

Abstract
Recognition of regions on the surface of one protein, that are similar to a binding site of another is crucial for the prediction of molecular interactions and for functional classifications. We first describe a novel method, SiteEngine, that assumes no sequence or fold similarities and is able to recognize proteins that have similar binding sites and may perform similar functions. We achieve high efficiency and speed by introducing a low-resolution surface representation via chemically important surface points, by hashing triangles of physico-chemical properties and by application of hierarchical scoring schemes for a thorough exploration of global and local similarities. We proceed to rigorously apply this method to functional site recognition in three possible ways: first, we search a given functional site on a large set of complete protein structures. Second, a potential functional site on a protein of interest is compared with known binding sites, to recognize similar features. Third, a complete protein structure is searched for the presence of an a priori unknown functional site, similar to known sites. Our method is robust and efficient enough to allow computationally demanding applications such as the first and the third. From the biological standpoint, the first application may identify secondary binding sites of drugs that may lead to side-effects. The third application finds new potential sites on the protein that may provide targets for drug design. Each of the three applications may aid in assigning a function and in classification of binding patterns. We highlight the advantages and disadvantages of each type of search, provide examples of large-scale searches of the entire Protein Data Base and make functional predictions.
Keywords: binding sites similarity, 3D database searches, protein function prediction, pharmacophore, computer-aided drug design
Abbreviations: RMSD, root-mean-square deviation; ALBP, adipocyte lipid-binding protein; HFABP, heart muscle fatty acid-binding protein; MFB2, Manduca sexta fatty acid-binding protein; BFABP, brain fatty acid-binding protein; SARS, severe acute respiratory syndrome
1. Introduction
Molecular recognition is one of the central processes in molecular biology. Comparison and detection of binding sites is a key step in the prediction of potential interactions. Since proteins function by interacting with other molecules, similarity in the binding patterns of proteins is closely related to similarity in their biological functions. There are two potential ways to infer the function of a novel protein. The first is to recognize a sequence or fold similarity with a protein(s) whose function is known. However, a similar fold does not necessarily imply a similar function. For example, proteins with the same fold, like TIM barrels, can have multiple functions.1 On the other hand, proteins with different folds, like subtilisin and trypsin, can share the same function. The alternative approach, implemented in our method, is to investigate the physico-chemical patterns and shape of the protein molecular surface. Proteins are assumed to perform similar functions if they share similar binding patterns and recognize similar binding partners, even if they have different sequences and (overall) fold homology.
Identification of regions on the surface of one protein that resemble a specific binding site of another is especially important for the following three applications.
-
1.
Functional analysis and classification: recognition of similarity in binding pattern to a well known protein may help in gaining a better understanding of its function and activation mechanism. These are crucial for the development of targeted drug leads like inhibitors. Functional annotation of newly determined structures can be a significant contribution to the Structural Genomics initiative.
-
2.
Potential ligands and ligand fragments: analysis of ligands bound to proteins with similar binding sites may provide hints of chemical groups that can be used to develop a drug for the protein target. The method can be used for lead generation and optimization as well as for de novo drug design.
-
3.
Prediction of side-effects: proteins with similar binding sites may bind the same drug and therefore may potentially cause side-effects. Thorough investigation of such proteins during the drug design process is important for the development of more specific drug leads.
Other methods that are commonly used for suggestion of new ligands or ligand fragments and for predictions of side-effects are alignment of small molecules2., 3. and docking.4., 5., 6., 7., 8., 9. These techniques model the interactions of the receptor with specific ligands and therefore do not analyze all potential interactions that a specific binding site may form. This is particularly important, since a single protein-binding site may have several binding patterns. Not only can the same binding site bind different ligands with different functional groups, but there is also evidence that at least in some enzymes a single compound can bind in different ways.10., 11., 12. A wide variety of methods have been developed for protein structural alignment.13 Most existing methods describe a protein structure by its Cα atoms and seek to maximize the overall similarity of the structures. However, when there is no fold similarity between the aligned structures, these methods usually do not provide a biologically significant alignment. Analysis of the similarities between binding sites can complement these techniques, ensuring full exploration of available structural data.
Several methods have been developed to identify specific three-dimensional patterns of amino acid side-chains. Artymiuk et al.14 represented each side-chain by pseudo-atoms and used a subgraph-isomorphism algorithm15 to identify the spatially conserved patterns. This algorithm (ASSAM) was recently enhanced to include additional constraints such as: the secondary structures, the solvent accessibility and the disulfide bridges.16 Wallace et al.17., 18. have introduced “coordinate templates”. These allow recognition of the “catalytic triads” that are typical for some of the protein families, like serine proteases, triacylglycerol lipases, ribonucleases and lysozymes. Using atomic representation, the geometric hashing technique19., 20., 21. was applied to efficiently compare a query protein to the template of the catalytic triad. This algorithm (TESS) has been recently updated by JESS,22 which is flexible and unconstrained by the template syntax. Binkowski et al.23 have recently presented an elegant approach to assess the similarity of sequence patterns of surface pockets and voids, which are conveniently organized in CASTp.24 Jones et al.25 have reviewed the methods for recognition of functional sites.
However, methods that recognize patterns of residues that are conserved in their 3D positions and in their amino acid identities are not always applicable. There are biological examples of proteins that can bind the same binding partners without sharing any conserved patterns of amino acid residues.26., 27. Rosen et al.28 searched for a site on the protein surface that resembles a specific, known active site. The molecular surface was represented using sparse critical points defined by Lin et al.29., 30. The translation and rotation invariant characteristics of pairs of critical points were used as a key for the geometric hashing procedure. In addition, the reliability of surface comparisons in searches for active sites was examined. It was concluded that although pure geometric surface matching is capable of finding biologically correct solutions, utilizing additional chemical “labeling” information is required to correctly rank and analyze the obtained solutions.
Kinoshita et al.31., 32. performed clique detection33 on the vertices of the triangulated solvent-accessible surface.34 They constructed a database of binding sites, eF-site,31 and used a structure of a complete protein structure to search it. However, the number of vertices in their surface representation is too large and it is too sensitive to conformational flexibilities. One of their conclusions was that other representative surface points may be more effective for robust and accurate comparisons.
An important contribution was recently published by Schmitt et al.35 They have defined generic pseudocenters that efficiently encode the physico-chemical properties important for molecular interaction. Each amino acid residue of a protein is represented as a set of such centers. Assuming that small molecule binding sites are detected in cavities, they constructed a database of binding sites Cavebase, which is integrated with Relibase.36 The clique detection algorithm was used to retrieve cavities that are similar to a specific query cavity. The solutions were ranked according to the similarity of property-based surface patches.
Here, we present a novel method, SiteEngine, that is capable of handling large protein structures in a matter of seconds. Unlike other methods that use the computationally expensive clique detection algorithm (NP-hard),37., 38. our heuristic algorithm is based on efficient hashing and matching of triangles of centers of physico-chemical properties. It introduces a low-resolution representation by chemically important surface points and performs fast scoring of all possible solutions, while retaining the correct ones. Successive scoring schemes, which are applied to smaller numbers of candidate solutions, perform a thorough exploration of the overall similarity of the surfaces as well as of local shapes of the chemically similar regions. We apply SiteEngine to a set of biological applications. First, we introduce a benchmark dataset, which is used to construct two surface description databases: one of complete protein structures and the other of binding sites. We compare between various searching applications that can be performed for recognition of functional sites. Each application is illustrated by examples of successful recognition of specific types of protein binding sites such as estradiol binding, adenine and ATP binding. For each example, we further make some specific predictions by providing a list of proteins recognized to share functional similarities with the query. We provide examples of classification of fatty acid-binding proteins and serine proteases and show the capability of the method to recognize the known similarity of the binding sites as well as of the catalytic residues. At the next stage, we apply SiteEngine to search a non-redundant dataset of all known protein structures. We describe the binding sites that are recognized to be the most similar to our query binding sites and discuss the quality of the predictions obtained. Since SiteEngine searches a complete structure of each protein in a matter of seconds, we find it to be well suited for such large-scale applications.
2. Functional Sites Recognition Algorithm
The method is developed toward the following three search applications: (1) searching a given functional site on the surfaces of different proteins stored in a database; (2) comparing a given functional site to a dataset of binding sites; (3) searching a complete protein structure for the presence of an a priori unknown functional site, similar to known sites.
These applications involve two types of comparisons: (i) searching a surface of a complete protein for a given functional site; (ii) comparison between two functional sites. However, from the algorithmic standpoint, the second type of comparison is essentially the same as searching for a given binding site on a protein surface, which is limited to a certain region of interest. Therefore, here we describe the algorithmic approach of SiteEngine for searching a complete protein structure for a region similar to a given binding site. The input to the algorithm consists of a binding site of one protein and of a complete structure of another, where the binding sites are defined by the surface description of the relevant regions. The structure of the complete molecule is searched for the presence of a region, which is similar to the input-binding site. The output is a transformation that superimposes the input-binding site on the recognized region and a score that measures the similarity between them. The main stages of the algorithm are summarized in Figure 1(a) .
Figure 1.
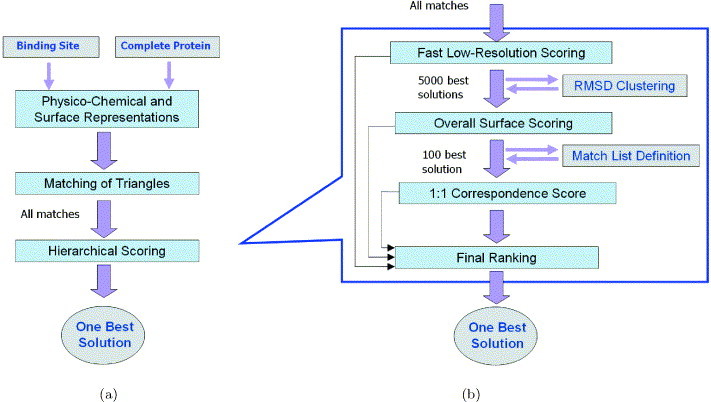
An overview of the algorithm. (a) The general flow. (b) A more detailed presentation of the hierarchical scoring stage.
2.1. Structure representation
Given the atomic coordinates of a protein structure, the first step is to calculate the physico-chemical properties of its residues. Each amino acid is assigned a set of 3D points, which are denoted as pseudocenters.35 Each pseudocenter represents an interaction center of one of the following physico-chemical properties: hydrogen-bond donor, hydrogen-bond acceptor, mixed donor/acceptor, hydrophobic aliphatic and aromatic(pi) contacts. The rules for the representation of each amino acid as a set of such centers follow Schmitt et al.35 However, unlike their definition, we do not consider a peptide bond as an aromatic property and we do not estimate the directionality of the H-bonding property. Figure 2(a) shows an example of a representation of cavity-flanking residues. In addition, we consider the pseudocenters of H-bonding properties of the side-chains of Arg, Lys and His to be positively charged, and those of Asp and Glu to be negatively charged. We have observed that these modifications lead to a slight improvement in experimental results. From the algorithmic standpoint, the similarity of charges is not a prerequisite for matching and is considered only at the scoring stage.
Figure 2.
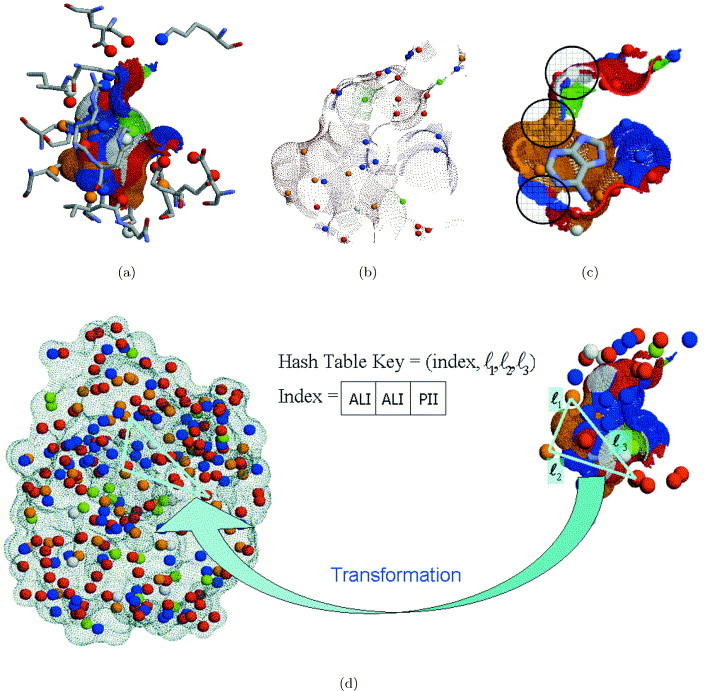
Physico-chemical representation of a molecule and of its surface. (a) The pseudocenters extracted from the cavity-flanking residues. Hydrogen-bond donors are colored in blue, acceptors in red, donors/acceptors in green, hydrophobic aliphatic in orange and aromatic in white. The surface points are colored according to the physico-chemical property of their corresponding atoms. (b) The low-resolution representation by centers of physico-chemical patches (patch centers), which are depicted as spheres. (c) The calculation of the shape function, measured in a sphere located at the patch center. The Figure shows the cross-section of an active site binding an adenine molecule. (d) The process of hashing and matching of triangles of pseudocenters.
A representation by pseudocenters is very efficient and suitable for algorithms like the geometric hashing.20 However, it is not sufficient for accurate representation and prediction of receptor–ligand interactions, especially in the case of hydrophobic aliphatic and aromatic contacts. Therefore, for each pseudocenter, we consider the surface region created by the atoms that contribute to the pseudocenter property. This provides physico-chemical labeling of the surface regions, so that only surface patches with similar properties will be matched. We use a smooth molecular surface as implemented by Connolly34., 39. and a Distance Transform grid, as implemented by Duhovny et al.40
For each chemically labeled surface patch we estimate the patch center by a surface point nearest to its center of gravity (see Figure 2(b) and (c)). Each patch center is used to estimate the average curvature of its surface patch by calculation of the solid angle shape function.40., 41., 42. In this calculation, a sphere of a certain radius is placed at the patch center. The average curvature is approximated by the fraction of the sphere inside the solvent-excluded volume of the protein. The radius of the sphere determines the region in which the curvature is approximated. We perform two calculations with different definitions of the radius of the sphere. In the first calculation, we consider a minimum radius sphere bounding the surface patch represented by the patch center. In the second calculation, the radius is user defined (by default, 6.0 Å for hydrophobic regions and 3 Å for others). An average of the two values is used to represent the shape of each surface patch.
2.2. Matching
At this stage, we calculate all possible transformations that will superimpose the input-binding site to a similar region of the surface of the other molecule. The algorithm is based on the matching of almost congruent triangles defined by triplets of pseudocenters. Figure 2(d) shows the hashing and matching procedures.
Each triplet of non-ordered non-colinear pseudocenters of the complete molecule is considered. Triplets that form triangles with side lengths within a predefined range are stored in a hash table. A key to the hash table consists of the three parameters of side lengths of a triangle and of an additional physico-chemical index, which encodes the properties of the triangle nodes (see Figure 2(d)). The physico-chemical index is represented by six bits, two for the encoding of the property of each node. This encoding is not unique due to the existence of centers with mixed donor/acceptor property. These can function both as hydrogen-bond donors and acceptors. To overcome this problem, we encode each such node twice, once as a donor and once as an acceptor.
Each triplet of ordered non-colinear pseudocenters of the query site is considered. Triplets that form triangles with side lengths within a predefined range are used to construct a hash key. This key is used to access the hash table and retrieve all congruent triangles of the complete molecule. The construction of the hash table ensures that we will match only triangles with nodes at similar spatial locations and with similar physico-chemical properties. In addition, we require that the values of the shape function of the corresponding nodes of the triangles will be similar up to a user-defined threshold. Each pair of matched triangles defines a transformation, which represents a potential solution (superimposition). Each candidate transformation is immediately scored by the low-resolution score and only transformations that received a relatively high score are retained.
The matching stage of our algorithm performs hashing of geometrical entities in a way similar to well known algorithms such as Geometric Hashing43 and Pose Clustering.44 These methods select transformations which have received the highest number of votes, e.g. in Pose Clustering a transformation that was identified by the highest number of matching triangles. The hashing stage of SiteEngine is extremely efficient, due to the consideration of the physico-chemical properties of the pseudocenters in addition to the geometrical constraints. As a result, we create less false-positive transformations and therefore greatly reduce the overall number of candidate solutions. We can score each candidate transformation and avoid any loss of competitive solutions due to the low number of votes. This approach allows identification of all candidate transformations that consist of at least three matching pseudocenters. The later stages of our scoring scheme will favor solutions with the highest number of matching pseudocenters (see 1:1 correspondence score).
2.3. Scoring
We implement a hierarchical scoring scheme, detailed in Figure 1(b). The first scheme, which is applied to all potential solutions, is calculated based on a low-resolution representation of the molecules and is therefore highly efficient. As the number of potential solutions is reduced to a smaller subset, the resolution of the molecular representation is increased leading to more precise calculations. The details of the implementation and the default parameters are provided in the Application 1, Application 2, Application 3, Application 4, Application 5, Application 6 .
Application 1.
Application 1
Application 2.
Application 2
Application 3.
Application 3
Application 4.
Application 4
Application 5.
Application 5
Application 6.
Application 6
2.3.1. Fast low-resolution scoring
The goal of this scoring scheme is to provide the initial ranking of candidate transformations and to filter out biologically unreasonable ones. The main idea is to select a small, chemically meaningful representative set of surface points and use them to efficiently estimate the potential surface similarity of the aligned surface patches. We select these points to be a set of patch centers, i.e. centers of physico-chemical surface patches of the input-binding site. We apply the candidate transformation and consider the local environment to which each patch center is transformed. First, we check whether the given patch centers are transformed to surface regions in the other molecule. Second, we check whether the physico-chemical environment to which it is transformed is similar to the one in the original molecule. Third, we compare the shape of the region to which it is transformed with the shape measured at the given patch center. Similarity in each of these attributes will increase the calculated score.
We found it sufficient to consider only the 5000 highest-ranking solutions. Transformations which superimpose the pseudocenters of the input-binding site so that the root-mean-square deviation (RMSD)45 between them is lower than a predefined threshold (3 Å), are considered to belong to the same cluster. For each cluster the best scoring transformation is selected.
2.3.2. Overall surface scoring
This scoring scheme is applied to a smaller number of the retained candidate transformations. It can therefore examine them more thoroughly using a higher level of resolution of molecular representation. Each candidate transformation is applied to each surface point. Then, as in the low-resolution score, we compare the properties of each surface point with the properties of the environment in the other molecule to which this point is transformed. Here too, similarity of both chemical and geometrical properties is scored higher than the similarity of only one of these. Since the number of considered surface points is much higher, they are divided into different categories by an approach similar to the one described by Duhovny et al.40., 42. The surface points of the input-binding site are divided into three categories according to their distance from the surface of the molecule on which it is superimposed. Each category counts the number of surface points within distance thresholds of 1 Å, 2 Å and 3 Å, respectively. In addition, in each category we calculate the number of points with the same physico-chemical property and charge, and add them to the counter of that category. We calculated a weighted sum of the counters of the three categories. The closer the category is to the surface the higher the weight that it receives.
2.3.3. The 1:1 correspondence score
As described in Figure 1(b), for each retained candidate transformation, we determine a 1:1 correspondence (match list) between the sets of pseudocenters of the two molecules. The obtained 1:1 correspondence is used for two purposes, to improve each candidate transformation by the least-squares fitting method46 and to score the similarity of the environments of the corresponding pseudocenters.
The match list is defined by calculating the maximum weight matching in a bipartite graph.38., 47. The bipartite graph is constructed in the following way. (1) The nodes of the graph are the pseudocenters of the two molecules. (2) An edge is added between each pair of pseudocenters that have similar (up to a threshold) spatial locations, physico-chemical properties and shape functions. (3) Each edge is assigned a weight that represents the similarity between the corresponding pseudocenters together with their local environments. It measures the distance, the charge compatibility of the H-bonding properties and the similarity of the local shapes of hydrophobic aliphatic regions. The maximum weight match47 in this graph provides a 1:1 correspondence between subsets of pseudocenters of the two molecules. The obtained match represents a set of pairs of pseudocenters of the two molecules, so that the points of each pair are the most similar in their geometrical and physico-chemical properties.
At the next stage, we calculate the score of the obtained 1:1 correspondence. This score consists of two parts: first, we calculate a score, which estimates the goodness-of-fit between the corresponding pseudocenters of the two molecules. Second, for each pair of centers with hydrophobic aliphatic or aromatic properties we perform a more thorough comparison of the corresponding surface patches. There are two factors that we consider to be important in this context: (1) the size of the overlap region between the patches superimposed by the candidate transformation; and (2) the shape of the common overlap region.
2.3.4. Final scoring and ranking
For each potential solution the final score is the combination of all the scores calculated by the algorithm. When performing extensive database searches it is difficult to consider more than one solution for each comparison. In these applications, we select only one solution with the highest value of the final score that maximizes the similarity with the searched pattern. We ignore the other solutions obtained for the same comparison. However, in other applications the number of output solutions is user defined and can be much larger.
2.4. Complexity and running times
The overall complexity of our algorithm is dominated by the complexity of the matching and low-resolution scoring stage. The worst case theoretical complexity of an algorithm is O(n 3 m 4). In practice, this bound is much lower, since there is a limited number of congruent triangles with similar physico-chemical properties. In addition, since we are interested only in triangles that represent potential binding patterns, we limit the side lengths of the considered triangles to be within a limited predefined range. Therefore, the practical running times of the method are proportional to O(nm 2). A sample of the algorithm running times is given in Table 1, Table 2, Table 3, Table 4 . The time measurements are done on a standard PC workstation (3.0 GHz Xeon processors, 4 GB memory) and do not include the time required for the construction of surfaces and grids, since these can be done in a preprocessing stage.
Table 1.
Recognition of adenine-binding sites by searching the database of whole proteins
| Rank | PDB | Protein | Fold | Sequence similarity (%) | Match score | Ligand | RMSD | Run time (seconds) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1atp | cAMP-dependent PK, catalytic subunit | Protein kinase-like | 100 | 100 | ATP∗ | 0.01 | 7.6 |
| 2 | 1csn | Casein kinase-1, CK1 | Protein kinase-like | 18 | 64 | ATP∗ | 0.03 | 7.5 |
| 3 | 1phk | γ-Subunit of glycogen phosphorylase kinase (Phk) | Protein kinase-like | 24 | 59 | ATP∗ | 0.3 | 7.2 |
| 4 | 1hck | Cyclin-dependent PK | Protein kinase-like | 23 | 53 | ATP∗ | 0.7 | 7.9 |
| 5 | 2src | c-src Tyrosine kinase | Protein kinase-like | 13 | 49 | ATP∗ | 0.9 | 10.2 |
| 6 | 1mu2 | HIV-1 reverse transcriptase | DNA/RNA polymerases | 8 | 47 | None | N/A | 17.3 |
| 7 | 1mjh | “Hypothetical” protein MJ0577 | Adenine nucleotide alpha hydrolase-like | 11 | 44 | ATP∗ | N/A | 5.7 |
| 8 | 1nsf | Hexamerization domain of N-ethylmalemide-sensitive fusion (NSF) protein | P-loop containing nucleotide triphosphate hydrolases | 15 | 43 | ATP∗ | N/A | 7 |
| 9 | 1g5y | Retinoid-X receptor alpha (RXR-alpha) | Nuclear receptor ligand-binding domain | 13 | 43 | REA | N/A | 7 |
| 10 | 1jd0 | Carbonic anhydrase | Caxbonic anhydrase | 13 | 43 | AZM | N/A | 6.8 |
| 11 | 1b4v | Cholesterol oxidase of GMC family | FAD/NAD(P)-binding domain | 14 | 43 | FAD∗ | N/A | 9.1 |
| 12 | 1mbm | NSP4 proteinase | Trypsin-like serine proteases | 11 | 55 | None | N/A | 13 |
| 13 | le6w | 3-Hydroxyacyl-CoA dehydrogenase | NAD(P)-binding Rossmann-fold domains, | 8 | 43 | NAD∗ | N/A | 12.9 |
| 14 | 3ert | Estrogen receptor alpha | Nuclear receptor ligand-binding domain | 14 | 42 | OHT | N/A | 10 |
| 15 | la27 | Human estrogenic 17beta-hydroxysteroid dehydrogenase | NAD(P)-binding Rossmann-fold domains | 13 | 42 | EST | N/A | 11 |
A list of proteins whose binding sites were recognized to be similar to an adenine-binding site of a cAMP-dependent protein kinase (1atp) is presented. The proteins are listed in the order of decreasing similarity to the query-binding site. The name of the ligand present in the located binding site is provided. Marked by ∗ are the entries that are known to bind adenine.
Table 2.
Recognition of estradiol-binding sites by searching the database of whole proteins
| Rank | PDB | Protein | Fold | Sequence similarity (%) | Match score | Ligand | Run time (seconds) |
|---|---|---|---|---|---|---|---|
| 1 | 1lhu | Sex hormone-binding globulin | Concanavalin A-like lectins/glucanases | 100 | 100 | EST∗ | 7 |
| 2 | 1qkt | Estrogen receptor alpha | Nuclear receptor ligand-binding domain | 16 | 45 | EST∗ | 8.6 |
| 3 | 1e8x | Phoshoinositide 3-kinase (P13K) helical domain | Alpha–alpha superhelix | 6 | 43 | ATP | 16.5 |
| 4 | 1gx9 | β-Lactoglobulin | Lipocalins | 17 | 43 | REA | 7.6 |
| 5 | 1ere | Estrogen receptor alpha | Nuclear receptor ligand-binding domain | 16 | 42 | EST∗ | 8 |
| 6 | 1l2i | Estrogen receptor alpha | Nuclear receptor ligand-binding domain | 16 | 42 | ETC∗ | 8.3 |
| 7 | 1a52 | Estrogen receptor alpha | Nuclear receptor ligand-binding domain | 18 | 42 | EST∗ | 8 |
| 8 | 1fby | Retinoid-X receptor alpha (RXR-alpha) | Nuclear receptor ligand-binding domain | 16 | 41 | REA | 8.2 |
| 9 | 1b4v | Cholesterol oxidase of GMC family | FAD/NAD(P)-binding domain | 8 | 41 | FAD | 9.9 |
| 10 | 3ert | Estrogen receptor alpha | Nuclear receptor ligand-binding domain | 16 | 40 | OHT∗ | 8.3 |
| 11 | 1equ | Estrogen receptor alpha | Nuclear receptor ligand-binding domain | 12 | 40 | EQU∗ | 8.9 |
| 12 | le6w | 3-Hydroxyacyl-CoA dehydrogenase | NAD(P)-binding Rossmann-fold domains | 4 | 40 | EST∗ | 14.3 |
| 13 | 1atp | cAMP-dependent PK, catalytic subunit | Protein kinase-like | 11 | 39 | ATP | 9.3 |
| 14 | 1err | Estrogen receptor alpha | Nuclear receptor ligand-binding domain | 17 | 39 | RAL∗ | 8.3 |
| 15 | 1ftp | Fatty acid-binding protein | Lipocalins | 14 | 39 | None | 7 |
A list of proteins whose binding sites were recognized to be similar to that of a sex hormone-binding globulin (1lhu) is presented. The proteins are listed in the order of decreasing similarity to the query binding site. In all cases, the program has successfully located the bindings sites. The name of the ligand present in the located binding site is provided. Marked by ∗ are the entries that are known to bind estradiol.
Table 3.
Recognition of ATP-binding sites by searching the database of active sites
| Rank | PDB | Protein | Fold | Sequence similarity (%) | Match score | Ligand | Run time (seconds) |
|---|---|---|---|---|---|---|---|
| 1 | 1mjh | Hypothetical protein MJ0577 | Adenine nucleotide alpha hydrolase-like | 100 | 100 | ATP | 4 |
| 2 | 9ldt | Lactate dehydrogenase | NAD(P)-binding Rossman-fold domain | 6 | 36 | NAD | 7.8 |
| 3 | 1atp | cAMP-dependent PK, catalytic subunit | Protein kinase-like (PK-like) | 8 | 35 | ATP | 6.4 |
| 4 | 1b4v | Cholesterol oxidase of GMC family | FAD/NAD(P)-binding domain | 11 | 34 | FAD | 6.8 |
| 5 | 1a27 | Human estrogenic 17beta-hydroxysteroid dehydrogenase | NAD(P)-binding Rossman-fold domain | 12 | 34 | FAD | 9.6 |
| 6 | 1nsf | Hexamerization domain of N-ethylmalemide-sensitive fusion (NSF) protein | P-loop containing nucleotide triphosphate hydrolases | 10 | 34 | ATP | 5.8 |
| 7 | 1a82 | Dethiobiotin synthetase | P-loop containing nucleotide triphosphate hydrolases | 5 | 34 | ATP | 6.3 |
| 8 | 1hsh | HIV-1 protease | Acid proteases | 6 | 33 | MK1 | 8.3 |
| 9 | 1e8x | Phoshoinositide 3-kinase (P13K) helical domain | Alpha–alpha superhelix | 6 | 33 | ATP | 7 |
| 10 | 1a49 | Pyruvate kinase | PIK beta-barrel domain-like | 10 | 32 | ATP | 6.4 |
| 11 | 2src | c-src Tyrosine kinase | Protein kinase-like | 10 | 32 | ATP | 7.5 |
| 12 | 1csn | Casein kinase-1, CK1 | Protein kinase-like | 14 | 32 | ATP | 6 |
| 13 | 1hck | Cyclin-dependent PK | Protein kinase-like | 10 | 31 | ATP | 6.1 |
| 14 | 1zin | Adenylate kinase | P-loop containing nucleotide triphosphate hydrolases | 6 | 31 | ATP | 6.8 |
| 15 | 1bx4 | Adenosine kinase | Ribokinase-like | 5 | 31 | ATP | 5.6 |
A list of proteins whose binding sites were recognized to be similar to an ATP-binding site of “hypothetical” protein MJ0577 (1mjh) is presented. The proteins are listed in the order of decreasing similarity to the query binding site. The name of the ligand present in the located binding site is provided.
Table 4.
Searching the database of binding sites with a complete protein structure of a fatty acid-binding protein (1lib)
| Rank | PDB | Protein | Fold | Sequence similarity (%) | Match score | Ligand | RMSD | Run time (seconds) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1lib | Adipocyte lipid-binding protein (ALBP) | Lipocalins | 100 | 100 | None∗ | N/A | 7 |
| 2 | 1lid | Adipocyte lipid-binding protein (ALBP) | Lipocalins | 100 | 72 | OLA∗ | 0.2 | 14.2 |
| 3 | 1lie | Adipocyte lipid-binding protein (ALBP) | Lipocalins | 100 | 70 | PLM∗ | 0.07 | 10.4 |
| 4 | 1hms | Heart muscle fatty acid-binding protein (HFABP) | Lipocalins | 64 | 63 | OLA∗ | 0.3 | 13 |
| 5 | 1b56 | Epidermal fatty acid-binding protein (EFABP) | Lipocalins | 51 | 62 | PLM∗ | 0.3 | 6.4 |
| 6 | 1pmp | Myelin P2 (P2) | Lipocalins | 64 | 61 | OLI∗ | 0.1 | 12 |
| 7 | 1qjg | Ketosteroid isomerase | Cystatin-like | 18 | 59 | EQU | N/A | 2.2 |
| 8 | 1hwr | HIV-1 protease | Acid proteases | 15 | 47 | 216 | N/A | 12.5 |
| 9 | 2lbd | Retinoic acid receptor gamma (RAR-gamma) | Nuclear receptor ligand-binding domain | 11 | 46 | REA∗ | N/A | 1.6 |
| 10 | 1com | Chorismate mutase | Bacillus chorismate mutase-like | 14 | 46 | PRE | N/A | 5.9 |
| 11 | 1flj | Rat (Rattus norvegicus), isozyme II | Carbonic anhydrase | 10 | 45 | GTT | N/A | 5.7 |
| 12 | 1cqq | Human rhinovirus type 2 | Trypsin-like serine proteases | 14 | 43 | AG7 | N/A | 17.9 |
| 13 | 1ftp | Locus muscle fatty acid-binding protein (L-MFABP) | Lipocalins | 42 | 42 | None∗ | N/A | 18.9 |
| 14 | 1opa | Cellular retinol-binding protein II (CRBPII) | Lipocalins | 37 | 40 | None∗ | N/A | 13.3 |
| 15 | 1ecm | Chorismate mutase domain of P-protein | Chorismate mutase 11 | 9 | 40 | TSA | N/A | 6.1 |
| 16 | 2cbr | Cellular retinoic-acid binding protein II (CRABP-II) | Lipocalins | 37 | 37 | A80∗ | 1.3 | 14.2 |
Marked by ∗ are ligands/binding sites known to bind/be similar to the query protein.
3. Results
In the section below, we show the experimental results obtained by applying the method to two datasets. First, we introduce a benchmark dataset that is used for a thorough evaluation of the method. We show the usefulness of the method for three types of searching applications as well as for biological classifications. Then, we proceed to apply the method to large-scale database searches of the non-redundant dataset constructed from the entire Protein Data Bank (PDB). We analyze and compare the results obtained on the two datasets.
3.1. Benchmark data set
A representative protein data set that was constructed to evaluate the performance of the algorithm is detailed in Table 5 . Two main criteria have motivated the selection of the proteins for the data set. First, we desired to include many structurally diverse proteins that can bind the same ligand. We have selected the adenine-binding proteins as a classical example of such a case.27., 48. We included in our data set the proteins used in the study by Kuttner et al.48 Thirty-three of these proteins are complexed with ATP and 11 with other adenine-containing ligands. Other functional families that were included are structurally diverse proteins that can bind estradiol, equilin and retinoic acid. Second, our motivation was to include representatives of important and well-studied structural families so that we will be able to check the classification capabilities and the consistency of our method. We have selected seven different protein families: HIV-1/HIV-2, HIV protease, anhydrase, antibiotics, fatty acid-binding proteins, chorismate mutases and serine proteases. In order to verify the tolerance of the method to local binding site flexibility, we have intentionally included several structures of homologous proteins, that are unbound or complexed with different ligands.
Table 5.
The data set
| Functional family | Total | Number of folds | PDB codes |
|---|---|---|---|
| Adenine-binding proteins | 34 | 18 | 1a49 1a82 1ads 1atp 1ayl 1b4v 1b8a 1bx4 1byq 1csc 1csn 1e2q 1e8x 1f9a 1fmw 1g5t 1gn8 1hck 1hpl 1j7k 1jjv 1kay 1kp2 1kpf 1mjh 1mmg 1nhk 1nsf 1phk 1qmm 1yag 1zin 2src 9ldt48 |
| Serine proteases | 24 | 4 | 1abi 1acb 1arb 1cho 1cse 1ela 1elc 1hah 1hne 1pek 1ppf 1sbn 1sga 1sgc 1tgs 1whs 1ysc 2alp 2lpr 3prk 3sga 3tec 4sgb 4tgl18., 28. |
| Fatty acid-binding proteins | 15 | 1 | 1b56 1cbs 1ftp 1hms 1ifc 1kqw 1lib 1lid 1lie 1mdc 1opa 1opb 1pmp 2cbr 2ifb |
| Estradiol-binding proteins | 11 | 4 | 1a27 1a52 1e6w 1ere 1err 1fds 1jgl 1l2i 1lhu 1qkt 3ert |
| Chorismate mutases | 7 | 1 | 1com 1csm 1dbf 1ecm 1fnj 1fnk 4csm28 |
| Retinoic acid-binding protein-like | 6 | 3 | 1fby 1fem 1g5y 1gx9 1tyr 2lbd |
| Anhydrases | 6 | 1 | 1azm 1flj 1jd0 1keq 1kop 1znc |
| Antibiotics | 6 | 1 | 1alq 1bt5 1dcs 1exm 1ghp 1rxf |
| HIV-1 protease | 6 | 1 | 1b60 1hsg 1hsh 1hwr 1kzk 1pro |
| HIV-1/HIV-2 | 4 | 1 | 1har 1mml 1mu2 1vrt |
| Viral proteinase | 4 | 1 | 1cqq 1lvo 1mbm 1q2w |
| Equilin binding proteins | 3 | 3 | 1equ 1oh0 1qjg |
| Total | 126 |
The list of the protein structures used for the method verification.
3.2. Database architecture
The proteins of the data set, listed in Table 5, were preprocessed to construct two types of databases:
-
1.
Database of complete protein structures: this database contains the complete protein structures with pre-calculated surfaces. Since in many cases the binding site is located between different peptide chains of a protein (e.g. HIV), for each structure we stored all chains whose coordinates appear in the PDB file.
-
2.
Database of protein-binding sites: this database stores only the binding sites extracted from the protein–ligand complexes. Each binding site is represented by a surface region around the ligand (surface points of a protein which are closer than 4.0 Å to the surface of a ligand). Binding sites of ligands that contained less than seven non-hydrogen atoms were not considered. Proteins from the data set that have no ligand were not represented in this type of database. The only exception are four proteins from the fatty acid-binding family, for which the binding sites are extracted by comparing to corresponding homologous structures of the dataset.
We present three types of searching applications that can be performed on these databases:
-
1.
Application I: searching the database of complete protein structures with a binding site. A query that is used to search the database is a binding site of a specific protein of interest. The search will provide a list of regions from different proteins, that are similar to the query site.
-
2.
Application II: searching the database of binding sites with a binding site. A known binding site of a protein of interest can be used to search for other binding sites that share the same structural and physico-chemical features.
-
3.
Application III: searching the database of binding sites with a complete protein structure. The query protein structure is searched for regions on its surface that can be similar to known binding sites.
Whether a certain binding site is used as a query or stored in the database, it is defined exactly in the same manner as described in the previous section. Below, we present results obtained by applying our method to each type of application.
3.3. Evaluation of the results
For each search example, we present a list of solutions that are ranked highest according to the value of the match score (detailed below). These solutions represent the proteins that are recognized to be most similar to the query. Below, we describe the calculation of the RMSD, which provides some measurement for the quality of the results obtained.
3.3.1. Match score
This score represents a portion of the binding pattern of interest found to match during the search. The score is presented as a percentage. We define Native_Score(B,P B) as the final score calculated by SiteEngine when a binding site (B) is searched in its native protein (P B). Since in this case all of the query features are matched, the score represents the maximal possible match (100%). When a binding site (B) is searched in a protein/binding site (M), the obtained final score of its best solution will be referred to as the Search_Score(M,B). This score will never exceed the Native_Score(B,P B) of the same binding site. Calculations of the Match_Score differ according to the type of the search application. In Applications I and II the Match_Score represents the portion of the query-binding site matched during the search. Therefore, it is calculated as a simple normalization of each comparison to a database protein/binding site (M) by the Native_Score of the query (B):Match_Score(M)=Search_Score(M,B)Native_Score(B,PB)×100As a result, this score provides a ranking of the database proteins/binding sites according to the percentage of the query pattern that they match. When the database is searched with a complete protein structure (Application III) the Match_Score represents how much of the database binding site (B) matches the query protein (M):Match_Score(B)=Search_Score(B,M)Native_Score(B,PB)×100In this application, this score provides a ranking of the database binding sites, according to the percentage of their features recognized in the query.
3.3.2. RMSD
When the compared proteins share an overall fold, we calculate the RMSD45 in a manner which is commonly used in unbound docking algorithms.4 Although SiteEngine aligns binding sites and no ligand information is used, the RMSD deviation calculated between the ligands can provide some insight regarding the results obtained. The RMSD is calculated between the locations of the ligand present in the binding site of the query. It is calculated between two possible locations for this ligand, one obtained when the query-binding site is superimposed by SiteEngine on the database molecule and the other obtained by aligning the Cα atoms9 of the two molecules. However, when the compared molecules do not have the same overall fold, this calculation cannot be performed. In addition, when the proteins do share the same fold, but manifest high structural variabilities, the alignment between the Cα atoms is not straightforward and can be misleading. Therefore, this measure is not always applicable and in many cases, instead of providing this value we state ”N/A”.
3.4. Searching the benchmark dataset
3.4.1. Recognition of adenine-binding sites by searching the database of complete protein structures
An adenine-binding site extracted from a cAMP-dependent protein kinase (1atp) was used to search the database of complete protein structures. The query-binding site was defined by protein surface points whose distance from an adenine ring of an ATP ligand is under 4.0 Å. Table 1 presents the highest ranking solutions. The dataset contained five proteins which share the same “protein kinase-like” fold as the query. As expected, all five are recognized as top ranking solutions and the query-binding site is correctly aligned to the binding sites of these proteins. Due to the similarity of the fold, we are able to calculate the RMSD between the locations of the ligands obtained by these solutions. As can be seen, in all cases the RMSD is less than 1.0 Å. The running times measured in this test case emphasize the ability of the method to handle large protein structures. For example, the longest running time (17.3 seconds) was observed for a 980 residue molecule (1mu2) represented by 1714 pseudocenters. The ability to search the complete surface of a molecule of this size highlights the speed of our method. Ranked 7 was a “hypothetical” protein MJ0577.49 Its ATP-binding site was correctly recognized when searching for an adenine-binding site. Figure 3 presents the alignment obtained between the molecules. As depicted in Figure 3(a) there is no fold similarity between the proteins, however, our method correctly recognizes the similarity between the binding sites. The ATP molecules and the complete structure of the cAMP-dependent protein kinase (1atp) is depicted for illustration only and were not used in the search. Figure 3(b) presents the pseudocenters that are identified to be similar.
Figure 3.
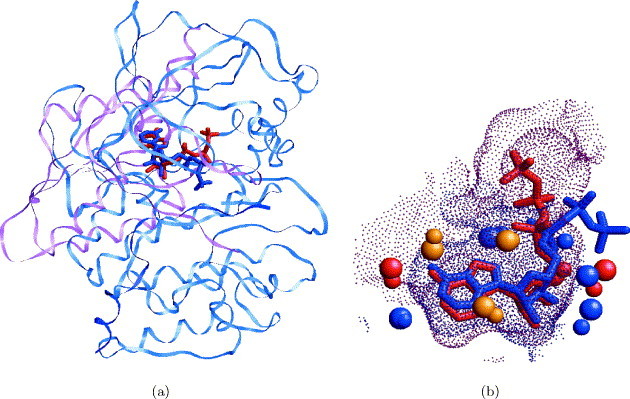
Recognition of similarity between the binding sites of cAMP-dependent protein kinase (1atp) and hypothetical protein MJ0577 (1mjh). (a) The proteins of a cAMP-dependent protein kinase (blue) and hypothetical protein MJ0577 (pink) are superimposed using the transformation of the solution. The ATP molecules from 1atp are colored blue and from 1mjh are colored red. The structures of the whole cAMP-dependent protein kinase (1atp) and of the ATP ligands are depicted for illustration only and were not used during the search. (b) A closer view of the active sites of the molecules. The surfaces of the active sites are represented as small dots and are colored red for 1mjh and blue for 1atp. The recognized centers of interaction (pseudocenters) are represented as spheres and are colored according to their physico-chemical properties as in Figure 2. The pseudocenters of 1atp are larger. The ATP molecules are colored as in (a).
3.4.2. Recognition of estradiol-binding sites by searching the database of complete protein structures
The constructed dataset contains 11 proteins that are known to bind estradiol. These proteins belong to four different folds: concanavalin A-like lectins/glucanases (1), immunoglobulin-like beta-sandwich (2), NAD(P)-binding Rossmann-fold domain (4), nuclear receptor ligand-binding domain (5). The dataset contains a total of seven structures that were complexed with estradiol, while the rest were crystallized with other small molecules.
A binding site of a sex hormone-binding globulin (1lhu) was used to search the data set. All 11 data set proteins that are known to bind estradiol are recognized within the 30 best solutions. Table 2 presents the 15 highest-ranking solutions. As expected, the top-ranking solution is the correctly recognized binding site of the protein of the sex hormone-binding globulin. Figure 4 presents two of the estradiol-binding sites correctly recognized by the algorithm. Figure 4(a) presents an estrogen alpha receptor whose binding site is ranked second and is recognized to be the most similar to the binding site of a sex hormone-binding globulin. Figure 4(b) presents a 17beta-hydrosteroid dehydrogenase that is ranked 29th. Its binding site is successfully located and is correctly recognized as estradiol binding, but it is identified to be less similar to that of a sex hormone-binding globulin.
Figure 4.
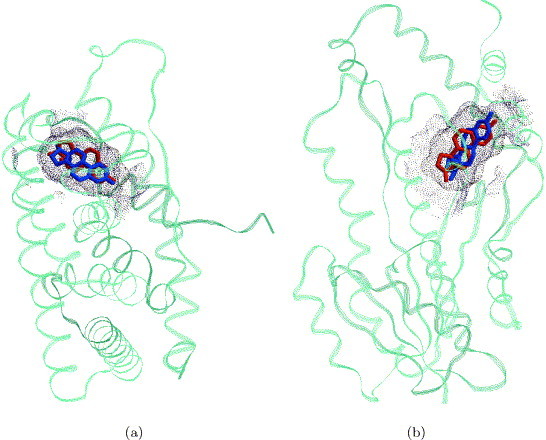
Highest and lowest-ranking solutions obtained in searching the data set for estradiol binding sites. (a) An estrogen alpha receptor (1qkt), colored cyan, was successfully recognized as estradiol-binding. Its binding site, depicted by blue dots, was identified as the most similar to that of a sex hormone-binding globulin (1lhu), depicted by red dots. The ligands from the complexes 1qkt and 1lhu are depicted for verification only and are colored in blue and red, respectively. (b) The binding site of a 17beta hydrosteroid dehydrogenase (1fds), colored cyan, was successfully recognized as estradiol-binding. Its binding sites, depicted by blue dots, was ranked 29 and identified as the less similar to that of a sex hormone-binding globulin (1lhu), depicted by red dots. The ligands from the complexes 1fds and 1lhu are depicted for verification only and are colored in blue and red, respectively.
Ranked third and fourth are the binding sites of two proteins which are not known to bind estradiol and are considered to be “false-positive” solutions. Figure 5 represents an analysis of a binding site of phoshoinositide 3-kinase (1e8x) that is ranked third. The protein that was used for the alignment is in a complex with ATP. However, there is another structure of the same protein in a complex with wortmannin (PDB code 1e7u), which has structural similarity to estradiol. Figure 5(a)–(c) show the similarity between wortmannin (C23H24O8) and estradiol (C18H24O2). Figure 5(d) presents the alignment between the surfaces of the binding sites of phoshoinositide 3-kinase (1e8x) and a sex hormone-binding globulin (1lhu). Ranked fourth is a binding site of a beta-lactoglobulin complexed with retinoic acid. As in the previous case, the alignment obtained between the binding sites provides a good superimposition between the hydrophobic ligands of estradiol and retinoic acid and places the retinoic acid in the estradiol-binding pocket as would be done with a docking program.4., 5., 9.
Figure 5.
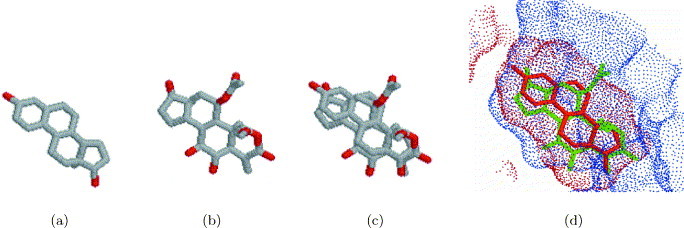
Binding site of phoshoinositide 3-kinase (PI3K): recognition of similarity to an estradiol-binding site of sex hormone-binding globulin. The binding site of phoshoinositide 3-kinase (PI3K) was ranked third when compared to an estradiol-binding site of sex hormone-binding globulin. (a) Estradiol molecule (Cl8H24O2) from a complex with 1lhu. Carbon atoms are colored gray and oxygen atoms are red. (b) Wortmannin molecule (C23H24O8) from a complex with PI3K in 1e7u. The atom coloring is as in (a). (c) The superimposition between the estradiol from 1lhu and wortmannin from 1e7u obtained by the superimposition of binding sites. (d) The superimposition between the surfaces of the binding sites of 1lhu (red dots) and 1e8x (blue dots). The alignment of ligands is the same as in (c), estradiol is depicted in red and wortmannin in green.
It is important to note that the binding sites of all estradiol-binding proteins are correctly recognized in spite of the fact that five of them were not complexed with estradiol. Some of these ligands are very different from estradiol both in their size and in chemical structure. However, these differences in binding partners as well as the local flexibility that is required to accommodate them did not prevent the successful recognition of the functional similarities made by SiteEngine.
3.4.3. Searching the database of binding sites to predict the function of a hypothetical protein
A hypothetical protein MJ0577 from a hyperthermophile Methanococcus jannaschii was crystallized as part of a Structural Genomics project with the goal of functional recognition.49 We have extracted its ATP-binding site and searched the database of binding sites to recognize those that are most similar to it. Table 3 lists the highest-ranking solutions of this search. All highest-ranking solutions bind ligands similar to ATP. The only exception is an HIV-1 protease, which was also recognized by Schmitt et al.35 to have a binding niche similar to an ATP-binding site of cAMP-dependent protein kinase. The measured running times in all comparisons, are less than ten seconds, showing the ability of the method to perform efficient, large-scale database searches.
3.4.4. Classification of protein-binding sites
Wallace et al.18 derived 3D coordinate templates representing the Ser-His-Asp “catalytic triads” that are typical for some of the protein families, like serine proteases, lipases and lysozymes. These templates were used to classify a representative set of 225 enzymes into four structural groups, up to three subgroups each. Rosen et al.28 selected 24 enzymes to represent this classification. In order to test our ability to recognize the catalytic triads and to classify the protein-binding sites, we have included these 24 protein structures in our data. We have randomly selected three proteins from the three most populated subgroups and used their binding sites to search the data set of complete protein structures. The selected structures were alpha-chymotrypsin (1acb), thermitase (3tec) and serine protease B (4sgb). The protein–protein interfaces of these proteins were defined by the surface points of a protein which are closer than 4.0 Å to the surface of their protein binding partner. These were used to search the database of complete protein structures (Application I). The results are fully consistent with the classification defined by Wallace et al. and members of the same subgroup as the query is always top-ranking.
3.4.5. Recognition of catalytic residues
Subtilisin-like and trypsin-like folds are the most common examples of proteins with different fold that can perform the same function. Proteins of these two folds share the same Ser-His-Asp catalytic triad and are included in the 24 proteins that represent the classification made by Wallace et al. Our data set contained 16 proteins of the trypsin-like fold and five proteins of the subtilisin-like. When the database of complete protein structures was searched with the protein–protein interface of thermitase (3tec) the first member of trypsin-like fold (1ela) was ranked 8. When it was searched with the interface of serine protease B (4sgb) the first member of the subtilisin-like fold (3tec) was ranked 11. The catalytic residues of histidine and serine, common to the proteins of these different folds, are correctly superimposed by our method with an alignment quite similar to the one presented by Schmitt et al.35 Similar to them, the catalytic aspartate residue was not considered in the calculations, since it is not surface exposed. However, the alignment calculated by SiteEngine, provides a good superimposition of all three residues of the catalytic triad, including the aspartate. Figure 6(a) presents the alignment of two protein–protein complexes obtained during the search with an interface of thermitase (3tec), which is a member of the subtilisin-like fold. Ranked 10, is a member of the trypsin-like fold, β-trypsin (2ptc) complexed with a pancreatic trypsin inhibitor. In spite of the fold differences between these proteins, the similarity in the histidine and serine catalytic residues was correctly recognized. The binding partners of these proteins, although not considered by SiteEngine, were correctly superimposed by the transformation of the solution.
Figure 6.
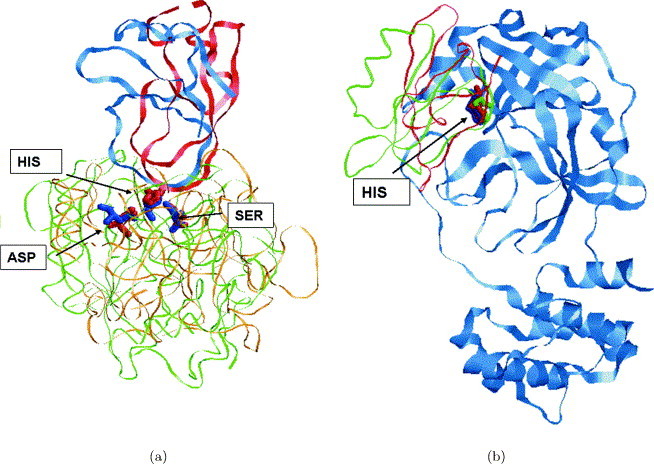
Recognition of catalytic residues. (a) When the dataset of complete protein structures was searched with the protein–protein interface of thermitase (3tec, colored green), which has a subtilisin-like fold, ranked 10 was β-trypsin (2ptc, colored orange), which has a trypsin-like fold. The catalytic residues (ordered from right to left) Ser225, His71, Asp38 of 3tec (colored blue) and Ser195, His57, Asp102 of 2ptc (colored red) are displayed in ball-and-stick. SiteEngine has recognized the similarity between the residues of serine (right) and histidine (middle). The binding partners pancreatic trypsin inhibitor from 2ptc and eglin C from 3tec are colored in red and blue, respectively. No information regarding these binding partners was considered by SiteEngine. (b) When the dataset of complete protein structures was searched with the protein–protein interfaces of thermitase (3tec) and of serine protease B (4sgb) the structure of SARS-coronavirus main protease (1q2w), colored blue, was ranked 11th and 17th, respectively. The Figure presents the catalytic histidine residues (in ball-and-stick) of these proteins, when they are superimposed on 1q2w by the transformation calculated by SiteEngine. The catalytic His41 of 1q2w is colored blue, His71 of 3tec is green and His57 of 2ptc is red. The recognized alignment places the binding partners eglin c of 3tec (green) and potato inhibitor PCI-1 of 4sgb (red) in the catalytic site of SARS-coronavirus main protease.
Another interesting result, which was obtained by these searches, is the striking similarity of the catalytic histidine residues that was recognized between these proteins and SARS-coronavirus main protease. Our dataset contained a recently determined structure of SARS-coronavirus main protease (1q2w), which is related to the severe acute respiratory syndrome (SARS) disease. SARS-coronavirus main protease, which cleaves the polyproteins of SARS-coronavirus, is responsible for the virus replication and therefore for the disease.50., 51. The protein of SARS-coronavirus main protease (1q2w) was ranked 11 when the dataset was searched with the protein–protein interface of thermitase (3tec) and 17th when searched with serine protease B (4sgb). In contrast to serine proteases which have a Ser-His-Asp catalytic triad, SARS-coronavirus main protease functions through a catalytic dyad, Cys-His. Our method has successfully detected the spatial similarity between the histidine residues common to all these proteins. Figure 6(b) presents a superimposition of the complexes of 3tec and 4sgb on the structure of lq2w by the transformation calculated by SiteEngine. As can be seen, the solution obtained provides a good alignment of the catalytic histidine residues of the three proteins. In addition, the binding partners of these proteins (eglin C from 3tec and potato inhibitor PCI-1 from 4sgb) are placed in the catalytic-binding site of the SARS-coronavirus main protease.
3.4.6. Searching the database of binding sites with a complete protein structure of a fatty acid-binding protein
The goal of this type of database search is to locate potential binding sites of a protein, for which this information is still unavailable. In order to verify our method, we have searched the database of binding sites with a complete structure of a fatty acid-binding protein (1lib). The location of the binding site in this protein is well known and the database of binding sites contained six binding sites that are very similar to it. SiteEngine was able to correctly select them from the database of binding sites. Table 4 presents the highest-ranking solutions. As can be seen, the six highest-ranking solutions are the binding sites that are known to be similar to the query protein. We have calculated the RMSD45 between the ligands of these proteins when they are superimposed by two transformations: one defined by the superimposition of the Cα atoms and the other defined by SiteEngine. As can be seen, the RMSD in all cases is very low and even existing techniques like docking4., 5. consider such transformations to be a success. As detailed below, binding sites of fatty acid-binding proteins that did not receive a high rank are known to exhibit a different binding pattern than the query protein. However, all database binding sites that were extracted from fatty acid-binding proteins were correctly superimposed on the region of the query protein known as its binding site. To visualize these results, Figure 7(a) presents the superimposition of the ligands from these binding sites on the query protein. No ligand information was used during the search and each ligand is superimposed onto the structure of the query protein using the transformation obtained by the alignment between the database-binding site and the query protein. As can be seen, in all cases the ligands are successfully placed in the actual binding site of the query protein.
Figure 7.
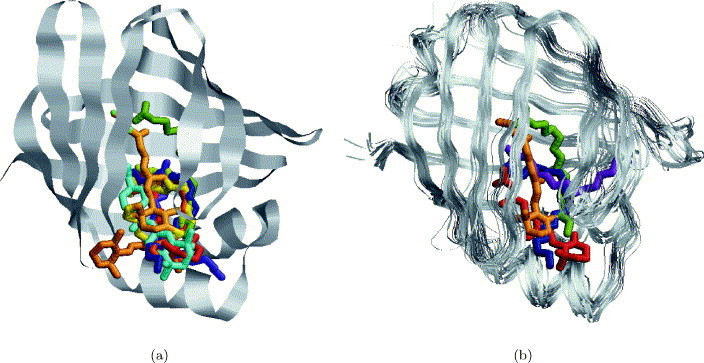
(a) Searching the database of binding sites with a complete structure of a fatty acid-binding protein (1lib). The Figure presents the ligands from all the fatty acid-binding proteins which were included in the data set listed in Table 5. Each ligand is superimposed onto the structure of the query protein using the transformation obtained by the alignment between the database binding site and the query protein. (b) Classification of fatty acid-binding proteins. The Figure presents the structural alignment via MultiProt55., 56. between 42 fatty acid-binding proteins used in the study. Some examples of ligands are depicted to represent the diversity of the binding modes. In green is the palmitic acid molecule from a complex with an IFABP protein (2ifb). In blue is the oleic acid molecule from a complex with the ALBP (1lid). In red is a retinol molecule from a complex with CRABP-II (1cbs) and in orange is a retinoic acid molecule from a complex with CRBPII protein (1opb). In purple is a palmitic acid molecule from MFB2 protein (1mdc), that has the same binding motif as ALBP, but exhibits a very high degree of flexibility of the binding site region.
3.4.7. Classification of fatty acid-binding proteins
Motivated by the previous example, we have applied our program to analyze the function and classify the fatty acid-binding proteins.52., 53. For this study, we took all the crystal structures classified by SCOP54 as members of the “fatty acid-binding protein-like” family. Table 6 lists the PDB codes of these 43 structures and their classification to domains as defined by SCOP. Figure 7(b) presents the structural alignment between the 43 structures as performed by MultiProt.9., 55., 56. As can be seen, the structures and the ligands are very similar; however, the ligand conformations and the binding patterns are very different. We have tested the ability of our program to classify members of this family according to the binding site motifs. We have selected four representative proteins that form the four most highly populated domains of this family: an adipocyte lipid-binding protein (1lid), an intestinal fatty acid-binding protein (2ifb), a cellular retinoic acid-binding protein (1cbs) and a cellular retinol-binding protein II (1opb). The results of all four searches are summarized in Table 7 , where each column ranks the 43 members of the family in decreasing order of similarity to the query-binding sites.
Table 6.
The data set of fatty acid-binding proteins
| Domain name and abbreviation | Total | PDB codes |
|---|---|---|
| Adipocyte lipid-binding protein (ALBP) | 12 | 1a18 1a2d 1ab0 1acd 1adl 1alb 1lib 1lic 1lid 1lie 1lif 2ans |
| Brain fatty acid-binding protein (BFABP) | 2 | 1fdq 1fe3 |
| Cellular retinoic acid-binding protein I (CRABP-I) | 3 | 1cbi 1cbr 2cbr |
| Cellular retinoic acid-binding protein II (CRABP-II) | 4 | 1cbq 1cbs 2cbs 3cbs |
| Cellular retinol-binding protein II (CRBPII) | 5 | 1crb 1kqw 1kqx 1opa 1opb |
| Cellular retinol-binding protein III (CRBPIII) | 1 | 1ggl |
| Cellular retinol-binding protein IV (CRBPIV) | 1 | 1lpj |
| Epidermal fatty acid-binding protein (EFABP) | 1 | 1b56 |
| Intestinal fatty acid-binding protein (IFABP) | 6 | 1dc9 1icm 1icn 1ifb 1ifc 2ifb |
| Liver fatty acid-binding protein (LFABP) | 1 | 1lfo |
| Heart muscle fatty acid-binding protein (HFABP) | 4 | 1hmr 1hms 1hmt 2hmb |
| Locus muscle fatty acid-binding protein (L-MFABP) | 1 | 1ftp |
| Manduca sexta fatty acid-binding protein (MFB2) | 1 | 1mdc |
| Myelin P2 (P2) | 1 | 1pmp |
PDB codes and domain name abbreviations of high-resolution crystal structures of proteins classified by SCOP54 as members of “fatty acid-binding protein-like” family.
Table 7.
Classification of fatty acid-binding proteins
| Similarity to 1lid |
Similarity to 2ifb |
Similarity to 1opb |
Similarity to 1cbs |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rank | PDB | Domain | RMSD | PDB | Domain | RMSD | PDB | Domain | RMSD | PDB | Domain | RMSD |
| 1 | 1lid | ALBP | 0.1 | 2ifb | IFABP | 0.1 | 1opb | CRBPII | 0.4 | 1cbs | CRABP-II | 0.0 |
| 2 | 11if | ALBP | 0.1 | 1ifb | IFABP | 0.2 | 1kqw | CRBPII | 0.2 | 2cbs | CRABP-II | 0.1 |
| 3 | 1lic | ALBP | 0.2 | 1icm | IFABP | 0.2 | 1opa | CRBPII | 0.8 | 3cbs | CRABP-II | 0.2 |
| 4 | 1adl | ALBP | 0.1 | 1icn | IFABP | 0.1 | 1crb | CRBPII | 0.8 | 1cbq | CRABP-II | 0.2 |
| 5 | 1lie | ALBP | 0.1 | 1dc9 | IFABP | 0.2 | 1kqx | CRBPII | 1.4 | 2cbr | CRABP-I | 0.2 |
| 6 | 1pmp | P2 | 0.2 | 1ifc | IFABP | 0.2 | 1lpj | CRBPIV | 0.6 | 1cbr | CRABP-I | 0.1 |
| 7 | 1ab0 | ALBP | 0.2 | 1opb | CRBPII | 1.9 | 2hmb | HFABP | 3.0 | 2hmb | HFABP | 0.8 |
| 8 | 1lib | ALBP | 0.2 | 1a2d | ALBP | 0.9 | 1hmt | HFABP | 1.4 | 1opa | CRBPII | 0.5 |
| 9 | 1a2d | ALBP | 0.04 | 1opa | CRBPII | 2.3 | 1hmr | HFABP | 3.6 | 1lie | ALBP | 1.3 |
| 10 | 1alb | ALBP | 0.2 | 2ans | ALBP | 0.6 | 1pmp | P2 | 2.6 | 1hms | HFABP | 1.2 |
| 11 | 2ans | ALBP | 0.2 | 1mdc | MFB2 | 1.1 | 1hms | HFABP | 3.1 | 1hmt | HFABP | 0.8 |
| 12 | 1a18 | ALBP | 0.1 | 1pmp | P2 | 1.9 | 1cbq | CRABP-II | 2.0 | 1adl | ALBP | 1.5 |
| 13 | 1acd | ALBP | 0.2 | 2hmb | HFABP | 2.2 | 1ab0 | ALBP | 2.2 | 1hmr | HFABP | 1.1 |
| 14 | 1hmt | HFABP | 0.04 | 1a18 | CRABP-II | 0.6 | 1lic | ALBP | 2.2 | 1lic | ALBP | 1.2 |
| 15 | 1fdq | BFABP | 0.4 | 1fe3 | BFABP | 1.6 | 1adl | ALBP | 4.0 | 1dc9 | IFABP | 2.1 |
| 16 | 1hms | HFABP | 0.1 | 1lie | ALBP | 6.6 | 1lie | ALBP | 3.1 | 1ftp | L-MFABP | 2.3 |
| 17 | 2hmr | HFABP | 0.2 | 1crb | CRBPII | 4.6 | 3cbs | CRABP-II | 6.5 | 1crb | CRBPII | 3.0 |
| 18 | 1fe3 | BFABP | 0.3 | 1fdq | BFABP | 1.4 | 1lif | ALBP | 2.4 | 1ifc | IFABP | 1.6 |
| 19 | 2hmb | HFABP | 0.4 | 1lfo | LFABP | 11 | 1cbs | CRABP-II | 3.2 | 1lif | ALBP | 2.0 |
| 20 | 1crb | CRABP | 0.5 | 1cbs | CRABP-II | 5.9 | 2cbs | CRABP-II | 6.8 | 1a18 | ALBP | 0.9 |
The ranking of the 43 proteins listed in Table 6 in the decreasing order of similarity to four different query-binding sites is presented. Each entry lists the PDB code and the SCOP domain and the RMSD between the ligands present in the query-binding sites. The query-binding sites (from 1lid, 2ifb, 1opb and 1cbs) represent four different binding motifs exhibited by the members of this family.
The first test was to search the data set with a binding site extracted from the adipocyte lipid-binding protein. According to Banaszak et al.53 adipocyte lipid-binding protein (ALBP) as well as myelin P2 (P2), heart muscle fatty acid-binding protein (HFABP) and Manduca sexta fatty acid-binding protein (MFB2) interact with their bound fatty acid using the P2 motif. As observed in Table 7, all 12 members of the ALBP family and the only structure of myelin P2 (P2) are top ranking. They are followed by the members of the heart muscle fatty acid-binding protein (HFABP) that have the same binding motif as the query-binding site. Members of the brain fatty acid-binding protein (BFABP), share the same “U-shape” fatty acid-binding mode as ALBP and HFABP57 and therefore were correctly recognized to be similar to the query. The only member of M. sexta fatty acid-binding protein (MFB2) was ranked 34. Figure 7 depicts the flexibility of the ligands of ALBP and MFB2 and provides an explanation for such a low rank.
When the data set was searched with the binding sites of the intestinal fatty acid-binding protein (2ifb), the cellular retinol-binding protein (1cbs) and the cellular retinol-binding protein II (1opb) the results were the same. The proteins were correctly classified and the top-ranking solutions were all the members of the same domain as the query.
In this test case the surfaces of complete protein structures were searched for the presence of the binding site of interest. In order to show that the query site was successfully located on the surface of each protein, we present the values of the RMSD which were calculated between the locations of the ligands oleic acid, palmitic acid, retinol and retinoic acid present in the query-binding sites extracted from 1lid, 2ifb, lopb, and lcbs, respectively. For each pairwise alignment the RMSD is calculated between the location of the ligand obtained by SiteEngine and the location of the same ligand obtained by the alignment of the backbones (Cα atoms) of the complete structures. Although no ligand information was used by SiteEngine, it can be seen that the RMSD values in most of the cases are very low. The extreme exception is the alignment of the binding site of 2ifb to the structure of 1lfo. The structure of the liver fatty acid-binding protein is very different from the rest of the family members due to the fact that more than one fatty acid is bound.58 The structural alignment by Cα atoms leads to an RMSD of 8.2 Å between the oleate ligand of 1lfo and palmitic acid of 2ifb. SiteEngine aligns these ligands with an RMSD of only 5.5 Å and it correctly detects the primary-binding site of 1lfo. It must be noted that these results were achieved in spite of the fact that only one best solution was considered for each pairwise alignment.
3.5. Analysis of the results obtained
In the absence of an overall fold or sequence similarity between the proteins, assessing the correctness of the obtained results is not straightforward. In these cases, there is no exact definition for the similarity between two binding sites. A query-binding region may contain features that are not essential for the binding, which may differ in proteins with exactly the same function. The absence of an exact definition of the pattern we are looking for makes the evaluation of such partial solutions even more complicated. Considering the superimposition between the ligands obtained by an alignment between unrelated proteins can also be misleading. Similar binding sites may accommodate ligands that differ in their size and shape and it is not clear what should be the correct superimposition between them. Figure 8(a) presents an alignment between the ATP ligands of the hexamerization domain of N-ethylmaleimide sensitive factor (1nsf) and cAMP-dependent protein kinase (1atp) obtained by the alignment of the corresponding binding sites. As can be seen in the Figure, the superimposition of the ligand molecules achieves a good alignment between the ribose parts of the ATP molecules while the orientation of the adenine moieties is different. However, when the binding sites are artificially superimposed using the transformation that aligns the adenine moieties, the distance between the phosphate tails of the ATP molecules is approximately 20 Å and only six pseudocenters are identified to be similar (as opposed to nine pseudocenters identified by SiteEngine). Therefore, it is not straightforward to identify which solution is the correct one, especially due to the fact that the adenine moiety is known to exhibit two different binding modes.10 Figure 8(b) presents an alignment between the NAD and ATP ligands of lactate dehydrogenase (9ldt) and the hypothetical protein MJ0577(1mjh) obtained by the alignment of the corresponding binding sites. Once again, the alignment between the binding sites of the proteins provides an alignment between the ribose parts of the NAD and ATP ligand molecules. However, the adenine ring of the ATP is aligned to the nicotinamide ring of NAD (and not to an adenine ring as expected). This solution provides the alignment of 13 similar centers of interaction shared by these binding regions. Moreover, there are two residues (1mjh, His40, Val142; 9ldt, His195, Val32) that are present in both binding sites and have the same spatial locations as well as identity of the amino acid. Since SiteEngine is a software tool it recognizes regions that maximize the similarity; however, it cannot assess the biological significance of the obtained predictions. These need to be further verified by physical experiments and human expertise.
Figure 8.
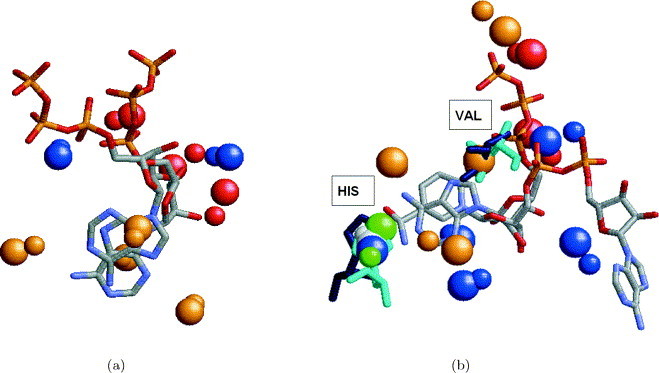
Alignment between the ligands induced by the alignment of the binding sites. (a) Alignment between the ATP ligands of hexamerization domain of N-ethylmaleimide sensitive factor (1nsf) and cAMP-dependent protein kinase (1atp) obtained by the alignment of the corresponding binding sites. The colored spheres represent the centers of interaction recognized by the program. The coloring of the spheres is as in Figure 2. The spheres of 1nsf are smaller. It can be seen that the solution provides a good alignment between the ribose parts of the two ATP molecules at the expense of the alignment between the adenine moieties. (b) Alignment between the NAD and ATP ligands of lactate dehydrogenase (9ldt) and hypothetical protein MJ0577 (1mjh) obtained by the alignment of the corresponding binding sites. The solution provides a good alignment between the ribose parts of the NAD and ATP ligand molecules. However, the adenine ring of ATP is aligned to a nicotinamide ring of NAD, and not to its adenine ring as expected. As can be seen, the resulting alignment provides a superimposition of 13 functional groups, that are depicted as balls. Moreover, it provides an alignment of two conserved residues His40 and Val142 of 1mjh (colored cyan) and His195, Val32 of 9ldt (colored blue) that are located in similar spatial locations.
4. Evaluation of Applications
We have applied SiteEngine to three types of applications. Below, we discuss the main advantages and disadvantages of each type (see Table 8).
Table 8.
Types of searching applications
| Application I: searching a database of complete protein structures with a binding site | Application II: searching a database of binding sites with a binding site | Application III: searching a database of binding sites with a complete protein structure |
|---|---|---|
| A. Advantages | ||
| 1. Can recognize new regions that can function as binding sites | 1. Reduced run time and storage space | 1. Can be used to recognize an unknown binding site of a protein of interest |
| 2. The database structures can be unbound. No information is missed | 2. Less false positives, since we compare only known binding sites | |
| 3. All of the solutions are relevant, due to their similarity to a specific region of interest | ||
| B. Disadvantages | ||
| 1. The binding site of a protein of interest must be known | 1. The binding site of a protein of interest must be known | 1. Large amount of information may be missed |
| 2. Some solutions may align regions which are not binding sites | 2. Large amount of information may be missed | 2. The query is not focused. Some solutions may align regions which are not binding sites |
| 3. Development of a reliable ranking scheme is not straightforward | ||
Advantages and disadvantages of three types of applications for recognition of functional sites.
4.1. Application I: searching the database of complete protein structures with a binding site
This type of database search is the most general and reliable. All of the available information is utilized and we can recognize totally new regions that can function as binding sites. It can be used to suggest a list of proteins that may bind ligands similar to the ligands of the protein of interest and may lead to side-effects. We have illustrated this application by two searches performed with the estradiol-binding site of the sex hormone-binding globulin and with the adenine-binding site of the cAMP-dependent protein kinase. In both cases, the highest-ranking solutions contained a list of unrelated proteins that can perform the same function as the query site. An additional application of this type of search is the classification of binding patterns. This was illustrated by the examples of serine proteases and fatty acid-binding proteins.
4.2. Application II: searching the database of binding sites with a binding site
Constructing a database of binding sites may significantly reduce the time and space required to perform large-scale searches. Searches of this type are more focused, since they consider regions that are already known to function as binding sites. Less potential solutions are considered, which allows a more careful examination of each. This type of application is limited to the comparison of regions that are already known to serve as binding sites. However, it may be useful in suggesting ligands or ligand fragments for applications such as structure-based drug design. Searching with the ATP-binding site of the hypothetical protein MJ0577 (1mjh) provides an example of how this type of search can assist in the recognition of function and can contribute to structural genomics projects.
4.3. Application III: searching the database of bindings sites with a complete protein structure
The advantage of such an approach is in the recognition of new a priori unknown regions in a protein of interest that can function as binding sites. However, this makes the search extremely unfocused and some of the solutions may align surface regions that have no functional significance. When looking for binding sites which are located in cavities, an alternative strategy may be to extract potential binding pockets, using existing cavity detection methods40., 59., 60., 61., 62., 63., 64. and use them to perform a more focused search. We have illustrated this application by searching the database with a complete structure of a fatty acid-binding protein. SiteEngine has successfully selected from the database of binding sites those that are known to be similar to the query and suggested a good alignment between them. However, the ranking of binding sites of different size according to their similarity to different regions on the surface of the complete protein is not straightforward. In this work, the ranking was done according to how much of the database-binding site was matched during the search. However, some binding sites received a high rank due to their small size and the fact that some surface patterns have a high probability of appearance on the surface of any protein structure. These considerations must be taken into account when developing more reliable searches of this type.
Searches of the first type are advantageous over applications II and III, since they explore the whole surfaces of complete protein structures. This application is not limited to the set of known binding sites and it can recognize new regions that can function as such. In addition, the construction of a database of binding sites is not straightforward and the results of applications II and III are influenced by the selected definition of a binding site. Since SiteEngine is robust enough to search complete protein structures with speed almost equal to comparisons between binding sites, we conclude that, whenever possible, application I is the preferred option.
4.4. Searching the entire PDB
Following the successful performance of the SiteEngine method on our benchmark dataset, we applied it to large-scale searches against a non-redundant dataset constructed from the entire PDB.65 The evaluation described in the previous section showed that application I is the most general and reliable of all applications. This application searches a database of complete proteins and utilizes all the available information of the proteins structures stored in the PDB. Below, we repeat the searches of the previous sections on the non-redundant ASTRAL66., 67., 68. dataset. This dataset consists of all known protein structures that have less than 40% sequence identity. Following removal of some low-resolution structures that contain only the coordinates of the Cα atoms, a total of 4375 protein structures were searched by our method. The details regarding the top ranking solutions of all the searches are provided in the Supplementary Material.
4.5. Recognition of adenine-binding sites by searching the entire PDB
The adenine-binding site extracted from cAMP-dependent protein kinase (1atp) was used to search the ASTRAL database of complete protein structures. As expected, most of the top-ranking solutions are the catalytic sites of other protein kinases. Although these proteins have different sequences, they share the same “protein kinase-like” fold. The 13 best solutions are the adenine binding sites of these proteins. In total, there are 17 such sites among the 30 top-ranking solutions. These binding sites were correctly located on the surfaces of these proteins and the performance of SiteEngine in these cases was similar to the five top-ranking solutions presented in Table 1. It is interesting to note that only five of the recognized adenine-binding sites are complexed with adenine, while the rest are unbound or accommodate other ligands. Ranked 19 is the correctly recognized binding site of a replication factor C (1iqp) that accommodates an ADP molecule. Figure 9(a) shows the correct recognition of this site and Figure 9(b) presents an additional binding site of d-Ala-d-Ala ligase (1iow) that accommodates an ADP molecule and is ranked 31. As can be seen the transformation calculated by SiteEngine provides a perfect alignment between the ATP substrate of the query site to the ADP of the recognized region. In spite of the difference between the overall structures, these proteins were recognized to have similar shapes of the binding sites and share 12 functional groups located in similar spatial locations. However, the biological “correctness” of many other of top-ranking solutions cannot be verified. Some of these were regions of such proteins as photosynthetic reaction centre (1dxr, ranked 14), pyruvate phosphate dikinase (1kbl, ranked 15) and the gamma subunit of DNA polymerase (1jr3, ranked 18). Figure 9(e) presents one of these solutions that we consider to be a false-positive. The region that is recognized on the surface of photosynthetic reaction centre (1dxr) received a high rank due to the similarity of its hydrophobic patches to the query. Some of such false-positive solutions might be filtered out by an additional requirement of the presence of certain features that are required to bind adenine. However, currently there is no automatic way to define such a set of features based on the protein structure alone.
Figure 9.
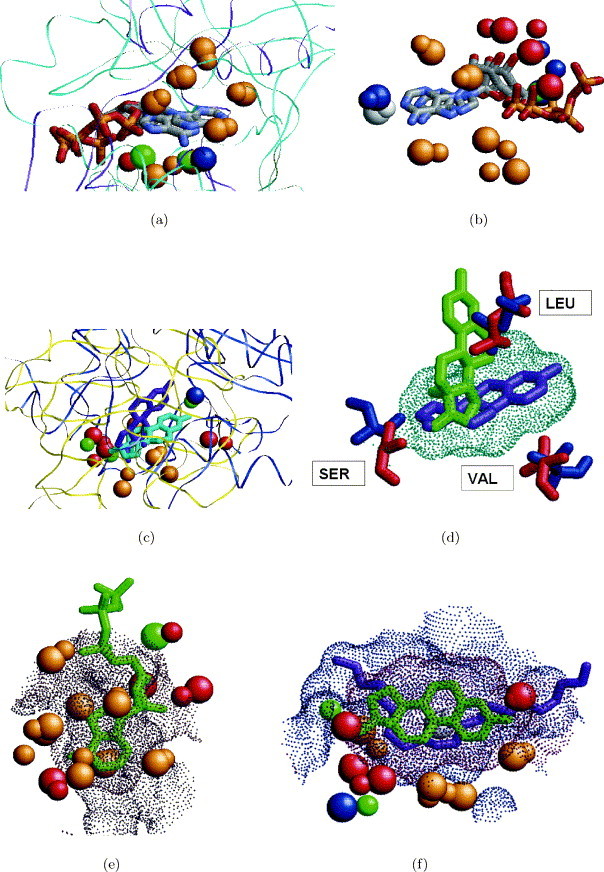
(a) Recognized similarity (ranked 19) of an ADP-binding site of a replication factor C (1iqp, colored purple) to the adenine-binding site of cAMP-dependent protein kinase (1atp, colored cyan). The Figure presents the obtained superimposition of the ligand molecules as well as of the functional groups. The functional groups that are shared by these sites are represented as spheres colored according to their physico-chemical properties as in Figure 2 (the spheres of 1atp are larger). (b) Recognized similarity (ranked 31) of functional groups and the alignment of ligands of an ADP-binding site of d-Ala-d-Ala ligase (1iow) to the adenine-binding site of cAMP-dependent protein kinase (1atp). (c) Recognized similarity (ranked 38) of the estradiol-binding sites of estrogen sulfotransferase (1aqu, blue) and sex hormone-binding globulin (1lhu, yellow). The estradiol ligand of 1lhu is cyan and that of 1aqu is purple. (d) Recognized similarity (ranked 43) of a binding site of a tropinone reductase (2ae2) to an estradiol-binding site of sex hormone-binding globulin (1lhu, depicted as cyan dots). The estradiol ligand of 1lhu is purple and an estradiol molecule that is placed in the binding site of 2ae2 according to its structural homologue (1fds) is shown in green. The two binding sites are recognized to share three residues that have the same spatial location and identity of the amino acid. The residues of 2ae2 are blue and those of 1lhu are red. (e) A false-positive solution that recognizes a similarity of the adenine-binding site cAMP-dependent protein kinase to a region of photosynthetic reaction centre (1dxr). The ATP molecule of 1atp is green. The surfaces of the two binding sites are depicted as dots (1atp, red; 1dxr, blue) and the functional groups are depicted as spheres. (f) A presumably false-positive solution of similarity of a CXE-binding site of an outer membrane protein NspA (1p4t) to an estradiol-binding site of sex hormone-binding globulin (1lhu). The binding sites are depicted as dots (1lhu, red; lp4t, blue) and the ligands as sticks (1lhu, green; 1p4t, purple).
4.6. Recognition of estradiol-binding sites by searching the entire PDB
The estradiol-binding site of a sex hormone-binding globulin (1lhu) was used to search the ASTRAL database of complete protein structures. The two top-ranking solutions are trivial and are the estradiol-binding sites of the query protein (ranked 1) and of another sex hormone-binding globulin (1d2s, ranked 2). In contrast to our benchmark dataset, the non-redundant ASTRAL dataset contains almost no proteins complexed with estradiol. Although most of the top-ranking regions are indeed binding sites, we do not have the information regarding their ability to bind estradiol. One such case is the CXE (pentaethylene glycol monodecyl ether) binding site of an outer membrane protein NspA (1p4t) that is ranked 3 and is illustrated in Figure 9(f). As can be seen, these binding sites have similar surfaces and physico-chemical environments. In total, there are six regions of membrane proteins within the 20 top-ranking solutions. Since we do not have the biological expertise to evaluate these results, we consider them to be false-positives.
The only dataset protein that is complexed with estradiol is the estrogen sulfotransferase (1aqu), which has a different fold than the query and is ranked 39. Figure 9(c) presents the obtained superimposition of the estradiol molecules as well as the correctly recognized similarity of the protein regions that accommodate them. Ranked 44 is a tropinone reductase (2ae2) that belongs to the same family of tyrosine-dependent oxidoreductases as 17beta hydrosteroid dehydrogenase (1fds), which is complexed with estradiol (depicted in Figure 4(b)). The structural alignment of the Cα atoms of proteins 2ae2 and 1fds (performed by MultiProt9., 55., 56.), provides a superimposition of the estradiol molecule of 1fds upon the structure of 2ae2. Remarkably, the estradiol molecule is placed on the same region that was recognized by SiteEngine as the potential estradiol-binding site. Figure 9(d) shows the three residues (1lhu Ser42, Val112, Leu131, and 2ae2 Ser146, Val197, Leu213) that are shared by tropinone reductase (2ae2) and the query. These residues have the same spatial location and identity. As can be seen, the conformations of the estradiol molecules are different. This may be due to a flexible loop (residues 212–222) at the binding site of 2ae2, which has a different conformation in 1fds.
4.7. Recognition of similarity of catalytic residues of protein-binding sites
Here, we verify the ability of SiteEngine to recognize similarities of the catalytic residues of serine proteases,17., 18., 28., 35. which have become a standard benchmark for evaluation of such methods. In none of its stages did the algorithm consider the information regarding the identity of the amino acid residues. However, the alignment is considered correct only if it superimposes the corresponding catalytic residues.
First, the ASTRAL dataset was searched with the binding site of thermitase (subtilisin-like fold, 3tec). The five top-ranking solutions are other proteins of the subtilisin-like fold. Ranked seven and eight are the binding sites of members of the trypsin-like fold, which share the same Ser-His-Asp catalytic triad. The similarity of the corresponding functional groups created by the triads is correctly recognized and the alignments of these solutions are very similar to that presented in Figure 6(a). However, there is one solution that emphasizes a limitation of our method. In addition to the five top-ranking solutions, the ASTRAL dataset contained a kexin protein (1ot5), which is also classified as a member of the subtilisin-like fold. The binding site of this protein is correctly located by SiteEngine and its catalytic triad is correctly matched to the query. However, it is ranked only 94. This low rank is due to a deviation of almost 7 Å of a flexible loop (3tec, 106–119) that is present in the binding site. Since SiteEngine does not explicitly address the flexibility of protein molecules, it considers the corresponding loop regions to be unmatched.
We proceed to search the ASTRAL dataset with a binding site of a member of trypsin-like fold (4sgb). Eleven out of 15 top ranking solutions are correctly recognized binding sites of proteins of the trypsin-like fold that share the Ser-His-Asp catalytic triad. Ranked seven is a 3C cysteine protease (1cqq). This protein is a member of the same SCOP family as SARS-coronavirus (1q2w) and the alignment obtained is similar to that presented in Figure 6(b). Ranked 15 is the catalytic site of the first member of subtilisin-like fold (1dtw). As before, the corresponding residues of the two catalytic triads are correctly matched by the transformation of SiteEngine. However, there are two unexpected solutions that received an extremely low rank. One is the epidermolytic (exfoliative) toxin A (1agj), which is ranked 2594. Although classified as a member of the trypsin-like fold, it has a different binding site as confirmed by the crystallographic studies.69 A similar result was obtained for a serine-carboxyl proteinase PSCP (1ga6) that is ranked 2345. Although its overall structure belongs to a subtilisin-like fold, its binding site is different and it functions through a Glu-Asp-Ser catalytic triad.70
4.8. Inferring the function of novel proteins
Almost half of the proteins from M. jannaschii, whose structures were determined as a part of the Structural Genomics project, are classified as functionally unknown “hypothetical” proteins.71 In this section, we use the ASTRAL dataset to show two examples of how SiteEngine can assist in functional annotation of these proteins.
First, we repeat the example of our previous section and use an ATP-binding site from MJ0577 to search the ASTRAL dataset of complete protein structures. As expected, the top-ranking solution is the query-binding site recognized in its native protein. The next two top-ranking solutions correctly recognize the similarity of the query to AMP-binding sites of ETFP subunits (1o97 and 1efv) that belong to the same SCOP superfamily54 as the query. Figure 10(a) presents the alignment of the binding site of 1o97 to the query. As can be seen, the binding sites are extremely similar and the ligands are perfectly aligned. The recognized similarity of the binding sites in addition to the similarity of the overall structures of these proteins can suggest similarity of their functions. Figure 10(b) presents another example, ranked 7, where SiteEngine has recognized a similarity to an ADP-binding site of arsenite-translocating ATPase ArsA (1ihu). Although the alignment of an ADP substrate of this protein to the ATP of the query is not very good, we have obtained a good alignment of the binding sites as well as of some secondary structure elements. While this protein is classified to a different fold than the query, the similarity, of the secondary structure elements superimposed by the transformation of SiteEngine suggests that the obtained alignment is correct and can assist in deciphering the function of this protein. Another surprising result of this search is the consistency of our presumably false-positive results to what was observed on our benchmark dataset. Six out of 15 top-ranking solutions of this search are oxidoreductases. The alignments obtained in these cases are similar to the one presented in Figure 8(b). However, once again, we are unable to confirm the biological correctness of this result.
Figure 10.
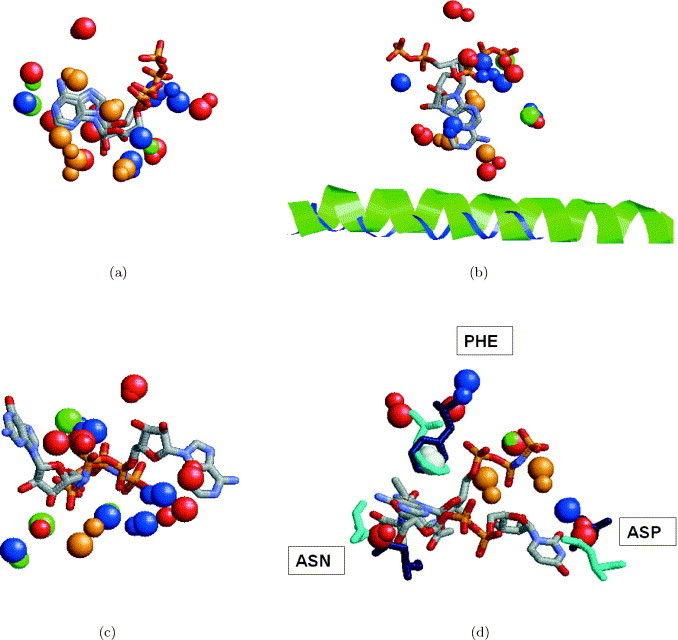
(a) The similarity of the functional groups (depicted as spheres) of an AMP-binding site of ETFP subunit (1o97) to that of the hypothetical protein MJ0577. This alignment is ranked 3 and provides a perfect superimposition of the corresponding substrates. (b) The similarity (ranked 7) of an ADP-binding site of arsenite-translocating ATPase ArsA (1ihu) to an ATP-binding site of the hypothetical protein MJ0577. Although the proteins belong to different overall folds, the similarity of some their secondary structures (e.g. the depicted helices) support the correctness of the obtained solution. (c) The similarity a G protein Gialpha1 (1cip, ranked 26) to a bound form of the ANP-binding site of the hypothetical protein MJ0226 (2mjp). SiteEngine recognized the similarity of the phosphate-binding regions of these sites. (d) Similarity (rank 30) of an UMA-binding site of UDP-N-acetylmuramoyl-l-alanine: d-glutamate (MurD) ligase (2uag) to an unbound form of the ANP-binding site of the hypothetical protein MJ0226 (1b78). The residues shared by these sites are colored blue (1b78) and cyan (2uag).
In the next example we applied the method to recognize functional sites similar to an ANP-binding site of the hypothetical protein MJ0226 with an unknown function. This example is especially challenging, since the binding site of this protein is very unusual. The binding mode of its ANP substrate is different from the binding mode in other proteins and its main interaction with the protein is by its phosphate groups, while its adenine ring is exposed and is pointing outwards from the protein surface. Here, we have performed two tests, in which the ASTRAL dataset was searched with both a bound (2mjp) and an unbound (1b78) form of this protein.
As expected the first two solutions are the trivial recognition of the binding sites of the proteins themselves. In both cases, these two solutions were followed by a hypothetical protein YggV (1k7k). Although extremely similar to the query, this protein is also a part of the Structural Genomics project and its function is unknown. When searched with a bound form of MJ0226 (2mjp), ranked 4 is a binding site of autoinducer-2 production protein LuxS (1j6w) and ranked 5 is a tandem phosphatase domain of RPTP LAR (1lar). Ranked 6 is a binding site of adenylate kinase with the substrate-mimicking inhibitor Ap5A. This inhibitor can be considered as an ATP molecule coupled to an AMP molecule via the additional phosphate group and its interaction with the protein shows the pathway of phosphoryl transfer.72 SiteEngine has recognized a high degree of similarity of the regions that interact with the phosphate groups, which are aligned by the calculated transformation. A similar alignment is also recognized with the GNP-binding site of a G protein gialphal (1cip, ranked 27), presented in Figure 10(c). This result seems consistent with the suggestion by Hwang et al.71 that this protein can function similarly to the signal sorters of G-proteins. When searched with an unbound form of MJ0226 (1b78), the ranking of the results (other than the top three) is slightly different due to the differences of the surfaces of the bound and unbound forms. Ranked 4 is a copper amine oxidase (1ivw), ranked 5 is a tryptophan indol-lyase (1ax4) and ranked 6 is a farnesyl diphosphate synthase (1uby). Figure 10(d) illustrates the result that is ranked 31, in which we recognize similarity to a UDP-N-acetylmuramoyl-l-alanine-d-glutamate (MurD) ligase (2uag). The obtained alignment of the binding sites provides a superimposition of the ANP substrate of the query to a nucleotide precursor UDP-N-acetylmuramoyl-l-alanine (UMA). As can be seen, the rings of the two ligands participate in similar aromatic interactions with a phenyl residue that is present in both binding sites. In total, these binding sites share three residues (1b78 Asn19, Asp73, Phe149, and 2uag Asn138, Asp35, Phe422) that have the same spatial location and identity.
4.9. Comparison of the experimental results
In this section, we compare the results obtained on the two datasets used in this work. The ASTRAL and the benchmark datasets are constructed for different purposes and contain structures with different PDB codes. The benchmark dataset was constructed for the purpose of a thorough evaluation of the method on a set of well-studied examples. Consequently, it contains redundancies that are important for the verification of the consistency and for the analysis of false-negatives. On the other hand, the ASTRAL dataset contains no proteins with similar sequences and structures. Applying the method to such a representative dataset is important to show the large-scale applicability of the method. However, in many cases the proteins of our benchmark dataset were not selected as representative structures and therefore were not included in the ASTRAL dataset. One such example is the set of proteins that are complexed with estradiol. There are 12 such proteins in our benchmark dataset and only one in the ASTRAL. Representatives that are selected for the same protein families do not necessarily bind estradiol, since they may contain mutations that can influence the functional region and can interfere with the binding. This has an impact on the obtained ranking and on our ability to evaluate the results.
When searching for regions similar to the adenine-binding site of cAMP-dependent protein kinase (1atp), the five top-ranking solutions obtained on our benchmark dataset (see Table 1) are within the ten top-ranking solutions obtained on the ASTRAL dataset. The hypothetical protein MJ0577 (1mjh), which is ranked eighth on the benchmark dataset is 263rd on the ASTRAL. In both datasets these ranks are within the 6% of the best solutions† . Hexamerization domain (1nsf) is ranked eighth (best 8%) on the benchmark dataset and its homologue (1d2n) that binds adenine is ranked 111th (best 3%) on the ASTRAL. Two additional examples are oxidoreductases, which are represented by the PDB codes 1b4v and le6w in the benchmark dataset and by 1gos and 1b16 in the ASTRAL. These are ranked 11th and 13th (best 10%) on the benchmark dataset and 214th (best 5%) and 41st (best 1%), respectively, on the ASTRAL.
When searching for regions similar to the ATP-binding site of the hypothetical protein MJ0577 (1mjh) the results are similar to the above. The lactate dehydrogenase (9ldt) that is ranked second on the benchmark dataset (see Table 3) is represented by li0z in the ASTRAL and is ranked eighth. Another example is the hexamerization domain (1nsf) that is ranked sixth on the benchmark dataset and is within the 5% of the best solutions. In the ASTRAL dataset, the representative structure of the same family (1d2n) is ranked 339th, which is the upper 8% of the best solutions. Another successful example is the members of the nitrogenase iron protein-like family (1a82, represented by lihu in the ASTRAL dataset) that are ranked seventh on both datasets. In general, there is a similarity of the rankings obtained on the two datasets and results that are within the 10% of the best solutions of the benchmark dataset are within the 10% of the best solutions of the ASTRAL dataset.
5. Summary and Conclusions
Recognition of functional sites in protein structures is extremely important for various biological applications, such as prediction of function and ligand binding. We have presented a novel method, SiteEngine, that in a matter of seconds can search large protein surfaces to recognize such sites and make predictions. We used a benchmark dataset to evaluate the performance of the method for three types of search applications. These experiments have shown that searching the database of complete protein structures is the most general and reliable application. Therefore, we have proceeded to use this application to search a non-redundant database constructed from the entire PDB. Below, we analyze its main advantages and weaknesses.
One of the main advantages of the method is its speed, which is obtained due to the following factors: (1) introduction of a low-resolution surface representation via chemically important surface points; (2) hashing and matching triangles of physico-chemical properties; (3) application of hierarchical scoring schemes for a thorough exploration of global and local similarities.
The biological significance of the results obtained by the method is the outcome of the following factors: (a) consideration of both physico-chemical and geometrical properties of a protein molecule; (b) consideration of both discrete (pseudocenters and patch centers) and continuous (surfaces and shapes) representations of the protein molecule; (c) development of a set of scoring schemes that score each type of potential interaction differently according to its main chemical characteristics; (e) scoring each candidate solution, without any specific pre-requirement regarding the size of the matched region.
However, SiteEngine is a software tool and therefore is limited in the quality of its biological predictions. It recognizes geometrically and chemically similar regions that belong to totally unrelated proteins. However, these similarities do not necessarily imply similarity in the binding partners and in the biological functions. SiteEngine can provide a list of proteins that are most likely to behave similarly to a binding site of interest. However, it cannot assess the biological significance of the recognized similarity.
As in many other applications in structural biology, the, major bottleneck of the method is scoring. In the current version of SiteEngine there is no implicit treatment of electrostatic potentials that have a strong impact on the interaction. Addition of such consideration may help to filter the false-positive solutions like the one presented in Figure 5. Additional weaknesses of the method are the requirement of high-resolution protein structures and addressing protein molecules as rigid bodies. Protein flexibility is addressed only through a set of thresholds that allow a certain variability in the locations. These are definitely insufficient for efficient searches of binding sites that can bind large flexible molecules.
Other limitations that influence the quality of the results are implied by the screening applications and are general to the problem. One is the absence of a clear definition of what exactly is a functional site and what are the features that define it. When the binding site is defined by its contacts with the smaller ligand, a significant amount of information may be missed. As a result, essential features might be ignored and the extracted pattern might be partially aligned to other functionally different binding sites. There is no simple automatic solution to this problem. One possibility is the construction of a database of consensus binding patterns, common to all proteins with the same function. Another problem is assessing the statistical significance of the obtained results. These are strongly influenced by the number of functional sites of the same type present in the searched database. Although the ASTRAL dataset provides a non-redundant coverage of protein structures, it contains many redundancies of functional sites. In order to provide a truly representative statistical evaluation it is essential to consider a non-redundant dataset of functional sites, the construction of which is a future challenge. In order to efficiently address these problems there is a need for methods for multiple structural alignments between binding sites. In the future, we intend to utilize the insights we have gained in the present method for the development of such algorithms.
Acknowledgements
We thank Maxim Shatsky and Dina Schneidman for useful discussions and for contribution of software to this project. We thank Dr Shuo Liang Lin for particularly valuable ideas and for critical reading of the manuscript. We thank Drs David Zanuy, Buyong Ma and K. Gunasekaran for useful suggestions. This research has been supported, in part, by the “Center of Excellence in Geometric Computing and its Applications” funded by the Israel Science Foundation (administered by the Israel Academy of Sciences). The research of H.J.W. and A.S.-P. is partially supported by the Hermann Minkowski-Minerva Center for Geometry at Tel Aviv University. The research of R.N. has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number NO1-CO-12400. The content of this publication does not necessarily reflect the view or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the US Government. The publisher or recipient acknowledges right of the US Government to retain a non-exclusive, royalty-free license in and to any copyright covering the article. Funded, in part, by the NCI under contract NO1-CO-12400.
Edited by J. Thornton
Footnotes
Supplementary data associated with this article can be found at doi: 10.1016/j.jmb.2004.04.012
Calculated as the obtained rank relative to the size of the dataset.
Supplementary Material.
Supplementary Material for this paper comprising details of implementation and the top ranking solutions of searches of the ASTRAL dataset performed with the binding sites of a c-AMP-dependent protein kinase (1atp), a sex hormone-binding globulin (1lhu), MJ0577 (1mjh), MJ0226 (2mjp and 1b78) is available on Science Direct
References
- 1.Nagano N., Orengo C.A., Thornton J.M. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 2002;321:741–765. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- 2.Dror O., Shulman-Peleg A., Nussinov R., Wolfson H.J. Predicting molecular interactions in silico: I. A guide to pharmacophore identification and its applications for drug design. Curr. Med. Chem. 2004;11:71–90. doi: 10.2174/0929867043456287. [DOI] [PubMed] [Google Scholar]
- 3.Lemmen C., Lengauer T. Computational methods for the structural alignment of molecules. J. Comput. Aided Mol. Des. 2000;14:215–232. doi: 10.1023/a:1008194019144. [DOI] [PubMed] [Google Scholar]
- 4.Schneidman-Duhovny D., Nussinov R., Wolfson H. Predicting molecular interactions in silico II: protein–protein and protein–drug docking. Curr. Med. Chem. 2004;11:91–107. doi: 10.2174/0929867043456223. [DOI] [PubMed] [Google Scholar]
- 5.Halperin I., Ma B., Wolfson H.J., Nussinov R. Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins: Struct. Funct. Genet. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 6.Taylor R.D., Jewsbury P.J., Essex J.W. A review of protein–small molecule docking methods. J. Comput. Aided Mol. Des. 2002;16:151–166. doi: 10.1023/a:1020155510718. [DOI] [PubMed] [Google Scholar]
- 7.Abagyan R., Totrov M. High-throughput docking for lead generation. Curr. Opin. Chem. Biol. 2001;5:375–382. doi: 10.1016/s1367-5931(00)00217-9. [DOI] [PubMed] [Google Scholar]
- 8.Langer T., Hoffmann R.D. Virtual screening: an effective tool for lead structure dicovery. Curr. Pharm. Des. 2001;7:509–527. doi: 10.2174/1381612013397861. [DOI] [PubMed] [Google Scholar]
- 9.Shatsky M., Dror O., Schneidman-Duhovny D., Nussinov R., Wolfson H.J. BioInfo3D: a suite of tools for structural bioinformatics. Nucl. Acids Res. 2004 doi: 10.1093/nar/gkh413. In the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phillips C.L., Ullman B., Brennan R.G., Hill C.P. Crystal structures of adenine phosphoribosyltransferase from Leishmania donovani. EMBO J. 1999;18:3533–3545. doi: 10.1093/emboj/18.13.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma B., Shatsky M., Wolfson H.J., Nussinov R. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milne G.M.A. Pharmacophore and drug discovery. In: Schleyer P.v R., Allinger N.L., Clark T., Gasteiger J., Kollman P.A., Schaefer H.F., III, Schreiner P.R., editors. Encyclopedia of Computational Chemistry. Wiley; Chichester: 1998. pp. 2046–2056. [Google Scholar]
- 13.Eidhammer I., Jonassen I., Taylor W.R. Structure comparison and structure patterns. J. Comput. Biol. 2001;7:685–716. doi: 10.1089/106652701446152. [DOI] [PubMed] [Google Scholar]
- 14.Artymiuk P.J., Poirrette A.R., Grindley H.M., Rice D.W., Willett P. A graph-theoretic approach to the identification of three-dimensional patterns of amino acid side-chains in protein structures. J. Mol. Biol. 1994;243:327–344. doi: 10.1006/jmbi.1994.1657. [DOI] [PubMed] [Google Scholar]
- 15.Ullmann J.R. An algorithm for subgraph isomorphism. J. Assoc. Comput. Mach. 1976;23:31–42. [Google Scholar]
- 16.Spriggs R.V., Artymiuk P.J., Willett P. Searching for patterns of amino acids in 3d protein structures. J. Chem. Inf. Comput. Sci. 2003;43:412–421. doi: 10.1021/ci0255984. [DOI] [PubMed] [Google Scholar]
- 17.Wallace A.C., Borkakoti N., Thornton J.M. TESS: a geometric hashing algorithm for deriving 3D coordinate templates for searching structural databases. Application to enzyme active sites. Protein Sci. 1997;6:2308–2323. doi: 10.1002/pro.5560061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallace A.C., Laskowski R.A., Thornton J.M. Derivation of 3D coordinate templates for searching structural databases: application to Ser-His-Asp catalytic triads in the serine proteinases and lipases. Protein Sci. 1996;5:1001–1013. doi: 10.1002/pro.5560050603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamdan, Y. & Wolfson, H. J. (1988). Geometric hashing: a general and efficient model-based recognition scheme. Proc. IEEE Int. Conf. Computer Vision, pp. 238–249, IEEE Computer Society Press.
- 20.Nussinov R., Wolfson H.J. Efficient detection of three-dimensional structural motifs in biological macromolecules by computer vision techniques. Proc. Natl Acad. Sci. USA. 1991;88:10495–10499. doi: 10.1073/pnas.88.23.10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachar O., Fischer D., Nussinov R., Wolfson H.J. A computer vision based technique for 3-D sequence independent structural comparison. Protein Eng. 1993;6:279–288. doi: 10.1093/protein/6.3.279. [DOI] [PubMed] [Google Scholar]
- 22.Barker J.A., Thornton J.M. An algorithm for constraint-based structural template matching: application to 3D templates with statistical analysis. Bioinformatics. 2003;19:1644–1649. doi: 10.1093/bioinformatics/btg226. [DOI] [PubMed] [Google Scholar]
- 23.Binkowski T., Adamian L., Liang J. Inferring functional relationship of proteins from local sequence and spatial surface patterns. J. Mol. Biol. 2003;232:505–526. doi: 10.1016/s0022-2836(03)00882-9. [DOI] [PubMed] [Google Scholar]
- 24.Binkowski T., Naghibzadeh S., Liang J. CASTp: computed atlas of surface topography of proteins. Nucl. Acids Res. 2003;31:3352–3355. doi: 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones S., Thornton J.M. Searching for functional sites in protein structures. Curr. Opin. Chem. Biol. 2004;8:3–7. doi: 10.1016/j.cbpa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Moodie S.L., Mitchell J.B.O., Thornton J.M. Protein recognition of adenylate: an example of a fuzzy recognition template. J. Mol. Biol. 1996;263:486–500. doi: 10.1006/jmbi.1996.0591. [DOI] [PubMed] [Google Scholar]
- 27.Denessiouk K.A., Rantanen V., Johnson M. Adenine recognition: a motif present in ATP-,CoA-,NAD-,NADP-, and FAD-dependent proteins. Proteins: Struct. Funct. Genet. 2001;44:282–291. doi: 10.1002/prot.1093. [DOI] [PubMed] [Google Scholar]
- 28.Rosen M., Lin S., Wolfson H.J., Nussinov R. Molecular shape comparisons in searches for active sites and functional similarity. Protein Eng. 1998;11:263–277. doi: 10.1093/protein/11.4.263. [DOI] [PubMed] [Google Scholar]
- 29.Lin S., Nussinov R., Fischer D., Wolfson H.J. Molecular surface representation by sparse critical points. Proteins: Struct. Funct. Genet. 1994;18:94–101. doi: 10.1002/prot.340180111. [DOI] [PubMed] [Google Scholar]
- 30.Lin S., Nussinov R. Molecular recognition via face center representation of a molecular surface. J. Mol. Graph. 1996;14:78–90. doi: 10.1016/0263-7855(96)00030-6. [DOI] [PubMed] [Google Scholar]
- 31.Kinoshita K., Furui J., Nakamura H. Identification of proteins functions from a molecular surface database, eF-site. J. Struct. Funct. Genomics. 2001;2:9–22. doi: 10.1023/a:1011318527094. [DOI] [PubMed] [Google Scholar]
- 32.Kinoshita K., Nakamura H. Identification of protein biochemical functions by similarity search using the molecular surface database ef-site. Protein Sci. 2003;12:1589–1595. doi: 10.1110/ps.0368703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bron C., Kerbosch J. Finding all cliques of an undirected graph. Commun. ACM. 1973;16:575–577. [Google Scholar]
- 34.Connolly M. Analytical molecular surface calculation. J. Appl. Crystallog. 1983;16:548–558. [Google Scholar]
- 35.Schmitt S., Kuhn D., Klebe G. A new method to detect related function among proteins independent of sequence or fold homology. J. Mol. Biol. 2002;323:387–406. doi: 10.1016/s0022-2836(02)00811-2. [DOI] [PubMed] [Google Scholar]
- 36.Hendlich M., Bergner A., Gunther J., Klebe G. Relibase: design and development of a database for comprehensive analysis of protein–ligand interactions. J. Mol. Biol. 2003;326:607–620. doi: 10.1016/s0022-2836(02)01408-0. [DOI] [PubMed] [Google Scholar]
- 37.Garey M.R., Johnson D.S., editors. Computers and Intractability: A Guide to the Theory of NP-Completeness. Freeman and Co; New York: 1979. [Google Scholar]
- 38.Cormen T.H., Leiserson C.E., Rivest R.L. Introduction to Algorithms. MIT Press, McGraw-Hill; New York: 1990. [Google Scholar]
- 39.Connolly M. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–713. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- 40.Duhovny D., Nussinov R., Wolfson H.J. Efficient unbound docking of rigid molecules. In: Guigo R., Gusfield D., editors. Proc. ALGO 02. Algorithms in Bioinformatics. Lecture Notes in Computer Science. vol. 2452. LNCS, Springer; 2002. pp. 185–200. (Proc. ALGO 02. Algorithms in Bioinformatics. Lecture Notes in Computer Science). [Google Scholar]
- 41.Connolly M.L. Measurement of protein surfaces shape by solid angles. J. Mol. Graph. 1986;4:3–6. [Google Scholar]
- 42.Duhovny, D. (2003). Active sites detection and docking. Master's thesis School of Computer Science, Tel-Aviv University.
- 43.Wolfson H.J., Rigoutsos I. Geometric hashing: an overview. IEEE Comput. Sci. Eng. 1997;11:263–278. [Google Scholar]
- 44.Stockman G. Object recognition and localization via pose clustering. J. Comput. Vis. Graphics Image Processing. 1987;40:361–387. [Google Scholar]
- 45.Kaindl K., Steipe B. Metric properties of the root-mean-square deviation of vector sets. Acta Crystallog. sect. A. 1997;53:809. [Google Scholar]
- 46.Kabsch W. A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallog. sect. A. 1978;34:827–828. [Google Scholar]
- 47.Mehlhorn K., Näher S. The LEDA Platform of Combinatorial and Geometric Computing. Cambridge University Press; Cambridge: 1999. [Google Scholar]
- 48.Kuttner Y.Y., Sobolev V., Raskind A., Edelman M. A consensus-binding structure for adenine at the atomic level permits searching for the ligand site in a wide spectrum of adenine-containing complexes. Proteins: Struct. Funct. Genet. 2003;52:400–411. doi: 10.1002/prot.10422. [DOI] [PubMed] [Google Scholar]
- 49.Zarembinski T.I., Hung L.W., Mueller-Dieckmann H.J., Kim K.K., Yokota H., Kim R., Kim S.H. Structure-based assignment of the biochemical function of a hypothetical protein: a test case of structural genomics. Proc. Natl Acad. Sci. USA. 1998;95:15189–15193. doi: 10.1073/pnas.95.26.15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anand K., Ziebuhr J., Wadhwani P., Mesters J., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 52.Gunasekaran K., Hagler A.T., Gierasch L.M. Sequence and structural analysis of cellular retinoic acid-binding proteins reveals a network of conserved hydrophobic interactions. Proteins: Struct. Funct. Genet. 2004 doi: 10.1002/prot.10520. In the press. [DOI] [PubMed] [Google Scholar]
- 53.Banaszak L., Winter N., Xu Z., Bernlohr D., Cowan S., Jones T. Lipid-binding proteins: a family of fatty acid and retinoid transport proteins. Advan. Protein Chem. 1994;230:89–151. doi: 10.1016/s0065-3233(08)60639-7. [DOI] [PubMed] [Google Scholar]
- 54.Murzin A., Brenner S., Hubbard T., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 55.Shatsky M., Nussinov R., Wolfson H.J. MultiProt—a multiple protein structural alignment algorithm. In: Guigo R., Gusfield D., editors. Proc. ALGO 02. Algorithms in Bioinformatics. Lecture Notes in Computer Science. vol. 2452. LNCS, Springer; 2002. pp. 235–250. (Proc. ALGO 02. Algorithms in Bioinformatics. Lecture Notes in Computer Science). [Google Scholar]
- 56.Shatsky M., Nussinov R., Wolfson H.J. A method for simultaneous alignment of multiple protein structures. Proteins: Struct. Funct. Genet. 2004 doi: 10.1002/prot.10628. In the press. [DOI] [PubMed] [Google Scholar]
- 57.Balendiran G.K., Schnutgen F., Scapin G., Borchers T., Xhong N., Lim K. Crystal structure and thermodynamic analysis of human brain fatty acid-binding protein. J. Biol. Chem. 2000;275:27045–27054. doi: 10.1074/jbc.M003001200. [DOI] [PubMed] [Google Scholar]
- 58.Thompson J., Winter N., Terwey D., Bratt J., Banaszak L. The crystal structure of the liver fatty acid-binding protein. a complex with two bound oleates. J. Biol. Chem. 1997;272:7140–7150. doi: 10.1074/jbc.272.11.7140. [DOI] [PubMed] [Google Scholar]
- 59.Kuntz L., Blaney J., Oatley S., Langridge R., Ferrin T. A geometric approach to macromolecule–ligand interactions. J. Mol. Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 60.Brady G., Stouten P. Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des. 2000;14:383–401. doi: 10.1023/a:1008124202956. [DOI] [PubMed] [Google Scholar]
- 61.Laskowski R.A. SURFNET: a program for visualizing molecular surfaces, cavities and intermolecular interactions. J. Mol. Graph. Model. 1995;13:323–330. doi: 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- 62.Liang J., Edelsbrunner H., Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Masuya M., Doi J. Detection and geometric modeling of molecular surfaces and cavities using digital mathematical morphology operations. J. Mol. Graph. Model. 1995;13:331–336. doi: 10.1016/0263-7855(95)00071-2. [DOI] [PubMed] [Google Scholar]
- 64.Meier R., Ackermann F., Hermann G., Posch S., Sagerer G. Segmentation of molecular surfaces based on their convex hull. Proc. Int. Conf. Image Processing. 1995:552–555. [Google Scholar]
- 65.Berman H., Westbrook J., Feng Z., Gilliland G., Bhat T., Weissig H. The Protein Data Bank. Nucl. Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brenner S., Koehl P., Levitt M. The astral compendium for sequence and structure analysis. Nucl. Acids Res. 2000;28:254–256. doi: 10.1093/nar/28.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chandonia J.M., Walker N.S., Lo Conte L., Koehl P., Levitt M., Brenner S. Astral compendium enhancements. Nucl. Acids Res. 2002;30:260–263. doi: 10.1093/nar/30.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chandonia J.M., Hon G., Walker N.S., Lo Conte L., Koehl P., Levitt M., Brenner S. The astral compendium in 2004. Nucl. Acids Res. 2004;32:189–192. doi: 10.1093/nar/gkh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cavarelli J., Prevost G., Bourguet W., Moulinier L., Chevrier B., Delagoutte B. The structure of Staphylococcus aureus epidermolytic toxin a, an atypic serine protease, at 1.7 Å resolution. Structure. 1997;5:813–824. doi: 10.1016/s0969-2126(97)00235-9. [DOI] [PubMed] [Google Scholar]
- 70.Wlodawer A., Li M., Dauter Z., Gustchina A., Uchida K., Oyama H. Carboxyl proteinase from Pseudomonas defines a novel family of subtilisin-like enzymes. Nature Struct. Biol. 2001;8:442–446. doi: 10.1038/87610. [DOI] [PubMed] [Google Scholar]
- 71.Hwang K.Y., Chung J.H., Kim S.H., Han Y.S., Cho Y. Structure-based identification of a novel ntpase from Methanococcus jannaschii. Nature Struct. Biol. 1999;6:691–696. doi: 10.1038/10745. [DOI] [PubMed] [Google Scholar]
- 72.Abele U., Schulz G.E. High-resolution structures of adenylate kinase from yeast ligated with inhibitor ap5a, showing the pathway of phosphoryl transfer. Protein Sci. 1995;4:1262–1271. doi: 10.1002/pro.5560040702. [DOI] [PMC free article] [PubMed] [Google Scholar]