

Graphical abstract
Keywords: Camphor, Imine derivatives, Antivirals, Influenza, Genotoxicity, Cytotoxicity
Abstract
The synthesis and biological evaluation of a novel series of dimeric camphor derivatives are described. The resulting compounds were studied for their antiviral activity, cyto- and genotoxicity. Compounds 3a and 3d in which the quaternary nitrogen atoms are separated by the C5H10 and С9H18 aliphatic chain, exhibited the highest efficiency as an agent inhibiting the reproduction of the influenza virus A(H1N1)pdm09. The cytotoxicity data of compounds 3 and 4 revealed their moderate activity against malignant cell lines; compound 3f had the highest activity for the CEM-13 cells. These results show close agreement with the data of independent studies on toxicity of these compounds, in particular that the toxicity of compounds strongly depends on spacer length.
1. Introduction
Functionalization of natural compounds exhibiting native biological activity is one of the most efficient approaches for the synthesis of biologically active substances in medicinal chemistry. Symmetric nitrogen-containing molecules bearing at least two natural fragments linked with a spacer have become widely used for medical applications. The type of natural fragments is rather diverse: alkaloids, steroids, mono- or diterpenes, etc. Thus, dimeric and trimeric compounds, which exhibited a strongly pronounced antitumor effect, have been synthesized based on artemizinine.1 Hybrids of steroid framework have been studied as analogues of juvenile hormones.2 The strategy of bivalent ligands has been successfully implemented for searching for acetylcholinesterase inhibitors. The alkaloids tarcine3 and galanthamine4 were used as starting natural compounds, and their bis-derivatives appeared promising agents for treating Alzheimer’s disease. There are some examples in the literature of dimeric molecules acquiring properties that are completely different from those of the precursor molecules.5 Indeed, symmetric derivatives of the alkaloid camphotecine enhance solubility of a compound, reduce its toxicity, and increase its specific interaction with an enzyme.6 Meanwhile, symmetric compounds containing two quaternary nitrogen atoms separated by different size spacers are generally known to exhibit high biological activity.7 These agents have been most widely used as myorelaxants.8, 9 Furthermore, many of them have been tested as antimalarial drugs.10, 11 However, very few examples of synthesis of dimeric compounds based on natural molecules linked to quaternary nitrogen atoms via spacers have been published. For example, ammonium derivatives of the diterpenoids steviol and isosteviol exhibited antimicrobial properties12 and dimeric steroids can acts as a catalyst13 Hence, synthesis of compounds containing several natural fragments linked with spacer groups, involving the insertion of two quaternized nitrogen atoms, is a promising trend in chemistry of biologically active substances.
In the present study we describe the biological activity of novel class of chemical compounds—dimeric camphor derivatives. Among other types of activity, we have investigated their ability to suppress the reproduction of influenza virus. Several examples of use of similar cage structures as anti-viral compounds are well known. Adamantane derivatives amantadine and rimantadine were the first antivirals against influenza. Isoborneol derivatives were also shown to possess antiviral activity against influenza virus.14, 15 Based on the amino derivatives of isoborneol, anti-influenza drug deitiforin was licensed in the former Soviet Union for influenza treatment. Its mechanism of activity was supposed to be similar to that of rimantadine. Several attempts were undertaken to optimize the molecule of amantadine and to overcome the virus’ resistance.16, 17, 18, 19 In particular, adamantane-based spiro-derivatives have been tested against influenza virus and demonstrated high inhibiting activity. In this regard, activity of spiro[piperidine-2,2′-adamantane] appeared comparable with that of amantadine while other 2-alkyl-2-aminoadamantanes appeared less effective. Moreover, none of them demonstrated an activity against influenza B virus thus suggesting the specific target for their activity that is absent in influenza B viruses.
Although being adamantane-based cage structures, compounds called bananins (derivatives of 1-[3-hydroxy-5-(hydroxymethyl)-2-methyl-4-pyridinyl]-2,8,9-trioxaadamantane-3,5,7-triol)20, 21 have not been tested against influenza virus. These compounds were shown to inhibit SARS coronavirus-specific helicase (SCV). In the cell-free system they were effective inhibitors of the ATPase activity of the SCV helicase with IC50 values in the range 0.5–3 μM.
Another group of anti-influenza compounds is represented by cage structures close to camphor scaffold–pinanamines. It was previously shown that (1R,2R,3R,5S)-3-pinanamine appeared more potent than amantadine in inhibiting amantadine-susceptible influenza virus.22 Moreover, although at relative high concentrations, some of pinanamine derivatives were able to inhibit amantadine-resistant mutant bearing S31N mutation, the fact suggesting the principal possibility to overcome the resistance of influenza viruses to adamantane derivatives using cage scaffold-based compounds. The target of adamantane derivatives is virus-specific proton channel M2. Initially, these compounds were supposed to interact with the internal channel of M2 protein. In general, in their protonated form these compounds are considered to block the tetrameric M2 ion channel pore, formed by its transmembrane domain and hence, its proton transport function. Amino acid substitutions conferring rimantadine-resistant phenotype are localized at positions 27, 28, 31 and 34. In 2008–2009, NMR solution revealed another adamantane-binding site around the amino acid position 44.23, 24 This study suggested that rimantadine bound to the outside of the M2 protein helices facing the lipid bilayer with residue D44 participating in a hydrogen bond interaction with rimantadine. It was postulated that the high-affinity binding site corresponded to the M2 ion channel pore; whereas the secondary, low-affinity binding site could be attributed to the lipid face of the pore. These two studies proposed different sites of and different models for the interaction of adamantane drugs with M2 first, anion channel pore-binding model and, second, the lipid-facing binding model.25 In the later case, the drug was shown to inhibit the channel from outside by an allosteric mechanism by stabilizing its closed state.
A common feature of all M2 inhibitors known so far is the presence of a primary amine group linked with a hydrophobic scaffold.26, 27 The existence of an external binding site for adamantane derivatives broadens the set of modifications of compounds for suppressing ion channel activity of M2. Narrow pore of the channel restricts the size and charge of compounds that must fit the pore in order to inhibit it effectively. On the other hand, when developing the compounds interacting with amino acids and/or lipid bilayer from outside the channel, one may use a much wider set of side substituents. In this case, other factors become important for binding, in particular, the ability of a compound to interact simultaneously with lipids of membrane bilayer and external amino acids of the transmembrane region of M2.
2. Results and discussion
2.1. Chemistry
(+)-Camphor 1, an abundant monoterpenoid with a bicyclic framework structure, was selected to be the starting compound. Imine 2 was synthesized via the interaction between compound 1 and N,N-diethylethane-1,2-diamine under conditions of azeotropic removal of water from toluene in the presence BF3·Et2O with the yield of 94% (Scheme 1 ). In order to synthesize symmetric dimeric molecules, dihalogenides of different size and structural type were used as spacers. When interacting with compound 2 in boiling acetonitrile, they gave rise to the target compounds 3a–g. The resulting salts 3a–g containing two quaternary nitrogen atoms were isolated using silica gel column chromatography. In order to study the effect of the structure of compounds 3a–g on their biological activity, spacer-free analogues—compounds 4a–b containing a single quaternary nitrogen atom—have been synthesized.
Scheme 1.

Reagents and conditions: (a) PhMe, BF3·Et2O (1–5 mol %), reflux (Dean–Stark), 12 h; (b) Br-R-Br (0.50 equiv), CH3CN, K2CO3, reflux 8–24 h; (c) Hal-R′ (3 equiv), CH3CN, reflux 4–8 h.
The NOESY data and DFT quantum chemical calculation for compound 2 gave grounds to propose a structure with the E configuration of the imino groups for all the target compounds. Thus, the spectrum of imine 2 contains NOE cross peaks between proton signals at carbon atoms C-(2) and C-(11) (the calculated distance between the nearest protons of the specified groups is ∼2.4 Å for E and ∼4.4 Å for Z configurations). Cross peaks are also observed between Me-10 and H-4c, 5c, as well as between Me-9 and H-2c, which enables the unambiguous assignment of their signals (Fig. 1 ). It follows from the calculated data that the E configuration is 5 kcal/mol more energetically profitable than the Z configuration. Let us mention that the similar camphor imines have previously also been classified as E isomers.28
Figure 1.
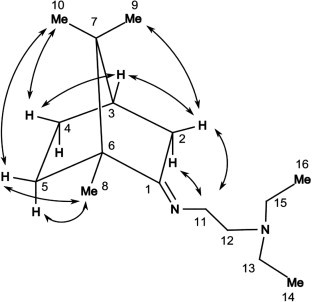
Main NOE cross peaks in the NOESY spectrum of compound 2.
Because of insufficient solubility of compounds 3a–g and 4b in CDCl3, their NMR spectra were recorded in MeOH-d 4. The substitution of a proton at the exo position 2 of the bornane framework by a deuterium atom was observed. The deuteration degree varied from sample to sample, which can presumably be attributed to different solvent exposure times and slightly varied acidity of the medium for the samples. The substitution at the vinyl position of imines in MeOH-d 4 solution is observed rather frequently and occurs due to the presence of the tautomeric equilibrium with the enamine form.29 Broadened signals for certain carbon atoms (and the signal for carbon atom C-(1) in the imino group in particular) were observed in the 13С NMR spectra of compounds 3a–g and 4a, b. Equimolar amounts of triethylamine were added to all the samples dissolved in MeOH-d 4 in order to ‘freeze’ the chemical exchange and narrow down the NMR spectral lines.
2.2. Study of biological activity
2.2.1. Antiviral activity
The obtained compounds 3a–g and 4a, b have been studied as potential antiviral agents. Natural compounds are considered to be the most prospective source for development of novel antivirals.30 In USA, about half of novel drugs approved for clinical use in the past decades are natural compounds and their chemically modified derivatives.31
There are few antivirals against influenza; drug resistance of the virus has been reported for most of them. The best-known etiotropic drugs for influenza treatment are adamantane derivatives amantadine (Symmetrel®, 1-aminoadamantane) and rimantadine (α-methyl-1-adamantylmethylamine hydrochloride). These compounds block the viral ion channel M2, thus preventing proton flow into the virion and further cleavage of hemagglutinin and fusion of membranes of the viral envelope and lysosomal vacuole. The study of activity of compounds 3a–g and 4a, b against influenza virus A/California/07/09 (H1N1)pdm09 revealed their high efficacy as inhibitors of virus reproduction. The values of antiviral activity and selectivity of these compounds exceeded those for reference compounds amantadine and rimantadine.
As can be seen from the results obtained, the most active substances were symmetric derivatives with an aliphatic spacer. It should be noted that the toxicity of compounds strongly depends on spacer length. In this regard, compounds 3b, c with –C6H12- and –C8H16-linkers, respectively, possess lowest toxicity (CTD50 = 1712 and 553 μM, correspondingly). Compound 3d appeared to be the most effective, having low EC50 and high CTD50 and SI; the latter exceeded SI for reference compounds 10-fold and more (Table 1 ).
Table 1.
Activity of camphor derivatives against influenza virus A(H1N1)pdm09
| Compound | CTD50a (μM) | EC50b (μM) | SIc |
|---|---|---|---|
| 1 | 3289.5 ± 216.0 | 1644.7 ± 144.4 | 2 |
| 2 | 806.8 ± 75.2 | 10.0 ± 0.6 | 81 |
| 3a | 1281.8 ± 114.4 | 14.7 ± 1.1 | 87 |
| 3b | 1712.3 ± 169.8 | 81.2 ± 6.3 | 21 |
| 3c | 552.5 ± 49.1 | 13.2 ± 1.2 | 42 |
| 3d | 577.6 ± 46.5 | 6.9 ± 0.4 | 82 |
| 3e | 285.3 ± 26.4 | 55.3 ± 3.7 | 5 |
| 3f | 127.5 ± 11.6 | 59.9 ± 4.4 | 2 |
| 3g | 1446.4 ± 138.2 | 43.4 ± 2.6 | 33 |
| 4a | 1886.8 ± 176.1 | 40.4 ± 3.8 | 47 |
| 4b | 1784.9 ± 154.4 | 139.8 ± 9.4 | 13 |
| Rimantadine | 335.2 ± 26.8 | 67.0 ± 4.9 | 5 |
| Amantadine | 284.1 ± 21.4 | 64.2 ± 4.7 | 4 |
| Deitiforin | 1266.2 ± 81.5 | 208.6 ± 15.4 | 6 |
CTD50, cytotoxic concentration; the concentration leading to 50% death of cells.
EC50 effective concentration; the concentration affording 50% inhibition of virus replication.
SI, selectivity index, ratio CTD50/EC50.
In this study, we used adamantane derivatives amantadine and rimantadine as well as the isoborneol derivative deitiforin. These compounds, similar to those synthesized in the course of our study, are based on cage structure (adamantane for amantadine and rimantadine; isoborneol for deitiforin and compounds 3a–g and 4a, b). As indicated previously, the influenza A virus contains a proton-selective ion channel (M2) that is the target of the adamantane family of drug inhibitors. Despite structure similarity of camphor derivatives to rimantadine and deitiforin, some of them appeared active against pandemic influenza strain that is resistant to adamantane derivatives. This suggests that the target site for our compounds should include amino acids differing from those interacting with rimantadine (27, 28, 31 and 34). Indeed, molecular modeling of interaction of dimeris camphor derivative 3a with M2 revealed that it preferentially binds with two adjacent chains of M2 tetramer (Fig. 2 ) in the region of amino acids 40–54. This model allows a speculation of why linkers of specific length confers the symmetric dimeric derivatives of camphor higher activity comparing to monomers. As M2 is a tetramer consisting of four peptides, there are four sites in the M2 molecule able to interact with camphor derivative as it is presented in the figure. Combination of two camphor moieties in one molecule might increase a probability of M2 binding and increase the strength of such binding, especially if these two moieties are linked with each other by linker of appropriate length allowing to reach two binding sites. In other words, if two camphor moieties are linked, binding of one moiety to the M2 target increases a probability of binding of second moiety to neighboring binding site of M2 comparing to two independent molecules. This suggestion undoubtedly should be a subject for further study.
Figure 2.
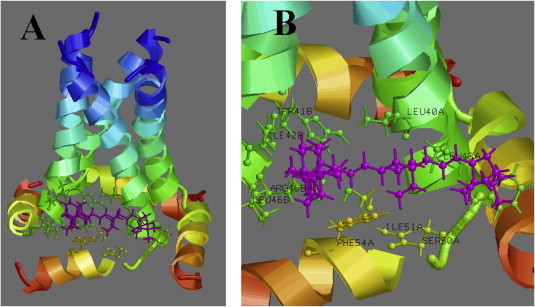
Interaction of compound 3a with intracellular domain of influenza virus M2 protein. (A) General view, all four monomers of M2 are presented, (B) close view, interaction with specific amino acids of two adjacent chains can be seen.
2.2.2. Evaluation of toxic and mutagenic effects of the compounds on bacterial biosensor cells
The global practice of studying the toxic and mutagenic effects of various compounds on the organism is mainly based on detection of immune and physiological disorders in the organism or on manifestation of new phenotypic features in a number of lower organisms. However, this kind of research is very time-consuming, requires expensive equipment to be used, and does not necessarily enable unambiguous interpretation of the results. The rapid methods for detecting organic contaminants and heavy metals32, 33 in environmental objects as well as for determining the total toxicity34, 35, 36, 37 and genotoxicity38, 39, 40, 41 of the analyzed compounds have been proposed over the past 30 years; recombinant whole-cell bacterial biosensors based on Escherichia coli and other microorganism species are used as test cultures. When designing whole-cell biosensors, the reporter protein genes are linked to the regulatory elements of the genes, which are characterized by a dose-dependent response to toxic and genotoxic chemical compounds, to external factors (exposure to UV light, radiation, temperature changes), or to physiological signals. Either green fluorescent protein (GFP) isolated from the jellyfish Aequorea victoria or luciferase from Vibrio fischeri is used as a reporter protein.
The two previously designed whole-cell biosensor test kits based on E. coli cells containing pRAC-gfp and pET36b-gfp plasmids were used in this work to evaluate possible toxic and mutagenic properties of the compounds 2–4 under study. GFP was used as a reporter protein in these test kits. The expression of the GFP protein gene in cells of the biosensor carrying the pRAC-gfp plasmid is controlled by the regulatory region of the recA gene of Proteus mirabilis, which is linked to it. It has been demonstrated that this E. coli strain can be used as an indicator strain ensuring the detection of mutagens of both physical and chemical nature.42 The designed whole-cell biosensor can presumably also be used to study the genotoxic effect of various compounds, novel drugs, dietary supplements, and foodstuffs.
Another variant of a whole-cell biosensor is based on the E. coli strain BL-21(DE3) carrying pET36b-gfp plasmid that ensures isopropyl-β-d-1-thiogalactopyranoside (IPTG)-induced expression of the gfp gene, which is contained in it and is regulated by bacteriophage T7 promoter. The toxic effect of any compound under study on biosensor cells is accompanied by inhibition of macromolecular synthesis reactions, including the protein synthesis reaction and, in particular, IPTG-induced synthesis of GFP. The total toxic effect of the compounds under study on a cell can be estimated from the decrease in induced synthesis of GFP.
Table 2 lists the results of determining the possible mutagenic and toxic effects of compounds 2–4 on cells of E. coli biosensor strains BL-21(DE3) carrying the pRAC-gfp or pET36b-gfp plasmid.
Table 2.
Toxic and genotoxic effects of the compounds studied in the cells of E. coli biosensor strains
| Designation of compound | The concentration of the compound (μM) in the culture medium of biosensor | The value of the fluorescence of GFP synthesized in biosensor cells (conventional units) |
Inhibition/induction of the synthesis of the reporter protein GFP (%) |
||
|---|---|---|---|---|---|
| Biosensor E. coli/pRAC-gfp | Biosensor E. coli/pET36b-gfp | E. coli/pRAC-gfp | E. coli/pET36b-gfp | ||
| 1 | 2 | 3 | 4 | 5 | 6 |
| 2 | 150 | 171.4 ± 14.5 | 512.7 ± 32.4 | — | — |
| 3a | 150 | 168.9 ± 13.5 | 546.9 ± 65.6 | — | — |
| 3b | 150 | 186.2 ± 22.9 | 495.5 ± 15.2 | — | — |
| 3c | 150 | 81.7 ± 3.6 | 165.5 ± 8.1 | 57% | 67% |
| 3d | 150 | 95.9 ± 4.3 | 172.3 ± 6.2 | 51% | 65% |
| 3e | 150 | 56.9 ± 3.1 | 110.7 ± 7.2 | 70% | 78% |
| 3f | 150 | 49.3 ± 2.1 | 51.3 ± 4.6 | 74% | 89% |
| 3g | 150 | 170.87 ± 8.9 | 466.46 ± 4.1 | — | — |
| 4a | 150 | 189.21 ± 17.7 | 535.72 ± 307 | — | — |
| 4b | 150 | 176.9 ± 16.9 | 547.9 ± 40.8 | — | — |
| ∗Controls | — | ∗1188.4 ± 7.4. | ∗2495.3 ± 18.9 | — | — |
| ∗∗Control | (10 μg/ml NА) | ∗∗2913.29 ± 61.3 | — | Induction 500% | — |
The fluorescence values of GFP synthesized by the cells of two types of whole-cell biosensors (E. coli/pRAC-gfp and E. coli/pET36b-gfp) and intended for assessing the possible mutagenic and toxic effects of the analyzed compounds are listed in columns 3 and 4. In controls ∗1 and ∗∗2, water (negative control) or nalidixic acid at concentration of 10 μg/l (positive control), respectively, was added to the E. coli/pRAC-gfp cell cultures instead of the analyzed compounds. In control ∗∗2, water was introduced to the E. coli/pET36b-gfp cell culture instead of the analyzed compounds; after 1.5 h, IPTG was also added to final concentration of 1 mM.
As follows from the data listed in Table 2, compounds 2, 3a, 3b, 3g, 4a and 4b have neither toxic nor mutagenic effects on bacterial indicator cells E. coli/pET36b-gfp and E.coli/pRAC-gfp. Moreover, none of the analyzed compounds exhibits a genotoxic effect on cells of the biosensor strain E. coli/pRAC-gfp. However, compounds 3c, 3d, 3e and 3f turned out to be highly toxic for two types of indicator cells. There is a direct dependence between a rise in toxicity of the symmetric dimeric molecules of (+)-camphor derivatives under study and an increasing length of aliphatic spacers. Compounds 3e and 3f exhibited the highest toxicity. These compounds are of an unquestionable interest in terms of putative application as components of drugs for suppressing malignant tumor development and as antiviral and antibacterial agents (Table 3 ).
Table 3.
Cytotoxic activity of synthetic camphor derivatives against lymphoblastoid cell lines
| Compound | Cytotoxicity (CTD50, μM) against cell line |
||
|---|---|---|---|
| CEM-13 | U-937 | MT-4 | |
| 3a | >100 | 95.8 ± 5.6 | 57.5 ± 3.8 |
| 3b | 60.2 ± 5.4 | 82.1 ± 4.4 | 48.3 ± 3.2 |
| 3c | 71.1 ± 6.2 | 67.2 ± 5.9 | 69.8 ± 2.7 |
| 3d | 66.0 ± 4.8 | 58.6 ± 3.4 | 66.0 ± 5.3 |
| 3e | 22.4 ± 0.6 | 53.6 ± 2.2 | 66.1 ± 4.4 |
| 3f | 6.7 ± 0.1 | 15.9 ± 1.1 | 40.4 ± 3.9 |
| 3g | >100 | 54.9 ± 2.6 | 40.5 ± 3.0 |
| 4a | >100 | 58.6 ± 4.6 | >100 |
| 4b | >100 | >100 | >100 |
2.2.3. Cytotoxicity
The cytotoxic activity of the synthesized compounds was determined by measuring the concentration inhibiting tumor cell viability by 50% (CTD50). The CTD50 was determined using the conventional MTT assay, which allows to estimate the number of survived cells. The results demonstrate that the compounds exhibited a moderate activity with respect to model cancer cell lines (within micromolar range); compound 3f possessed the highest activity. Moreover, when comparing the cytotoxicity of compounds regarding four cell lines used in the study (CEM-13, U-937, MT-4 and MDCK), it should be noted that MDCK cells appeared much more resistant to toxic action of camphor derivatives. This may be explained by (i) different conditions of cultivation of these cell lines and (ii) selective toxicity of the compounds studied against lymphoblastoid cells comparing to ones of epithelial origin. Further studies are therefore required to get additional data to evaluate the properties of symmetric camphor derivatives as potential cytotoxic anti-cancer agents.
3. Conclusion
A series of symmetric compounds having two camphor fragments in their framework, two imine groups and two quaternary nitrogen atoms separated by spacers of different size have been synthesized. The antiviral activity, cyto- and genotoxicity of the resulting compounds have been studied. Compound 3d, in which the quaternary nitrogen atoms are separated by the С9H18 aliphatic chain, exhibited the highest efficiency as an agent inhibiting the reproduction of the influenza virus (strain A/California/07/09 (H1N1)pdm09). The therapeutic index of this compound is more than 10-fold higher than those of the comparative drugs, amantadine and rimantadine. It is worth mentioning that the synthesized camphor imines are stable crystalline compounds that undergo neither destruction nor polymerization for a long period. The cytotoxicity data of compounds 3 and 4 demonstrate that they exhibit a moderate anticancer activity; compound 3f had the highest activity for the CEM-13 cell line. These results show close agreement with the data of independent studies on genotoxicity and cytotoxicity of these compounds. Compounds 3e and 3f exhibit the highest toxicity. After further detailed study, these compounds can be of interest in terms of their putative application as components of drugs for suppressing malignant tumor development.
4. Experimental section
4.1. General chemical methods
Reagents and solvents were purchased from commercial suppliers and used as received. Dry solvents were obtained according to the standard procedures. Optical rotation: polAAr 3005 spectrometer; CHCl3 soln 1H and 13C NMR spectra were recorded with a Bruker AV-400 (1H: 400.13 MHz, 13C:100.78 MHz), DRX-500 (1H: 500.13 MHz, 13C: 125.76 MHz) and AV-600 (1H: 600.30 MHz, 13C:150.95 MHz) in CDCl3, CD3OD or DMSO-d 6; chemical shifts δ in ppm rel to residual [δ(CHCl3) 7.24, δ(CDCl3) 76.90 ppm; δ(CHD2OD) 3.31, δ(CD3OD) 49.00; δ(CHD2SOCD3) 2.50, δ(CD3SOCD3) 39.50], J in Hz; assignments on a routine basis by a combination of 1D and 2D experiments (COSY, COLOC, HSCQ, HMBC). NOESY spectrum of 2 was recorded with mixing time 0.350 s (500 MHz). For quantum chemical calculations, we used the cluster of the Information Computation Center, Novosibirsk State University. Both most stable conformers for E- and Z-isomers of compound 2 were optimized by DFT (functional PBE,43 basis L1 (Λ01,44 cc-pVDZ analog), using the PRIRODA program45). HR-MS: DFS Thermo Scientific spectrometer in a full scan mode (15–500 m/z, 70 eV electron impact ionization, direct sample administration). Column chromatography (CC) was performed on silica gel (60–200 l, Macherey–Nagel). The HPLC/MS system consisting of an Agilent 1200 liquid chromatograph and a micrOTOF-Q hybrid quadrupole time-of-flight mass spectrometer (Bruker) were used to verify the structure of diquaternized compounds. Operating parameters of mass detection. Ionization method: atmospheric pressure electrospray ionization (API-ES). Scanning ions in the m/z range = 80–3000. Drying gas (nitrogen) flow: 4 l/min; its temperature: 190 °С, pressure at nebulizer: 1.0 bar. Solution of a compound in methanol was fed into the spray chamber of the mass spectrometer by introducing 5 μl of the solution into the solvent flow (MeOH, 0.1 ml/min) using an autosampler. Spectral and analytical investigations were carried out at Collective Chemical Service center of Siberian Branch of the Russian Academy of Sciences.
The purity of the target compounds was determined by gas chromatography methods. All of the target compounds reported in this paper have a purity of no less than 95%.
Numeration of atoms in the compounds is given for assigning the signals in the NMR spectra and does not coincide with that for the names according to the nomenclature of compounds (see Supplementary data). Specific rotation is expressed as (deg ml) (g dm)–1; concentration is expressed as (g) (100 ml)–1.
4.2. (E)-N1,N1-Diethyl-N2-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)ethane-1,2-diamine 2
(+)-Camphor (26 mmol) and N 1,N 1-diethylethane-1,2-diamine (30 mmol) were mixed in toluene with catalytic BF3·Et2O (1–5 mol %), followed by azeotropic removal water with Dean–Stark for 15 h. Then the reaction mixture was washed with saturated NaCl solution. The organic layer was separated. The aqueous layer was extracted with CH2Cl2 (3 × 10 ml). The combined organic phases were dried over Na2SO4. The solvent was evaporated. The residue was separated by vacuum distillation. Т = 100–102 °С at 1 torr. As a result we have obtained 2 in 94% yield (4.7 g, 24.4 mmol). =18.7 (с 1.1, СHCl3) 1 H NMR (500 MHz, CDCl3, δ, ppm, J/Hz): 0.65 (3H, s, Me-9), 0.81 (3H, s, Me-10), 0.84 (3H, s, Me-8), 0.92 (6H, t, J 14,13 = 7.1, Me-14 and Me-16), 1.08 (1H, ddd, 2 J = 12.3, J 4 endo , 5 endo = 9.3, J 4 endo , 5 exo = 4.2, H-4endo), 1.24 (1H, ddd, 2 J = 12.3, J 5 endo , 4 endo = 9.3, J 5 endo , 4 exo = 4.5, H-5endo), 1.54 (1H, ddd, 2 J = J 5 exo , 4 exo = 12.3, J 5 exo , 4 endo = 4.2, H-5exo), 1.73 (1H, d, 2 J = 16.9, H-2endo), 1.73 (1H, ddddd, 2 J = J 4 exo , 5 exo = 12.3, J 4 exo , 5 endo = J 4 exo , 3 = 4.5, J 4 exo , 2 exo = 3.2, H-4exo), 1.81 (1H, dd, J 3, 2 exo = J 3, 4 exo = 4.5, H-3), 2.24 (1H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 2.45 and 2.45 (each 2H, q, J 13, 14 = 7.1, H-13 and H-15), 2.55 (2H, t, J 12, 11 = 7.6, H-12), 3.18 and 3.25 (each 1H, dt, 2 J = 12.1, J 11, 12 = 7.6, H-11). 13 C NMR (125 MHz, CDCl3, δ, ppm): 181.90 s (C-1), 53.22 t (C-12), 53.20 c (C-6), 50.73 t (C-11), 47.22 t (C-13 and C-15), 46.64 s (C-7), 43.61 d (C-3), 35.26 t (C-2), 31.89 t (C-5), 27.22 t (C-4), 19.29 q (C-9), 18.70 q (C-10), 11.71 q (C-14 and C-16), 11.14 q (C-8). HR-MS: 250.2402 (M+ C16H30N2; calcd 250.2404)
4.3. General method for synthesis of compounds 3a–g
A solution of 2 0.5 g (2 mmol) and anhydrous CH3CN (10 ml) was treated with dihalogenid (1 mmol) and heated on a bath at 70–75 °C for different time from 10 to 30 h depending on dihalogenide structure. The solvent was removed at reduced pressure. The resulting precipitate was chromatographed over silica gel ((CHCl3 + NH4OH)/MeOH eluent, (100:0 → 0:100)).
4.3.1. N1,N1,N5,N5-Tetraethyl-N1,N5-bis(2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)ethyl)pentane-1,5-diaminium bromide 3a
The reaction mixture was refluxed for 10 h. This gave compound 3a (0.30 g, 0.4 mmol, 40%). Mp = 137–140 °С. It was found that there is a 50% deuterium-substitution in the second exo-position in the compounds spectra of NMR. The signals of deuterium-substitution compound different from unsubstituted are marked by asterisk ∗. NMR 1 H (500 MHz, CD3 OD + NEt3, δ, ppm, J/Hz): 0.78 (6H, s, Me-9), 0.95 (6H, s, Me-8), 0.97 (6H, s, Me-10), 1.27–1.35 (4H, m, H-4endo, H-5endo), 1.36 (12H, t, J 14, 13 = 7.1, Me-14 and Me-16), 1.45–1.53 (2H, m, H-19), 1.69–1.77 (2H, m, H-5exo), 1.85–1.94 (6H, m, H-18 and H-4exo), 1.98 (2H, br s, H-2endo ∗), 1.99–2.02 (4H, m, H-3 and H-2endo), 2.01 (2H, d, J 3, 4 exo = 4.5, H-3∗), 2.50 (2H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 3.36–3.42 (4H, m, H-17), 3.49 (8H, q, J 13, 14 = 7.1, H-13 and H-15), 3.52–3.56 (4H, m, H-12), 3.56–3.68 (4H, m, H-11). NMR 13 C (125 MHz, CD3 OD + NEt3, δ, ppm): 188.22 s (C-1), 58.96 t (C-17), 58.69 t (C-12), 55.35 s (C-6), 55.24 t (C-13 and C-15), 48.44 s (C-7), 46.57 t (C-11), 45.32 d (C-3), 45.23 d (C-3∗), 36.89 t (C-2), 36.61 dt (J C, D = 19.8, CHD∗-2), 33.13 t (C-5), 28.06 t (C-4), 28.03 t (C-4∗), 24.39 t (C-19), 22.70 t (C-18), 20.00 q (C-9), 19.26 q (C-10), 11.75 q (C-8), 8.14 q (C-14 and C-16). ESI: m/z [M−2Br]2+ 285.279, (calcd for C37H70N4) 285.280.
4.3.2. N1,N1,N6,N6-Tetraethyl-N1,N6-bis(2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)ethyl)hexane-1,6-diaminium bromide 3b
The reaction mixture was refluxed for 15 h. This gave compound 3b (0.35 g, 0.47 mmol, 47%). Mp = 220 °С. It was found that there is a 80% deuterium-substitution in the second exo-position in the compounds spectra of NMR. The signals of deuterium-substitution compound different from unsubstituted are marked by asterisk ∗. NMR 1 H (500 MHz, CD3 OD + NEt3, δ, ppm, J/Hz): 0.78 (6H, s, Me-9), 0.94 (6H, s, Me-8), 0.97 (6H, s, Me-10), 1.27–1.35 (4H, m, H-4endo, H-5endo), 1.35 (12H, t, J 14, 13 = 7.1, Me-14 and Me-16), 1.49–1.54 (4H, m, H-19), 1.68–1.76 (2H, m, H-5exo), 1.77–1.86 (4H, m, H-18), 1.86–1.95 (2H, m, H-4exo), 1.99 (2H, br s, H-2endo ∗), 1.98–2.00 (2H, m, H-3), 2.00 (2H, d, J 3, 4 exo = 4.5, H-3∗), 2.00 (2H, d, 2 J = 16.9, H-2endo), 2.50 (2H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 3.34–3.40 (4H, m, H-17), 3.49 (8H, q, J 13, 14 = 7.1, H-13 and H-15), 3.52–3.56 (4H, m, H-12), 3.65–3.68 (4H, m, H-11). NMR 13 C (125 MHz, CD3 OD + NEt3, δ, ppm): 188.11 s (C-1), 59.18 t (C-17), 58.62 t (C-12), 55.29 s (C-6), 55.16 t (C-13 and C-15), 48.39 s (C-7), 46.59 t (C-11), 45.26 d (C-3), 45.17 d (C-3∗), 36.88 t (C-2), 36.60 dt (J C, D = 19.8, CHD∗-2), 33.09 t (C-5), 28.02 t (C-4), 27.99 t (C-4∗), 26.93 t (C-19), 22.76 t (C-18), 20.01 q (C-9), 19.27 q (C-10), 11.71 q (C-8), 8.18 q (C-14 and C-16). (ESI): m/z [M−2Br]2+ 292.787, (calcd for C38H72N4) 292.288.
4.3.3. N1,N1,N8,N8-Tetraethyl-N1,N8-bis(2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylideneamino)ethyl)octane-1,8-diaminium bromide 3с
The reaction mixture was refluxed for 10 h. This gave compound 3c (0.60 g, 0.77 mmol, 77%). Mp = 167–170 °С. It was found that there is a 20% deuterium-substitution in the second exo-position in the compounds spectra of NMR. The signals of deuterium-substitution compound different from unsubstituted are marked by asterisk ∗. NMR 1 H (500 МHz, CD3 OD + NEt3, δ, ppm, J/Hz): 0.78 (6H, s, Me-9), 0.94 (6H, s, Me-8), 0.98 (6H, s, Me-10), 1.25–1.32 (4H, m, H-4endo, H-5endo), 1.34 (12H, t, J 14, 13 = 7.1, Me-14 and Me-16), 1.38–1.50 (8H, m, H-19 and H-20), 1.70–1.82 (6H, m, H-18 and H-5exo), 1.88–1.94 (2H, m, H-4exo), 1.94–2.00 (4H, m, H-2endo, H-2endo ∗, H-3, H-3∗), 2.48 (2H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 3.30–3.35 (4H, m, H-17), 3.45 (8H, q, J 13, 14 = 7.1, H-13 and H-15), 3.48–3.54 (4H, m, H-12), 3.54–3.64 (4H, m, H-11). NMR 13 C (125 МHz, CD3 OD + NEt3, δ, ppm): 188.17 s (C-1), 59.36 t (C-17), 58.60 t (C-12), 55.36 s (C-6), 55.02 t (C-13 and C-15), 48.44 s (C-7), 46.54 t (C-11), 45.32 d (C-3), 45.23 d (C-3∗), 36.80 t (C-2), 36.53 dt (1 J C, D = 19.7, CHD-2∗), 33.12 t (C-5), 30.20 t (C-20), 28.08 t (C-4), 28.05 t (C-4∗), 27.37 t (C-19), 22.91 t (C-18), 19.96 q (Me-9), 19.25 q (Me-10), 11.71 q (Me-8), 8.00 q (Me-14 and Me-16). (ESI): m/z [M−2Br]2+ 306.28, (calcd for C40H76N4) 306.30, [M−Br]+ 693.47, (calcd for C40H76BrN4) 691.52.
4.3.4. N1,N1,N9,N9-Tetraethyl-N1,N9-bis(2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)ethyl)nonane-1,9-diaminium bromide 3d
The reaction mixture was refluxed for 25 h. This gave compound 3d (0.68 g, 0.86 mmol, 86%). Mp = 162–164 °С. It was found that there is a 60% deuterium-substitution in the second exo-position in the compounds spectra of NMR. The signals of deuterium-substitution compound different from unsubstituted are marked by asterisk ∗. NMR 1 H (500 МHz, CD3 OD + NEt3, δ, ppm, J/Hz): 0.78 (6H, s, Me-9), 0.94 (6H, s, Me-8), 0.97 (6H, s, Me-10), 1.26–1.33 (4H, m, H-4endo, H-5endo), 1.34 (12H, t, J 14, 13 = 7.1, Me-14 and Me-16), 1.36–1.46 (10H, m, H-19, H-20 and H-21), 1.70–1.80 (6H, m, H-18 and H-5exo), 1.86–1.94 (2H, m, H-4exo), 1.94–2.03 (4H, m, H-2endo, H-2endo ∗, H-3, H-3∗), 2.48 (2H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 3.30–3.35 (4H, m, H-17), 3.45 (8H, q, J 13, 14 = 7.1, H-13 and H-15), 3.48–3.54 (4H, m, H-12), 3.54–3.64 (4H, m, H-11). NMR 13 C (125 МHz, CD3 OD + NEt3, δ, ppm): 188.17 s (C-1), 59.39 t (C-17), 58.59 t (C-12), 55.35 s (C-6), 55.01 t (C-13 and C-15), 48.43 s (C-7), 46.54 t (C-11), 45.31 d (C-3), 45.23 d (C-3∗), 36.80 t (C-2), 36.52 dt (1 J C, D = 19.7, CHD-2∗), 33.12 t (C-5), 30.47 t (C-21), 30.23 t (C-20), 28.08 t (C-4), 28.04 t (C-4∗), 27.45 t (C-19), 22.91 t (C-18), 19.96 q (Me-9), 19.25 q (Me-10), 11.69 q (Me-8), 7.99 q (Me-14 and Me-16). (ESI): m/z [M−2Br]2+ 313.813, (calcd for C41H78N4) 313.311, [M−Br]+ 707.512, (calcd for C41H78BrN4) 705.540.
4.3.5. N1,N1,N10,N10-Tetraethyl-N1,N10-bis(2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene) amino)ethyl)decane-1,10-diaminium bromide 3e
The reaction mixture was refluxed for 15 h. This gave compound 3e (0.57 g, 0.71 mmol, 71%). Mp = 167–170 °С. NMR 1 H (600 МHz, CD3OD + NEt3, δ, ppm, J/Hz): 0.78 (6H, s, Me-9), 0.94 (6H, s, Me-8), 0.98 (6H, s, Me-10), 1.26–1.32 (4H, m, H-4endo, H-5endo), 1.33 (12H, t, J 14, 13 = 7.1, Me-14 and Me-16), 1.36–1.46 (12H, m, H-19, H-20 and H-21), 1.70–1.79 (6H, m, H-18 and H-5exo), 1.89–1.94 (2H, m, H-4exo), 1.96 (2H, д, 2 J = 16.9, H-2endo), 2.01 (2H, dd, J 3, 2 exo = J 3, 4 exo = 4.5, H-3), 2.48 (2H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 3.31–3.35 (4H, m, H-17), 3.44 (8H, q, J 13, 14 = 7.1, H-13 and H-15), 3.48–3.54 (4H, m, H-12), 3.54–3.61 (4H, m, H-11). NMR 13 C (150 МHz, CD3 OD + NEt3, δ, ppm): 188.19 s (C-1), 59.35 t (C-17), 58.54 t (C-12), 55.36 s (C-6), 54.95 t (C-13 and C-15), 48.45 s (C-7), 46.51 t (C-11), 45.30 d (C-3), 36.79 t (C-2), 33.11 t (C-5), 30.56 t (C-21), 30.35 t (C-20), 28.08 t (C-4), 27.49 t (C-19), 22.90 t (C-18), 19.96 q (Me-9), 19.26 q (Me-10), 11.71 q (Me-8), 7.96 q (Me-14 and Me-16). (ESI): m/z [M−2Br]2+ 320.30, (calcd for C42H80N4) 320.31.
4.3.6. N1,N1,N12,N12-Tetraethyl-N1,N12-bis(2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)ethyl)dodecane-1,12-diaminium bromide 3f
The reaction mixture was refluxed for 30 h. This gave compound 3f (0.48 g, 0.6 mmol, 60%). It was found that there is a 50% deuterium-substitution in the second exo-position in the compounds spectra of NMR. The signals of deuterium-substitution compound different from unsubstituted are marked by asterisk ∗. NMR 1 H (500 МHz, CD3 OD + NEt3, δ, ppm, J/Hz): 0.78 (6H, s, Me-9), 0.94 (6H, s, Me-8), 0.97 (6H, s, Me-10), 1.26–1.33 (4H, m, H-4endo, H-5endo), 1.33 (12H, t, J 14, 13 = 7.1, Me-14 and Me-16), 1.36–1.46 (8H, m, H-19 and H-20), 1.70–1.80 (6H, m, H-18 and H-5exo), 1.86–1.94 (2H, m, H-4exo), 1.94–2.03 (4H, m, H-2endo, H-2endo ∗, H-3, H-3∗), 2.48 (2H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 3.30–3.35 (4H, m, H-17), 3.44 (8H, q, J 13, 14 = 7.1, H-13 and H-15), 3.48–3.54 (4H, m, H-12), 3.54–3.64 (4H, m, H-11). NMR 13 C (125 МHz, CD3 OD + NEt3, δ, ppm): 188.13 s (C-1), 59.40 t (C-17), 58.59 t (C-12), 55.33 s (C-6), 54.99 t (C-13 and C-15), 48.42 c (C-7), 46.55 t (C-11), 45.30 d (C-3), 45.21 d (C-3∗), 36.80 t (C-2), 36.52 dt (1 J C, D = 19.7, CHD-2∗), 33.11 t (C-5), 30.60 t (C-21 and C-22), 30.32 t (C-20), 28.07 t (C-4), 28.04 t (C-4∗), 27.47 t (C-19), 22.89 t (C-18), 19.96 q (Me-9), 19.26 q (Me-10), 11.69 q (Me-8), 7.98 q (Me-14- and Me-16). (ESI): m/z [M−2Br]2+ 334.333, (calcd for C44H84N4) 334.334.
4.3.7. (R,R,E)-N,N′-(1,4-Phenylenebis(methylene))bis(N,N-diethyl-2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)ethanaminium) bromide 3g
The reaction mixture was refluxed for 15 h. This gave compound 3g (0.52 g, 0.68 mmol, 68%). Mp = 173–175 °С NMR 1 H (500 MHz, CD3 OD + NEt3, δ, ppm, J/Hz): 0.81 (6H, s, Me-9), 0.96 (6H, s, Me-8), 0.98 (6H, s, Me-10), 1.28–1.40 (4H, m, H-4endo, H-5endo), 1.49 (12H, t, J 14, 13 = 7.1, Me-14 and Me-16), 1.71–1.79 (2H, m, H-5exo), 1.90–1.97 (2H, m, H-4exo), 2.01 (2H, d, 2 J = 16.9, H-2endo), 2.04 (2H, dd, J 3, 2 exo = J 3, 4 exo = 4.5, H-3), 2.53 (2H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 3.42 and 3.43 (each 4H, q, J 13, 14 = 7.1, H-13 and H-15), 3.50–3.56 (4H, m, H-12), 3.65–3.80 (4H, m, H-11), 4.78 (4H, s, H-17), 7.81 (4H, s, H-19 and H-20). NMR 13 C (125 MHz, CD3 OD + NEt3, δ, ppm): 188.53 s (C-1), 134.89 d (C-19 and C-20), 131.59 s (C-18), 61.93 t (C-17), 58.34 t (C-12), 55.51 s (C-6), 54.98 t (C-13 and C-15), 48.52 s (C-7), 46.77 t (C-11), 45.35 d (C-3), 36.98 t (C-2), 33.14 t (C-5), 28.09 t (C-4), 20.01 q (C-9), 19.25 q (C-10), 11.79 q (C-8), 8.50 q (C-14 and C-16). (ESI): m/z [M−2Br]2+ 302.276, (calcd for C40H68N4) 302.272.
4.4. N,N-Diethyl-N-methyl-2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)ethanaminium iodide 4a
A solution of 2 (1 g, 4 mmol) and anhydrous CH3CN (10 ml) was treated with excess of iodomethane and heated on a bath at 70–75 °C for 2 days. The solvent was removed at reduced pressure. The resulting precipitate was chromatographed over silica gel (CHCl3/MeOH eluent, (100:0 → 0:100)). This procedure gave 4a (0.80 g, 2 mmol, 50%). NMR 1 H (400 МHz, DMSO-d 6, δ, ppm, J/Hz): 0.70 (3H, s, Me-9), 0.85 (3H, s, Me-8), 0.89 (3H, s, Me-10), 1.17–1.23 (2H, m, H-4endo, H-5endo), 1.23 (6H, t, J 14, 13 = 7.1, Me-14 and Me-16), 1.58–1.70 (1H, m, H-5exo), 1.75–1.86 (1H, m, H-4exo), 1.88 (1H, d, 2 J = 16.9, H-2endo), 1.95 (1H, dd, J 3, 2 exo = J 3, 4 exo = 4.5, H-3), 2.39 (1H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 2.98 (3H, s, Me-17), 3.35 (4H, q, J 13, 14 = 7.1, H-13 and H-15), 3.41–3.55 (4H, m, H-12 and H-11). NMR 13 C (100 МHz, DMSO-d 6, δ, ppm): 183.92 s (C-1), 59.84 t (C-12), 56.17 t (C-13 and C-15), 53.54 s (C-6), 47.13 q (Me-17), 46.88 s (C-7), 44.95 t (C-11), 43.20 d (C-3), 35.23 t (C-2), 31.61 t (C-5), 26.78 t (C-4), 19.33 q (Me-9), 18.73 q (Me-10), 11.24 q (Me-8), 7.68 q (Me-14 and Me-16). (ESI): m/z [M−I]+ 265.261, (calcd for C17H33N2) 265.264.
4.5. N,N,N-Triethyl-2-((E)-((1R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)ethanaminium bromide 4b
A solution of 2 1 g (4 mmol) and anhydrous CH3CN (10 ml) was treated with excess bromoethane and heated on a bath at 70–75 °C for 5 days. The solvent was removed at reduced pressure. The resulting precipitate was chromatographed over silica gel (CHCl3/MeOH eluent, (100:0 → 0:100)). This procedure gave 4b (0.63 g, 1.8 mmol, 45%). It was found that there is a 60% deuterium-substitution in the second exo-position in the compounds spectra of NMR. The signals of deuterium-substitution compound different from unsubstituted are marked by asterisk ∗. NMR 1 H (500 МHz, CD3OD + NEt3, δ, ppm, J/Hz): 0.78 (3H, s, Me-9), 0.94 (3H, s, Me-8), 0.97 (3H, s, Me-10), 1.25–1.32 (2H, m, H-4endo, H-5endo), 1.34 (9H, tt, J 14, 13 = 7.2, 3 J 14, N = 1.8, Me-14, Me-16 and Me-18), 1.70–1.77 (1H, m, H-5exo), 1.88–2.02 (3H, m, H-4exo, H-2endo and H-3), 2.48 (1H, ddd, 2 J = 16.9, J 2 exo , 3 = 4.5, J 2 exo , 4 exo = 3.2, H-2exo), 3.43 (6H, q, J 13, 14 = 7.2, H-13, H-15 and H-17), 3.47–3.51 (2H, m, H-12), 3.51–3.65 (2H, m, H-11). NMR 13 C (125 МHz, CD3 OD + NEt3, δ, ppm): 188.15 s (C-1), 57.98 tt (1 J C, N = 2.8, C-12), 55.35 s (C-6), 54.49 tt (1 J C, N = 2.8, C-13, C-15, C-17), 48.41 s (C-7), 46.43 t (C-11), 45.32 d (C-3), 45.23 d (C-3∗), 36.77 t (C-2), 36.49 dt (1 J C, D = 19.9, CHD-2∗), 33.08 t (C-5), 28.07 t (C-4), 28.04 t (C-4∗), 19.93 q (Me-9), 19.25 q (Me-10), 11.62 q (Me-8), 7.89 (Me-14, Me-16 and Me-18). (ESI): m/z [M−Br]+ 279.277, (calcd for C18H35N2) 279.280.
4.6. Viruses and cells
Influenza virus A/California/07/09 (H1N1)pdm09 from the collection of viruses of the Research Institute of Influenza, St.-Petersburg, Russia, was used. The virus was cultivated in 10–12 day-old chicken embryos for 48 h at +37 °C. MDCK cells (ATCC CCL 34) in a minimal essential medium (MEM, PAA, Austria, Cat.# E15-825) were seeded on 96-well plates (Orange Scientific No. 5530100) and incubated at 37 °C in 5% CO2 until a confluent monolayer formed. To cultivate the virus, 5% albumin, trypsin (1 μg/ml), and 16 mM HEPES (pH 7.6) were added.
4.6.1. Toxicity studies
The microtetrazolium test (MTT) was used to study the cytotoxicity of the compounds (Mossman, 1983). Briefly series of twofold dilutions of each compound (1000–4 μg/ml) in MEM were prepared. MDCK cells were incubated for 48 h at 37 °C in 5% CO2 in the presence of the dissolved substances. The degree of destruction of the cell monolayer was then determined with the microtetrazolium test (MTT). The cells were washed twice with saline, and a solution of 3-(4,5-dimethylthiazolyl-2) 2,5-diphenyltetrazolium bromide (ICN Biochemicals Inc., Aurora, Ohio) (0.5 μg/ml) in saline was added to the wells. After 1 h incubation the wells were washed and the formazan residue dissolved in DMSO (0.1 ml per well). The optical density of cells was then measured on a Victor 2 1440 multifunctional reader (Perkin Elmer, Finland) at a wavelength of 535 nm and plotted against the concentration of the compounds. Each compound concentration was tested in three parallels. The 50% cytotoxic dose (CTD50) of each compound (i.e., the compound concentration that causes the death of 50% of cells in a culture, or decreases the optical density twice as compared to the control wells) was calculated from the data obtained.
4.6.2. Determination of the antiviral activity
The compounds in appropriate concentrations were incubated with MDCK cells for 1 h at 37 °C. The cell culture was then infected with 10-fold dilutions of the virus (10−1–10−6). The plates were incubated for 48 h at 37 °C in the presence of 5% CO2. The infection activity of the virus was evaluated in a hemagglutination reaction with chicken erythrocytes. A virus-containing solution (100 μl) was placed in the wells of a round-bottom plate. An equal amount of a 1% suspension of chicken erythrocytes in saline was added. The reaction was evaluated after 60 min incubation at room temperature. Each concentration of the compounds was tested in three parallels. A virus titer was considered as reciprocal to decimal logarithm of maximum dilution that caused complete agglutination of erythrocytes and was expressed as logarithms of the 50% experimental infection dose (log10 EID50). The antiviral activity of the compounds was estimated by the decrease in the virus titer as compared with the control. The 50% effective concentration (EC50) of the drug, that is, the concentration at which the virus production decreased by a factor of two (a virus titer per 0.3 log10 EID50) and the selectivity index (the ratio of CTD50 to EC50) were calculated from the data obtained.
4.7. Genotoxicity activity assays
Assessment of genotoxicity and toxicity of the compounds under study using whole-cell biosensors.
The possible mutagenic and toxic effects of terpenoids on the cells of E. coli biosensor strains BL-21(DE3) carrying pRAC-gfp or pETm-gfp plasmid were determined in accordance with the following scheme. The overnight cultures of E. coli biosensor strains were reseeded into vials with fresh LB culture and grown on a shaker at +37 °С until the optical density D 600 = 0.4–0.5 was reached. Different amounts of the analyzed terpenoid were then introduced into the vials; the cultures were grown for additional 1.5 h. Next, the vials containing the biosensor strain carrying the pRAC-gfp plasmid were incubated at 4–6 °С for 1.5 h to ensure the maturation of the GFP chromophore.
At the final stage, the optical density of the cell suspension for both types of biosensors was determined in each vial at 600 nm; the fluorescence spectrum was recorded on an RF-5301PC spectrofluorophotometer (Shimadzu, Japan) in the range of 500–600 nm at the excitation wavelength λ exit = 491 nm.
A 20% decrease in the average fluorescence intensity as compared to the control was the toxicity criterion for an analyzed compound. A 20% and 50% decrease indicates that the compound is toxic and extremely toxic, respectively. Biosensor strain cells carrying the pETm-gfp plasmid, which had not been treated with a compound under study but in which the 1 mM IPTG-induced GFP gene expression had been stimulated, were used as the control. Fluorescence in the induced control cells is supposed to increase by at least 100% as compared to that in uninduced biosensor cells.
4.8. Cell viability assays
The human cancer cells of the MT-4, CEM-13 (the cells of T-cellular human leucosis), and U-937 (human monocytes) were used in this study. The cells were cultured in the RPMI-1640 medium that contained 10% embryonic calf serum, l-glutamine (2 mmol/L), gentamicin (80 μg/ml), and lincomycin (30 mg/ml) in a CO2 incubator at 37 °C. The compounds were dissolved in DMSO and added to the cellular culture at the required concentrations. Cells were placed on 96-well microliter plates and cultivated at 37 °C in 5% CO2/95% air for 72 h. Three wells were used for each concentration. The cells which were incubated without the compounds were used as a control. The cell viability was assessed through an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-phenyl-2H-tetrazolium bromide] conversion assay. 1% MTT was added to each well. Four hours later DMSO was added and mixed for 15 min. Finally, the optical density values were monitored at 570 nm.
4.9. Computer modeling
The molecular docking for modeling the interaction between compounds under investigation and influenza virus M2 protein (protein database code 2LOJ) was done by Hex online server (http://hexserver.loria.fr/).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.08.014.
Supplementary data
Supplementary data.
References and notes
- 1.Mankil J., Sangmin L., Jungyeob H., Kyunghoon L., Hanjo K., Soo K. J. Med. Chem. 2003;46:987. [Google Scholar]
- 2.Svobodov H., Ryšav H., Pavlнk M., Šaman D., Drašar P., Wimmer Z. Bioorg. Med. Chem. 2010;18:8194. doi: 10.1016/j.bmc.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Rydberg E.H., Brumshtein B., Greenblatt H.M., Wong D.M., Shaya D., Williams L.D., Carlier P.R., Pang Y.P., Silman I., Sussman J.L. J. Med. Chem. 2006;49:5491. doi: 10.1021/jm060164b. [DOI] [PubMed] [Google Scholar]
- 4.Guillou C., Renko M.A., Gras D.Z., Thal C.E. Bioorg. Med. Chem. Lett. 2000;10:637. doi: 10.1016/s0960-894x(00)00059-7. [DOI] [PubMed] [Google Scholar]
- 5.Hadjipavlou-Litina D., Magoulas G.E., Bariamis S.E., Drainas D., Avgoustakis K., Papaioannou D. Bioorg. Med. Chem. 2010;18:8204. doi: 10.1016/j.bmc.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 6.Riva E., Comi D., Borrelli S., Colombo F., Danieli B., Borlak J., Gia O.M., Dalla L.V. Bioorg. Med. Chem. 2010;18:8660. doi: 10.1016/j.bmc.2010.09.069. [DOI] [PubMed] [Google Scholar]
- 7.Tischer M., Pradel G., Ohlsen K., Holzgrabe U. ChemMedChem. 2012;7:22. doi: 10.1002/cmdc.201100404. [DOI] [PubMed] [Google Scholar]
- 8.Salakhutdinov N.F., Tolstikov G.A. Mini-Rev. Med. Chem. 2010;10:1248. doi: 10.2174/13895575110091248. [DOI] [PubMed] [Google Scholar]
- 9.Lee C. Br. J. Anaesthesia. 2001;87:755. doi: 10.1093/bja/87.5.755. [DOI] [PubMed] [Google Scholar]
- 10.Salom-Roig X.J., Hamzé A., Calas M., Henri J.V. Comb. Chem. High Throughput Screening. 2005;8:49. doi: 10.2174/1386207053328219. [DOI] [PubMed] [Google Scholar]
- 11.Tischer M., Sologub L., Pradel G., Holzgrabe U. Bioorg. Med. Chem. 2010;18:2998. doi: 10.1016/j.bmc.2010.03.067. [DOI] [PubMed] [Google Scholar]
- 12.Korochkina M.G., Nikitashina A.D., Khaybullin R.N., Petrov K.A., Strobykina I.Yu., Zobov V.V., Kataev V.E. Med. Chem. Commun. 2012;3:1449. [Google Scholar]
- 13.Guthrie J.P., Cullimore P.A., McDonald R.S., O‘Leary S. Can. J. Chem. 1982;60:747. [Google Scholar]
- 14.Kiselev O.I., Mishin V.P., Eroshkin V.I., Kozeletskaia K.N., Usova E.V., Rudenko V.I., Chupakhin O.N. Mol Biol (Mosk) 1994;28:1009. [PubMed] [Google Scholar]
- 15.Kazinets O.N., Amvros’eva T.V., Rusiaev V.A. Vopr. Virusol. 2002;47:22. Russian. [PubMed] [Google Scholar]
- 16.Scholtissek C., Quack G., Klenk H.D., Webster R.G. Antiviral Res. 1998;37:83. doi: 10.1016/s0166-3542(97)00061-2. [DOI] [PubMed] [Google Scholar]
- 17.Kolocouris N., Kolocouris A., Foscolos G.B., Fytas G., Neyts J., Paldako E., Balzarini J., Snoeck R., Andrei G., De Clercq E. J. Med. Chem. 1996;39:3307. doi: 10.1021/jm950891z. [DOI] [PubMed] [Google Scholar]
- 18.Tataridis D., Fytas G., Kolocouris A., Fytas C., Kolocouris N., Foscolos G.B., Padalko E., Neyts J., De Clercq E. Bioorg. Med. Chem. Lett. 2007;17:692. doi: 10.1016/j.bmcl.2006.10.092. [DOI] [PubMed] [Google Scholar]
- 19.Kolocouris A., Spearpoint P., Martin S.R., Hay A.J., López-Querol M., Sureda F.X., Padalko E., Neyts J., De Clercq E. Bioorg. Med. Chem. Lett. 2008;18:6156. doi: 10.1016/j.bmcl.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z., Huang J.D., Wong K.L., Wang P.G., Zhang H.J., Tanner J.A., Spiga O., Bernini A., Zheng B.J., Niccolai N. FEBS J. 2011;278:383. doi: 10.1111/j.1742-4658.2010.07961.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanner J.A., Zheng B.J., Zhou J., Watt R.M., Jiang J.Q., Wong K.L., Lin Y.P., Lu L.Y., He M.L., Kung H.F., Kesel A.J., Huang J.D. Chem. Biol. 2005;12:303. doi: 10.1016/j.chembiol.2005.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu W., Zeng S., Li C., Jie Y., Li Z., Chen L. J. Med. Chem. 2010;53:3831. doi: 10.1021/jm901664a. [DOI] [PubMed] [Google Scholar]
- 23.Schnell J.R., Chou J.J. Nature. 2008;451:591. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pielak R.M., Schnell J.R., Chou J.J. Proc. Natl. Acad. Sci. U.S.A. 2009;106:7379. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du Q.S., Huang R.B., Wang C.H., Li X.M., Chou K.C. J. Theor. Biol. 2009;259:159. doi: 10.1016/j.jtbi.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Wang J., Ma C., Balannik V., Pinto L.H., Lamb R.A., DeGrado W.F. ACS Med. Chem. Lett. 2011;2:307. doi: 10.1021/ml100297w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao X., Li C., Zeng S., Hu W. Eur. J. Med. Chem. 2011;46:52. doi: 10.1016/j.ejmech.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Gamble D.L., Hems W.P., Ridge B. J. Chem. Soc., Perkin Trans. 1. 2001:248. [Google Scholar]
- 29.Boyd D.R., Jennings W.B., Warin L.C. J. Org. Chem. 1986;51:992. [Google Scholar]
- 30.Newman D.J., Cragg G.M. J. Nat. Prod. 2012;75:311. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carter G.T. Nat. Prod. Rep. 2011;28:1783. doi: 10.1039/c1np00033k. [DOI] [PubMed] [Google Scholar]
- 32.Liu X., Germaine K.J., Ryan D., Dowling D.N. Sensors. 2010;10:1377. doi: 10.3390/s100201377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Behzadian F., Barjeste H., Hosseinkhani S., Zarei A.R. Curr. Microbiol. 2011;62:690. doi: 10.1007/s00284-010-9764-5. [DOI] [PubMed] [Google Scholar]
- 34.Woutersen M., Belkin S., Brouwer B., Wezel A.P., Heringa M.B. Anal. Bioanal. Chem. 2011;400:915. doi: 10.1007/s00216-010-4372-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yagi K. Appl. Microbiol. Biotechnol. 2007;73:1251. doi: 10.1007/s00253-006-0718-6. [DOI] [PubMed] [Google Scholar]
- 36.Robbens J., Dardenne F., Devriese L., De Coen W., Blust R. Appl. Microbiol. Biotechnol. 2010;88:1007. doi: 10.1007/s00253-010-2826-6. [DOI] [PubMed] [Google Scholar]
- 37.Shin H.J. Appl. Microbiol. Biotechnol. 2011;89:867. doi: 10.1007/s00253-010-2990-8. [DOI] [PubMed] [Google Scholar]
- 38.Quillardet P., Huisman O., D’Ari R., Hofnung M. Proc. Natl. Acad. Sci. U.S.A. 1982;79:5971. doi: 10.1073/pnas.79.19.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kostrzynska M., Leung K.T., Lee H., Trevors J.T. J. Microbiol. Methods. 2002;48:43. doi: 10.1016/s0167-7012(01)00335-9. [DOI] [PubMed] [Google Scholar]
- 40.Chen Z., Lu M., Zhuang G., Wang H. Anal. Chem. 2011;83:3248. doi: 10.1021/ac200426x. [DOI] [PubMed] [Google Scholar]
- 41.Biran A., Ben Yoav H., Yagur-Kroll S., Pedahzur R., Buchinger S., Shacham-Diamand Y., Reifferscheid G., Belkin S. Anal. Bioanal. Chem. 2011;400:3013. doi: 10.1007/s00216-011-5007-2. [DOI] [PubMed] [Google Scholar]
- 42.Lavrinenko I.A., Lavrinenko V.A., Ryabchenko A.V., Beklemishev A.B. Bull. Exp. Biol. Med. 2006;141:33. doi: 10.1007/s10517-006-0086-3. [DOI] [PubMed] [Google Scholar]
- 43.Perdew J.P., Burke K., Ernzerhof M. Phys. Rev. Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 44.Laikov D.N. Chem. Phys. Lett. 2005;416:116. [Google Scholar]
- 45.Laikov D.N., Ustynyuk Yu.A. Russ. Chem. Bull. 2005;54:820. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data.