Fig. 2.
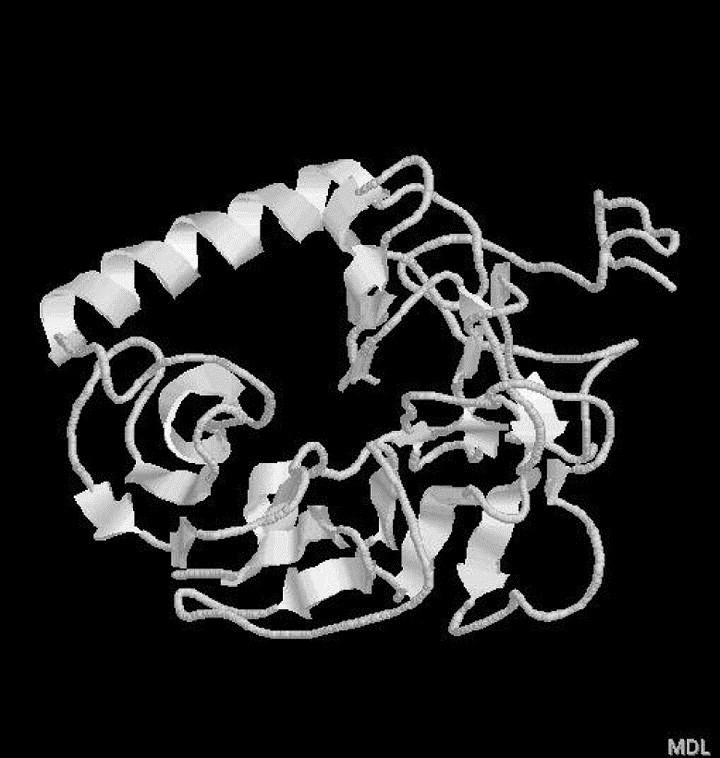
3D model for S1 (75-609), which is basically composed of one long α helix and six three-stranded β-sheets arranged in a propeller fashion.
The non-canonical interactions in S1 protein structure were identified by NCI program and the result shows that there are three pairs of main chain-side chain interactions: Trp 171 (donor) and Phe 179 (acceptor), Ala 210 (donor) and Phe 29 (acceptor), and Leu 209 (donor) and Trp 171 (acceptor). Among these interactions, Trp171 accept one N–H⋯π bond from Leu209 and donates one N–H⋯π bond to Phe179, which forms a sandwich-like interaction, as existed in human rac1 [18] and SARS-CoV main protease [4]. These noncanonical bindings fix the helix Trp171 locate to the two β-sheets Phe179 and Leu209 locate, hence stabilizes the structure of S1 protein, but probably reduces the active site cavity for ligand binding (Fig. 3). Indeed, the non-canonical interactions have been shown to be important for the stability of protein structure [19], [20], [21] and ligand recognition [22].
Fig. 3.
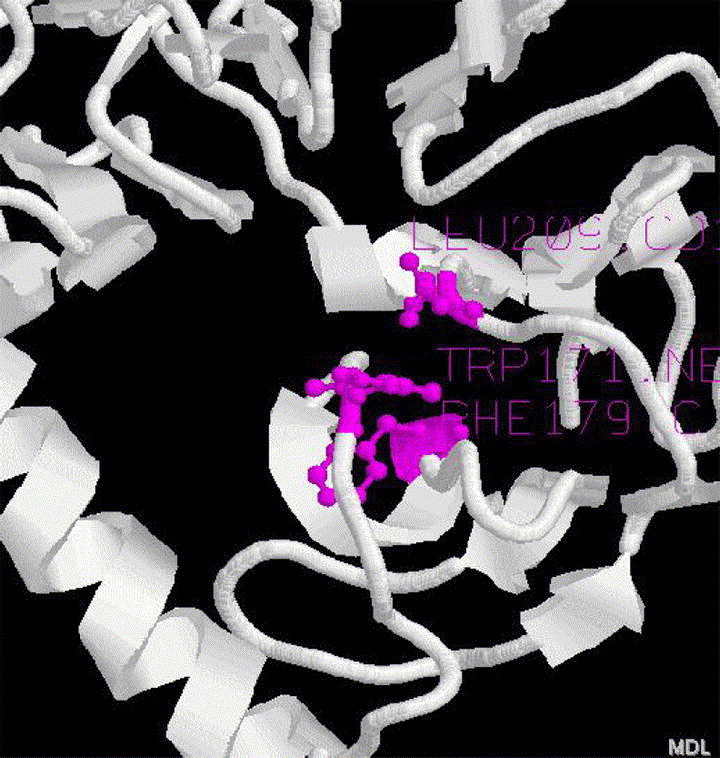
Non-canonical interactions in the structure of S1 (75-609). The residue pairs involved are: Trp171 and Phe179, Leu209 and Trp171, represented by blue ball-stick (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.).
In order to extract more information from the predicted structure of S1 protein, we employed VAST, DALI and CE programs to search its structure neighbors and found two interesting neighbors: 1INY (neuraminidase from influenza A virus) and 1B9T (neuraminidase from influenza B virus). In fact, both type A and B neraminidases are composed of six four-stranded anti-parallel β-sheets arranged in a propeller fashion [23], [24] and resemble the structure of S1 stated above: basically composed of one long α helix and six three-stranded β-sheets arranged in a propeller fashion. The common feature of these structures is that they share the same active center, as seen in their superpositions (Fig. 4). The structure alignment between 1INY, 1B9T and S1 is shown in Fig. 5 . The result demonstrates that there are a number of similar structure patterns showing conservative residues (bold representation). Indeed, their functions are also similar: neuraminidase is an influenza virus glycoprotein embedded in the viral envelope, its function is to facilitate the release of progeny virions from infected cells to other cells [23], [24], while SARS-CoV S1 is also responsible for the entry of SARS-CoV into cells, as stated above.
Fig. 4.
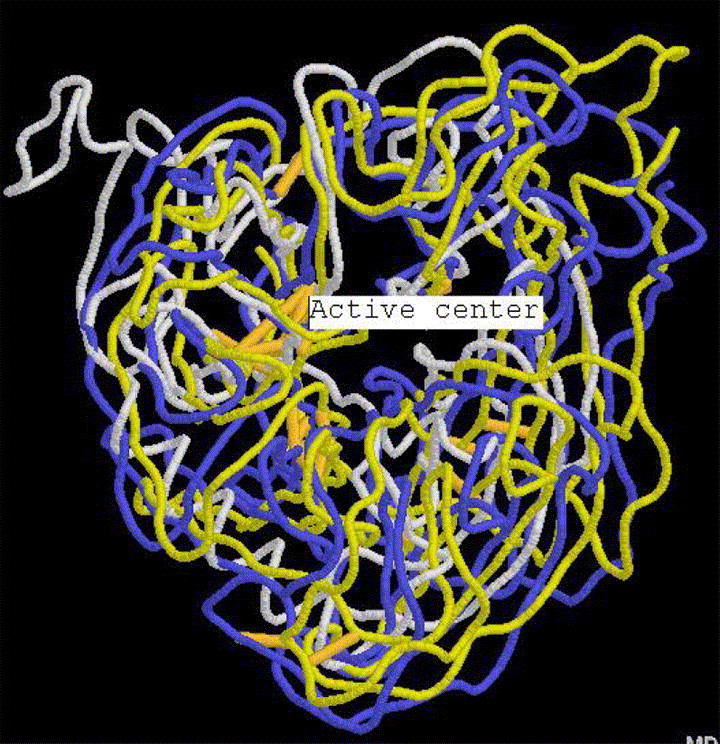
The superposition of S1 (white) with its structure neighbors: 1INY (neuraminidase from influenza A virus, blue) and 1B9T (neuraminidase from influenza B virus, yellow) (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.).
Fig. 5.

The structure alignment between S1 and its structure neighbors: 1INY (neuraminidase from influenza A virus) and 1B9T (neuraminidase from influenza B virus). The bold residues indicate conservative residues. The residues marked as ‘#’ make contact with inhibitors.
Naturally, such a structure similarity between SARS-CoV S1 protein and influenza virus neuraminidase leads us to consider the possible application of neuraminidase inhibitors to SARS therapy. For example, Epana and RAI inhibitors have been shown to exhibit great potency for type A/B influenza viruses [23], [24]. Fig. 6, Fig. 7 shows the structures of docking the two inhibitors from influenza virus to SARS-CoV S1 protein. It can be seen that these compounds can bind to the active center of S1 in the same way as they did in neuraminidase, the residues that make contact with the inhibitors (RMSD≤5) are marked as ‘#’ in Fig. 5 and represented by blue ball-stick in Fig. 6, Fig. 7, among them are 10 conservative residues.
Fig. 6.
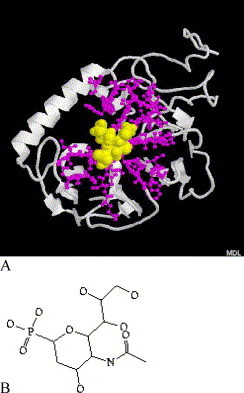
(A) The interactions between S1 (white cartoon) and Epana inhibitor (yellow spacefill) from influenza A virus neuraminidase, the binding pockets are represented by blue ball-stick. (B) The chemical structure of Epana inhibitor (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.).
Fig. 7.
(A) The interactions between S1 (white cartoon) and RAI inhibitor (yellow spacefill) from influenza B virus neuraminidase, the binding pockets are represented by blue ball-stick. (B) The chemical structure of RAI inhibitor (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.).
Furthermore, we also speculate that influenza virus anti-neuraminidase antibody could be applied to SARS-CoV S1 protein after appropriate modification, such as, Nc10 and Nc41 anti-influenza virus neuraminidase antibodies [25], [26]. Indeed, a very recent publication confirmed our speculation, Sui et al. [27] identified an anti-S1 human monoclonal antibody 80R, which potently neutralizes SARS-CoV infection and efficiently inhibits syncytia formation through blocking of S1 binding to its receptor angiotensin-converting enzyme 2 (ACE2) on host cells. The sequence alignments between Nc10/Nc41 and 80R show more than 50% identity (Fig. 8).
Fig. 8.

The sequence alignment between anti-SARS-CoV-S1 human monoclonal antibody 80R and influenza virus anti-neuraminidase antibodies Nc10/Nc41.
In summary, our modeling exercise on S1 protein find a structure similarity between SARS-CoV S1 protein and influenza virus neuraminidase. In fact, there are clinically similar symptoms for influenza virus and SARS-CoV, both can cause a fever, cough, pains, pneumonia and death [28]. Moreover, our study suggests that the influenza virus neuraminidase inhibitors and anti-neuraminidase antibody could be a starting point for screening anti-SARS drug, vaccine and antibody.
References
- 1.Chou K.C, Wei D.Q, Zhong W.Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jenwitheesuk E, Samudrala R. Identifying inhibitors of the SARS coronavirus proteinase. Bioorg. Med. Chem. Lett. 2003;13(22):3989–3992. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiong B, Gui C.S, Xu X.Y, Luo C, Chen J, Luo H.B, Chen L.L, Li G.W, Sun T, Yu C.Y, Yue L.D, Duan W.H, Shen J.K, Qin L, Shi T.L, Li Y.X, Chen K.X, Luo X.M, Shen X, Shen J.H, Jiang H.L. A 3D model of SARS-CoV 3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol. Sinica. 2003;24:497–504. [PubMed] [Google Scholar]
- 4.Zhang X.W, Yap Y.L. Exploring the binding mechanism of the main proteinase in SARS-associated coronavirus and its implication to anti-SARS drug design. Bioorg. Med. Chem. 2004;12:2219–2223. doi: 10.1016/j.bmc.2004.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W, Moore M.J, Vasilieva N, Sui J, Wong S.K, Berne M.A, Somasundaran M, Sullivan J.L, Luzuriaga K, Greenough T.C, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S, Guo T, Ji X, Sun Z. Bioinformatical study on the proteomics and evolution of SARS-CoV. Chin. Sci. Bull. 2003;48:1277–1287. doi: 10.1007/BF03184163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X.W, Yap Y.L. Structural similarity between HIV-1 gp41 and SARS-CoV S2 proteins suggests an analogous membrane fusion mechanism. J. Mol. Struct. THEOCHEM. 2004;677:73–76. doi: 10.1016/j.theochem.2004.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 9.Sali A, Blundell T.L. Comparative protein modeling by satisfaction of spatial restrains. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 10.Wallner B, Elofsson A. Can correct protein models be identified? Protein Sci. 2003;12:1073–1086. doi: 10.1110/ps.0236803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laskowski R.A, MacArthur M.W, Moss D.S, Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 12.Babu M.M. NCI: a server to identify non-canonical interactions in protein structures. Nucleic Acids Res. 2003;31:3345–3348. doi: 10.1093/nar/gkg528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shindyalov I.N, Bourne P.E. Protein structure alignment by incremental combinatorial extention (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 14.Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson J.D, Higgins D.G, Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 17.Jeon H, Meng W, Takagi J, Eck M.J, Springer T.A, Blacklow S.C. Implications for familial hypercholesterolemia from the structure of the LDL receptor YWTD-EGF domain pair. Nat. Struct. Biol. 2001;8(6):499–504. doi: 10.1038/88556. [DOI] [PubMed] [Google Scholar]
- 18.Hirshberg M, Stockley R.W, Dodson G, Webb M.R. The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nat. Struct. Biol. 1997;4:147–152. doi: 10.1038/nsb0297-147. [DOI] [PubMed] [Google Scholar]
- 19.Babu M.M, Singh S, Balaram P. A C–H⋯O hydrogen bond stabilized polypeptide chain reversal motif at the C terminus of helices in proteins. J. Mol. Biol. 2002;322:871–880. doi: 10.1016/s0022-2836(02)00715-5. [DOI] [PubMed] [Google Scholar]
- 20.Fabiola G.F, Krishnaswamy S, Nagarajan V, Pattabhi V. C–H⋯O hydrogen bonds in beta sheets. Acta Crystallogr. Sec. D. 1997;53:316–320. doi: 10.1107/S0907444997000383. [DOI] [PubMed] [Google Scholar]
- 21.Senes A, Ubarretxena-Belandia I, Engelman D.M. The C–H⋯O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc. Natl Acad. Sci. USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kryger G, Silman I, Sussman J.L. Structure of acetylcholinesterase complexed with E2020: implications for the design of new anti-alzheimer drugs. Structure. 1999;7:297–307. doi: 10.1016/s0969-2126(99)80040-9. [DOI] [PubMed] [Google Scholar]
- 23.Finley J.B, Atigadda V.R, Duarte F, Zhao J.J, Brouillette W.J, Air G.M, Luo M. Novel aromatic inhibitors of influenza virus neuraminidase make selective interactions with conserved residues and water molecules in the active site. J. Mol. Biol. 1999;293(5):1107–1119. doi: 10.1006/jmbi.1999.3180. [DOI] [PubMed] [Google Scholar]
- 24.White C.L, Janakiraman M.N, Laver W.G, Philippon C, Vasella A, Air G.M, Luo M. A sialic acid-derived phosphonate analog inhibits different strains of influenza virus neuraminidase with different efficiencies. J. Mol. Biol. 1995;245:623–634. [PubMed] [Google Scholar]
- 25.Malby R.L, McCoy A.J, Kortt A.A, Hudson P.J, Colman P.M. Three-dimensional structures of single-chain Fv-neuraminidase complexes. J. Mol. Biol. 1998;279(4):901–910. doi: 10.1006/jmbi.1998.1794. [DOI] [PubMed] [Google Scholar]
- 26.Tulip W.R, Varghese J.N, Laver W.G, Webster R.G, Colman P.M. Refined crystal structure of the influenza virus N9 neuraminidase-NC41 Fab complex. J. Mol. Biol. 1992;227:122–148. doi: 10.1016/0022-2836(92)90687-f. [DOI] [PubMed] [Google Scholar]
- 27.Sui J, Li W, Murakami A, Tamin A, Matthews L.J, Wong S.K, Moore M.J, Tallarico A.S, Olurinde M, Choe H, Anderson L.J, Bellini W.J, Farzan M, Marasco W.A. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl Acad. Sci. USA. 2004;101(8):2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oxford J, Balasingam S, Lambkin R. A new millennium conundrum: how to use a powerful class of influenza anti-neuraminidase drugs (NAIs) in the community. J. Antimicrob. Chemother. 2004;53(2):133–136. doi: 10.1093/jac/dkh037. [DOI] [PMC free article] [PubMed] [Google Scholar]