

Abstract
Involvement of viruses in human neurodegenerative diseases and the underlying pathologic mechanisms remain generally unclear. Human respiratory coronaviruses (HCoV) can infect neural cells, persist in human brain, and activate myelin-reactive T cells. As a means of understanding the human infection, we characterized in vivo the neurotropic and neuroinvasive properties of HCoV-OC43 through the development of an experimental animal model. Virus inoculation of 21-day postnatal C57BL/6 and BALB/c mice led to a generalized infection of the whole CNS, demonstrating HCoV-OC43 neuroinvasiveness and neurovirulence. This acute infection targeted neurons, which underwent vacuolation and degeneration while infected regions presented strong microglial reactivity and inflammatory reactions. Damage to the CNS was not immunologically mediated and microglial reactivity was instead a consequence of direct virus-mediated neuronal injury. Although this acute encephalitis appears generally similar to that induced by murine coronaviruses, an important difference rests in the prominent spongiform-like degeneration that could trigger neuropathology in surviving animals.
Keywords: Coronavirus, Encephalitis, Inflammation, Neurodegenerative disease, Spongiform-like degeneration, Viral meningoencephalomyelitis
Introduction
Although the etiology of most neuroautoimmune, neuroinflammatory, and/or neurodegenerative diseases remains unclear, virus infections could directly trigger neurodegeneration or initiate a CNS-directed inflammatory process leading to central nervous system (CNS) damage, or a combination of both. Indeed, Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS) could actually represent infectious diseases Calne et al., 1986, Kristensson, 1992, Kirk and Zhou, 1996, Allen et al., 1996, Hayase and Tobita, 1997, Klein et al., 1999, Boucher et al., 2001, Jubelt and Berger, 2001, Giraud et al., 2001, Sola et al., 2002. Moreover, psychiatric disorders were also investigated as a possible consequence of viral infections Waltrip et al., 1995, Lewis, 2001. The vertebrate CNS was long thought to be inaccessible to cells of the immune system or to viruses. However, the presence of virus in the CNS is more frequent than expected and viral detection in the cerebrospinal fluid of patients suggests the ability of viruses to cross the blood-brain barrier Koskiniemi and Vaheri, 1989, Georgsson, 1994. In fact, neuroinvasive viruses can damage the CNS and produce neurological disease in sensitive hosts, due to the misdirected immune response of the host (virus-induced immunopathology) and/or viral replication in cells of the brain (virus-induced cytopathology). Nevertheless, primary infections of the brain are not common and viruses are the leading cause of encephalitis, which results from either direct infection (acute encephalitis) or the immune response to an infection (postinfectious encephalitis or acute demyelinating encephalomyelitis). In acute encephalitis, viral replication occurs in the brain tissue itself, causing destructive lesions of the gray matter: this was reported after herpes simplex, rabies, or some arbovirus infections Rupprecht et al., 2002, Shoji et al., 2002. Therefore, the knowledge of infectious agents involved in neurological diseases and mechanisms underlying the induction of neuropathology by these pathogens will be invaluable for preventing and developing novel clinical interventions.
Coronaviruses are enveloped positive-stranded RNA viruses that infect multiple species of mammals, including man, causing diseases that range from encephalitis to enteritis. Human coronaviruses (HCoV) are recognized respiratory pathogens responsible for up to 35% of common colds McIntosh, 1996, Myint, 1994 and also involved in nocosomial infections (Sizun et al., 2000). They have occasionally been associated with other pathologies, such as pneumonia, meningitis, and enteritis Riski and Hovi, 1980, Resta et al., 1985. Moreover, HCoV have the ability to replicate and persist in human neural cells Bonavia et al., 1997, Arbour et al., 1999a,b) and to have neuroinvasive properties Burks et al., 1980, Murray et al., 1992, Stewart et al., 1992, Arbour et al., 2000. This has stimulated research on their possible involvement in neurological disorders. Of the two known HCoV serotypes, designated OC43 and 229E, HCoV-OC43 is antigenically related to murine coronaviruses (MHV). Given that, under certain conditions, MHV causes experimental CNS inflammatory demyelination that pathologically resembles MS Bailey et al., 1949, Lampert et al., 1973, Weiner, 1973, Wang et al., 1990, the related human coronavirus represents a logical target for investigation.
In the present study, we report the development of a mouse model to characterize in vivo HCoV-OC43-mediated neuropathogenesis. We describe the acute disease induced by HCoV-OC43 infection, which resulted from neuronal infection and loss. This animal model constitutes a tool to study neuroinvasive and neurovirulence properties of a common cold virus and the mechanisms underlying the development of a diffuse vacuolating meningoencephalitis, an emerging medical problem Shoji et al., 2002, Whitley and Gnann, 2002.
Results
Inoculation routes and virus doses
BALB/c and C57BL/6 mice were selected in view of their relative susceptibility to both respiratory and enteric strains of MHV (Barthold and Smith, 1987). We tried different inoculation routes to establish the neurotropic and neuroinvasive properties of HCoV-OC43 in mice. An intraperitoneal inoculation with a virus dose of 105 TCID50 revealed that HCoV-OC43 virus infection could be lethal until 8 days postnatal (DPN) and the same doses were nonlethal at 21 DPN. With an intraoral inoculation of 104–105 TCID50 of virus, we were unable to reveal the presence of virus or virus gene products in any tissue tested (brain, spinal cord, heart, lung, liver, and spleen), even by RT-PCR. Mice were susceptible to intranasal (IN) inhalation of the HCoV-OC43 solution at 104–105 TCID50. This infection was lethal in 1-week-old C57BL/6 mice. However, 21 DPN mice infected this way did not show clinical signs of pathology, but 4 of the 8 animals were found positive for viral RNA by RT- PCR analyses. Viral RNA was mainly found in the CNS but some mice also showed virus RNA in the spleen (data not shown). Therefore, virus could spread from the periphery to the CNS after IN inhalation.
Having shown HCoV-OC43 neuroinvasive properties, we chose for the remaining experimentation to use intracerebral inoculation (IC) so as to favor a CNS infection. The IC route results in a more reproducible infection, a better control of viral doses introduced into the brain. The correlation between viral infectious dose and 100% mortality in the two strains of mice after inoculation is shown in Table 1. Mice became less susceptible with age and were resistant at 35 DPN for C57BL/6 and at 28 DPN for BALB/c mice. We then determined the optimal experimental conditions to obtain a sublethal dose that still allowed virus replication and virus-induced CNS pathology in 21-day-old mice. Viral dose was administered under deep anesthesia and was determined to be 10 μl of a virus solution containing 10 TCID50 for C57BL/6 and 105 TCID50 for BALB/c mice aged 21 DPN. Under these conditions, inoculated mice developed signs of acute disease characterized by loss of weight, apathy, ruffled fur, humped posture, and wasting (Figs. 1A and B). Animals showed atrophy of skeletal muscles and occasionally exhibited paralysis of their forelimbs. During the second week postinfection, some of the animals recovered and clinical signs totally disappeared. For others, pathological signs increased and led to death. The infected animals became anorexic, inactive, and dehydrated, increasing percentages of mortality. We established survival curves for each mouse strain (Fig. 1C). Eighty percent of the C57BL/6 mice died within the first 15 days postinfection and only 20% of BALB/c mice died during this period, even after receiving a higher viral dose. Moreover, mice inoculated with supernatants from cell cultures infected with brain tissue from affected mice developed the same disease, demonstrating that the virus was responsible for pathology.
Table 1.
Determination of infectious viral lethal dose for killing 100% of the infected animals, using two strains of mice, after IC inoculation.
| DPN | C57BL/6 mice | BALB/c mice |
|---|---|---|
| 8 | 1–10 | 1–10 |
| 15 | 5–50 | 100–1000 |
| 21 | 50–100 | 100,000 |
| 28 | 100–1000 | Nonlethal |
| 35 | Nonlethal | Nonlethal |
Note. This dose (expressed in TCID50) is in function of mouse age (days postnatal; DPN) at the time of inoculation.
Fig. 1.
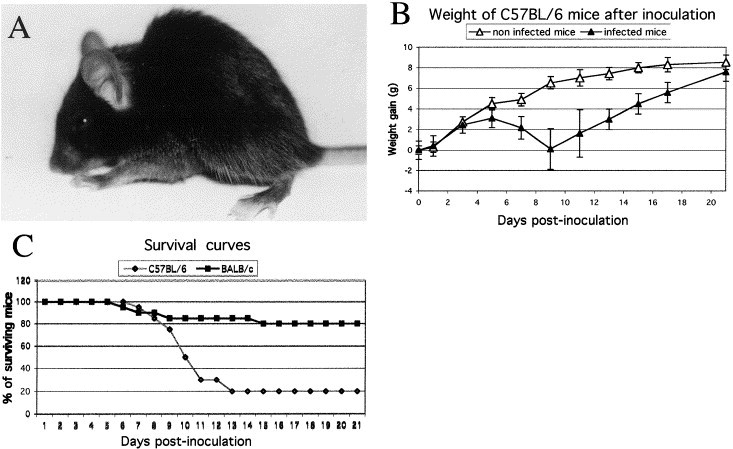
Characterization of HCoV-OC43 infection. (A) Typical posture of an infected mouse at an advanced stage of disease when the animal presented highly pronounced humped back with restrained mobility (9 DPN). (B) C57BL/6 mice were weighed every 2 days after infection to estimate weight variations. HCoV-OC43-infected mice gained weight normally during the first 5 days after infection, after which they all lost weight during the acute phase of the disease. The more affected mice lost more weight more rapidly than less affected mice and died during this period. After 9 days postinfection, mice which survived gained weight to reach the weight of control animals around 21 days postinfection. (C) Survival curves of mice after HCoV-OC43 infection. BALB/c mice received a higher dose than the C57BL/6 mice, 10,000 TCID50 versus 10 TCID50. However, C57BL/6 were less resistant, with 80 versus 20% of death after infection.
Viral RNA and infectious virus
Viral RNA could be detected in the brain as early as 24 h postinfection, and after 2 to 3 days in the spinal cord. All C57BL/6 mice were positive for HCoV-OC43 RNA during the first 11 days postinfection and during the first 9 days for BALB/c mice (Figs. 2A and B). A screening of viral replication was performed by RT-PCR in a variety of tissues and results obtained indicated that infection was restricted to the CNS during the first 9 days postinfection. After that, in the most affected mice, viral RNA was also found in heart, spleen, and lungs, and at lower levels in liver and muscles between the 11th and 13th days postinfection in C57BL/6, suggesting a viremic spread or transneuronal transmission (Fig. 3B). The presence of HCoV-OC43 RNA was detectable in the brain until 11 days postinfection for 40% of BALB/c mice and until 15 days postinfection for 25% of C57BL/6 mice. No viral RNA could be found from tissues harvested after these times postinfection. It was also confirmed that the RT-PCR assay designed to specifically detect HCoV-OC43 was indeed specific and could not have detected an enzootic MHV strain (Fig. 3C).
Fig. 2.
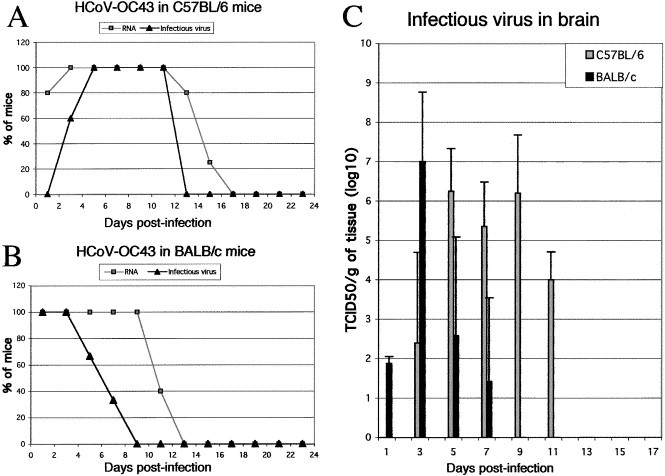
HCoV-OC43 infectious virus and RNA in the CNS of 21 DPN mice. (A) 100% of brains from C57BL/6 mice inoculated ic with 10 TCID50 of HCoV- OC43 were positive for viral RNA between 3 and 11 days postinfection. Only 25% of the surviving mice were found positive after 15 days postinfection and RNA was not found thereafter. Infectious virus appeared later and disappeared before elimination of viral RNA. Between 5 and 11 days postinfection, 100% of brains contained infectious virus. (B) Detection of HCoV-OC43 RNA in the brain of BALB/c mice inoculated ic with 105 TCID50 of HCoV-OC43 revealed that 100% of these mice were positive until 9 days postinfection. Infectious virus was detectable in all mice only during the first 3 days postinfection and gradually fewer mice were found positive. (C) Histogram representing the amount of infectious virus detected in five brains from the two strains of mice at different intervals postinfection. The limit of the detection assay was 100.5 TCID50.
Fig. 3.
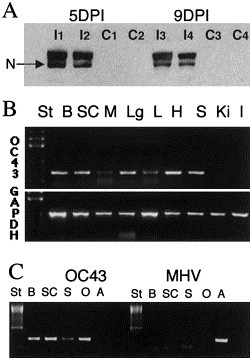
Detection of proteins and RNA in brains of C57BL/6 mice inoculated ic with HCoV-OC43 at 21 DPN. (A) Western blot analysis at 5 and 9 days postinfection, revealing the presence of the HCoV-OC43 N proteins in inoculated mice (I1, I2, I3, and I4 mice) and its absence in control mice (C1, C2, C3, and C4). (B) RT-PCR analysis revealed at 13 days postinfection that virus had spread from the CNS (B: brain; SC: spinal cord) to other peripheric organs, such as heart (H), lungs (Lg), spleen (S), and to a lesser extent, the liver (L) and muscles (M). HCoV-OC43 virus was never found in kidneys (Ki) nor in intestine (I). The quality of RNA extraction was estimated using GAPDH RNA detection. (C) RT-PCR analysis demonstrating that HCoV-OC43 and not an enzootic MHV strain was detected. HCoV-OC43 primers recognized HCoV-OC43 mRNA in B, SC, or S tissues of an infected mouse as well as in control RNA (O: HCoV-OC43-infected cells) but not MHV-A59 mRNA (A: MHV-infected cells). On the other hand, on the same sample, MHV primers recognized only MHV-A59 mRNA but not HCoV-OC43 mRNA.
Infectious virus appeared around 3 days postinfection and could be isolated from the CNS of C57BL/6 mice during the first 2 weeks postinfection (Fig. 2A). The highest levels of infectious virions were found between 5 and 9 days postinfection (Fig. 2C). In BALB/c mice, virus was detectable at 1 day postinfection and reached the highest titer around 3 days postinfection (Figs. 2B and C). The highest infectious titers observed were 108 TCID50/g for brain and 106 TCID50/g for spinal cord extracts. No infectivity could be detected at and after 13 days postinfection for C57BL/6 and 9 days postinfection for BALB/c mice.
Viral proteins were found in the brain and spinal cord of C57BL/6 mice between 5 and 11 days postinfection (Fig. 3A) and were undetectable after 10 days postinfection. We detected two forms of the N protein, as already noted in 8 DPN HCoV-OC43-inoculated mice (Jacomy and Talbot, 2001) or after MHV-4 infection (Talbot et al., 1984).
Blood collected at different time points after infection revealed that serum contained antibodies specific for HCoV-OC43. Humoral immunity started to appear at 1 week postinfection and increased during the first month postinfection, and antiviral antibodies were still present at 4 months postinfection, as shown by indirect immunofluorescence on infected HRT-18 cells (data not shown). No immunofluorescent cells were seen with serum obtain from control mice.
Histology
Histochemical labeling of viral distribution at different times after infection revealed that virus infection initiated by ic inoculation was quickly disseminated throughout the CNS. Cells positive for viral antigens were first observed at 3 days postinfection in the gray matter of the brains of C57BL/6 mice. At this time, microglial activation was still undetectable as assessed by Mac-2 immunostaining. At 1 week postinfection, HCoV-OC43 had spread to all CNS regions, predominantly in the entire cerebral cortex, the striatum, the hippocampus, the hypothalamus areas, the colliculus superior, and the brain stem, including the spinal cord (Fig. 4, Fig. 5). The cerebellum was frequently spared, but Purkinje cells were found positive for virus in some animals. Astrogliosis revealed by GFAP staining increased and activated microglial cells started to appear along the ventricles (Figs. 4F, G, and H). Activated microglial cells were not observed in the CNS of noninfected control mice at any time during investigation, as monitored by the absence of staining for Mac-2, a marker not expressed in nonreactive microglia (Walther et al., 2000). In the spinal cord at 7 days postinfection, an HCoV-OC43-specific MAb labeled sensory and motor neurons, and microgliosis and astrogliosis were also detected in infected regions (Fig. 5).
Fig. 4.
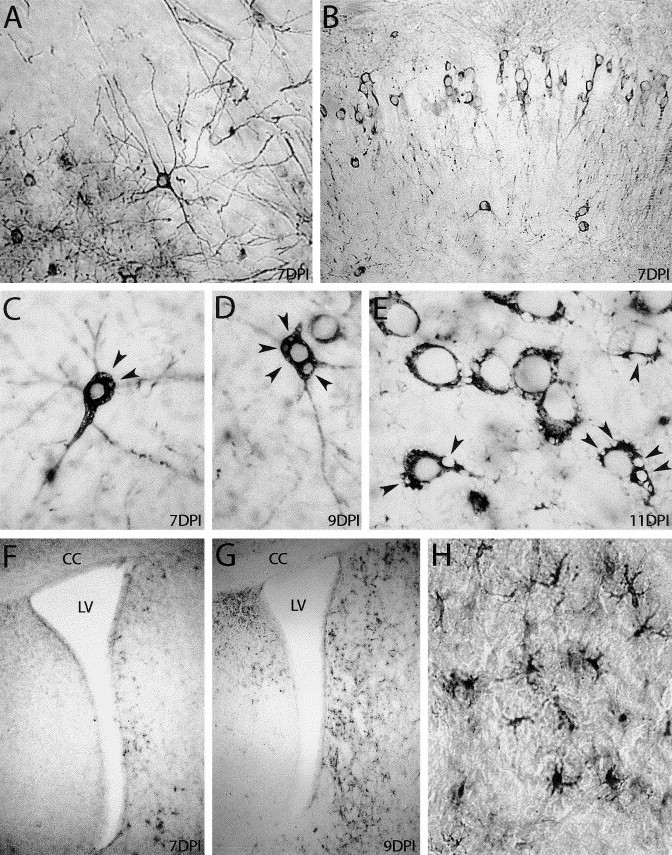
Immunocytochemical staining of infected (21 DPN) C57BL/6 mice brains. HCoV-OC43-infected neurons stained in black in the cerebral cortex (A) and in the CA1 layer of the hippocampus (B), 1 week after virus inoculation. In C, D, and E, HCoV-OC43-positive neurons exhibited empty vacuoles (arrowheads) in the cytoplasm at 7 days postinfection, which increased at 9 and 11 days postinfection. Panels F, G, and H show reactive microgliosis in the brain. At 7 days postinfection (F), Mac-2-stained cells started to appear along the lateral ventricle (LV) and became more numerous at 9 days postinfection (G). H shows a magnification of microglial cells observed in G (CC: corpus callosus). A, B, H: original magnification: ×200; F,G: magnification: ×40; C, D, E: ×400.
Fig. 5.
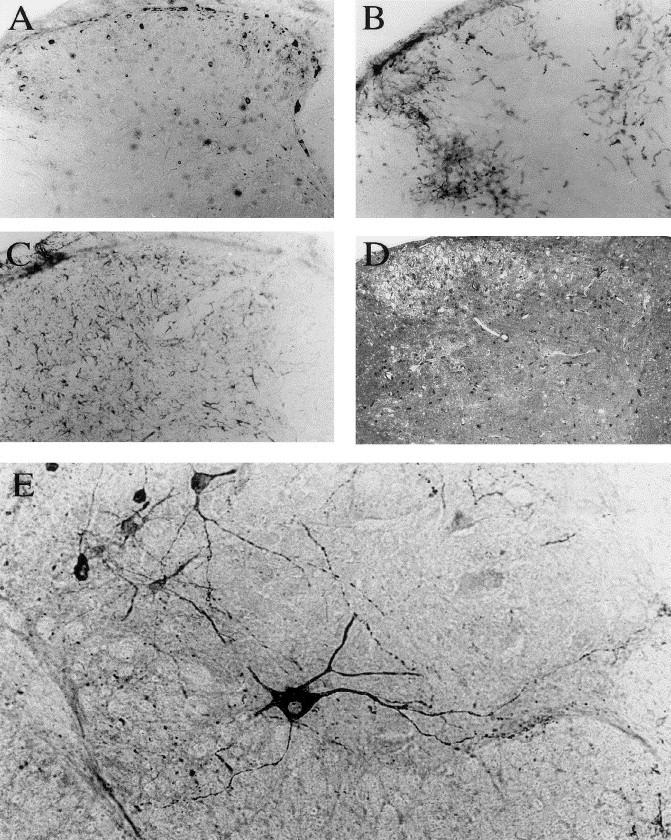
Immunocytochemical staining of the spinal cord of a C57BL/6 mouse (21 DPN) 1 week after HCoV-OC43 infection. (A, B, and C) Evidence of inflammatory reaction on three consecutive sections of the cervical dorsal horn. (A): Antiviral MAb. (B) Mac-2 microglia/macrophages cell marker. (C): GFAP astrocytic cell marker. (D): spongiosis aspect of gray matter revealed by toluidine blue staining. (E): Infection of motor neurons by HCoV-OC43 in the ventral horn of the lumbar spinal cord. Magnification: ×100 before enlargement.
The progression of the infection was accompanied by identical neuropathologic features in the two strains of mice: neurons exhibited severe signs of pathology, most of them showing necrosis and vacuolation. This started by the development of small and round empty vacuoles in the cytoplasm, which increased in size (Figs. 4C, D, and E). These spongiform-like lesions were seen primarily within the neuronal cell bodies, the neuropil being generally unaffected (Fig. 6C). This feature was never observed in noninfected brain (Fig. 6A). Ultrastructurally, numerous cells presented cytoplasm disorganization without lysis of the cellular membranes. Degenerative changes included cytoplasm rarefaction, dilatation of the rough endoplasmic reticulum (RER), and disaggregation of polyribosomes leading to the appearance of free ribosomes. Hematoxylin-eosin staining also revealed the presence of degenerated neurons with picknotic or small densely stained nuclei and eosinophilic cytoplasm (Figs. 6C, E, H, and I). At an advanced stage of disease, loss of neurons was pronounced and was particularly evident in CA1 and CA3 hippocampal layers (Fig. 6D–I).
Fig. 6.
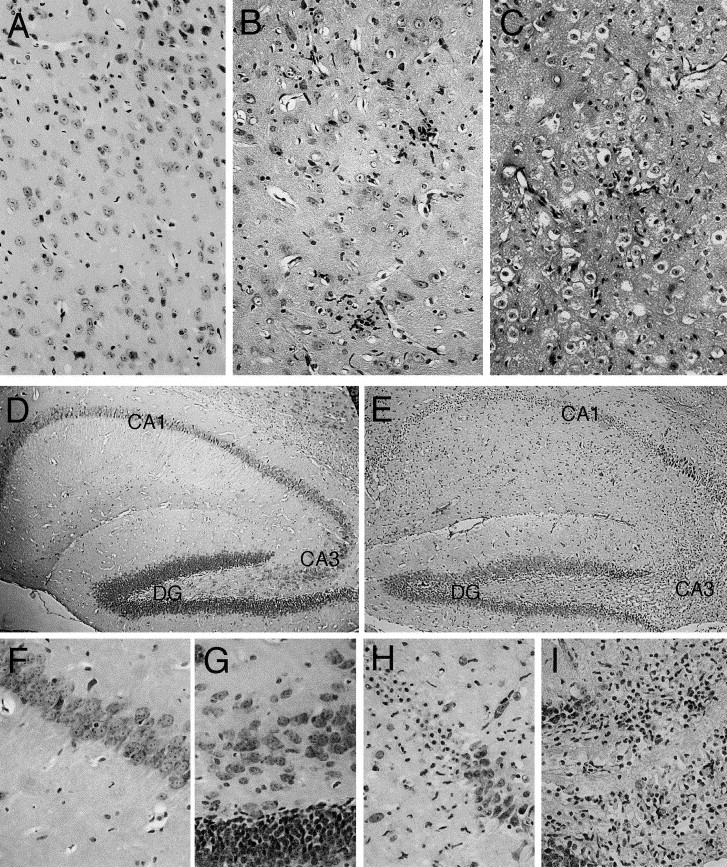
Cytotoxic effect of HCoV-OC43 infection in the CNS of C57BL/6 mice (21 DPN) at 11 days postinfection (hematoxylin and eosin staining) (A, B, and C) Overview of the thalamus (original magnification: ×200). (A) Control mice showing no degenerative changes. B (less affected animal) and C (more affected animal) illustrate infiltrating cells and spongiform-like lesions (characterized by vacuolation in neuronal cell bodies) and degenerative neurons with small densely stained nuclei or eosinophilic cytoplasm. (D) Normal hippocampus from control mice, with magnification of CA1 (F) and CA3 (G) hippocampus layers. (E) After infection, degenerative changes appeared mostly in CA1 (H) and CA3 (I) hippocampal layers; degenerated neurons exhibited picnotic nuclei and vacuolated cells were present in CA3 hippocampal layers (DG: Dentate Gyrus). (D and E) Magnification: ×40; F, G, H, and I: original magnification: ×200.
Histological examination of the brain or spinal cord revealed scattered infiltration of inflammatory cells, starting by mononuclear cell infiltrations (Fig. 6B) and perivascular cuffing. Some macrophage-mediated elimination (neuronophagia) was also encountered. In the spinal cord, viral particles observed 7 days postinfection at the electron microscopic level were mostly localized in the cell cytoplasm, closely associated with the Golgi apparatus or in extracellular spaces (Fig. 7). Viral replication and transneuronal passage occurred in a stepwise fashion that utilized existing cellular processes. When HCoV-OC43 replication and spread reached maximal levels, around 9 days postinfection, astrogliosis and microgliosis progressively increased in all infected regions of the CNS until the death of the animal (Figs. 4G and H). Therefore, a correlation between pathological signs of disease observed in mice and morphological injury of the brain was apparent.
Fig. 7.
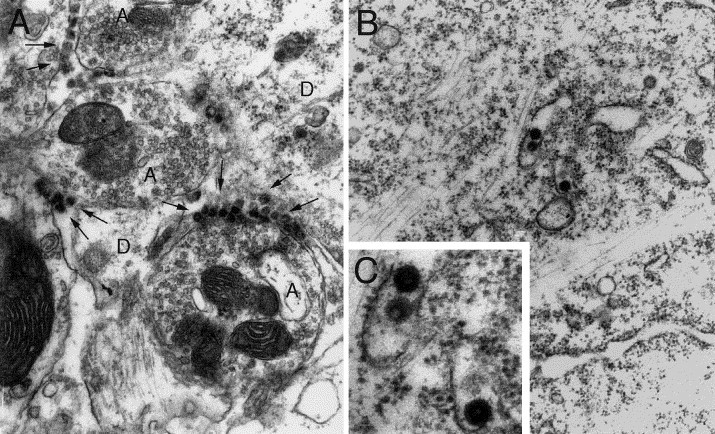
Detection of HCoV-OC43 infection at the electron microscopic level, in the spinal cord of C57BL/6 mice infected 7 days previously. (A) Numerous viral particles (arrows) were seen between the axons (A) and the dendrites (D). (B) Virus particles found in the endoplasmic reticulum of the cytoplasm (original magnification: ×20,000). (C) Magnification of B to show virions. Viral spikes around virions are typical of coronaviruses. No retroviral particles were observed (original magnification: ×40,000).
Immunosuppression by CsA in C57BL/6
Clearance of MHV from the CNS appears to involve T cells (Sussman et al., 1989) and age- acquired resistance to virus could be abolished in immunosuppressed animals (Zimmer and Dales, 1989). Therefore, we examined the effect of immunosuppression on HCoV-OC43- mediated neuropathogenesis. The immunosuppressive effects of cyclosporin A (CsA) are clearly established (Borel et al., 1976). As CsA causes a specific reversible inhibition of immunocompetent lymphocytes (preferentially T cells) and inhibits gene transcription for certain cytokines, in particular IL-2 Kupiec-Weglinski et al., 1984, Elliott et al., 1984, Shevach, 1985, we investigated whether CsA could modify the course of the acute HCoV-OC43 infection on the development of CNS lesions or on viral replication.
It is known that CsA injected into mice at 50 mg/kg/day induces neurotoxicity (hypocellular and disorganized organs), whereas CsA at 12.5 mg/kg/day induces no abnormalities and spread to all organs (Boland et al., 1984). Therefore, CsA doses were selected to avoid cytotoxic effects and mortality in mice and were in accordance with immunosuppression- inducing doses described in the literature Bolton et al., 1982, Pasick et al., 1992. Control mice treated with CsA at a daily dose of 20 mg/kg did not show any apparent adverse effects: they gained weight normally and did not present ruffled fur or lethargy. For CsA-treated and untreated mice, the kinetics of weight loss was similar after HCoV-OC43 infection. Nevertheless, immunosuppression by CsA slightly precipitated the disease but increased mortality (Fig. 8). This was more pronounced in mice treated with CsA at 20 mg/kg/day where 100% of mice died, whereas only 80% of oil-treated mice succumbed to HCoV-OC43 infection.
Fig. 8.
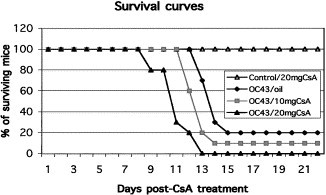
Survival curves of infected mice treated with cyclosporin A (CsA). Group of 10 C57BL/6 mice treated with cyclosporin A at 20 mg/kg/day (OC43/20 mgCsA) and at 10 mg/kg/day (OC43/10 mgCsA) became more susceptible to HCoV-OC43 infection, with 100 and 90% of death after infection versus 80% in non-CsA-treated animal. Infected C57BL/6 mice treated with oil alone (OC43/oil) presented similar survival curves as previously reported, with 80% of death after infection. Noninfected HCoV-OC43 mice treated with CsA at 20 mg/kg/day (Control/20 mgCsA) illustrated that CsA was not toxic under these conditions.
Discussion
Infection of mice by HCoV-OC43 was dependent on a number of variables, including dose, route of inoculation, age of the host, and its genetic background. Indeed, our results show striking susceptibility differences between two strains of mice: BALB/c mice were more resistant than C57BL/6. Moreover, resistance increased with age in the two strains of mice. This suggests that susceptibility to human coronavirus neuropathogenesis may be linked to genetic factors. Our study also confirms that human coronaviruses have neuroinvasive properties in mice, which was first shown in newborn mice (Barthold et al., 1990), and that such neuroinvasion is possible even after maturation of the immune system (King et al., 1992), which is consistent with their detection in human brain Burks et al., 1980, Murray et al., 1992, Stewart et al., 1992, Arbour et al., 2000. Even though our study does not confirm a specific route of entry into the CNS, a transneuronal route already demonstrated for MHV Lavi et al., 1988, Barthold et al., 1990, Perlman et al., 1990a constitutes a likely possibility.
Twenty-one days postnatal mice infected by ic inoculation of HCoV-OC43 developed signs of acute disease characterized by apathy, hunched posture, ruffled fur, and tremors, comparable to pathological signs described after MHV infection (Kristensson et al., 1986). Following IC inoculation, some mice developed disseminated infection, and mortality seemed to be related to the amount of infectious virus in the CNS. Mice inoculated IN with HCoV-OC43 did not show pathological signs, despite evidence of a disseminated infection. This was also previously observed after IN inoculation with MHV-A59, which caused only very mild pathologic symptoms and mortality even with very high viral doses (Lavi et al., 1986). The clinical signs of pathology after HCoV-OC43 IC inoculation coincided with the peak in virus yields observed at approximately 5 to 9 days postinfection for C57BL/6 mice. This indicates that virus replication in the CNS apparently played a major role in the establishment of the pathology.
Infected mice showed extensive inflammatory responses characterized by mononuclear perivascular cuffing, neuronophagia, and a great number of reactive glial cells in the infected regions. To investigate whether infiltration of inflammatory cells contributed to neurodegeneration or if infectious virus was directly responsible for vacuolating lesions and neuronal death, we evaluated the effect of treating animals with cyclosporin A, a powerful immunosuppressant drug. With the dose used (10 or 20 mg/kg/day), CsA is known to be distributed extensively throughout the body and not to cause neurotoxicity in mice (Boland et al., 1984). Immunosuppression precipitated human coronavirus-induced disease and increased the percentage of acute death (80 vs 100%). Under CsA treatment, neurons also presented vacuolation and degeneration. Therefore, the pathology observed following HCoV- OC43 infection was likely not immunologically mediated, unlike that induced by MHV-A59 and MHV-JHM Sussman et al., 1989, Wang et al., 1990, although experiments with immunodeficient mice of the same genetic background will be needed to definitely address this question with HCoV-OC43. Moreover, macrophage/microglial reactivity was delayed when related to infection and appeared only when the virus was present in most parts of the brain. The inflammatory response and macrophage/microglial cell recruitment seem to be strongly correlated with virus clearance, as was also demonstrated after MHV infection (Sussman et al., 1989). Some strains of MHV, including A59 and JHM, are neuroinvasive in rodents, eliciting either an acute encephalitis or a chronic paralytic disease (for review, see Perlman, 1998). Unlike the slow neurodegenerative disease caused by MHV Bailey et al., 1949, Lampert et al., 1973, Weiner, 1973, Lavi et al., 1984, Wang et al., 1990, HCoV-OC43 resulted in a productive and cytotoxic infection of neuronal cells in the CNS, which led to neurodegeneration.
Having previously demonstrated a persistent infection of HCoV-OC43 in primary murine CNS cell cultures and in the CNS of mice inoculated at 8DPN (Jacomy and Talbot, 2001), and given the observations that MHV antigens or RNA were still detectable in the spinal cord several weeks after infection Woyciechowska et al., 1984, Perlman et al., 1990b, we expected to detect a persistent infection in the CNS of infected 21 DPN mice. However, RT- PCR analysis revealed that viral RNA could not be detected after the second week postinfection, suggesting a nonpersistent infection of HCoV-OC43 virus in 21 DPN mice.
Histological analysis of infected 21 DPN mice showed virus spread throughout the brain and spinal cord, as we had previously described for 8 DPN mice (Jacomy and Talbot, 2001). Neurons remained the major cellular targets for virus, which was probably disseminated from neurons to neurons by a cell-to-cell transport, as was described after MHV infection Lavi et al., 1988, Sun and Perlman, 1995. Nevertheless, even though neurons were susceptible to MHV infection, oligodendrocytes and astrocytes represented the major infected cell types Perlman and Ries, 1987, Sun and Perlman, 1995, Sun et al., 1995. This may explain why the pathology observed in mice was different after infection with these two antigenically related coronaviruses: MHV can cause demyelination Bailey et al., 1949, Lampert et al., 1973, Weiner, 1973, Wang et al., 1990 with a spongy state observed in the white matter (Lavi et al., 1984), whereas HCoV-OC43 encephalitis is accompanied by vacuolating degeneration of the gray matter. The latter lesions were not detected in a previous study Pearson and Mims, 1983, Barthold et al., 1990, probably because mice were younger and died within 3 to 7 days postinfection. Spongiform cellular changes were occasionally reported after MHV infection. For example, vacuolation was observed in the subthalamic- nigral region after ic inoculation of MHV-A59 (Fishman et al., 1985), and foci of vacuolation were observed in the hypothalamus, cerebellar peduncles, and pons regions after IN inoculation of MHV-S (Barthold and Smith, 1983). Nevertheless, these degenerative changes were not commonly observed after MHV infection and were restricted to small cell populations, even after infection with 300- to 600-fold higher virus doses into C57BL/6 mice.
Interestingly, the appearance of clear round vacuoles and neuronal death represents a hallmark of CNS degeneration observed in prion diseases (Prusiner, 1998). Nevertheless, mitochondrial disease (McKelvie et al., 1991), Leigh's disease Kimura et al., 1991, Agapitos et al., 1997, Pick's disease (Deruaz et al., 1993), or Alzheimer's disease Duffy et al., 1988, Budka et al., 1995 also display spongiform CNS lesions, which are independent of the prion protein. Pathways and causal start points of transmissible spongiform encephalopathies or acute encephalitis remain unknown. Occasionally, viral infections of the CNS were described to induce spreading spongiosis, such as in human T cell leukemia virus associated myelopathy (HTLV-1) or in HIV encephalitis Rhodes, 1987, Goldwater and Paton, 1989. In rodents, mutants of VSV virus (Rabinowitz et al., 1976) and Moloney murine leukemia virus (Mo-MuLV) Gardner et al., 1973, Czub et al., 1994 were shown to experimentally induce spongiosis. Histologically, vacuolar degeneration induced by HCoV-OC43 was mainly restricted to the neuronal cell bodies, whereas that caused by prion or retrovirus first affected the neuropil. Moreover, the inflammatory response was very limited in prion encephalopathy, whereas HCoV-OC43 induced extensive brain inflammation. These indicate different mechanisms underlying vacuolation and neuronal death.
Interestingly, noninflammatory neuropathologies have been considered evidence against a viral etiology. Infections by opportunistic pathogens such as respiratory or enteric virus in immuosuppressed patients (HIV, transplantation, and cancer chemotherapy) may also cause CNS pathology. It has been reported that immunocompromised patients have increased incidences of malignancies induced by viral infection (Penn, 1987). Therefore, severe cases of encephalitis have devastating effects on the brain and spinal cord functions. Given our previous observations of HCoV-OC43 persistence in human brain Stewart et al., 1992, Arbour et al., 2000, we propose that respiratory pathogens with a neurotropic and neuroinvasive potential could be associated with neurodegenerative disease in susceptible individuals. This animal model of human coronavirus neuropathogenesis may prove helpful in the characterization of coronavirus-induced neurodegeneration in surviving infected animals and of transneuronal virus spread to the CNS. Moreover, susceptibilities of endothelial cells and leukocytes to viral infection need to be investigated as a possible alternate route of virus entry into the CNS. Finally, possible explanations for our observations of striking differences in susceptibility to HCoV-OC43 infection of two strains of mice remain to be investigated.
Even though mice are not the natural host for HCoV-OC43 infections, they may provide data that, together with studies using human neural cell cultures and post-mortem brain tissue, may contribute to our understanding of the underlying mechanisms and neuropathological consequences of coronavirus infections in humans.
Materials and methods
Virus, mice, and inoculations
The OC43 strain of HCoV was originally obtained from the American Type Culture Collection (ATCC, Rockville, MD), plaque-purified, and grown on the human rectal carcinoma cell line HRT-18 as previously described (Mounir and Talbot, 1992). HCoV- OC43 virus stocks (106 TCID50/ml) were kept at −80°C.
To determine the susceptibility of mice to HCoV-OC43 infection, two different strains of MHV-seronegative female mice (BALB/c and C57BL/6, from Jackson Laboratories, Bar Harbor, ME, U.S.A.) were inoculated. Inoculations were performed on mice at various days postnatal (1, 8, 15, 21, 28, 35 DPN) using 10 μl of various dilutions of the initial virus stock and using different inoculation routes: intraperitoneal, intraoral, intranasal, and intracerebral, to investigate HCoV-OC43 infection parameters, in particular its neuroinvasive property. The viral dose was administrated IC under deep anesthesia of ketamine-xylamine (ketamine at 200 mg/kg and xylamine at 10 mg/kg). In the present study, we chose to infect 21 DPN mice with an IC inoculation of 10 μl containing 10 TCID50 of HCoV-OC43 for C57BL/6 and 10,000 TCID50 for BALB/c mice. Twenty mice of each strain were used to establish survival curves. Every 2 days postinfection, five animals from two groups of infected mice of each strain were sacrificed and processed for detection of viral RNA, viral proteins, and infectious virus. Moreover, two infected mice of each strain were perfused every 2 days for histological analysis during the first month postinfection. For each experiment, age-matched control animals, which had received a virus-free solution containing culture medium from the HRT-18 cell line, were used.
To confirm that HCoV-OC43 was responsible for the observed pathology, we infected HRT-18 cell cultures with brain homogenates, prepared as described below, from infected mice. Cell-free supernatants harvested 4 days later were confirmed to contain infectious virus and 10 μl was reinoculated IC into other animals.
Immunosuppression by cyclosporin A
Cyclosporin A (Sigma Chemical Co., St. Louis, MO) was rehydrated in pure ethanol as specified by the manufacturer. It was then dissolved before use in olive oil to favor free diffusion of hydrophobic cyclosporin molecules through the plasma membrane into the cytoplasm (Handschumacher et al., 1984), and heated for 2 h at 60°C. Mice received a subcutaneous injection of 10 or 20 mg/kg/day. This drug was administered 1 day prior to virus inoculation and daily thereafter for 10 days. Three groups of 10 female C57BL/6 mice were infected with 10 TCID50 of HCoV-OC43 and 8 control females were inoculated with HRT-18 medium. Two groups of HCoV-OC43-infected mice received a single daily injection of CsA, either 10 or 20 mg/kg/day. Four control mice and the third group of HCoV-OC43- infected mice received injection of olive oil alone. The four remaining control mice were treated with CsA at 20 mg/kg/day, to verify the absence of CsA toxicity.
Infectious virus assays
Brain and spinal cords were dissected, homogenized in 10% (w/v) sterile PBS, centrifuged at 4°C, 20 min at 1000 g; then supernatants were immediately frozen at −80°C and stored until assayed. The extracts were processed for the presence and quantification of infectious virus by an indirect immunohistochemistry assay, as previously described (Bonavia et al., 1997). HCoV-OC43-susceptible HRT-18 cells were inoculated with serial logarithmic dilutions of each tissue sample in a 96-well Linbro plate (ICN Biomedical Canada Ltd., Costa Mesa, CA). After 4 days of incubation at 33°C in 5% (v/v) CO2, cells were washed in PBS and fixed with 0.3% (v/v) hydrogen peroxide (H2O2) in methanol for 30 min. After washing with PBS, they were incubated for 2 h at 37°C in 1/1000 dilution of an ascites fluid from mouse MAb 1–10C.3, directed against the nucleocapsid protein of HCoV-OC43 (Arbour et al., 1999b). Afterwards, cells were washed in PBS and HRP goat anti-mouse immunoglobulins (DAKO, Diagnostics Canada Inc., Mississauga, ON) were added and incubated for 2 h at 37°C. Antibody complexes were detected by incubation in 3.3′-diaminobenzidine tetrahydrochloride solution (DAB, Sigma), with 0.01% (v/v) H2O2.
Immunohistochemistry
Mice were perfused by intraventricular injection of 4% (v/v) paraformaldehyde, under deep ketamine-xylazine anesthesia. Brains and spinal cords were removed and tissue blocks were left in the fixative for 24 h. Coronal sections from brain and segments from cervical and lumbar spinal cord were sectioned at a thickness of 40 μm with a Lancer vibratome. Serial sections were collected in 0.05 M Tris-buffered saline (TBS) and were then incubated overnight with primary antibodies, as previously described (Jacomy and Bosler, 1996). For viral antigens, we used 1/1000 dilutions of ascites fluids of the 4-E11.3 hybridoma that secretes monoclonal antibodies specific for the nucleocapsid protein of the serologically related hemagglutinating encephalomyelitis virus of pigs (Bonavia et al., 1997). Astrocytes were identified with a rabbit anti glial fibrillary acidic protein antibody (GFAP, DAKO) diluted 1/500, microglia/macrophages by an ascites fluid of the rat Mac-2 antibody (ATCC) diluted 1/1000. Then, sections were rinsed and processed with Vectastain ABC Kit (Vector Laboratories, Burlingame, CA). Labeling was revealed with 0.03% (w/v) DAB solution (Sigma) and 0.01% (v/v) H2O2, which yielded a dark brown product. Some sections were counterstained with the classical Cresyl violet stain. To further investigate histological changes occurring in mouse brains, half hemispheres from control and infected animals were paraffin-embedded and 10-μm sections were stained with hematoxylin-eosin. This was performed by the Pathology Department, Animal Resources Centre, McGill University (Montréal, Québec, Canada).
Samples for electron microscopy were postfixed for 2 h with 2% (v/v) osmium tetraoxide in 0.1 M phosphate buffer at pH 7.5, dehydrated in graded ethanol series, and Epon-embedded as previously described (Jacomy and Bosler, 1996). One-micron sections were stained with toluidine blue and examined by light microscopy. Subsequent ultrathin sections were collected on collodion-coated single-slot grids, stained with lead citrate, and examined with transmission electron microscope.
Detection of antiviral antibodies
Blood from infected or control mice were collected at 1, 2, 3, or 4 weeks and at 2, 3, and 4 months postinfection. Sera were collected and kept at −20°C until use for the detection of antibodies against HCoV-OC43 by indirect immunofluorescent labeling of infected HRT-18 cells. Briefly, HRT-18 cells cultured on 12-well slides were infected by HCoV-OC43 and fixed 4 days later in cold methanol and then kept at −20°C until needed. At the time of the assays, slides were incubated 1 h at room temperature with serum from control and infected mice, diluted 1/100, 1/500, and 1/1000. After several washes in PBS, slides were incubated 1 h at 37°C with Alexa Fluor 488 F(ab′)2 fragments of goat anti-mouse IgG (H+L), at a dilution of 1/15,000 (Molecular Probes, Inc., Eugene, OR) and observed under a fluorescence microscope.
Western blot analyses
Tissues were homogenized in SUB buffer, containing 8 M urea, 0.5% (w/v) SDS, and 2% (v/v) β-mercaptoethanol and then centrifuged for 15 min at 4°C, in a microfuge at 13,000 g and supernatants were collected, as previously described (Jacomy et al., 1999). Samples (5 μg total protein) were fractionated on a 7.5% polyacrylamide gel (SDS–PAGE) and either visualized by Coomassic blue staining or transferred to nitrocellulose for Western blot analysis. Nitrocellulose membranes were preincubated in 5% (w/v) skimmed milk powder in TS buffer (0.05 M Tris, pH 7.4, 0.15 M NaCl) and then incubated overnight at 4°C with 4E11.3 antiviral MAB. After several washes with TS buffer containing 0.05% (v/v) Tween 20, membranes were incubated 1 h with peroxidase-conjugated anti-mouse IgG diluted 1/1000 (DAKO). Bands were visualized using a Western blot chemoluminescent kit (Super Signal, Pierce, Rockford, MD).
Preparation of RNA and RT-PCR
Tissues were dissected every 2 days postinfection. Total RNA was extracted by homogenization in Trizol (Gibco-BRL, Burlington, CA). For RT-PCR, one pair of HCoV-OC43 primers was designed to amplify a region containing 305 nucleotides (primers O1 and O3) of the gene coding for the N protein (Arbour et al., 1999b). The target sequences were specific to HCoV-OC43 and did not amplify MHV. The suitability of RNA for RT-PCR amplification was verified by an RT-PCR specific for a housekeeping gene encoding glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using a pair of GAPDH primers amplifying a region containing 833 nucleotides (Arbour et al., 1999b). One pair of MHV primers was also designed to amplify a conserved region of the MHV N protein gene. Primers were 5′-CCTCTACTGTAAAACCTGATATGG-3′ and 5′-CTAATTTAGATCCAAAGAAGAAGC-3′, corresponding to nucleotides 677–700 and 868–991, respectively. Approximately 5 μg of RNA was reverse transcribed with Expand Moloney murine leukemia virus reverse transcriptase (Gibco-BRL) and the cDNA products were incubated in 20 pmol of each sense and antisense primers, 2.5 mM MgCl2, 1× PCR buffer (10 mM Tris–HCl, pH 8.3; 50 mM KCl), and 0.4 mM of each deoxynucleotide triphosphate, heated at 94°C for 5 min and 60°C (HCoV-OC43) or 50°C (GAPDH and MHV) for 5 min. After the addition of Expand high-fidelity PCR system DNA polymerase (rTaq, 5000 U/μl; Amersham Pharmacia Biotech Inc., Baie d'Urfé, QC), 30 amplification cycles of 2 min at 72°C, 1 min at 95°C, and 2 min at 60°C (HCoV-OC43) or 50°C (GAPDH and MHV) were performed. Ten microliters of this reaction mix was loaded onto a 1.2% (w/v) agarose gel containing 5 μl ethidium bromide.
Acknowledgements
We thank Annie Boucher, INRS-Institut Armand-Frappier, for the critical review of the manuscript and Francine Lambert for excellent technical assistance. We also thank Dr. Serge Dea (who tragically passed away on January 3, 2003), INRS-Institut Armand-Frappier, for the generous gift of the 4-E11.3 antibody, and Dr. Yves Robitaille, McGill University, for constructive comments on neuropathology. We also thank the McGill University Animal Resources Centre for their help with some histology. This work was supported by Grant MT-9203 from the Canadian Institutes of Health Research (Institute of Infection and Immunity).
References
- Agapitos E., Pavlopoulos P.M., Patsouris E., Davaris P. Subacute necrotizing encephalopathy (Leigh's disease): a clinicopathologic study of ten cases. Gen. Diag. Pathol. 1997;142:341–355. [PubMed] [Google Scholar]
- Allen I.V., McQuaid S., McMahon J., Kirk J., McConnell R. The significance of measles virus antigen and genome distribution in the CNS in SSPE for mechanisms of viral spread and demyelination. J. Neuropathol. Exp. Neurol. 1996;55:471–480. doi: 10.1097/00005072-199604000-00010. [DOI] [PubMed] [Google Scholar]
- Arbour N., Côté G., Lachance C., Tardieu M., Cashman N.R., Talbot P.J. Acute and persistent infection of human neural cell lines by human coronavirus OC43. J. Virol. 1999;73:3338–3350. doi: 10.1128/jvi.73.4.3338-3350.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour N., Day R., Newcombe J., Talbot P.J. Neuroinvasion by human respiratory coronaviruses and association with multiple sclerosis. J. Virol. 2000;74:8913–8921. doi: 10.1128/jvi.74.19.8913-8921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour N., Ekandé S., Côté G., Lachance C., Chagnon F., Tardieu M., Cashman N.R., Talbot P.J. Persistent infection of human oligodendrocytic and neuroglial cell lines by human coronavirus 229E. J. Virol. 1999;73:3326–3337. doi: 10.1128/jvi.73.4.3326-3337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey O.T., Pappenheimer A.M., Cheever F.S., Daniels J. A murine virus (JHM) causing disseminated encephalomyelitis with extensive destruction of myelin. II. Pathology. J. Exp. Med. 1949;90:195–212. doi: 10.1084/jem.90.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthold S.W., Smith A.L. Mouse hepatitis virus S in weanling Swiss mice following intranasal inoculation. Lab. Anim. Sci. 1983;33:355–360. [PubMed] [Google Scholar]
- Barthold S.W., Smith A.L. Response of genetically susceptible and resistance mice to intranasal inoculation with mouse hepatitis virus JHM. Virus Res. 1987;7:225–239. doi: 10.1016/0168-1702(87)90030-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthold S.W., de Souza M.S., Smith A.L. Susceptibility of laboratory mice to intranasal and contact infection with coronaviruses of other species. Lab. Anim. Sci. 1990;40:481–485. [PubMed] [Google Scholar]
- Boland J., Atkinson K., Britton K., Darveniza P., Johnson S., Biggs J. Tissue distribution and toxicity of cyclosporin A in the mouse. Pathology. 1984;16:117–123. doi: 10.3109/00313028409059087. [DOI] [PubMed] [Google Scholar]
- Bolton C., Allsopp G., Cuzner M.L. The effect of cyclosporin A on the adoptive transfer of experimental allergic encephalomyelitis in the Lewis rat. Clin. Exp. Immunol. 1982;47:127–132. [PMC free article] [PubMed] [Google Scholar]
- Bonavia A., Arbour N., Wee Yong V., Talbot P.J. Infection of primary cultures of human neural cells by human coronavirus 229E and OC43. J. Virol. 1997;71:800–806. doi: 10.1128/jvi.71.1.800-806.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel J.F., Feurer C., Gubler H.U., Stahelin H. Biological effects of Cyclosporin A: a new antilymphocytic agent. Agents Actions. 1976;6:468–475. doi: 10.1007/BF01973261. [DOI] [PubMed] [Google Scholar]
- Boucher A., Denis F., Duquette P., Talbot P.J. Generation from multiple sclerosis patients of long-term T-cell clones that are activated by both human coronavirus and myelin antigens. Adv. Exp. Med. Biol. 2001;494:355–362. doi: 10.1007/978-1-4615-1325-4_53. [DOI] [PubMed] [Google Scholar]
- Budka H., Aguzzi A., Brown P., Brucher J.M., Bugiani O., Gullotta F., Haltia M., Hauw J.J., Ironside J.W., Jellinger K. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases) Brain Pathol. 1995;5:459–466. doi: 10.1111/j.1750-3639.1995.tb00625.x. [DOI] [PubMed] [Google Scholar]
- Burks J.S., DeVald B.L., Jankovsky L.D., Gerdes J.C. Two coronaviruses isolated from central nervous system tissue of multiple sclerosis patients. Science. 1980;209:933–934. doi: 10.1126/science.7403860. [DOI] [PubMed] [Google Scholar]
- Calne D.B., Eisen A., McGeer E., Spencer P. Alzheimer's disease, Parkinson's disease, and motoneuron disease: abiotrophic interaction between ageing and environment. Lancet. 1986;8:1067–1070. doi: 10.1016/s0140-6736(86)90469-1. [DOI] [PubMed] [Google Scholar]
- Czub S., Lynch W.P., Czub M., Portis J.L. Kinetic analysis of spongiform neurodegenerative disease induced by a highly virulent murine retrovirus. Lab. Invest. 1994;70:711–723. [PubMed] [Google Scholar]
- Deruaz J.P., Assal G., Peter-Favre C. A clinicopathological case of progressive aphasia. Rev. Neurol. (Paris) 1993;149:186–191. [PubMed] [Google Scholar]
- Duffy P., Mayeux R., Kupsky W. Familial Alzheimer's disease with myoclonus and “spongy change”. Arch. Neurol. 1988;45:1097–1100. doi: 10.1001/archneur.1988.00520340051011. [DOI] [PubMed] [Google Scholar]
- Elliott J.F., Lin Y., Mizel S.B., Bleackley R.C., Harnish D.G., Paetkau V. Induction of interleukin 2 messenger RNA inhibited by cyclosporin A. Science. 1984;226:1439–1441. doi: 10.1126/science.6334364. [DOI] [PubMed] [Google Scholar]
- Fishman P.S., Gass J.S., Swoveland P.T., Lavi E., Highkin M.K., Weiss S.R. Infection of the basal ganglia by a murine coronavirus. Science. 1985;30:877–879. doi: 10.1126/science.2992088. [DOI] [PubMed] [Google Scholar]
- Gardner M.B., Henderson B.E., Officer J.E., Rongey R.W., Parker J.C., Oliver C., Estes J.D., Huebner R.J. A spontaneous lower motor neuron disease apparently caused by endogenous type-C RNA virus in wild mice. J. Natl. Cancer Int. 1973;51:1243–1254. doi: 10.1093/jnci/51.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgsson G. Neuropathologic aspects of lentiviral infections. Ann. NY Acad. Sci. 1994;6:50–67. doi: 10.1111/j.1749-6632.1994.tb38895.x. [DOI] [PubMed] [Google Scholar]
- Giraud P., Beaulieux F., Ono S., Shimizu N., Chazot G., Lina B. Detection of enteroviral sequences from frozen spinal cord samples of Japanese ALS patients. Neurology. 2001;26:1777–1778. doi: 10.1212/wnl.56.12.1777. [DOI] [PubMed] [Google Scholar]
- Goldwater P.N., Paton J.C. Apparent non-involvement of prions in the pathogenesis of spongiform change in HIV-infected brain. J. Neuropathol. Exp. Neurol. 1989;48:184–186. doi: 10.1097/00005072-198903000-00005. [DOI] [PubMed] [Google Scholar]
- Handschumacher R.E., Harding M.W., Rice J., Drugge R.J., Speicher D.W. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science. 1984;226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- Hayase Y., Tobita K. Influenza virus and neurological diseases. Psychiatry Clin. Neurosci. 1997;51:181–184. doi: 10.1111/j.1440-1819.1997.tb02580.x. [DOI] [PubMed] [Google Scholar]
- Jacomy H., Bosler O. Intrinsic organization and monoaminergic innervation of the suprachiasmatic nucleus transplanted to adult rats. A light- and electronic- microscopic study. J. Neurocytol. 1996;25:659–673. doi: 10.1007/BF02284832. [DOI] [PubMed] [Google Scholar]
- Jacomy H., Talbot P.J. Susceptibility of murine CNS to OC43 infection. Adv. Exp. Med. Biol. 2001;494:101–107. doi: 10.1007/978-1-4615-1325-4_16. [DOI] [PubMed] [Google Scholar]
- Jacomy H., Zhu Q., Couillard-Despres S., Beaulieu J.M., Julien J.P. Disruption of type IV intermediate filament network in mice lacking the neurofilament medium and heavy subunits. J. Neurochem. 1999;73:972–984. doi: 10.1046/j.1471-4159.1999.0730972.x. [DOI] [PubMed] [Google Scholar]
- Jubelt B., Berger J.R. Does viral disease underlie ALS? Lessons from the AIDS pandemic. Neurology. 2001;57:945–946. doi: 10.1212/wnl.57.6.945. [DOI] [PubMed] [Google Scholar]
- Kimura S., Kobayashi T., Amemiya F. Myelin splitting in the spongy lesion in Leigh encephalopathy. Pediatr. Neurol. 1991;7:56–58. doi: 10.1016/0887-8994(91)90108-w. [DOI] [PubMed] [Google Scholar]
- King L.E., Morford L.A., Gibbons J.P., Fraker P.J. Flow cytometric analysis of the expression of murine B and T surface markers from the birth to adulthood. Immunol. Lett. 1992;31:73–78. doi: 10.1016/0165-2478(92)90013-e. [DOI] [PubMed] [Google Scholar]
- Kirk J., Zhou A.L. Viral infection at the blood-brain barrier in multiple sclerosis: an ultrastructural study of tissues from a UK Regional Brain Bank. Mult. Scler. 1996;1:242–252. [PubMed] [Google Scholar]
- Klein R., Mullges W., Bendszus M., Woydt M., Kreipe H., Roggendorf W. Primary intracerebral Hodgkin's disease: report of a case with Epstein–Barr virus association and review of the literature. Am. J. Surg. Pathol. 1999;23:477–481. doi: 10.1097/00000478-199904000-00014. [DOI] [PubMed] [Google Scholar]
- Koskiniemi M., Vaheri A. Effect of measles, mumps, rubella vaccination on pattern of encephalitis in children. Lancet. 1989;7:31–34. doi: 10.1016/s0140-6736(89)91683-8. [DOI] [PubMed] [Google Scholar]
- Kristensson K., Holmes K.V., Duchala C.S., Zeller N.K., Lazzarini R.A., Dubois-Dalcq M. Increased levels of myelin basic protein transcripts in virus-induced demyelination. Nature. 1986;322:544–547. doi: 10.1038/322544a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensson K. Potential role of viruses in neurodegeneration. Mol. Chem. Neuropathol. 1992;16:45–58. doi: 10.1007/BF03159960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupiec-Weglinski J.W., Filho M.A., Strom T.B., Tilney N.L. Sparing of suppressor cells: a critical action of cyclosporine. Transplantation. 1984;38:97–101. doi: 10.1097/00007890-198408000-00001. [DOI] [PubMed] [Google Scholar]
- Lampert P.W., Sims J.K., Kniazeff A.J. Mechanism of demyelination in JHM virus encephalomyelitis. Acta Neuropathol. 1973;24:76–85. doi: 10.1007/BF00691421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi E., Gilden D.H., Wroblewska Z., Rorke L.B., Weiss S.R. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology. 1984;34:597–603. doi: 10.1212/wnl.34.5.597. [DOI] [PubMed] [Google Scholar]
- Lavi E., Gilden D.H., Highkin M.K., Weiss S.R. The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab. Anim. Sci. 1986;36:130–135. [PubMed] [Google Scholar]
- Lavi E., Fishman P.S., Highkin M.K., Weiss S.R. Limbic encephalitis after inhalation of a murine coronavirus. Lab. Invest. 1988;58:31–36. [PubMed] [Google Scholar]
- Lewis D.A. Retroviruses and pathogenesis of schizophrenia. Proc. Natl. Acad. Sci. USA. 2001;98:4293–4294. doi: 10.1073/pnas.081075898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K. In: Virology. third ed. Fields B.N., Knipe D.M., editors. Raven Press; New York: 1996. pp. 1095–1103. [Google Scholar]
- McKelvie P.A., Morley J.B., Byrne E., Marzuki S. Mitochondrial encephalomyopathies: a correlation between neuropathological findings and defects in mitochondrial DNA. J. Neurol. Sci. 1991;102:51–60. doi: 10.1016/0022-510x(91)90093-m. [DOI] [PubMed] [Google Scholar]
- Mounir S., Talbot P.J. Sequence analysis of the membrane protein gene of human coronavirus OC43 and evidence for O-glycosylation. J. Gen. Virol. 1992;73:2731–2736. doi: 10.1099/0022-1317-73-10-2731. [DOI] [PubMed] [Google Scholar]
- Murray R.S., Brown B., Brian D., Cabirac G.F. Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann. Neurol. 1992;31:525–533. doi: 10.1002/ana.410310511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myint S.H. Human coronavirus—a brief review. Rev. Med. Virol. 1994;4:35–46. [Google Scholar]
- Pasick J.M., Wilson G.A., Morris V.L., Dales S. SJL/J resistance to mouse hepatitis virus-JHM-induced neurologic disease can be partially overcome by viral variants of S and host immunosuppression. Microb. Pathog. 1992;13:1–15. doi: 10.1016/0882-4010(92)90027-L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson J., Mims C.A. Selective vulnerability of neural cells and age-related susceptibility to OC43 virus in mice. Arch. Virol. 1983;77:109–118. doi: 10.1007/BF01309260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn I. Cancers following cyclosporine therapy. Transplantation. 1987;43:32–35. doi: 10.1097/00007890-198701000-00008. [DOI] [PubMed] [Google Scholar]
- Perlman S., Ries D. The astrocyte is a target cell in mice persistently infected with mouse hepatitis virus, strain JHM. Microb. Pathog. 1987;3:309–1449. doi: 10.1016/0882-4010(87)90064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S. Pathogenesis of coronavirus-induced infections. Review of pathological and immunological aspects. Adv. Exp. Med. Biol. 1998;440:503–513. [PubMed] [Google Scholar]
- Perlman S., Evans G., Afifi A. Effect of olfactory bulb ablation on spread of a neurotropic coronavirus into the mouse brain. J. Exp. Med. 1990;172:1127–1132. doi: 10.1084/jem.172.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Jacobsen G., Olson A.L., Afifi A. Identification of the spinal cord as a major site of persistence during chronic infection with a murine coronavirus. Virology. 1990;175:418–426. doi: 10.1016/0042-6822(90)90426-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S.B. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz S.G., Dal Canto M.C., Johnson T.C. Comparison of central nervous system disease produced by wild-type and temperature-sensitive mutants of vesicular stomatis virus. Infect. Immunol. 1976;13:1242–1249. doi: 10.1128/iai.13.4.1242-1249.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resta S., Luby J.P., Rosenfeld C.R., Siegel J.D. Isolation and propagation of a human enteric coronavirus. Science. 1985;229:978–981. doi: 10.1126/science.2992091. [DOI] [PubMed] [Google Scholar]
- Rhodes R.H. Histopathology of the central nervous system in the acquired immunodeficiency syndrome. Hum. Pathol. 1987;18:636–643. doi: 10.1016/s0046-8177(87)80365-9. [DOI] [PubMed] [Google Scholar]
- Riski N., Hovi T. Coronavirus infections of man associated with diseases other than the common cold. J. Med. Virol. 1980;6:259–265. doi: 10.1002/jmv.1890060309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht C.E., Hanlon C.A., Hemachudha T. Rabies re-examined. Lancet Infect. Dis. 2002;2:327–343. doi: 10.1016/s1473-3099(02)00287-6. [DOI] [PubMed] [Google Scholar]
- Shevach E.M. The effects of cyclosporin A on the immune system. Annu. Rev. Immunol. 1985;3:397–423. doi: 10.1146/annurev.iy.03.040185.002145. [DOI] [PubMed] [Google Scholar]
- Shoji H., Azuma K., Nishimura Y., Fujimoto H., Sugita Y., Eizuru Y. Acute viral encephalitis: the recent progress. Intern. Med. 2002;41:420–428. doi: 10.2169/internalmedicine.41.420. [DOI] [PubMed] [Google Scholar]
- Sizun J., Yu M.W., Talbot P.J. Survival of human coronaviruses 229E and OC43 in suspension and after drying on surfaces: a possible source of hospital-acquired infections. J. Hosp. Infect. 2000;46:55–60. doi: 10.1053/jhin.2000.0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola P., Bedin R., Casoni F., Barrozzi P., Mandrioli J., Merelli E. New insights into the viral theory of amyotrophic lateral sclerosis: study on the possible role of Kaposi's sarcoma-associated virus/human herpesvirus 8. Eur. Neurol. 2002;47:108–112. doi: 10.1159/000047961. [DOI] [PubMed] [Google Scholar]
- Stewart J.N., Mounir S., Talbot P.J. Human coronavirus gene expression in the brains of multiple sclerosis patients. Virology. 1992;191:502–505. doi: 10.1016/0042-6822(92)90220-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N., Grzybicki D.M., Castro R.F., Murphy S.P., Perlman S. Activation of astrocytes in the spinal cord of mice chronically infected with a neurotropic coronavirus. Virology. 1995;213:482–493. doi: 10.1006/viro.1995.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N., Perlman S. Spread of a neurotropic coronavirus to spinal cord white matter via neurons and astrocytes. J. Virol. 1995;69:633–641. doi: 10.1128/jvi.69.2.633-641.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman M.A., Shubin R.A., Kyuwa S., Stohlman S.A. T-cell-mediated clearance of mouse hepatitis virus strain JHM from the central nervous system. J. Virol. 1989;63:3051–3056. doi: 10.1128/jvi.63.7.3051-3056.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot P.J., Knobler R.L., Buchmeier M.J. Western and dot immunoblotting analysis of viral antigens and antibodies: application to murine hepatitis virus. J. Immunol. Methods. 1984;73:177–188. doi: 10.1016/0022-1759(84)90043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther M., Kuklinski S., Pesheva P., Guntinas-Lichius O., Angelov D.N., Neiss W.F., Asou H., Probstmeier R. Galectin-3 is upregulated in microglial cells in response to ischemic brain lesions, but not to facial nerve axotomy. J. Neurosci. Res. 2000;15:430–435. doi: 10.1002/1097-4547(20000815)61:4<430::AID-JNR9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Waltrip R.W., 2nd., Buschanan R.W., Summerfelt A., Breier A., Carpenter W.T., Bryant N.L., Rubin S.A., Carbone K.M. Borna disease virus and schizophrenia. Psychiatry Res. 1995;56:33–44. doi: 10.1016/0165-1781(94)02600-n. [DOI] [PubMed] [Google Scholar]
- Wang F.I., Stohlman S.A., Fleming J.O. Demyelination induced by murine hepatitis virus JHM strain (MHV-4) is immunologically mediated. J. Neuroimmunol. 1990;30:31–34. doi: 10.1016/0165-5728(90)90050-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner L.P. Pathogenesis of demyelination induced by mouse hepatitis virus. Arch. Neurol. 1973;28:298–303. doi: 10.1001/archneur.1973.00490230034003. [DOI] [PubMed] [Google Scholar]
- Whitley R.J., Gnann J.W. Viral encephalitis: familiar infections and emerging pathogens. Lancet. 2002;359:507–513. doi: 10.1016/S0140-6736(02)07681-X. [DOI] [PubMed] [Google Scholar]
- Woyciechowska J.L., Trapp B.D., Patrick D.H., Shekarchi I.C., Leinikki P.O., Sever J.L., Holmes K.V. Acute and subacute demyelination induced by mouse hepatitis virus strain A59 in C3H mice. J. Exp. Pathol. 1984;1:295–306. [PubMed] [Google Scholar]
- Zimmer M.J., Dales S. In vivo and in vitro models of demyelinating diseases XXIV. The infectious process in cyclosporin A treated Winstar Lewis rats inoculated with JHM virus. Microbial Pathog. 1989;6:7–16. doi: 10.1016/0882-4010(89)90003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]