

Highlights
-
•
Bats and birds are reservoirs of zoonotic viruses.
-
•
Their unique immune systems allow them to harbor a large variety of viruses.
-
•
Coronaviruses and influenza viruses are examples of interspecies transmission of viruses.
Keywords: coronavirus, influenza, RNA virus, virus evolution, emerging infectious disease
Abstract
As exemplified by coronaviruses and influenza viruses, bats and birds are natural reservoirs for providing viral genes during evolution of new virus species and viruses for interspecies transmission. These warm-blooded vertebrates display high species biodiversity, roosting and migratory behavior, and a unique adaptive immune system, which are favorable characteristics for asymptomatic shedding, dissemination, and mixing of different viruses for the generation of novel mutant, recombinant, or reassortant RNA viruses. The increased intrusion of humans into wildlife habitats and overcrowding of different wildlife species in wet markets and farms have also facilitated the interspecies transmission between different animal species.
Emergence of new viruses
Both mammalian and avian coronaviruses (CoV) have diverse host ranges. Phylogenetic dating of RNA-dependent RNA polymerase (RdRp) sequence divergence suggested that the most recent common ancestor (MRCA) of mammalian CoVs appeared around 7000–8000 years ago, whereas the MRCA of avian CoVs dates back to 10 000 years ago (Figure 1 ). These results are likely underestimations because they could not account for additional sequence diversity from undiscovered viruses. Nonetheless, the present estimates roughly coincide with the dispersal of the human population around the world about 50 000–100 000 years ago and greatly increased in the last 10 000 years during the first historic transition. During this transition, humans began various farming activities, such as forest clearing for agriculture and animal herding, leading to a significant shift in the ecology and population dynamics of viruses owing to the intrusion of wildlife habitats and intensive mixing of different animal hosts. Finally, the expansion of human travel and trading directly led to the spread of viruses to distant and isolated places. The migration of early humans over long distances was very limited and effectively a unidirectional ‘rare’ event. Eventually, improvements in transportation technology enabled distant trade missions in early Mesopotamia around 5000 years ago and possibly earlier in other regions [1]. These periodic yet infrequent visits might have enabled transmission of various disease agents to previously segregated non-immune populations, leading to a serial founder effect associated with a boom and bust cycle. However, further technological improvements, especially the development of aviation and the flight industry in the last century, have allowed this type of travel to occur at such high frequencies that multiple segregated host populations effectively have become a single large population. These changes coincided with increased breeding between different host populations (for both humans and domestic animals), which can significantly impact the genetic and immunological make-up of the host populations. When these occurrences are considered in the context of the high mutation rate of RNA viruses, they become a driving force for speciation and subsequent evolution of new viruses. Although phylogenetic analysis and dating of individual influenza genes and lineages have been reported previously, large sequence divergence and frequent reassortment led to difficulties in precisely dating and phylogenetic positioning the common ancestor of modern influenza viruses [2].
Figure 1.
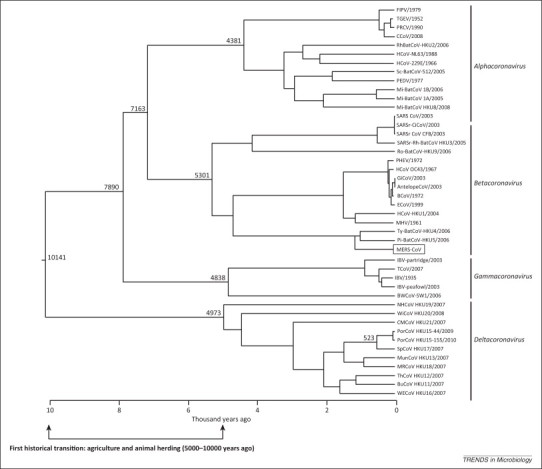
The divergence of coronaviruses into Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus is estimated to have occurred approximately 5000 years ago. This tree was generated by analyzing RNA-dependent RNA polymerase (RdRp) genes under the relaxed-clock model with an uncorrelated log-normal distribution in Bayesian evolutionary analysis sampling trees (BEAST) software. Values at branch points represent the estimated timing of divergence events in numbers of years before the present. Adapted from [45].
About 70% of the emerging pathogens infecting humans originate from animals. Most of these major outbreaks were due to RNA viruses as a result of their higher mutation rates compared with other types of microbes and their capability for unique genetic change, either by genetic recombination in positive-sense RNA viruses or genetic reassortment in RNA viruses with segmented genomes. Those with greatest impact on humans include the severe acute respiratory syndrome coronavirus (SARS-CoV), influenza virus, and HIV. Little was known about CoVs until the 2003 SARS epidemic, which caused 774 deaths among 8098 cases in over 30 countries [3]. The natural reservoir of the more ancestral bat SARS-CoV is the Chinese horseshoe bat (Rhinolophus sinicus), which may have transmitted the virus to other game mammals including Himalayan palm civets (Paguma larvata), raccoon dogs (Nyctereutes procyonoides), and Chinese ferret badgers (Melogale moschata) in wildlife markets in South China [4]. This finding sparked intense hunting for novel CoVs in humans and different animal species, especially in bats. The latest emerging novel human CoV, originally named human coronavirus EMC/2012 and later renamed Middle East respiratory syndrome coronavirus (MERS-CoV), which has caused 30 deaths among 54 cases in the Middle East, Europe, and Africa, is also phylogenetically closely related to the Tylonycteris bat CoV HKU4 (Ty-BatCoV-HKU4) and Pipistrellus bat CoV HKU5 (Pi-BatCoV-HKU5) discovered in bats in Hong Kong 5, 6, 7, 8 (http://www.who.int/csr/don/2013_06_05/en/index.html). The importance of birds as natural reservoirs of emerging influenza viruses is underscored by the persistent threat of avian influenza H5N1 since 1997 and the emergence of H7N9 in 2013 9, 10, 11, 12. The role of bats in the emergence of novel influenza viruses is less clear although influenza A H17 and H3N2 viruses have been discovered in Sturnira lilium recently and in Nyctalus noctula bats in Kazakhstan in 1970, respectively. We review the importance of bats and birds in the genesis of new virus mutants and interspecies jumping using CoVs and influenza viruses as examples.
Bats as natural reservoirs for emerging viruses
A number of unique ecological, biological, immunological, and genetic features make bats a favorable animal reservoir for the emergence of novel viruses. Bats have remarkable species diversity, with over 1240 species (20% of the nearly 5000 known species within Mammalia and only second to rodents) [9]. Because viruses are obligatory intracellular microbes, it is assumed that viruses have multiple independent evolutionary origins not separable from co-evolution of their hosts. This high biodiversity of bats makes them an important source of new viruses for interspecies jumping.
Bats are widely distributed in all continents except the polar regions and a few oceanic islands. Their roosting or hibernation environment ranges from natural habitats such as caves, rock crevices, bird nests, and tree cavities, to man-made structures like mines, tombs, buildings, and bridges, which bring them closer to humans and companion animals or livestock. Their unique ability among mammals to fly long distances (up to 2000 km) to locate suitable habitats also allows them to acquire or disseminate viruses [9]. Their habit of roosting in large colonies ranging from 10–200 000 bats and relative longevity of up to 35 years also provide abundant mating opportunities and thus exchange of viral genetic material and further viral spread to other species. Exposures to infected urine and aerosols generated during defecation have been suggested as possible routes of intraspecies and interspecies transmission of viruses from bats [9]. Bats may also transmit viruses to human and other animals via bites and scratches as in the case of rabies. Consumption and handling of undercooked bat meat is still practiced in China, Guam, and some parts of Asia.
Besides ecological and biological traits, bats also possess special immunological attributes that enhance their ability to serve as gene pools for emerging viruses (Table 1 ). Asymptomatic or persistent viral shedding with little evidence of pathology in bats is well reported [13]. One possible mechanism is the very early control of viral replication by their innate immune response involving the early recognition by pattern recognition receptors (PRRs) and interferons (IFNs) followed by partial control by the adaptive immune response [14]. Similar to other mammalian species, some bats possess the two major families of virus-sensing PRRs, namely the Toll-like receptors (TLRs), which are membrane-bound PRRs that detect single-stranded RNA (ssRNA) or double-stranded RNA (dsRNA), and retinoic acid inducible gene I (RIG-I)-like helicases (RLHs), which are cytosolic PRRs that detect dsRNA. Bat TLRs have been described in many fruit bat species and are highly similar to other mammalian TLRs 15, 16. In addition, cytoplasmic RLHs including RIG-I, melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) have been found in Pteropus alecto and their homologs in Myotis davidii [16]. These PRRs allow bats to recognize a similar range of pathogens as other mammalian species and in turn these pathogens are controlled by IFNs. Type I IFNs have been described in numerous fruit bat species [17], and type III IFNs have been found in Myotis lucifugus, P. alecto, and Pteropus vampyrus [18]. Notably, the type I IFNω family of genes are expanded in some bat species with up to a dozen members, and the type III IFN receptors are more widely distributed in tissues of P. alecto than in humans and mice [19]. Bats may also demonstrate a delayed or differential IFN response during in vitro infection by different kinds of viruses 17, 18.
Table 1.
Innate and adaptive immune systems in bats and birds
| Protein | Function | Bats | Birds | |
|---|---|---|---|---|
| Innate immunity | RIG-I | Cytosolic PRR; detects dsRNA | Present and functional in the black flying fox (Pteropus alecto; AEW46678); homolog also present in David's Myotis (Myotis davidii; ELK34300) [16] | Present and functional in mallard duck (ACA61272) and present in goose (ADV58759), but absent in chicken [33] |
| MDA5 | Cytosolic PRR; detects dsRNA | Present and functional in black flying fox (P. alecto; AEW46679); homolog also present in David's Myotis (M. davidii; ELK28159) | Present in mallard duck (GU936632), goose (AGC51036), and chicken (ADD83027) 33, 86 | |
| LGP2 | Cytosolic PRR; detects dsRNA | Present and functional in black flying fox (P. alecto); homolog also present in David's Myotis (M. davidii) | Present in chicken [34] | |
| TLR7 | Membrane-bound PRR; detects ssRNA | Present in black flying fox (P. alecto; ADO01609), Leschenault's rousette (Rousettus leschenaultia; BAH02556), and David's Myotis (M. davidii; ELK30184) 15, 85 | Present in chicken (ACR26250) and mallard duck (ABK51522) 35, 87, 88 | |
| TLR3 | Membrane-bound PRR; detects dsRNA | Present in black flying fox (P. alecto; ADO01605), Leschenault's rousette (R. leschenaultia; BAH02555), and David's Myotis (M. davidii; ELK23529) 15, 85 | Present in chicken (ABG79022) and muscovy duck (AFK29094) [89] | |
| TLR8 | Membrane-bound PRR; detects ssRNA | Present in black flying fox (P. alecto; ELK17709) and David's Myotis (M. davidii; ELK30183) [85] | Disrupted in chicken and duck genome [90] | |
| IFN-α | Type I IFN; inducible cytokine that regulates the antiviral response | Variable number of members present in multiple bat species, including black flying fox (P. alecto; α1: ELK15818, α5: ELK06973) and David's Myotis (M. davidii; α3: ELK29335); may be absent in other Myotis sp. [17] | At least 13 putative IFN-α genes identified in the chicken genome [91]; also identified in the mallard duck (ADU60335) and Gerylag goose (AFU54612), but exact number of members is not known | |
| IFN-β | Type I IFN; inducible cytokine that regulates the antiviral response | Present in multiple bat species, including black flying fox (P. alecto; ELK06976) and David's Myotis (M. davidii; ELK29334) | Present in chicken (NP_001020007) and mallard duck | |
| IFN-ω | Type I IFN; inducible cytokine that regulates the antiviral response | Multiple members present in bats, including black flying fox (P. alecto; ELK06971, ELK06975, ELK08131, ELK15817, and ELK15819) and David's Myotis (M. davidii; ELK26494, ELK26493, and ELK26495) | Absent | |
| IFN-λ | Type III IFN; inducible cytokine that regulates the antiviral response | Two members identified in black flying fox (P. alecto; λ1: AEF33950, λ2: AEF33949); also present in the closely related Malaysian flying fox (Pteropus vampyrus) [18] | At least one member present in chicken (ABU82742) [92] | |
| Adaptive immunity | Ig VH | Variable region of immunoglobulin heavy chain; contributes to diversity of antibody repertoire | At least 23 genes classified into five families were identified in the black flying fox (P. alecto; GQ427153:GQ427172) [18]; diverse genes in the VH3 family repertoire also reported for other bat species [93] | Single functional VH gene in chicken; 58 ψVH pseudogenes located upstream in IgH locus also contribute to repertoire diversity through gene conversion; similar organization in ducks and other birds [37] |
| Ig DH | Diversity region of Ig heavy chain; contributes to diversity of antibody repertoire | Diverse DH elements in the little brown bat (Myotis lucifugus) [93] | Approximately 15 DH elements in chicken | |
| Ig JH | Joining region of Ig heavy chain | At least 13 elements identified in the little brown bat (M. lucifugus) [93] | Single JH element in chicken | |
| Ig VL | Variable region of immunoglobulin light chain; contributes to diversity of antibody repertoire | λ and κ loci present in black flying fox (P. alecto) [94], representative of megabats; λ locus present in microbats, but κ locus is absent [94] | Single functional VL gene in single (λ) IgL locus in chicken; approximately 25 ψVL pseudogenes contribute through gene conversion [95] | |
| Ig JL | Joining region of Ig light chain | (Not studied) | Single JL element in chicken | |
| Ig Cδ | Constant region of IgD | Absent in black flying fox (P. alecto) and probably other megabats; present in microbats, such as the little brown bat (Myotis lucifugus; ADI96045, ADI96044) [96] | Absent |
However, important differences between the adaptive immune response of bats and other mammals have been observed. In the humoral response, bats have an antibody repertoire as diverse as those of humans and mice. At least 23 genes for immunoglobulin (Ig) VH and the λ and κ loci of Ig VL have been identified in P. alecto, and diverse Ig DH and Ig JH elements and the λ locus of Ig VL have been found in M. lucifugus and other microbats (Table 1). However, the amino acid sequence composition of the antigen-binding site (CDR3 region) of the expressed VH region in bats is different from those of humans and mice with a higher proportion of arginine and alanine residues, and lower proportion of tyrosine residues, and thus possibly a lower poly-reactivity [20]. Such antibodies have lower avidity and form a weaker association with antigens. Furthermore, the primary antibody response may be delayed from reaching a peak and the secondary antibody response may also be slow or delayed in bats. In adverse environmental situations such as prolonged exposure to low temperature at 8 °C simulating the state of hibernation, bats may temporarily fail to mount an antibody response but resume this ability 1 week after transferring to 24 °C. Importantly, bats may clear viral infection even in the absence of neutralizing antibody by other unknown mechanisms [14]. Bats appeared asymptomatic during bat–SARS-CoV infection despite a high viral load in their anal swabs with absent or low serum neutralizing antibody although their body weights were generally lower than the uninfected bats 4, 21. As for a cell-mediated immune response, those of bats are generally slower to peak after stimulation by T-cell mitogens. Environmental changes such as roost ecology and physiological alterations such as pregnancy also affect cell-mediated immunity of bats [14]. Further studies should be performed to ascertain if these quantitative and qualitative differences in the adaptive immune response of bats may account for their unique interaction with viruses leading to asymptomatic infection and viral persistence.
The discovery of influenza virus in bats challenges the notion that aquatic birds are the only source of all influenza A gene segments 9, 10. The recently discovered H17 influenza virus from Guatemala bats is unique in that all eight gene segments appear to be distinct from any known influenza A gene segments [10]. The hemagglutinin (HA) of this virus has unique structural features and exhibits receptor binding and fusogenic activities that are distinct from its counterparts in other influenza viruses [22]. Likewise, the neuraminidase (NA) of this virus, called N10, is phylogenetically distinct from the NAs of all influenza A and B viruses. The N10 is also structurally distinct with no enzymatic activity [23]. Additional surveillance is necessary to understand the ecology of influenza viruses in bats.
Waterfowl and shorebirds as a source of influenza virus genes
The high biodiversity of birds (over 10 000 species) allows avian influenza viruses to evolve in different host environments. Birds living near wetlands or aquatic environments, especially in the orders Anseriformes (aquatic waterfowls) and Charadriiformes (shorebirds), are considered to be the sources or gene pool for all influenza viruses 24, 25. Waterfowl feed on submerged water plants in marsh water that are readily contaminated by virus from excreta of infected waterfowls or other small animals. Birds are frequently infected by influenza A viruses. Re-infection during the same season with the same virus and coinfection with different viruses can occur [26]. Despite frequent infections, waterfowl usually remain asymptomatic even when infected by avian influenza viruses highly pathogenic in chicken [27]. Asymptomatic infections in ducks and geese often preceded devastating outbreaks in chickens. Influenza viruses persisting in asymptomatic waterfowls allow accumulation of mutations that may facilitate interspecies transmission [28]. Moreover, wild waterfowls often aggregate with domestic ducks and geese during feeding and roosting and therefore allow efficient mixing of different influenza viruses to form new subtypes. In the backyard farm setting, domestic ducks and geese are often mixed with chickens and other domestic poultries allowing interspecies transmission, which may eventually transmit into humans. Finally, migratory waterfowls can disseminate novel influenza viruses to very distant locations [9].
Similar to bats, birds have a unique immune system that is best studied in ducks (Table 1). Genes related to antiviral immunity, including major histocompatibility complex type I (MHC-I), interferon-induced protein with tetratricopeptide repeats 5 (IFIT5), oligoadenylate synthetase-like (OASL), CC chemokine ligand (CCL)19, CCL21, MDA5, TLR7, and IFN-α, are expressed in duck lungs as early as 1 day after H5N1 virus inoculation 29, 30, 31. Proinflammatory cytokines were lower in duck than in chicken embryonic fibroblasts infected with H5N1 virus [32]. Major differences in the immune system between ducks and chickens may account for the differential susceptibility to highly pathogenic avian influenza. Whereas chicken cells naturally lack RIG-I or RIG-I activity, duck RIG-I has a similar structure and function to its human ortholog. It was shown that transfection of duck RIG-I into a chicken embryonic fibroblast cell line augments the IFN response and suppresses viral replication [33]. Chicken cells apparently rely upon MDA5 and LGP2 to sense influenza virus infection [34]. Furthermore, the duck TLR7 differs from chicken in the ligand binding LRR domains [35]. When infected by influenza virus or stimulated with TLR7 agonists, chickens and ducks differentially activate the production of MDA5, TLR7, and IFN-α [31]. Notably, TLR8 and IFNω, which are present in bats, are either disrupted or not found in birds.
The adaptive immune system is particularly unique in ducks. Influenza virus elicits a short-lived antibody response in ducks [36]. Unlike mammals, ducks have three types of antibody, namely IgA, IgM, and IgY (human IgG counterpart). The predominant antibody response in infected ducks is the truncated form of IgY, which lacks the Fc region and does not exhibit hemagglutination inhibition, complement fixation, opsonization, and antibody-dependent cellular cytotoxicity. IgA is not produced until ducks are 2 weeks old and is expressed late after an infection [37]. T lymphocytes, although present, are not fully functional in young birds [38]. Although ducks have five MHC-I genes for antigen presentation, only two are expressed [39], which limits the repertoire of presentable peptides. These immunological features may allow waterfowl to shed large amount of viruses for a prolonged period [40]. In ducks infected by low pathogenic avian influenza virus, RIG-I, CCL19, and CCL21 are only slightly upregulated in intestinal tissues 29, 33. These unique features in the immune system of birds, especially in ducks, allow them to be the reservoir for influenza viruses.
Interspecies transmission of CoVs
Before the discovery of SARS-CoV, CoVs were considered as causative agents of respiratory tract infections, gastroenteritis, hepatitis, and encephalomyelitis in birds and mammals. The first two human CoVs, HCoV-229E and HCoV-OC43, primarily caused self-limiting upper respiratory tract infections such as the common cold [41]. The emergence of SARS-CoV represented a new era in the history of CoV research. The significant global medical, economical, and social impact of the 2003 outbreak regenerated research interest on CoVs. One important finding was the identification of bats as the natural animal reservoir and civet and other mammals as the intermediate amplifying hosts of SARS-CoV [4]. These discoveries revolutionized the ‘hunting’ strategy for novel CoVs and redefined the classification of CoVs based on their updated phylogeny and the critical role of bats in inter- and intra-species transmissions of CoVs. With the new wealth of knowledge on CoV phylogeny generated after the SARS epidemic, the Coronavirus Study Group of the International Committee for Taxonomy of Viruses changed the classification into four genera, Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus to replace the traditional antigenic groups of 1, 2, and 3 (Figure 2A) (http://www.ictvonline.org/virusTaxonomy.asp?bhcp=1). This remarkable diversity of species and genotypes, as well as the emergence of novel species capable of adapting to new hosts and ecological niches, is the result of environmental, viral, and host factors that favor interspecies transmission.
Figure 2.
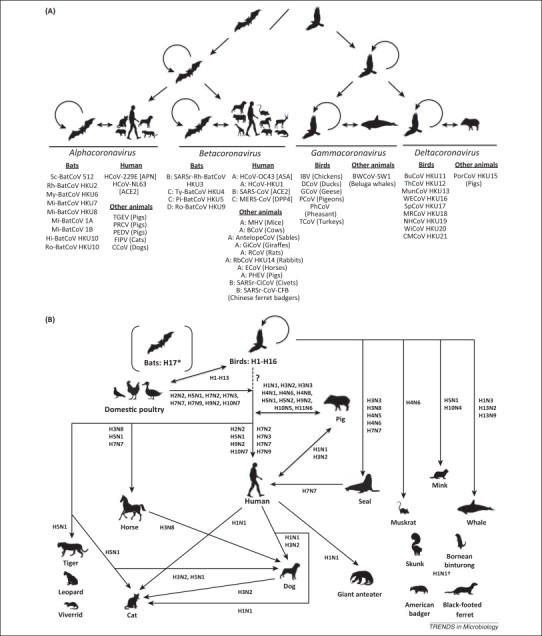
Bats and birds as probable gene sources for the evolution of (A) coronaviruses and (B) influenza viruses, based on epidemiological, virological, and phylogenetic evidence. ‘A’, ‘B’, ‘C’, and ‘D’ represent groups A, B, C, and D in Betacoronavirus. * denotes the undetermined role of bats as reservoirs for emerging influenza viruses. † denotes the undetermined source of A(H1N1)pdm09 virus found in American badger, black-footed ferret, Bornean binturong, and skunk.? with dotted line denotes the unproven direct wild bird-to-human transmission of influenza viruses. Figure 2A is adapted from [45]. Abbreviations: [], host receptor utilized by coronavirus; (), animal host; ACE2, angiotensin converting enzyme 2; AntlopeCoV, sable antelope coronavirus; APN, aminopeptidase N; ASA, 9-O-acetylated sialic acid; BCoV, bovine coronavirus; BuCoV HKU11, bulbul coronavirus HKU11; BWCoV-SW1, beluga whale coronavirus SW1; CCoV, canine coronavirus; CMCoV HKU21, common moorhen coronavirus HKU21; DCoV, duck coronavirus; ECoV, equine coronavirus; FIPV, feline infectious peritonitis virus; GCoV, goose coronavirus; GiCoV, giraffe coronavirus; HCoV-229E, human coronavirus 229E; MERS-CoV, Middle East respiratory syndrome coronavirus; HCoV-HKU1, human coronavirus HKU1; HCoV-NL63, human coronavirus NL63; HCoV-OC43, human coronavirus OC43; Hi-BatCoV HKU10, Hipposideros bat coronavirus HKU10; IBV, infectious bronchitis virus; MHV, murine hepatitis virus; Mi-BatCoV 1A, Miniopterus bat coronavirus 1A; Mi-BatCoV 1B, Miniopterus bat coronavirus 1B; Mi-BatCoV HKU7, Miniopterus bat coronavirus HKU7; Mi-BatCoV HKU8, Miniopterus bat coronavirus HKU8; MRCoV HKU18, magpie robin coronavirus HKU18; MunCoV HKU13, munia coronavirus HKU13; My-BatCoV HKU6, Myotis bat coronavirus HKU6; NHCoV HKU19, night heron coronavirus HKU19; PCoV, pigeon coronavirus; PEDV, porcine epidemic diarrhea virus; PhCoV, Pheasant coronavirus; PHEV, porcine hemagglutinating encephalomyelitis virus; Pi-BatCoV HKU5, Pipistrellus bat coronavirus HKU5; PorCoV HKU15, porcine coronavirus HKU15; PRCV, porcine respiratory coronavirus; RCoV, rat coronavirus; RbCoV HKU14, rabbit coronavirus HKU14; Rh-BatCoV HKU2, Rhinolophus bat coronavirus HKU2; Ro-BatCoV HKU9, Rousettus bat coronavirus HKU9; Ro-BatCoV HKU10, Rousettus bat coronavirus HKU10; Sc-BatCoV 512, Scotophilus bat coronavirus 512; TEGV, transmissible gastroenteritis virus; SARS-CoV, severe acute respiratory syndrome coronavirus; SARSr-CiCoV, SARS-related civet coronavirus; SARSr-CoV-CFB, SARS-related Chinese ferret badger coronavirus; SARSr-Rh-BatCoV HKU3, SARS-related Rhinolophus bat coronavirus HKU3; TCoV, turkey coronavirus; SpCoV HKU17, sparrow coronavirus HKU17; ThCoV HKU12, thrush coronavirus HKU12; Ty-BatCoV HKU4, Tylonycteris bat coronavirus HKU4; WECoV HKU16, white-eye coronavirus HKU16; WiCoV HKU20, wigeon coronavirus HKU20.
Environmental factors
Environmental factors have played a key role in the emergence of SARS-CoV. The open door policy and economic boom in China after 1978 is naturally associated with an increasing population and mobility as well as an increasing cultural demand for food and game animals in South China. The density of both animals and humans in South China provided an ideal incubator for brewing novel viruses that could adapt to new hosts. Phylogenetic analysis and epidemiological data revealed that SARS-CoV has likely emerged from its natural animal reservoir, the bats, to civets and other game food mammals during their caging in the overcrowded and unhygienic wildlife markets of South China before further crossing the species barrier to humans [4]. This chronological sequence is compatible with the deletion of a 29-nucleotide segment in Orf8 as a consistent sequence feature of human SARS-CoV isolates [3]. Interestingly, recent data have shown that although bat isolates are the ancestral host to SARS-CoVs, the isolates from small carnivores such as civet and raccoon dog all cluster within the human SARS-CoV sequences with the small carnivore sequences being terminal branches of the human intermediate. Thus, a direct bat-to-human transmission followed by bidirectional transmission between intermediate mammals and humans has also been suggested as the chronological dynamics in the emergence of SARS-CoV [42]. It should be cautioned that this interpretation of the phylogenetic tree may be biased by the limited sampling of SARS-CoV in animal reservoirs relative to human patients. Without further data from animal surveillance, the plausible scenario of the civet being the intermediate animal host of SARS-CoV in the early human cases could not be ruled out entirely. Nevertheless, both models support the crucial role of bats in these interspecies transmission events. This key finding of bats and other mammals as the natural and amplifying hosts of SARS-CoV led to the closure of most wildlife markets in South China followed by subsidence of the epidemic. Ensuing animal surveillance of bats and other animal species in this region led to the discovery of many more novel animal CoVs. Indeed, bat CoVs have been proposed to be the gene source of all Alphacoronavirus and Betacoronavirus, whereas avian CoVs are considered as the gene source of all Gammacoronavirus and Deltacoronavirus 43, 44, 45, 46.
Notably, only a small minority of the estimated 1240 bat and 10 000 avian species has been tested for CoVs so far. It is likely that many more CoVs could be discovered in bats, birds, and other animal hosts such as rodents, bovines, canines, felines, swine, and equines. Bats and birds provide a rich pool of virus species for interspecies exchange of genetic fragments and interspecies transmission. Phylogenetic analysis provided evidence for interspecies transmission events including the emergence of HCoV-OC43 from bovines to human, porcine CoV HKU15 likely from sparrow, canine coronavirus (CCoV) II and feline coronavirus (FCoV) II from recombination between early CCoV-I and FCoV-I, and transmissible gastroenteritis virus (TGEV) from CCoV-II [47]. Among bats, illustrative scenarios include the sharing of a common ancestor between Ghana bat CoV group I and HCoV-229E [48], and the recent transmission of bat-CoV-HKU10 from Leschenault's rousettes to Pomona leaf-nosed bats [49]. The recent emergence of the novel 2012 MERS-CoV may represent the latest example of interspecies transmission from bats to other animals and to humans as evidenced by its close phylogenetic relatedness with the Ty-batCoV-HKU4 and Pi-batCoV-HKU5, and other novel betacoronaviruses recently found in different bat species in Mexico, Ghana, and Europe 6, 7, 50, 51.
Viral and host factors
Besides the importance of abundant animal reservoirs as gene sources and favorable environmental conditions, three major viral factors are instrumental in allowing CoVs to cross species barriers [41]. First, their unusually high estimated mutation rate associated with RNA replication of nearly 2 × 10−6 is exceptional even among RNA viruses. The infidelity of RdRp of CoVs makes them especially plastic. Second, their unique random template switching mediated by a ‘copy-choice’ mechanism during RNA replication in which discontinuous RNA transcription and the presence of full length and subgenomic negative-strand RNAs lead to frequent strand switching and recombination between viral genomes and subgenomic replication complexes. This allows a high frequency of homologous RNA recombination to as high as 25% during mixed infection. Phylogenetic evidence of natural recombination has been found in both HCoV-HKU1 and HCoV-OC43 and animal CoVs such as bat-SARS-CoV and bat-CoV-HKU9. Third, CoVs have the largest genomes (26.4–31.7 kb) of all known RNA viruses, allowing extra plasticity in accommodating and modifying genes without too much loss of fitness. This is evidenced by the numerous unique open reading frames and protein functions encoded towards the 3′ end of the genome.
The importance of these viral factors in allowing CoVs to cross interspecies barriers are well illustrated in SARS-CoV. Comparative analyses between multiple isolates of human and civet SARS-CoVs showed that the virus has undergone rapid adaptation in different hosts especially with mutations at the receptor binding domain (RBD) of the spike (S) protein, which showed evidence of positive selection during inter- and intraspecies transmission events [42]. The RBD of the S protein of CoVs interacts with the cellular receptor and is intensely selected by the host antibody response. In SARS-CoV, the S1 fragment between amino acids 318 and 510 is the RBD and primarily binds to the human angiotensin converting enzyme 2 (ACE2) receptor as well as co-receptors such as C-type lectins like DC- and/or L-Sign for virus docking and entry [47]. Furthermore, the SARS-CoV RBD is capable of recognizing the ACE2 receptors of different animal species including bat, civet, mouse, and raccoon dog, thus allowing interspecies transmission of the virus. The RBD of the human and civet isolates differ by merely six amino acid residues of which four are located in the receptor-binding motif, which is the loop region of the RBD that contains 13 out of 14 residues that interface with the ACE2 receptor. The K479N and S487T mutations in the RBD of civet isolates increased the binding affinity to the human ACE2 receptor and were crucial in the virus’ adaptation to human [47]. The RBD of the emerging novel MERS-CoV has been characterized recently [52]. Sequence alignment of the RBD in SARS-CoV S protein with that of the corresponding region between amino acids 377 and 662 in MERS-CoV S protein showed that both fragments S1 and S2 share low homology with 14% identity and 38% similarity only, whereas the core domain consisting of β-sheets and α-helices have higher homology with 23% identity and 61% similarity. In contrast to SARS-CoV, the human ACE2 receptor is neither necessary nor sufficient for the replication of MERS-CoV [53]. Interestingly, HCoV-NL63 also binds to ACE2 but with a different part of S and causes only mild respiratory illnesses in most patients [54]. Other human CoV receptors, such as aminopeptidase N, a human and mammalian cell surface metalloprotease with multiple physiological functions that is expressed on intestinal, lung, and kidney epithelial cells, in the case of HCoV-229E, and 9-O-acetylated sialic acid, which is widespread in mammalian tissues in the case of HCoV-OC43, may account for the successful adaptation of these CoVs in humans after interspecies transmission from their ancestral animal hosts [55]. Given the wide tissue and species tropism of MERS-CoV, a receptor which is widely distributed in different human tissues and animal species is likely [56]. Indeed, dipeptidyl peptidase 4 (DPP4), a multifunctional type-II transmembrane glycoprotein of 766-amino-acid-long and with exopeptidase activity, which is expressed primarily on non-ciliated bronchial epithelium, the epithelial cells in kidney, small intestine, liver, prostate, and activated leukocytes, has been shown to be a functional receptor for MERS-CoV [57]. Despite the importance of the host cell receptor in determining whether interspecies transmission of an emerging virus can occur, many other cellular factors such as restriction factors, the innate immune response, and metabolic characteristics of the host cell that may abort or facilitate the viral replication have yet to be defined.
Interspecies transmission of influenza viruses
Most influenza viruses are host specific. H5N1 influenza viruses failed to be transmitted from experimentally infected poultry to pigs [58]. Naturally occurring avian influenza viruses in mammals are rare. Even different avian species have different susceptibilities to the same influenza virus. However, interspecies jumping occurs frequently (Figure 2B). Reassortant pandemic influenza viruses often have significant mortality and morbidity. The 1957 H2N2 and the 1968 H3N2 pandemic influenza A viruses are reassortments between circulating avian and human influenza viruses, whereas the A(H1N1)pdm09 virus is a reassortant between the circulating swine influenza viruses [59]. For the 1918 pandemic H1N1 virus, some investigators suggested that the 1918 pandemic influenza virus directly jumped from an avian source to human, whereas others postulated that the virus circulated in swine before human. All gene segments from these pandemic influenza viruses have been ultimately traced to an avian source.
Direct transmission of avian influenza viruses from birds and poultry to humans have been documented for H5N1, H7N2, H7N3, H7N7, H7N9, H9N2, H10N7 11, 12. Swine influenza viruses including the swine-origin influenza H3N2 [(H3N2)v] [60] and triple reassortant swine H1N1 [61] have been transmitted directly from pigs to humans. Based on phylogenetic and epidemiological analysis, many influenza viruses have also been directly transmitted between avian species, between avian and non-human mammals, and between different mammalian species (Figure 2B). Outbreaks due to interspecies jumping include the avian H5N1 outbreaks in zoos involving different mammalian species [62] and in cats [63], equine H3N8 and avian H3N2 outbreaks in dogs [64], and the avian H3N8 outbreak in seals [65].
Environmental factors
Although wild birds have been regarded as the source of all influenza viruses, most human and mammalian cases of avian influenza were acquired from domestic poultry. The 1997 Hong Kong outbreak of avian influenza A H5N1 was related to a high concentration of different live poultry species in wet markets and farms with high number of visitors and farms with poor biosecurity measures. Poultry-to-human transmission of the emerging avian H7N9 virus has also been confirmed [12]. Efforts have been made to prevent the transmission of avian influenza to humans [11]. Biosecurity measures have been adopted in farms to prevent the introduction of avian influenza viruses into poultry farms. Poultry has been vaccinated to prevent infection. Ducks, geese, and quails have been segregated from chickens in farms to prevent the spread of avian influenza viruses from these asymptomatic birds to chickens. Surveillance of poultry and wild birds allows early detection of avian influenza viruses. Poultry depopulation is used to control H5N1 or H7N9 outbreaks detected in farms or markets.
Viral and host factors
For an influenza virus to jump species, the viruses must be able to evade the host immunity of the donor and recipient species, and to infect, replicate, spread, and persist within the recipient species. For example, one of the major reasons for the successful emergence of A(H1N1)pdm09 virus in humans was the lack of pre-existing humoral immunity in the younger population and it was postulated that prior seasonal influenza H1N1 virus could not persist in humans due to the crossreactive antibody against the HA stalk that was elicited by the A(H1N1)pdm09 virus 66, 67. Viral mutations that facilitate interspecies jumping of the virus can be achieved by antigenic drift, antigenic shift, or non-homologous recombination. Reassortments require exchange of gene segments between different influenza viruses. This is especially common for pigs, in which classical swine H1N1 virus, human-like H3N2 virus, and avian-like H1N1 virus co-circulate. Prior to the 2009 pandemic influenza, it was believed that new human pandemic strains required a new combination of HA and NA subtypes arising from avian and swine species. However, in the 2009 pandemic, an antigenic shift from a human H1N1 subtype to a swine H1N1 subtype was sufficient. Non-homologous recombination, which is the exchange of RNA between two different RNA segments, has been proposed to be responsible for the mutant HA during the H7N3 avian influenza virus outbreak in British Columbia [68]. Unlike CoVs, homologous recombination has not been demonstrated for influenza viruses [69]. In addition to mutations in the HA and NA, many mutations in the internal genes such as polymerase B2 (PB2), polymerase A-X (PA-X), and nuclear export protein (NEP) are also important for interspecies transmission (Table 2 ).
Table 2.
Genetic changes of internal genes of influenza virus affecting interspecies transmission
| Protein | Effect of viral gene mutations that facilitate interspecies transmission | Host protein implicated in host restriction | Mutations responsible for interspecies jumping |
|---|---|---|---|
| PB2 | Improve polymerase activity and RNA replication in recipient species [11] Binding to the host protein importin necessary for the transport of PB2 into the nucleus [97] |
Importin [97] | 591K: avian-to-human adaptation of H5N1 virus [98] 627K: avian-to-human adaptation for H5N1and H7N9 viruses 11, 12 701N: avian-to-human adaptation for H5N1 [11], H7N9 [12], and H7N7 viruses [99]; avian to seal adaptation for H3N8 virus [65] Ability to bind importin-α7: avian-to-human adaptation [97] |
| PA | Improve polymerase activity and RNA replication in recipient species [100] | N/A | 552S: avian-to-human adaptation [100] |
| PB1-F2 | Improve polymerase activity and RNA replication in recipient species [101] | N/A | Truncated form of PB1-F2: avian-to-human and avian-to-swine adaptation [102] |
| PA-X | Changes in inflammatory, apoptotic, and T-lymphocyte signaling pathway [103] | N/A | Truncated form of PA-X: avian to human, swine, and canine and equine adaptation [103] |
| PA-N182 | Avian viruses may lack this protein [104]; however, its function is currently not known | N/A | Non-AUG codon at position 182: found mainly in avian and swine H9N2 virus [104] |
| Nucleoprotein | Binding to the host protein importin is necessary for the transport of viral nucleoprotein into the nucleus [97] Differential susceptibility to the host antiviral protein MxA [105] |
Importin [97] MxA [105] |
Nucleoprotein from 2009 pandemic H1N1 virus: avian-to-human adaptation [105] |
| NS1 | Binding to TRIM25, NS1 blocks RIG-I ubiquitination and type I IFN production in recipient species [106] | TRIM25 [106] | NS1 from 2009 pandemic H1N1 virus: avian-to-human adaptation [106] |
| NS2 (or NEP) | Improve polymerase activity and RNA replication in recipient species [107] | N/A | M16I: avian-to-human adaptation for H5N1 virus [107] |
Abbreviations: MxA = myxovirus resistance gene A; TRIM25 = tripartite motif 25.
Receptor binding specificity and receptor distribution in different animals
HA initiates infection by binding to the host cell surface sialic acid (SA) receptor. Out of 17 HA subtypes of influenza, only six have caused human infections, and only H1–H3 have caused pandemics [11]. Receptor binding specificity is determined by the amino acid sequence of HA. Several studies have shown that avian and human influenza viruses preferentially bind α2,3-SA that is expressed abundantly in the avian gastrointestinal tract and α2,6-SA that is found in the respiratory tract in humans, respectively. Animal hosts that carry both α2,3-SA and α2,6-SA abundantly in the upper respiratory tract, such as pigs [70] and quails [71], are considered to be susceptible to both avian and human influenza viruses and therefore potential mixing vessels for reassortant viruses. Ducks have only α2,3-SA in the intestine, but both α2,3-SA and α2,6-SA in the respiratory tract [72]. This is consistent with the observation that H5N1 virus shedding is higher in the trachea than in the cloaca [40], and why human viruses do not replicate in duck intestines.
Many mutations in the HA of H5N1 and H7N9 viruses have been associated with its change of affinity from α2,3-SA to α2,6-SA 11, 12. Currently, Egypt has the highest incidence of H5N1 in the world, and many viral polymorphisms enriched in the Egyptian strains enable them to bind better to α2,6-SA [11]. However, studies in recent years have challenged this simple model. First, it is now clear that other relatively pathogenic human influenza viruses, such as the 1918 and 2009 pandemic influenza viruses and some human H5N1 viruses, can bind strongly to both α2,3-SA and α2,6-SA [73]. The D222G mutation of the A(H1N1)pdm09 virus, which causes more severe disease, is associated with an increased affinity for α2,3-SA without loss of affinity for α2,6-SA [74]. The increased pathogenicity in humans can be explained by the enhanced affinity of these viruses to the α2,3-SA that lines the non-ciliated cuboidal bronchiolar cells and alveolar type II cells in human [59]. By contrast, strong affinity for α2,6-SA allows for efficient intraspecies transmission among humans. Second, the binding affinity can be affected by the length and topology of the receptors. Viruses that bind well to cone-like short α2,6-SA may not bind well to the umbrella-like long α2,6-SA [75]. Third, there are discrepant findings in the receptor distribution in different animals. Traditionally, it is widely accepted that both α2,3-SA and α2,6-SA are abundantly expressed in the trachea of pigs [76]. However, some recent studies have found that α2,6-SA is the predominant receptor type in the upper respiratory tract of pigs [77]. There are also controversies regarding the predominant receptor in the respiratory tract of chickens [78].
Glycosylation of the HA can mask the antigenic epitope, allowing escape from immune surveillance [79]. Furthermore, glycosylation may also help binding of HA to the host SA. A H7N7 virus causing a fatal human infection has an A125T substitution (H3 numbering), which generates a glycosylation site and is associated with increased binding to α2,3-SA [80]. Another potential limiting factor for interspecies transmission may be the specificity of host proteases required for proteolytic cleavage of HA0 into HA1 and HA2. For example, a seal influenza A virus H7N7 adapted and became pathogenic in chickens after acquiring basic amino acids at the HA cleavage site [81].
NA in interspecies transmission
It has been postulated that a shorter NA stalk length promotes the adaptation of influenza viruses from waterfowl to terrestrial poultry and humans as evident by the shortened NA stalk in the H5N1 and H7N9 epidemic viruses. In a poultry farm, short-stalk NA was found in only 2% of the reads in ducks using deep sequencing, whereas it was present in 100% of reads in turkeys [82]. Severe disease was observed in chickens infected with a recombinant H7N1 virus with short-stalk NA than those infected with the H7N1 virus with long-stalk NA. However, virus with long-stalk NA showed higher shedding in duck intestine than those with short-stalk NA [83]. A study of H9N2 virus has shown that short-stalk NA has higher NA activity than long-stalk NA [84]. The mechanism of the NA stalk length and interspecies transmission require further study.
Concluding remarks
Both bats and birds are warm-blooded sustained flying vertebrates with a very high metabolic rate, biodiversity, and also roosting or migratory behavior [85]. The development and functioning of their immune systems are far less studied than that of humans or rodents. Apart from restrictions posed by host immune response and host cell receptors, many other cellular factors governing the viral life cycles of emerging viral mutants are unknown. The recent discovery of a unique influenza virus in bats may open up a new dimension in the genesis of influenza virus beyond birds while many more CoVs remain to be discovered in bats and birds. These flying vertebrates are the reservoir of many viral families from which pathogenic human and mammalian viruses are derived. It will be of great interest to elucidate the structural and mechanistic basis for the differences between these animals and humans in their innate and adaptive immune systems using state-of-the-art technologies including next-generation sequencing, gene targeting, and reverse genetics (Box 1 ).
Box 1. Outstanding questions.
-
•
How do the innate and adaptive immune systems of bats and birds structurally and functionally differ from those of humans?
-
•
Why can bats and birds harbor a large variety of viruses without developing severe diseases?
Acknowledgments
This work was supported in part by the Providence Foundation Ltd, in memory of the late Lui Hac Minh, by Cheer Master Investments Ltd (Mr Yeung Sai Hong), and by the Health and Medical Research Fund of the Food and Health Bureau, Hong Kong Special Administrative Region, China.
References
- 1.Edens C. Dynamics of trade in the ancient mesopotamian “World System”. Am. Anthropol. New Ser. 1992;94:118–139. [Google Scholar]
- 2.Xu J. Evolutionary history and phylodynamics of influenza A and B neuraminidase (NA) genes inferred from large-scale sequence analyses. PLoS ONE. 2012;7:e38665. doi: 10.1371/journal.pone.0038665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng V.C. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007;20:660–694. doi: 10.1128/CMR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau S.K. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U.S.A. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan J.F. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 2012;65:477–489. doi: 10.1016/j.jinf.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo P.C. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 2007;81:1574–1585. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woo P.C. Genetic relatedness of the novel human group C betacoronavirus to Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5. Emerg. Microbes Infect. 2012;1:e35. doi: 10.1038/emi.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan J.F. The emerging novel Middle East Respiratory Syndrome Coronavirus: the “knowns” and “unknowns”. J. Formos. Med. Assoc. 2013;112:372–381. doi: 10.1016/j.jfma.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong S. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol. 2007;17:67–91. doi: 10.1002/rmv.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong S. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. U.S.A. 2012;109:4269–4274. doi: 10.1073/pnas.1116200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.To K.K. Avian influenza A H5N1 virus: a continuous threat to humans. Emerg. Microbes Infect. 2012;1:e25. doi: 10.1038/emi.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet. 2013;381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calisher C.H. Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker M.L. Antiviral immune responses of bats: a review. Zoonoses Public Health. 2013;60:104–116. doi: 10.1111/j.1863-2378.2012.01528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iha K. Molecular cloning and expression analysis of bat toll-like receptors 3, 7 and 9. J. Vet. Med. Sci. 2010;72:217–220. doi: 10.1292/jvms.09-0050. [DOI] [PubMed] [Google Scholar]
- 16.Cowled C. Molecular characterisation of RIG-I-like helicases in the black flying fox, Pteropus alecto. Dev. Comp. Immunol. 2012;36:657–664. doi: 10.1016/j.dci.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kepler T.B. Chiropteran types I and II interferon genes inferred from genome sequencing traces by a statistical gene-family assembler. BMC Genomics. 2010;11:444. doi: 10.1186/1471-2164-11-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou P. Type III IFNs in pteropid bats: differential expression patterns provide evidence for distinct roles in antiviral immunity. J. Immunol. 2011;186:3138–3147. doi: 10.4049/jimmunol.1003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou P. Type III IFN receptor expression and functional characterisation in the pteropid bat, Pteropus alecto. PLoS ONE. 2011;6:e25385. doi: 10.1371/journal.pone.0025385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker M.L. Immunoglobulin heavy chain diversity in Pteropid bats: evidence for a diverse and highly specific antigen binding repertoire. Immunogenetics. 2010;62:173–184. doi: 10.1007/s00251-010-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau S.K. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 2010;84:2808–2819. doi: 10.1128/JVI.02219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu X. Hemagglutinin homologue from H17N10 bat influenza virus exhibits divergent receptor-binding and pH-dependent fusion activities. Proc. Natl. Acad. Sci. U.S.A. 2013;110:1458–1463. doi: 10.1073/pnas.1218509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu X. Crystal structures of two subtype N10 neuraminidase-like proteins from bat influenza A viruses reveal a diverged putative active site. Proc. Natl. Acad. Sci. U.S.A. 2012;109:18903–18908. doi: 10.1073/pnas.1212579109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsen B. Global patterns of influenza a virus in wild birds. Science. 2006;312:384–388. doi: 10.1126/science.1122438. [DOI] [PubMed] [Google Scholar]
- 25.Webster R.G. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharp G.B. Coinfection of wild ducks by influenza A viruses: distribution patterns and biological significance. J. Virol. 1997;71:6128–6135. doi: 10.1128/jvi.71.8.6128-6135.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guan Y. A model to control the epidemic of H5N1 influenza at the source. BMC Infect. Dis. 2007;7:132. doi: 10.1186/1471-2334-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hatchette T.F. Influenza A viruses in feral Canadian ducks: extensive reassortment in nature. J. Gen. Virol. 2004;85:2327–2337. doi: 10.1099/vir.0.79878-0. [DOI] [PubMed] [Google Scholar]
- 29.Vanderven H.A. Avian influenza rapidly induces antiviral genes in duck lung and intestine. Mol. Immunol. 2012;51:316–324. doi: 10.1016/j.molimm.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleming-Canepa X. Expression of duck CCL19 and CCL21 and CCR7 receptor in lymphoid and influenza-infected tissues. Mol. Immunol. 2011;48:1950–1957. doi: 10.1016/j.molimm.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cornelissen J.B. Differential innate responses of chickens and ducks to low-pathogenic avian influenza. Avian Pathol. 2012;41:519–529. doi: 10.1080/03079457.2012.732691. [DOI] [PubMed] [Google Scholar]
- 32.Liang Q.L. Immune-related gene expression in response to H5N1 avian influenza virus infection in chicken and duck embryonic fibroblasts. Mol. Immunol. 2011;48:924–930. doi: 10.1016/j.molimm.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Barber M.R. Association of RIG-I with innate immunity of ducks to influenza. Proc. Natl. Acad. Sci. U.S.A. 2010;107:5913–5918. doi: 10.1073/pnas.1001755107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liniger M. Chicken cells sense influenza A virus infection through MDA5 and CARDIF signaling involving LGP2. J. Virol. 2012;86:705–717. doi: 10.1128/JVI.00742-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacDonald M.R. The duck toll like receptor 7: genomic organization, expression and function. Mol. Immunol. 2008;45:2055–2061. doi: 10.1016/j.molimm.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 36.Globig A. Consecutive natural influenza A virus infections in sentinel mallards in the evident absence of subtype-specific hemagglutination inhibiting antibodies. Transbound. Emerg. Dis. 2013;60:395–402. doi: 10.1111/j.1865-1682.2012.01357.x. [DOI] [PubMed] [Google Scholar]
- 37.Magor K.E. Immunoglobulin genetics and antibody responses to influenza in ducks. Dev. Comp. Immunol. 2011;35:1008–1016. doi: 10.1016/j.dci.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Causey D., Edwards S.V. Ecology of avian influenza virus in birds. J. Infect. Dis. 2008;197(Suppl. 1):S29–S33. doi: 10.1086/524991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon D.A. The MHC of the duck (Anas platyrhynchos) contains five differentially expressed class I genes. J. Immunol. 2005;175:6702–6712. doi: 10.4049/jimmunol.175.10.6702. [DOI] [PubMed] [Google Scholar]
- 40.Hulse-Post D.J. Role of domestic ducks in the propagation and biological evolution of highly pathogenic H5N1 influenza viruses in Asia. Proc. Natl. Acad. Sci. U.S.A. 2005;102:10682–10687. doi: 10.1073/pnas.0504662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woo P.C. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 2009;234:1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- 42.Bolles M. SARS-CoV and emergent coronaviruses: viral determinants of interspecies transmission. Curr. Opin. Virol. 2011;1:624–634. doi: 10.1016/j.coviro.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woo P.C. Molecular diversity of coronaviruses in bats. Virology. 2006;351:180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woo P.C. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 2009;83:908–917. doi: 10.1128/JVI.01977-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woo P.C. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012;86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lau S.K. Isolation and characterization of a novel Betacoronavirus subgroup A coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J. Virol. 2012;86:5481–5496. doi: 10.1128/JVI.06927-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graham R.L., Baric R.S. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 2010;84:3134–3146. doi: 10.1128/JVI.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfefferle S. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 2009;15:1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lau S.K. Recent transmission of a novel alphacoronavirus, bat coronavirus HKU10, from Leschenault's rousettes to pomona leaf-nosed bats: first evidence of interspecies transmission of coronavirus between bats of different suborders. J. Virol. 2012;86:11906–11918. doi: 10.1128/JVI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anthony S. Coronaviruses in bats from Mexico. J. Gen. Virol. 2013;94:1028–1038. doi: 10.1099/vir.0.049759-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Annan A. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013;19:456–459. doi: 10.3201/eid1903.121503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang S. A predicted receptor-binding and critical neutralizing domain in S protein of the novel human coronavirus HCoV-EMC. J. Infect. 2013;66:464–466. doi: 10.1016/j.jinf.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muller M.A. Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. mBio. 2012;3 doi: 10.1128/mBio.00515-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hofmann H. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein A. 9-O-acetylated sialic acids have widespread but selective expression: analysis using a chimeric dual-function probe derived from influenza C hemagglutinin-esterase. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7782–7786. doi: 10.1073/pnas.91.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan J.F. Differential cell line susceptibility to the emerging novel human betacoronavirus 2C EMC/2012: implications on disease pathogenesis and clinical manifestation. J. Infect. Dis. 2013;207:1743–1752. doi: 10.1093/infdis/jit123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raj V.S. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Londt B.Z. Failure to infect pigs co-housed with ducks or chickens infected experimentally with A/turkey/Turkey/1/2005 (H5N1) highly pathogenic avian influenza virus. Vet. Microbiol. 2013;162:944–948. doi: 10.1016/j.vetmic.2012.11.040. [DOI] [PubMed] [Google Scholar]
- 59.Cheng V.C. Two years after pandemic influenza A/2009/H1N1: what have we learned? Clin. Microbiol. Rev. 2012;25:223–263. doi: 10.1128/CMR.05012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lindstrom S. Human infections with novel reassortant influenza A(H3N2)v viruses, United States, 2011. Emerg. Infect. Dis. 2012;18:834–837. doi: 10.3201/eid1805.111922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shinde V. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005-2009. N. Engl. J. Med. 2009;360:2616–2625. doi: 10.1056/NEJMoa0903812. [DOI] [PubMed] [Google Scholar]
- 62.Amonsin A. Genetic characterization of H5N1 influenza A viruses isolated from zoo tigers in Thailand. Virology. 2006;344:480–491. doi: 10.1016/j.virol.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 63.Desvaux S. Highly pathogenic avian influenza virus (H5N1) outbreak in captive wild birds and cats, Cambodia. Emerg. Infect. Dis. 2009;15:475–478. doi: 10.3201/eid1503.081410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crispe E. Infection of dogs with equine influenza virus: evidence for transmission from horses during the Australian outbreak. Aust. Vet. J. 2011;89(Suppl. 1):27–28. doi: 10.1111/j.1751-0813.2011.00734.x. [DOI] [PubMed] [Google Scholar]
- 65.Anthony S.J. Emergence of fatal avian influenza in New England harbor seals. mBio. 2012;3 doi: 10.1128/mBio.00166-12. e00166–00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pica N. Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. U.S.A. 2012;109:2573–2578. doi: 10.1073/pnas.1200039109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miller E. Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet. 2010;375:1100–1108. doi: 10.1016/S0140-6736(09)62126-7. [DOI] [PubMed] [Google Scholar]
- 68.Pasick J. Intersegmental recombination between the haemagglutinin and matrix genes was responsible for the emergence of a highly pathogenic H7N3 avian influenza virus in British Columbia. J. Gen. Virol. 2005;86:727–731. doi: 10.1099/vir.0.80478-0. [DOI] [PubMed] [Google Scholar]
- 69.Boni M.F. Homologous recombination is very rare or absent in human influenza A virus. J. Virol. 2008;82:4807–4811. doi: 10.1128/JVI.02683-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murcia P.R. Evolution of an Eurasian avian-like influenza virus in naive and vaccinated pigs. PLoS Pathog. 2012;8:e1002730. doi: 10.1371/journal.ppat.1002730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamada S. Adaptation of a duck influenza A virus in quail. J. Virol. 2012;86:1411–1420. doi: 10.1128/JVI.06100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuchipudi S.V. Differences in influenza virus receptors in chickens and ducks: Implications for interspecies transmission. J. Mol. Genet. Med. 2009;3:143–151. doi: 10.4172/1747-0862.1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng B. D225G mutation in hemagglutinin of pandemic influenza H1N1 (2009) virus enhances virulence in mice. Exp. Biol. Med. (Maywood) 2010;235:981–988. doi: 10.1258/ebm.2010.010071. [DOI] [PubMed] [Google Scholar]
- 74.Chan K.H. Wild type and mutant 2009 pandemic influenza A (H1N1) viruses cause more severe disease and higher mortality in pregnant BALB/c mice. PLoS ONE. 2010;5:e13757. doi: 10.1371/journal.pone.0013757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chandrasekaran A. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat. Biotechnol. 2008;26:107–113. doi: 10.1038/nbt1375. [DOI] [PubMed] [Google Scholar]
- 76.Ito T. Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J. Virol. 1998;72:7367–7373. doi: 10.1128/jvi.72.9.7367-7373.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Londt B.Z. The infectivity of pandemic 2009 H1N1 and avian influenza viruses for pigs: an assessment by ex vivo respiratory tract organ culture. Influenza Other Respir. Viruses. 2013;7:393–402. doi: 10.1111/j.1750-2659.2012.00397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu J.E. Expression patterns of influenza virus receptors in the respiratory tracts of four species of poultry. J. Vet. Sci. 2011;12:7–13. doi: 10.4142/jvs.2011.12.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang C.C. Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18137–18142. doi: 10.1073/pnas.0909696106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang H. Structure and receptor complexes of the hemagglutinin from a highly pathogenic H7N7 influenza virus. J. Virol. 2012;86:8645–8652. doi: 10.1128/JVI.00281-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li S.Q. Generation of seal influenza-virus variants pathogenic for chickens, because of hemagglutinin cleavage site changes. J. Virol. 1990;64:3297–3303. doi: 10.1128/jvi.64.7.3297-3303.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Croville G. Field monitoring of avian influenza viruses: whole-genome sequencing and tracking of neuraminidase evolution using 454 pyrosequencing. J. Clin. Microbiol. 2012;50:2881–2887. doi: 10.1128/JCM.01142-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoffmann T.W. Length variations in the NA Stalk of an H7N1 influenza virus have opposite effects on viral excretion in chickens and ducks. J. Virol. 2012;86:584–588. doi: 10.1128/JVI.05474-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun Y. Amino acid 316 of hemagglutinin and the neuraminidase stalk length influence virulence of H9N2 influenza virus in chickens and mice. J. Virol. 2013;87:2963–2968. doi: 10.1128/JVI.02688-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang G.J. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2013;339:456–460. doi: 10.1126/science.1230835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karpala A.J. Characterization of chicken Mda5 activity: regulation of IFN-beta in the absence of RIG-I functionality. J. Immunol. 2011;186:5397–5405. doi: 10.4049/jimmunol.1003712. [DOI] [PubMed] [Google Scholar]
- 87.Downing T. The differential evolutionary dynamics of avian cytokine and TLR gene classes. J. Immunol. 2010;184:6993–7000. doi: 10.4049/jimmunol.0903092. [DOI] [PubMed] [Google Scholar]
- 88.Volmer C. Immune response in the duck intestine following infection with low-pathogenic avian influenza viruses or stimulation with a Toll-like receptor 7 agonist administered orally. J. Gen. Virol. 2011;92:534–543. doi: 10.1099/vir.0.026443-0. [DOI] [PubMed] [Google Scholar]
- 89.Jiao P.R. Molecular cloning, characterization, and expression analysis of the Muscovy duck Toll-like receptor 3 (MdTLR3) gene. Poult. Sci. 2012;91:2475–2481. doi: 10.3382/ps.2012-02394. [DOI] [PubMed] [Google Scholar]
- 90.MacDonald M.R. Genomics of antiviral defenses in the cuk, a natural host of influenza and hepatitis B viruses. Cytogenet. Genome Res. 2007;117:195–206. doi: 10.1159/000103180. [DOI] [PubMed] [Google Scholar]
- 91.Kaiser P. A genomic analysis of chicken cytokines and chemokines. J. Interferon Cytokine Res. 2005;25:467–484. doi: 10.1089/jir.2005.25.467. [DOI] [PubMed] [Google Scholar]
- 92.Han X. Molecular cloning and characterization of chicken interferon-gamma receptor alpha-chain. J. Interferon Cytokine Res. 2008;28:445–454. doi: 10.1089/jir.2007.0135. [DOI] [PubMed] [Google Scholar]
- 93.Bratsch S. The little brown bat, M. lucifugus, displays a highly diverse V H, D H and J H repertoire but little evidence of somatic hypermutation. Dev. Comp. Immunol. 2011;35:421–430. doi: 10.1016/j.dci.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 94.Papenfuss A.T. The immune gene repertoire of an important viral reservoir, the Australian black flying fox. BMC Genomics. 2012;13:261. doi: 10.1186/1471-2164-13-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ratcliffe M.J. Antibodies, immunoglobulin genes and the bursa of Fabricius in chicken B cell development. Dev. Comp. Immunol. 2006;30:101–118. doi: 10.1016/j.dci.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 96.Butler J.E. The two suborders of chiropterans have the canonical heavy-chain immunoglobulin (Ig) gene repertoire of eutherian mammals. Dev. Comp. Immunol. 2011;35:273–284. doi: 10.1016/j.dci.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 97.Gabriel G. Differential use of importin-alpha isoforms governs cell tropism and host adaptation of influenza virus. Nat. Commun. 2011;2:156. doi: 10.1038/ncomms1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamada S. Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS Pathog. 2010;6:e1001034. doi: 10.1371/journal.ppat.1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jonges M. Comparative analysis of avian influenza virus diversity in poultry and humans during a highly pathogenic avian influenza A (H7N7) virus outbreak. J. Virol. 2011;85:10598–10604. doi: 10.1128/JVI.05369-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mehle A. Reassortment and mutation of the avian influenza virus polymerase PA subunit overcome species barriers. J. Virol. 2012;86:1750–1757. doi: 10.1128/JVI.06203-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen C.J. Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J. Virol. 2010;84:10051–10062. doi: 10.1128/JVI.00592-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zell R. Prevalence of PB1-F2 of influenza A viruses. J. Gen. Virol. 2007;88:536–546. doi: 10.1099/vir.0.82378-0. [DOI] [PubMed] [Google Scholar]
- 103.Shi M. Evolutionary conservation of the PA-X open reading frame in segment 3 of influenza A virus. J. Virol. 2012;86:12411–12413. doi: 10.1128/JVI.01677-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Muramoto Y. Identification of novel influenza A virus proteins translated from PA mRNA. J. Virol. 2013;87:2455–2462. doi: 10.1128/JVI.02656-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zimmermann P. The viral nucleoprotein determines Mx sensitivity of influenza A viruses. J. Virol. 2011;85:8133–8140. doi: 10.1128/JVI.00712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rajsbaum R. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012;8:e1003059. doi: 10.1371/journal.ppat.1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Manz B. Adaptive mutations in NEP compensate for defective H5N1 RNA replication in cultured human cells. Nat. Commun. 2012;3:802. doi: 10.1038/ncomms1804. [DOI] [PubMed] [Google Scholar]