

Abstract
Cell-penetrating peptides (CPPs) are short cationic peptides that have been extensively studied as drug delivery vehicles for proteins, nucleic acids and nanoparticles. However, the formulation of CPP-based therapeutics into different pharmaceutical formulations and their stability in relevant biological environments have not been given the same attention. Here, we show that a newly developed CPP, PepFect 14 (PF14), forms non-covalent nanocomplexes with short interfering RNA (siRNA), which are able to elicit efficient RNA-interference (RNAi) response in different cell-lines. RNAi effect is obtained at low siRNA doses with a unique kinetic profile. Furthermore, the solid dispersion technique is utilized to formulate PF14/siRNA nanocomplexes into solid formulations that are as active as the freshly prepared nanocomplexes in solution. Importantly, the nanocomplexes are stable and active in mediating RNAi response after incubation with simulated gastric fluid (SGF) that is highly acidic. These results demonstrate the activity of PF14 in delivering and protecting siRNA in different pharmaceutical forms and biological environments.
Abbreviations: CPP, Cell penetrating peptide; HPRT1, hypoxanthine phosphoribosyl transferase; ON, Oligonucleotide; PF, PepFect; MR, Molar ratio; NTA, Nanoparticle Tracking Analysis; RNAi, RNA interference; siRNA, Small interfering RNA
Keywords: Cell penetrating peptide, siRNA, Solid formulation, Acid stability, Gastric fluid
Graphical abstract

1. Introduction
Oligonucleotide (ON)-based therapeutics have recently gained much interest as very promising drug candidates. The ability to easily design ONs to specifically repair a pathological genetic defect either on the DNA or RNA level is a remarkable advantage compared to the expensive, time-consuming screening required for small molecule drugs. Additionally, the specific targeting of a particular gene can lead to fewer side effects. Among the therapeutic modalities, which include antisense, antigene and splice-switching ONs; siRNAs gained special interest. What makes the siRNA more appealing than the other approaches is that it cleaves target mRNA in a catalytic manner with the help of the RISC complex; thus, lower doses are required to achieve gene knockdown compared to the conventional antisense approaches [1]. However, as for other ON therapeutics, siRNAs are large hydrophilic molecules that cannot bypass the hydrophobic plasma membrane barrier to reach their target mRNA in the cytoplasm. This is why tremendous research efforts have been put for the development of delivery systems for siRNA. One class of vectors that could be promising in delivering siRNA therapeutics is cell-penetrating peptides (CPPs). CPPs are polybasic and/or amphipathic peptides, usually less than 30 amino acids in length that are able to traverse the plasma membrane even when coupled to large molecules or nanoparticles [2], [3]. The cargo can either be covalently linked or non-covalently complexed with the CPP. The non-covalent complexation strategy has the advantages of being more simple and versatile; thus, several CPPs have been utilized to deliver proteins, peptides, plasmids and ONs via non-covalent complexation [4], [5]. The group of Gilles Divita was the first to show that CPPs can be used to deliver ONs after non-covalent complexation using the MPG peptide [6]. Having net positive charge, MPG has been shown to form nanocomplexes with negatively charged single- and double-stranded ONs, which are efficiently internalized by cells. Since the initial publication, several CPPs have been developed that can form nanocomplexes with various nucleic acids and efficiently mediate their delivery in several in-vitro and in-vivo settings [4], [5], [7]. However, certain chemical modifications of the CPPs were needed to improve the nanoparticle formation capability and enhance membrane interaction upon non-covalent complexation. C-terminal cysteamide modification was shown to be crucial for CPP-mediated siRNA delivery using the MPG, PEP and CADY peptide families by increasing membrane association and stabilizing particle formation by the formation of peptide dimers [8], [9], [10], [11]. Acetylation of these peptides was also required to enhance stability [10]. Addition of hydrophobic moieties to CPPs has been shown to be an efficient mean to increase the efficiency of the nanocomplexes. Cholesteryl modification of polyarginines and MPG-8 was demonstrated to enhance their activity for delivery of siRNA in-vivo [12]. The group of Shiroh Futaki at Kyoto University was the first to demonstrate that stearylation of polyarginines could enhance their transfection efficiency by 100-fold [13]. In recent years, our group applied stearylation and other chemical modifications to the TP10 backbone leading to the development of the PepFect family of peptides (PFs) [14]. Stearylation enables the peptide to form more stable nanocomplexes with nucleic acids and drastically enhances the transfection efficiency compared to the non-stearylated counterparts [15], [16], [17], [18], [19]. Among the PepFect family, PF14 was shown to mediate efficient splice correction activity in various cell lines when complexed with splice-switching ONs. Furthermore, we found that the PF14/ON nanocomplexes can be dried into stable solid formulations via the solid-dispersion technique utilizing conventional pharmaceutical excipients [19]. Here, we show that PF14 is also capable of efficiently delivering siRNA into various cell lines both in the liquid and solid form. Most importantly, we show that PF14 solid formulations are stable in acidic simulated gastric conditions.
2. Materials and methods
2.1. PF14 and siRNAs synthesis
PF14 (Stearyl-AGYLLGKLLOOLAAAALOOLL-NH2) was synthesized and purified as described earlier [19]. Two siRNAs, targeting firefly luciferase and hypoxanthine phosphoribosyl transferase (HPRT1), were synthesized and purified at GE Healthcare (Uppsala, Sweden). Sequences for siRNAs are: Luciferase sense: 5′-ACGCCAAAACAUAAAGAAAG, and antisense: 5′-UUCUUUAUGUUUUUGGCGUCU, HPRT1 sense: 5′-GCCAGACUUUGUUGGAUUUGAAATT and antisense 5′-AAUUUCAAAUCCAACAAAGUCUGGCUU. An unrelated siRNA was synthesized by Sigma (Sweden) and had the sequence: 5′-CCGUGUGAAUCAUUGUCUU sense and 5′-CAGACAAUGAUUCACACGG antisense. All siRNAs were stored as 100 μM aliquots in − 20 °C.
2.2. Cell culture, transfections and luciferase assay
The luciferase stable cell lines, Baby Hamster Kidney cells (BHK-21), human hepatocellular carcinoma (HepG2) and human embryonic kidney (HEK 293) cells were used to test the efficiency of formulated siRNA on down-regulation of luciferase expression. Luciferase-stable BHK21 cells were generated by lipofection of pGL3 luciferase plasmid (Clontech) under selection with Hygromycin (Sigma). Luciferase-stable HEK 293 cells were kindly provided by Professor Paavo Honkakoski (University of Kuopio) and described previously in [20]. Luciferase-stable HepG2 cells were generated by lentiviral transduction under the control of CMV promoter. Human hepatocellular carcinoma cell line (HUH 7) was bought from ATCC (accession number: PTA-8561) and was used to test the knocking down of the endogenous gene HPRT1. All cell lines were grown in DMEM medium with glutamax (Invitrogen, Sweden) and supplemented with 10% fetal bovine serum (FBS) (Invitrogen). Jurkat cells (ATCC) were grown in 10% FBS-RPMI-1640 media, supplemented with 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate, and antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin). Cells were maintained at 37 °C, 5% CO2 in humidified incubators. One day prior transfection, cells were seeded at 5 × 104 cells per well (Jurkat cells were seeded at 2 × 105 per well) in a 24-well plate. Aqueous solutions of PF14/siRNA were added to the cells to give a final a concentration of siRNA ranging between 6 and 100 nM. Transfections were performed in both serum and serum-free conditions, where serum was added to the cells to the final concentration of 10% after 4 h of transfection. The known transfection reagents Lipofectamine 2000 (LF 2000) and Lipofectamine RNAimax® (LF RNAimax) (Invitogen) was used as a control. For ending the experiments, cells were either lysed using lysis buffer (0.1% triton in PBS) for subsequent luciferase and total protein measurements or trypsinized and saved as pellets in − 80 °C for isolation of RNA. For the lysed cells, luciferase activity was assessed using the corresponding substrate (Promega, Sweden) and monitored by GLOMAX® 96 micro-plate luminometer (Promega). Total protein was measured using the Lowry method (BioRad, USA) or the Micro BCA® protein assay kit (Thermo Scientific-Pierce Protein Research Products) according to the manufacturer's protocol.
2.3. Preparation of PF14/siRNA nanocomplexes in solution and in solid formulation
Nanocomplexes were formed by mixing PF14 and siRNA for 45–60 min at room temperature and meanwhile the cell medium was replaced in the wells with fresh serum-free or serum-containing medium (450 ml). Thereafter, complexes were added to each well. When using LF2000 and LF RNAiMAX (Invitrogen, Sweden), the complexes were prepared according to the manufacturer's protocol. For preparation of PF14/siRNA solid formulation, we used mannitol (Duchefa Biochemie, Holland) as a solid dispersion matrix. Mannitol solution was prepared at a concentration of 100 mg/ml in MQ water and added to the aqueous solution of the PF14/siRNA nanocomplexes to give a final concentration of 5%. The mixture was thereafter dried in Savant DNA speed-vac (model DNA 120–230) for 2 h, during which the temperature ranged between 55 and 60 °C. The dried powder was reconstituted in 50 μl water or simulated gastric fluid (SGF) then added to 450 μl of cell medium with or without serum for cell experiments or for further physical characterization.
2.4. RNA isolation and quantitative reverse transcriptase multiplex PCR (q RT-PCR)
Total RNA was isolated from the cell pellets using the RNeasy plus kit (QIAGEN, Sweden). RNA was analyzed using multiplex q RT-PCR to amplify both HPRT1 and GAPDH as an endogenous control and this assay was performed using the Quantifast® Multiplex RT-PCR kit (Qiagen). Sequences of primers and probes for HPRT1 and GAPDH (Sigma) were as follows: HPRT1-fwd: 5′-GAGCTATTGTAATGACCAGTC, HPRT1-rev: 5′-TGACCAAGGAAAGCAAAG, GAPDH-fwd: 5′-CTCCTGTTCGACAGTCAG, GAPDH-rev: 5′-GCCCAATACGACCAAATC, HPRT1 taqman probe: 5′-[JOE]TGACTCCGACCTTCACCTTCC[BHQ1] and GAPDH taqman probe: 5′-[6FAM]TGCCAGTGTCAATTATATCTTCCACAA[BHQ1] where JOE and 6-FAM are two fluorophores having different emission spectra and Black Hole Quencher ( BHQ1) was used as a quencher. Multiplex q RT-PCR reaction setup was done according to the Quantifast® kit protocol where 80 ng of RNA were used for all reactions and the final volume of each reaction was 25 μl. Standard curves were made in all runs using known amount of RNA and serially diluted in order to confirm the efficiency of PCR, which was about 100%. Cycling conditions of PCR were: 20 min at 50 °C for reverse transcription, 5 min at 95 °C for PCR initial activation step and 45 cycles, each of 2 steps: 15 s, 95 °C denaturation and 30 s, 60 °C for annealing/extension. Quantitative PCR was performed using the StepOnePlus® Real time PCR system (Applied Biosystems, Sweden) and the data was analyzed by the ∆∆Ct method using the StepOne® software version 2.2.
2.5. Nanoparticle Tracking Analysis (NTA) and zeta potential
Sizes of nanocomplexes either freshly prepared or as solid formulation were measured using the newly developed method known as Nanoparticle Tracking Analysis (NTA). Fresh PF14/siRNA nanocomplexes were measured both in water and in OptiMEM® (Invitrogen) with 10% FBS. Solid formulations were measured after dissolving in water or simulated gastric fluid (SGF) with or without pepsin and in OptiMEM® with 10% FBS. NTA measurements were performed with a NanoSight LM-10HS (NanoSight, Amesbury, UK) equipped with a sample chamber with a 405 nm laser. 0.5 ml of each sample was injected in the sample chamber with sterile disposable syringes. All measurements were performed at room temperature. NanoSight software was used for capturing and analyzing the data. The samples were measured for 40 s with manual shutter and gain adjustments. The mean size, mode and standard deviation (SD) were calculated together with the cumulative under size values (D50 and D90) for each sample. Concentration of the nanocomplexes in each sample was measured by the machine and a 10 sec video clip file showing the particles moving under Brownian motion was taken for each sample. Zeta potential measurements were carried out using DelsaNano-c (Beckman Coulter) according to the manufacturer's protocols.
2.6. Acid stability assessment
SGF was prepared according to the US pharmacopeia. Briefly, SGF is a solution of 0.2% (w/v) NaCl, 0.32% (w/v) purified pepsin (Sigma) and 0.7% (v/v) conc. HCl. It has a pH of about 1.2. The nanocomplexes in solid formulation were resuspended and incubated in 50 μl of SGF. The nanocomplexes in solution (50 μl) were mixed and incubated with 50 μl of double-strength SGF (0.4% NaCl, 0.64% pepsin and 1.4% HCl) to retain equivalent SGF conditions without dilution. After incubation, the mixtures were added to cells in serum free conditions for 4 h followed by addition of serum to a final concentration of 10%. In control wells, only SGF was added. After 24 h, the cells were lysed and luciferase assay together with the total protein measurements were performed as stated earlier. The down-regulation of luciferase activity was normalized to the expression levels in the control wells treated with SGF.
3. Results
3.1. PF14 mediates efficient RNAi in reporter cell-lines
We tested the ability of PF14/siRNA nanocomplexes to down-regulate luciferase expression in BHK21 cell-line, stably expressing luciferase, both in serum and in serum-free conditions. We screened several peptide/siRNA molar ratios (MRs) with different dose titrations. Transfection utilizing PF14 in serum-free medium resulted in 80% knock-down at MR30 at a dose of 50 nM siRNA, while the other MRs were less efficient (Fig. 1A). In serum-containing medium, PF14/siRNA nanocomplexes at MR40 mediated more than 90% knock-down utilizing 100 nM of siRNA (Fig. 1B). In both serum-free and serum containing media, PF14/siRNA nanocomplexes surpassed the activity of LF2000. Furthermore, we performed dose titration using MR30 in HepG2 cell-line stably expressing luciferase in comparison with LF2000 and LF RNAiMax, which is more optimized for siRNA delivery. PF14 nanocomplexes displayed superior activity in a dose-responsive manner at all the used doses (Fig. 1C). These results demonstrate that PF14 can efficiently deliver siRNA into the cells mediating a profound RNAi effect and gene knock-down at relatively low doses and in a dose-dependent manner.
Fig. 1.

PF14 mediates efficient RNAi in reporter cell-lines. BHK-21 cells and HepG2 (5 × 104) were seeded 24 h prior to experiments into 24-well plates. Cells were treated with PF14/siRNA nanocomplexes at three different concentrations at MRs 20:1, 25:1 and 30:1 for 4 h in serum-free medium followed by addition of serum to final concentration of 10% and incubated additionally for 20 h (A), and at MRs 30:1, 35:1 and 40:1 for 24 h in serum-containing medium (B). (C) For HepG2 cell-line, the cells were treated with PF14/siRNA nanocomplexes at different concentrations at MR30 in serum-containing medium. PF14 complexed with control (unrelated) siRNA was used at the highest siRNA dose and the highest MR in A, B and C. LF 2000® and LF RNAiMax® were used according to the manufacturer's protocol. Cells were lysed in 0.1% Triton X-100 and luciferase activity was measured. RNAi assay results are presented as percent of luciferase expression relative to untreated cells. The values represent the mean of at least three experiments performed in duplicate (mean ± SEM, n = 3).
3.2. PF14/siRNA nanocomplexes induce knock-down of endogenous genes with a unique kinetic profile
In order to learn more about the efficiency and the kinetics of gene down-regulation by the nanocomplexes, we assessed the silencing of an endogenous gene, HPRT1, in HUH7 cells. In dose–response experiments, we found that the concentration at which 50% of HPRT1 mRNA was depleted (EC50) was 12 nM and 8 nM for the serum-containing and serum-free conditions respectively (Fig. 2A). Additionally, we ran kinetics experiments using the dose of 50 nM, where the down-regulation upon transfection was recorded after 2 h and followed up to 4 days. PF14/siRNA nanocomplexes mediated extremely fast effect, since the depletion of HPRT1 mRNA reached 70% after only 2 h. This effect was significantly higher than the effect mediated by the well-known transfection reagent LF RNAimax®. Moreover, the nanocomplexes efficiently maintained silencing of HPRT1 mRNA for up to 4 days, which was the last time point tested. In line with this, PF14/siRNA nanocomplexes also mediated efficient knock-down of HPRT1 mRNA in the Jurkat cells, which is regarded as a hard-to-transfect cell-line (Supplementary Fig. 1).
Fig. 2.

PF14 induces knock-down of an endogenous gene with a unique kinetic profile. A. Dose–response curves for the down-regulation of HPRT1 by PF14/siRNA nanocomplexes in HUH7 cells in serum-containing medium (solid line) and in serum-free medium (dotted line). The concentration at which 50% of HPRT1 mRNA was depleted (EC50) was 12 nM and 8 nM for the serum and serum-free conditions respectively. B. Comparison between the efficiency of one selected dose (50 nM) of siRNA transfected by PF14 or by LF RNAimax® or naked siRNA without any transfection reagent. Control siRNA of unrelated sequence at the same concentration transfected with PF14 was also included. C and D: Kinetics of the HPRT1 down-regulation using a 50 nM dose of siRNA transfected with PF14 at serum-containing and serum-free conditions and with LF RNAimax®. C. represents the kinetics up to 24 h while D. shows the time-course study as monitored up to 4 days. Significant differences were detected in the % of HPRT1 mRNA remaining after 2 h using PF14 (in serum free) and LF RNAimax® (P < 0.001) and after 4 h (P < 0.01). Significant difference was seen as well between treatment with PF14 (in serum) and LF RNAimax® after 2 h (P < 0.05).
3.3. PF14 nanocomplexes can be formulated in solid form using the solid dispersion technique
Similar to the procedure used for formulating PF14 nanocomplexes with splice-switching ONs, we formulated PF14/siRNA into solid formulations via the solid dispersion technique. Upon screening several excipients with different concentrations, mannitol at the final concentration of 5% was the most optimal (data not shown). In this method, pre-formed nanocomplexes are mixed with certain volume of mannitol solution and subsequently the mixture is dried using speed-vac at 50–60 °C. Interestingly, after resuspension and addition to the cells, mannitol-based solid formulations demonstrated RNAi activity similar to the freshly prepared nanocomplexes (Fig. 3 ).
Fig. 3.
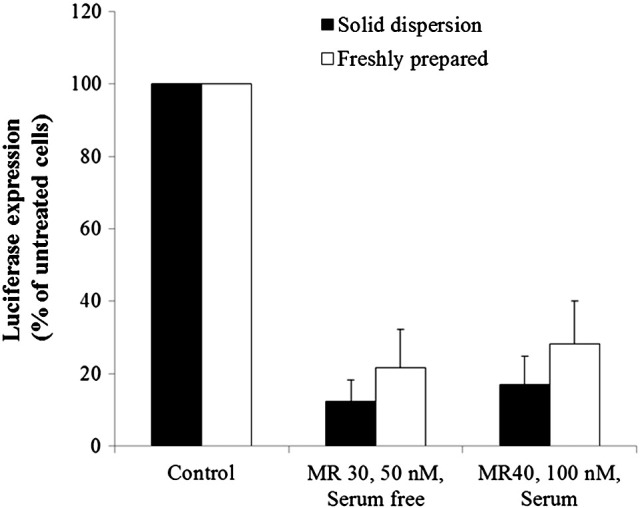
PF14/siRNA nanocomplexes can be formulated in solid form using the solid dispersion technique. Mannitol solution was prepared at final concentration of 100 mg/ml in MQ water. PF14/siRNA nanocomplexes were prepared in 25 μl final volume and mixed with the mannitol solution to the final concentrations of 5% mannitol in the reaction mixture. The final volume of the mixture was 50 μl. The mixture was then dried in speed-vac for 2 h, during which the temperature ranged between 55 and 60 °C. Before transfection, the dried powder was reconstituted in 50 μl of MQ water and added to the wells. HEK 293 luciferase-stable cells (5 × 104) were treated with reconstituted nanocomplexes in serum containing or serum-free medium. Cells were then lysed in 0.1% Triton X-100 and luciferase activity was measured. RNAi assay results are presented as percent of luciferase expression relative to untreated cells (control).
3.4. PF14/siRNA nanocomplexes are stable upon drying and resuspension
To ensure the presence of nanocomplexes and their stability after the relatively harsh solid dispersion procedure, we performed detailed particle size analysis using the NTA technique. NTA is a new system for sizing particles that was first commercialized in 2006. This technique combines laser light scattering microscopy with a CCD camera, which enables the visualization and recording of nanoparticles in solution [21]. The NTA software is then able to identify and track individual nanoparticles moving under Brownian motion and relates the movement to a particle size according to a formula derived from the Stokes-Einstein equation. This particle-by-particle methodology provides high-resolution particle size distributions compared to traditional light scattering and other ensemble techniques [21]. Additionally, it measures concentration and validates all data with video files of the particles moving under Brownian motion. The freshly prepared formulation showed a particle size mean of 125 nm when measured in water and it was insignificantly different from that measured in serum-containing medium. This demonstrates the tolerability of the particles to serum. When the solid formulation of PF14/siRNA was measured, it showed a mean particle size of 119 nm, which again was insignificantly different from the same formulation when measured in serum-containing medium. This similarity demonstrates the stability of the particles after the solid dispersion procedure (Fig. 4 ). Additionally, we measured the zeta potential of the nanocomplexes in the solid formulation to determine the surface properties that are crucial for activity. We found that, when resuspended in water, the nanocomplexes possess a positive zeta potential (22.74 ± 0.49 mV). However, when resuspended in the biorelevant OptiMEM® medium, the zeta potential is reversed to negative values; -12.98 ± 0.63 mV and − 8.54 ± 0.32 mV in the absence and presence of serum respectively. This trend follows the same pattern demonstrated before by PF14 nanocomplexes with splice-switching ONs [22] and highlights the effect of medium on the physical properties and hence the uptake mechanism of the nanocomplexes.
Fig. 4.

PF14 forms definite nanocomplexes with siRNA that are stable upon drying and resuspension. Nanocomplexes, both freshly prepared and the in solid formulation, were studied using the Nanoparticle Tracking Analysis (NTA) system. A. The freshly prepared nanocomplexes measured in water. B. The freshly prepared nanocomplexes measured in OptiMEM® with 10% FBS, C. the solid formulation measured in water, D. the solid formulation measured in OptiMEM® with 10% FBS. The graphs on the middle panels show the size distribution of the particles with the particle size plotted on the x-axis and the particles concentration (E6/ml) plotted on the y-axis. The graphs on the rightmost panels are 3D plots (size vs. intensity vs. concentration), while the leftmost panels show the corresponding NTA video frames. In middle panels for samples B and D, the yellow curves represent the size distribution measured for the vehicle alone which is OptiMEM® with 10% FBS. D50: the diameter where half of the population lies below, D90: the diameter where 90% of the population lies below.
3.5. PF14/siRNA solid formulation is stable in simulated gastric acidic conditions
The most widely used application of the solid formulation is for oral delivery. For therapeutics to be bioavailable after oral administration they have to survive the drastic acidic and enzymatic conditions in the stomach. Therefore, we aimed to test the stability of PF14/siRNA nanocomplexes in SGF, which was prepared according to the US pharmacopeia with and without pepsin. Interestingly, we found that the nanocomplexes retained most of their RNAi activity even when subjected to such drastic conditions for 30 min (Fig. 5A). The nanocomplexes were stable in SGF without pepsin for up to 2 h; however, in the presence of pepsin the RNAi effect decreased in a time dependent manner (Supplementary Fig. 2). This indicates that PF14 protects siRNA from acidic conditions for prolonged periods of time; however, further formulation approaches might be needed to enhance pepsin-resistance. We have confirmed the integrity of the nanocomplexes after incubation with SGF via NTA particle size analysis (Fig. 5B), which showed that they do not dissociate and remain intact in these conditions. The particle size increased slightly, which might also contribute to the modest decrease of activity.
Fig. 5.

Solid PF14/siRNA formulation is stable in simulated gastric acidic conditions.
A. PF14/siRNA solid formulations were prepared at MR30 using 50 nM siRNA, and then the solid dispersion method was applied using mannitol at a concentration of 5%. Freshly prepared nanocomplexes and solid formulations were then incubated with SGF with or without pepsin or with water for 30 min before addition to the cells. After incubation, HEK 293 luciferase stable cells (5 × 104) were treated with the nanocomplexes for 4 h in serum-free medium followed by addition of serum to final concentration of 10% and incubated additionally for 20 h. Cells were then lysed in 0.1% Triton X-100 and luciferase activity was measured. RNAi assay results are presented as percent of luciferase expression relative to control wells where only SGF was added. B. Solid formulations were reconstituted in water then incubated for 30 min in SGF with or without pepsin. Mean size of the particles was 149 nm in case of SGF and 183 nm in SGF with pepsin.
4. Discussion
ON-based therapeutics represent the most direct way to translate genomic data into therapeutics by targeting specific DNA or RNA sequences. However, the bulky and hydrophilic nature of ONs hinders their accessibility to intracellular targets across the plasma membrane. For these reasons, many delivery technologies were developed to carry ONs across the cellular membrane barrier. Much of the research focus was specifically toward developing siRNA delivery vectors because of the favorable pharmacodynamic profile of siRNA over other ONs. This is due to the catalytic nature of its gene silencing effect. Cationic lipid formulations have been extensively utilized as siRNA vehicles and widely used as transfection reagents in-vitro; e.g. Lipofectamine®. However, only a limited number have been successfully used in-vivo due to their poor colloidal stability and toxicological concerns [23], [24]. The new generation of lipid-based siRNA vectors like SNALPs [25] and Lipidoids [26] has probably overcome some of these limitations; however, the complex multicomponent nature of such systems could limit them from being widely used. Alternatively, complex-forming CPPs represent a single-step system for efficient delivery of siRNAs. We have previously shown that PepFect6 (PF6), a chemically modified CPP, can efficiently deliver siRNA to in-vitro in several hard-to-transfect cell-lines and in different organs following systemic delivery in mice without any associated toxicity [18]. PF6 and PF14 both belong to the PepFect chemically-modified CPP family developed in our lab. They are based on the TP10 peptide with added chemical modifications to enhance complex formation, stability, uptake and endosomal release [14], [17], [8]. In the case of PF14, it is N-terminally stearylated and contains several ornithine residues. Stearylation of CPPs enhances the complex formation capacity of the CPP with nucleic acids and can enhance endosomal escape [16], [17]. On the other hand, ornithines, being nonstandard amino acids, would render the peptide more resistant to serum proteases [19]. PF14 proved to be very efficient in the delivery of single-stranded ONs for splice correction in solution and in solid formulation [19]. In this study, we wanted to test the activity of PF14 in delivering siRNAs and whether the solid formulation approach would also be successfully applicable for this type of ONs as well. Furthermore, we wanted to extend the process of pharmaceutical development of such nanocomplexes by testing their stability in simulated gastric conditions; a model for oral administration.
Initially, we tested the ability of PF14 to mediate RNAi response in reporter cell-lines. Luciferase-expressing BHK21 and HepG2 cells were used. Interestingly, PF14/siRNA nanocomplexes mediated efficient RNAi knock-down of luciferase expression in a dose-dependent manner at different MRs. More than 80% down-regulation of luciferase expression was obtained at the best MRs (Fig. 1). The knock-down was achieved at relatively low doses of siRNA and the effect was maintained in the presence of serum. Next, we used the nanocomplexes to down-regulate an endogenous mRNA and quantified the response using q RT-PCR. PF14/siRNA nanocomplexes demonstrated a very low EC50 in both serum and serum-free media with more than 70% knock-down in the first 2 h, significantly surpassing LF RNAimax (Fig. 2). Surprisingly, the RNAi effect was maintained up to 4 days confirming the sustainable activity of the nanocomplexes. Endogenous gene knock-down was also demonstrated in the Jurkat suspension cell-line, which is known to be hard-to-transfect (Supplementary Fig. 1).
Having proven the efficiency in different cell-lines in the presence or absence of serum, we wanted to test the activity and stability of the nanocomplexes if formulated into a solid form, which can have several pharmaceutical benefits. Solid pharmaceutical forms are the most widely used drug formulations due to patient convenience and high stability profiles. They are usually used for oral and respiratory administration as tablets, capsules and powders for inhalation. Furthermore, they are used in powders for injection to enhance the stability of intravenous formulations and facilitate transport and storage. Active and stable solid formulations for siRNA could add new opportunities to the applicability of the RNAi technology and would be a proof-of-concept that delivery technologies can transform siRNA from being a powerful laboratory tool into a viable drug product. Utilizing the solid formulation, siRNAs against various pathogenic targets can be designed and utilized via the oral or pulmonary pathways. For example, siRNAs against hepatitis C virus (HCV), hypercholesterolemia and irritable bowel diseases can be utilized via the oral route and siRNAs against cystic fibrosis, influenza and severe acute respiratory syndrome (SARS) infections can be utilized via the pulmonary route. Here, we applied the technology that we used for PF14 formulation with single-stranded ONs; the solid dispersion technique. It is based on mixing the nanocomplexes with a solution of a soluble excipient, which will be the solid dispersant, then drying out the solvent; water in this case. In this way the nanocomplexes will uniformly distribute over the solid matrix. To evaporate water we dried the mixtures at 50–60 °C under vacuum and centrifugation utilizing speed-vac. Although this procedure is fairly harsh due to the relatively high temperature and pressure that the nanocomplexes are subjected to, it is much cheaper to scale-up if such formulations are to be mass produced compared to the lyophilization techniques. From the different excipients we tested (data not shown); mannitol at the concentration of 5% was the most active and mediated an RNAi effect that is not significantly different from the freshly-prepared nanocomplexes (Fig. 3). These results agree with our earlier data for PF14 nanocomplexes with single-stranded ONs and confirm that PF14 indeed forms stable nanocomplexes that can withstand formulation conditions.
To better visualize the nanocomplexes and study their physicochemical properties before and after formulation we ran NTA measurements, which possess better resolution than the conventional DLS techniques (Fig. 4). The mean particle size was not significantly different between the freshly prepared formulation and after solid dispersion either in water or serum-containing medium (Fig. 4). This demonstrates that the nanocomplexes remain intact in different media and can withstand the drying and dispersion conditions. However, the surface charge (zeta potential) was dependent on the dispersion medium. When dispersed in water, the nanocomplexes in solid formulation were positively charged, but upon dispersion in a biorelevant medium (OptiMEM®) with or without serum, the nanocomplexes became negatively charged. We have recently shown similar behavior for PF14 nanocomplexes with single-stranded ONs and that such nanocomplexes were taken-up into cells via the class A scavenger receptors [22]. The net negative charge of PF14/siRNA nanocomplexes implies that they recruit the same pathway.
Finally, we wanted to test the stability of the nanocomplexes in SGF and its suitability for oral administration. Interestingly, the nanocomplexes maintained the RNAi response after incubation in SGF for up to 30 min. even in the presence of pepsin (Fig. 5). The acid resistivity was maintained for a period up to 2 h, however, pepsin decreased the activity over longer periods of incubation (Supplementary Fig. 2.). Remarkably, this demonstrates that the nanocomplexes protect the siRNA form acidic conditions although it is natural phospho-diester RNA without any further chemical modifications.
5. Conclusions
In conclusion, we found that PF14 can be used as an efficient delivery vector for siRNA in various cell-lines. Furthermore, PF14/siRNA nanocomplexes can be formulated into a solid dosage form that is stable in simulated gastric conditions. These results open the door for testing oral CPP-based siRNA therapeutics in animal models, toward developing convenient ON-based drug products as a new class of therapeutics.
Acknowledgments
We would like to thank all the contributing funding agencies. This work was supported by Swedish Research Council (VR-NT); by SSF (Sweden-Japan); by Center for Biomembrane Research, Stockholm; by the EU through the European Regional Development Fund through the Center of Excellence in Chemical Biology, Estonia; by the DoRa Program of The European Social Fund; by Archimedes Foundation; by the Torsten and Ragnar Söderberg Foundation; and by the Egyptian Ministry of Higher Education (to E.M.Z.). We would like to also thank Professor Mamoun Muhammed (Royal Institute of Technology — KTH) for permission to use the DelsaNano-c instrument for zeta potential measurements.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2012.06.006.
Appendix A. Supplementary data
Supplementary materials
References
- 1.Whitehead K.A., Langer R., Anderson D.G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ezzat K., El Andaloussi S., Abdo R., Langel Ü. Peptide-based matrices as drug delivery vehicles. Curr. Pharm. Des. 2010;16:1167–1178. doi: 10.2174/138161210790963832. [DOI] [PubMed] [Google Scholar]
- 3.Verdurmen W.P., Brock R. Biological responses towards cationic peptides and drug carriers. Trends Pharmacol. Sci. 2011;32:116–124. doi: 10.1016/j.tips.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Deshayes S., Morris M., Heitz F., Divita G. Delivery of proteins and nucleic acids using a non-covalent peptide-based strategy. Adv. Drug Deliv. Rev. 2008;60:537–547. doi: 10.1016/j.addr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Mäe M., El Andaloussi S., Lehto T., Langel Ü. Chemically modified cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2009;6:1195–1205. doi: 10.1517/17425240903213688. [DOI] [PubMed] [Google Scholar]
- 6.Morris M.C., Vidal P., Chaloin L., Heitz F., Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997;25:2730–2736. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heitz F., Morris M.C., Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simeoni F., Morris M.C., Heitz F., Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weller K., Lauber S., Lerch M., Renaud A., Merkle H.P., Zerbe O. Biophysical and biological studies of end-group-modified derivatives of Pep-1. Biochemistry. 2005;44:15799–15811. doi: 10.1021/bi051535d. [DOI] [PubMed] [Google Scholar]
- 10.Gros E., Deshayes S., Morris M.C., Aldrian-Herrada G., Depollier J., Heitz F., et al. A non-covalent peptide-based strategy for protein and peptide nucleic acid transduction. Biochim. Biophys. Acta. 2006;1758:384–393. doi: 10.1016/j.bbamem.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Crombez L., Aldrian-Herrada G., Konate K., Nguyen Q.N., McMaster G.K., Brasseur R., et al. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009;17:95–103. doi: 10.1038/mt.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim W.J., Christensen L.V., Jo S., Yockman J.W., Jeong J.H., Kim Y.H., et al. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol. Ther. 2006;14:343–350. doi: 10.1016/j.ymthe.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Futaki S., Ohashi W., Suzuki T., Niwa M., Tanaka S., Ueda K., et al. Stearylated arginine-rich peptides: a new class of transfection systems. Bioconjug. Chem. 2001;12:1005–1011. doi: 10.1021/bc015508l. [DOI] [PubMed] [Google Scholar]
- 14.Lehto T., Ezzat K., Langel Ü. Peptide nanoparticles for oligonucleotide delivery. Prog. Mol. Biol. Transl. Sci. 2011;104:397–426. doi: 10.1016/B978-0-12-416020-0.00010-3. [DOI] [PubMed] [Google Scholar]
- 15.Mäe M., El Andaloussi S., Lundin P., Oskolkov N., Johansson H.J., Guterstam P., et al. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J. Control. Release. 2009;134:221–227. doi: 10.1016/j.jconrel.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 16.Lehto T., Abes R., Oskolkov N., Suhorutsenko J., Copolovici D.M., Mäger I., et al. Delivery of nucleic acids with a stearylated (RxR)4 peptide using a non-covalent co-incubation strategy. J. Control. Release. 2010;141:42–51. doi: 10.1016/j.jconrel.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 17.Lehto T., Simonson O.E., Mäger I., Ezzat K., Sork H., Copolovici D.M., et al. A peptide-based vector for efficient gene transfer in vitro and in vivo. Mol. Ther. 2011;19:1457–1467. doi: 10.1038/mt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Andaloussi S., Lehto T., Mäger I., Rosenthal-Aizman K., Oprea I.I., Simonson O.E., et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011;39:3972–3987. doi: 10.1093/nar/gkq1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ezzat K., El Andaloussi S., Zaghloul E.M., Lehto T., Lindberg S., Moreno P.M., et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011;39:5284–5298. doi: 10.1093/nar/gkr072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honkakoski P., Jaaskelainen I., Kortelahti M., Urtti A. A novel drug-regulated gene expression system based on the nuclear receptor constitutive androstane receptor (CAR) Pharm. Res. 2001;18:146–150. doi: 10.1023/a:1011068015301. [DOI] [PubMed] [Google Scholar]
- 21.Filipe V., Hawe A., Jiskoot W. Critical evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates. Pharm. Res. 2010;27:796–810. doi: 10.1007/s11095-010-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ezzat K., Helmfors H., Tudoran O., Juks C., Lindberg S., Padari K., et al. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012;3:1172–1180. doi: 10.1096/fj.11-191536. [DOI] [PubMed] [Google Scholar]
- 23.Jeong J.H., Park T.G., Kim S.H. Self-assembled and nanostructured siRNA delivery systems. Pharm. Res. 2011;28:2072–2085. doi: 10.1007/s11095-011-0412-y. [DOI] [PubMed] [Google Scholar]
- 24.Behlke M.A. Progress towards in vivo use of siRNAs. Mol. Ther. 2006;13:644–670. doi: 10.1016/j.ymthe.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmermann T.S., Lee A.C., Akinc A., Bramlage B., Bumcrot D., Fedoruk M.N., et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 26.Love K.T., Mahon K.P., Levins C.G., Whitehead K.A., Querbes W., Dorkin J.R., et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. U. S. A. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials