

Abstract
Clinical trials of gene therapy for cystic fibrosis suggest that current levels of gene transfer efficiency are probably too low to result in clinical benefit, largely as a result of the barriers faced by gene transfer vectors within the airways. The respiratory epithelium has evolved a complex series of extracellular barriers (mucus, lack of receptors, immune surveillance, etc.) aimed at preventing penetration of lumenally delivered materials, including gene therapy vectors. In addition, once in the cell, further hurdles have to be overcome, including DNA degradation, nuclear import and the ability to maintain long-term transgene expression. Strategies to overcome these barriers will be addressed in this review and include the use of: (i) clinically relevant adjuncts to overcome the extra- and intracellular barriers; (ii) less-conventional delivery routes, such as intravenous or in utero administration; (iii) more efficient non-viral vectors and ‘stealth’ viruses which can be re-administered; and (iv) new approaches to prolong transgene expression by means of alternative promoters or integrating vectors. These advances have the potential to improve the efficiency of gene delivery to the airway epithelium, thus making gene therapy a more realistic option for cystic fibrosis.
Keywords: Cystic fibrosis, Gene therapy, Barriers, Virus, Non-viral vector
1. Introduction: where are we in the clinic?
Cystic fibrosis (CF) is the most common recessively inherited lethal disease among Caucasian population, affecting approximately one in 2500 newborns. CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, and to date around 1000 different mutations have been identified. The underlying gene mutation leads to defective production of the CFTR protein, a cAMP-regulated chloride (Cl−) channel located in the apical membrane of epithelial cells. Although the organs affected in CF also include the pancreas, gut, liver and reproductive tract, the clinical picture is dominated by pulmonary disease, with recurrent cycles of infection leading to inflammation, bronchiectasis and, in greater than 90% of patients, death from respiratory failure. The isolation of the gene responsible for CF in 1989 suggested the feasibility of new therapeutic treatments based on CFTR gene transfer to patients with CF. The rationale for the development of gene therapy protocols relies on the fact that heterozygotes appear to be phenotypically normal, expression of CFTR is low and the dysfunctional epithelial lining cells in the most affected organ, the lung, are accessible through non-invasive techniques. Furthermore, although conventional treatments have increased life expectancy of CF patients to approximately 30 years, there is clearly an urgent need for a novel therapeutic approach.
Preclinical studies carried out soon after the isolation of the gene showed that both viral and non-viral gene transfer agents (GTAs) were able to correct the chloride ion transport defect in CF transgenic mice. The success of these studies led several groups to initiate clinical trials of gene transfer in CF patients and three classes of GTAs have been used so far: adenovirus, adeno-associated virus (AAV) and cationic lipids. Although the lung remains the most medically relevant target, many investigators chose to start their clinical studies by looking at gene transfer to the nasal mucosa. Nasal airway epithelia have a similar histology and the same CF-associated abnormalities in ion transport as pulmonary epithelia, but compared to lung, the nasal cavity has an easier access for both gene transfer and safety measurements and represents a reduced risk to the patient in case of the occurrence of side effects.
Nine clinical trials using recombinant adenovirus as a GTA have been published so far [1], [2], [3], [4], [5], [6], [7], [8], [9], with five involving administration of the virus to the lung epithelium [2], [6], [7], [8], [9]. The results reported are not always consistent and of the 30 noses assessed following a single application, approximately one-third showed some changes in chloride transport. No functional measurements were assessed in the lung studies, although some evidence of vector-derived CFTR mRNA was found. Common findings in all the studies published include (i) a dose-dependent mild local inflammation and (ii) the progressive lack of expression following repeated administration.
Two phase I, single administration, dose escalation trials using AAV as a GTA have been reported so far. AAV-CFTR was delivered to the maxillary sinus [10] or nebulised to the lungs of CF subjects [11]. In both cases AAV delivery was shown to be safe and vector DNA was found by PCR, up to 70 days in the maxillary sinuses [10] and 14–30 days in the airways [11]. When delivered to the maxillary sinus, some degree of dose-dependent chloride transport correction was observed. However, in both studies no vector-derived mRNA was detected.
Cationic lipids have been used in eight clinical trials [12], [13], [14], [15], [16], [17], [18], [19], with two of them involving nebulisation of the lipoplexes to the lower airways [16], [19]. Different lipoplexes were used including DC-Chol–DOPE [12], [14], [17], DOTAP [15], GL-67 [13], [16], [19] and EDMPC–cholesterol [18], with DNA doses ranging from 10 μg to 1.25 mg when administered to the nose and up to 42.2 mg when nebulised to the lungs [16]. Results were similar to those reported with adenoviral vectors and a correction towards normal values of the chloride defect was observed both in the nose (about 20%) [12], [14], [15], [16] and in the lung (25%) [16], lasting between 7 days and 3 weeks [16]. Interestingly, in one study naked DNA was reported to be as efficient as lipoplexes [13]. Unlike viruses, it has been reported that lipoplexes can be successfully re-administered without apparent loss of efficacy [17]. However, mild flu-like symptoms were noted in both lung trials following aerosolisation of liposome–DNA complexes and are probably related to the presence of unmethylated CpG motifs in bacterially derived plasmid DNA [16], [19].
In summary, within 13 years of the cloning of the CFTR gene tremendous progress has been made and proof-of-principle for correction of the basic chloride defect has been established within the target organ in vivo in CF subjects. However, in none of the clinical trials cited, has sodium hyperabsorption been altered. Each of the three GTAs used has achieved limited success, with none outshining the others. Interestingly, despite non-viral vectors being arguably less efficient than viruses in animal models and laboratory studies, clinical trials have shown that this might be different in CF subjects. More importantly, it is now generally accepted that the efficiency of in vivo gene transfer with currently available vectors needs to be increased. We will here review the progress made in improving GTAs and the hurdles to efficient transgene expression, which any vector has to overcome before entering the airway epithelial cells.
2. Extracellular barriers
The respiratory epithelium presents a particular challenge for GTAs, since one function of the upper respiratory tract is to keep foreign particles out of the lung. Airway epithelia have evolved a complex series of barriers to prevent penetration of lumenally delivered materials (including both viral and non-viral GTAs) into the cell or interstitial compartment. These barriers consist of: (i) a well-defined mucus layer that may bind inhaled vectors and remove them via mucus clearance mechanisms, (ii) a glycocalyx that may bind vectors and prevent binding to cell surface receptors and perhaps most importantly, and (iii) an apical cell membrane that is relatively devoid of viral receptors and growth/tropic receptors (Fig. 1 ). This series of barriers is complemented by epithelial tight junctions that are ‘moderately leaky’ to ions but quite ‘tight’ for larger solutes, thereby preventing penetration by current vectors from the lumenal surfaces into the interstitium. In addition, CF lungs are characterised by the presence of a discontinuous barrier of purulent secretions, which contain exogenous actin, DNA and inflammatory products that can modify the integrity of a lipoplex and limit gene delivery to the airways. CF sputum has been shown to retard the movement of particles having a size comparable with lipoplexes and to almost completely block 560-nm particles [20]. Furthermore, binding of negatively charged CF mucus components to the gene complexes may change their surface charge and size, resulting in a decreased transport of the lipoplex through the mucus and therefore in a decreased cellular uptake [21].
Fig. 1.
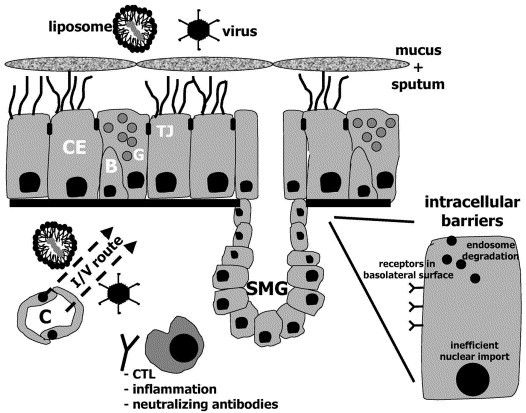
Extracellular and intracellular barriers limit gene transfer efficiency of viral and non-viral vectors to airway epithelial cells. Extracellular barriers include the presence of infected mucus and sputum, mucociliary clearance and tight junctions between the cells, which limit the access of viral vectors to receptors localised on the basolateral membrane. Alternatively, gene transfer agents could be delivered intravenously (I/V route), even if it is unlikely that airway epithelial cells will be targeted. Inflammation, CTL-mediated degradation of transduced cells and neutralizing antibodies can further limit transgene expression or re-administration of the gene transfer vectors. Hidden stem or progenitor cells could be targeted by lentiviral vectors. Once inside the cell, the genetic material has to overcome endosome and cytoplasmic degradation and to get into the nucleus. CE, ciliated cell; B, basal cell; G, goblet cell; TJ, tight junctions; C, capillary; SMG, submucosal gland.
Using an ex vivo sheep trachea model, we demonstrated that cationic lipid-mediated gene transfer, but not adenoviral vectors or recombinant Sendai virus [22], was inhibited by normal mucus, with removal of this layer significantly increasing transgene expression [23]. Similar results were obtained when cationic polymers, such as PEI, were used. However, we have recently shown that the mucus barrier can, in part, be overcome by treatment with mucolytic agents, such as Nacystelyn, when either a cationic lipid (GL-67 or EDMPC:Chol) or a cationic polymer (PEI) are used to deliver genes to airway epithelium [24].
Similarly, sputum and bronchoalveolar lavage fluid (BALF) recovered from CF patients have been shown to inhibit liposome- [25], adenovirus- [26] and AAV-mediated gene transfer efficiency [27]. However, when CF sputum was treated with recombinant human DNase, an increased liposome-mediated gene delivery was observed [25]. Recombinant human DNase has been shown to reduce sputum viscoelasticity, facilitate the transport of nanospheres though CF sputum [20] and, when mixed with lipoplexes or polyplexes, not to alter their ability to mediate gene delivery [28].
Recent findings have suggested that the glycocalyx may also represent another major obstacle to gene transfer to the airway epithelium. The glycocalyx is composed of many carbohydrate-bearing structures, including glycoproteins, glycolipids and proteoglycans. Treatment with agents to remove components of the glycocalyx, such as neuroaminidase, has been shown to enhance the susceptibility of polarised cells to transduction by Ad or AAV vectors [29].
Attempts have been made to circumvent barriers at the mucosal surface by delivering transgenes via the bloodstream. The intravenous route may make it possible to access the basolateral membrane of airway epithelial cells, characterised by a higher rate of endocytosis and an increased density of viral receptors. However, the difficulties in overcoming the large number of barriers between the vascular compartment and airway epithelial cells (endothelial cells, endothelial cell basement membrane, interstitium and epithelial basement membrane) make this route challenging. A large number of studies have been conducted to identify which cells in the lung are transduced after intravenous (i.v.) injection of liposome–DNA complexes. Most of these have concluded that the majority of the cells transfected are the pulmonary endothelial cells, with some studies reporting transfection of type I/II alveolar epithelial cells [30]. Only a few studies have reported airway epithelial cell transfection after i.v. injection of cationic lipid–DNA complexes [31], [32]. Recently, Koehler et al., have reported β-galactosidase expression in nasal and bronchial epithelium and tracheal submucosal glands for the first time, after i.v. injection of DODAC–DOPE liposomes. This may have been due to properties of this specific formulation (such as a longer circulation time) since similar results were not obtained when other GTAs such as DOTAP–DOPE, DC-Chol–DOPE. GL-67A and 22 kDa PEI were administered [32].
Independently from the route of administration, innate immune defence mechanisms and pre-existing immunity are likely to play a crucial role in defending the lung from foreign particles, including gene therapy materials. Airway macrophages are known to reduce the amount of GTA able to reach the epithelial cells either via a direct mechanism (phagocytosis) or, since they are antigen presenting cells, through stimulation of the host immune system. Pre-existing immunity from antibodies to wild-type viruses, such as adenovirus and AAV, could hinder their ability to target epithelial cells efficiently. A survey of normal and CF subjects has shown that virtually all subjects had antibodies to Ad5 and to AAV2 (the two most used viral vectors so far), although only 55 and 32%, respectively, were neutralising antibodies [33]. It has also been observed that individuals with a higher baseline anti-Ad neutralising antibody titre mounted a higher neutralising antibody response after vector administration [34].
3. Cell surface barriers
The early studies with model systems that employed poorly differentiated airway epithelial cells suggested that gene transfer efficiency for a variety of vectors would be high. However, with the advent of well-differentiated culture systems it became clear that (i) the airway lumen-facing columnar cell, the predominant cell type that must be transduced in vivo, is relatively resistant to viral and nonviral gene transfer and (ii) the apical surfaces of well-differentiated airway epithelial cells have a low basal and stimulated rate of endocytosis. In contrast, currently available cell cultures are composed of basal or poorly differentiated cells that are highly transducible, but do not mirror the airway lumen in vivo [35].
3.1. Both synthetic and viral vectors are unable to enter into polarised cells
With regard to synthetic vectors, Jiang et al. showed that GL-67-mediated gene transfer efficiency decreased progressively as the cells became more polarised and differentiated [36]. Similar reports demonstrated that much of the reduction in gene delivery was due to a decrease in binding of the cationic lipid to the surface of mature airway epithelial cells [37] and also to a decline in the rate of internalisation of the bound complexes into these cells [38]. The hypothesis is that reduction in binding may be due to differences in surface charge between poorly and well-differentiated cells. While poorly differentiated cells exhibited receptor-mediated endocytosis, pinocytosis and phagocytosis, well-differentiated cell were only capable of receptor-mediated endocytosis [37]. Some reports have indicated that cell proliferation may influence cationic lipid-mediated transfection activity, either by making epithelial cells themselves more susceptible to transfection or by temporary disruption of the tight junctions [38]. This would allow the lipoplexes to either access the basolateral surface of well-differentiated cells or to access more immature, less differentiated cells.
With regard to viral vectors, it has been demonstrated that the receptors for adenovirus [39], AAV-2 [40] and retrovirus are localised to the basolateral membrane, and are therefore not accessible when these vectors are delivered topically.
3.2. The challenge of limited efficiency
In principle, four general strategies may improve gene transfer efficiency to airway epithelia.
3.2.1. Tight junctions
The barrier function of epithelial tight junctions can be transiently disrupted so that vectors can access the basolateral membrane of target cells, which are rich in viral receptors and have a higher rate of endocytosis. This can be achieved by using Ca2+ chelator agents, such as EGTA [41], [42], non-ionic detergents, such as polidocanol [43], and antibodies able to block the function of proteins involved in the tight junction-complex, such as E-cadherin [44]. In addition, medium-chain fatty acids, such as sodium caprate, have been shown to be better agents for enhancing gene transfer than EGTA, via disruption of claudin-1, a major structural component of the tight junctions [45]. Croyle et al. recently found that a blend of sucrose, mannitol and Pluronic F68 enhances adenoviral-mediated gene expression in both large and small lung airways. This formulation was shown to increase tight junction permeability and allow the use of 1/2 log lower viral dose [46]. Airway instillation of perfluorochemical (PFC) liquid can also transiently open tight junctions, enhancing adenoviral- [47], AAV [48] and cationic lipid-mediated gene transfer [49], although this procedure can increase inflammation.
3.2.2. Modification of the host
The apical membrane can be modified so that it binds vectors. One approach has been to incorporate unnatural sugars into membrane glycoproteins and use them as a molecular handle on which a novel receptor is constructed. The artificial receptor enhanced adenoviral binding and gene transfer to cells that were normally relatively resistant to adenovirus infection [50]. This suggests the feasibility of a gene transfer strategy in which the biosynthetic machinery of the cell is used to engineer novel receptors on the cell surface.
3.2.3. Modification of the vector
The GTA can be modified in order to target a receptor on the apical membrane that has the capacity to both bind and internalise a vector. One of the first examples reported is the P2Y2-purinoreceptor, which is highly expressed on the apical surface of epithelial cells and is stimulated to internalise upon UTP activation. An increase in gene transfer efficiency was observed when adenoviral vectors conjugated to biotinylated UTP ligands were used to target the endogenous P2Y2-purinoreceptor on well-differentiated human airway epithelial cells, known to be refractory to adenovirus-mediated gene transfer [51]. Similar results were obtained when GTAs were retargeted to bind to the bradykinin receptor [52], the urokinase plasminogen activator receptor [53] and the serpin enzyme complex receptor (sec-R), all of which are expressed on the apical surface of airway epithelial cells. Administration of plasmid DNA carrying the CFTR cDNA condensed with poly-l-lysine sec-R ligand to CF mice produced a correction of the Cl− efflux, a more normal sodium channel activity and a reversal of nitric oxide synthase-2 downregulation [54].
Investigators are currently trying to characterise new ligands to target receptors present on the apical surface of airway epithelia. One method is to use a phage display library screened for peptides binding with high affinity to airway epithelial cells. This has been mainly done in vitro and several promising candidates have already been identified [55], but attempts to identify more relevant molecules by in vivo bio-panning are currently under evaluation.
An attractive alternative strategy is to evaluate systematically glycoproteins from other enveloped viruses for their ability to efficiently infect airway epithelia and to use these glycoproteins to pseudotype recombinant viral vectors. Some viruses have already been shown to infect the polarised airway epithelium from the apical surface, including respiratory pathogens, such as human coronavirus 229E [56], H1N1, H2N2 and H3N2 influenza A virus strains [57] and viruses from the Filoviridae family such as Marburg and Ebola virus. By using an Ebola-pseudotyped HIV vector, Kobinger et al. showed efficient and stable transduction of intact airway epithelium from the apical surface in vivo [58].
The modification of the vector can also be nonspecific. Given the low density of Ad-receptors on the apical membrane, several strategies to increase cell binding via a non-fibre-dependent pathway have been developed. Complexing Ad vectors with polycations [59] or incorporating Ad in calcium phosphate precipitates [60] have been shown to enhance gene transfer to airway epithelia in vitro and in vivo. These strategies are thought to neutralise the adverse charge interaction between negatively charged Ad particles and the negatively charged cell surface, resulting in improved binding and uptake of Ad vector.
Furthermore, it has been reported that both synthetic [22] and viral vector [61] mediated gene transfer to differentiated airway epithelia can be increased by a prolonged contact time.
3.2.4. Identification of new viral vectors
Improved binding and entry from the apical surface have been reported with both new viral vectors and new serotypes of existing vectors. It has recently been shown that, in contrast to AAV 2, AAV serotype 5 is able to infect human airway epithelia from the lumenal surface, suggesting that 2,3-linked sialic acid is either a receptor for AAV 5 or a necessary component of a receptor complex [62]. Recombinant Sendai virus, a single-stranded RNA paramyxovirus, is also able to infect airway epithelial cells from the lumenal surface. Its envelope proteins, F (fusion) and HN (haemagglutinin), have been shown to interact with cholesterol and sialic acid, respectively, both molecules known to be present on the apical membrane of airway epithelial cells [22]. Similarly, a recombinant respiratory syncytial virus has recently been developed because of its ability to infect ciliated cells via the lumenal membrane [63].
4. Intracellular barriers
Viral and nonviral vectors enter cells through endocytosis, a normal process for the internalisation and degradation of extracellular material.
4.1. Endosome
Plasmid-based vectors are quite susceptible to endosomal degradation and attempts have been made to enhance transgene escape from the endosome–lysosome pathway (Fig. 1). One approach involves using fusogenic lipids or peptides, such as GALA and HA-2 from influenza haemagglutinin, to disrupt the endosome membrane. The neutral lipid dioleoylphosphatidylethanolamine (DOPE) is generally employed as a fusogenic helper lipid in a cationic lipid–DNA complex. The second approach involves using a DNA delivery system with a high buffering capacity and the flexibility to swell when protonated. The rationale that endosomes can be ruptured if the pH drop in the late endosome is inhibited by the buffering capacity of the formulation led to the use of pH-sensitive liposomes and polyethylenimine as gene transfer agents [64]. Because of the great number of secondary amines, PEI behaves as a ‘proton sponge’, able to buffer the low pH in the endosomes, resulting in the inhibition of the low pH-activated nucleases. This leads to a large increase in the ionic concentration inside the endosome, finally resulting in osmotic swelling due to water entry and rupture of the organelle [65].
In contrast to plasmid DNA, viral vectors have evolved mechanisms to escape endosome degradation and for some of them it is a prerequisite for subsequent nuclear localisation of the virions. Hansen et al. have shown that the passage through the endosome acidic component exposes AAV to conditions that modify the capsid, allowing the virus to use the cytoskeleton for subsequent trafficking events to the nucleus [66]. Sendai virus uses the F protein to fuse its envelope with the cell plasma membrane, allowing the genetic material to be released directly into the cytoplasm and thus avoiding endosome degradation. This led to the use of HVJ-liposome as a new gene transfer agent in which UV-inactivated SeV particles are mixed together with lipids in a liposome formulation in order to allow the plasmid DNA to be introduced into the cytoplasm of the transfected cell [67].
4.2. Cytoplasm-based degradation pathways
Viral vectors and some synthetic vectors, such as PEI and HVJ-liposome, are characterised by the ability to escape endosome degradation. However, once released in the cytoplasm and before entering the nucleus, other hurdles are encountered. Synthetic vector-based strategies are characterised by low gene transfer efficiency because plasmid DNA is quickly degraded by Ca2+-sensitive cytosolic nucleases, with an apparent half-life of 50–90 min [68], [69]. Duan et al. showed that in polarised epithelial cells AAV capsid is ubiquinated after endocytosis and that this process is a barrier to rAAV transduction. Proteasome-dependent degradation of ubiquinated molecules represents a major pathway for disposal of both endogenous and foreign proteins. In vivo application of proteasome inhibitors in mouse lungs augmented rAAV-mediated gene transfer from undetectable levels to a mean of 10.4±1.6% of the epithelial cells in large bronchioles [70].
4.3. Nuclear import
Once within the cytoplasm, the transgene must be imported into the nucleus to be transcribed. For plasmid-based expression, nuclear import is a rate-limiting step and intracellular trafficking of pDNA, either naked or complexed to synthetic vectors, is largely uncharacterised (Fig. 1). During non-viral gene transfer, entry of exogenous DNA into the nucleus occurs only in cells that are actively dividing, i.e., when the nuclear envelope breaks down. This is consistent with the observation that well-differentiated, non-dividing airway epithelial cells show very low transfection efficiency. Pollard et al. showed that less than 1/1000 naked cDNA copies microinjected in the cytoplasm were effectively trafficked to the nucleus [71].
These results may be largely due to the inability of pDNA to effectively translocate through the nuclear pore complexes (NPCs). Each NPC is comprised of a large family of proteins (nucleoporins) forming a structure in which a central channel is surrounded by eight peripheral channels. It is thought that the peripheral channels, about 9–10 nm diameter, allow small solutes and proteins up to 50–60 kDa to freely diffuse in an out the nucleus. Larger proteins need a nuclear localisation signal (NLS) in order to be actively transported through the central channel of the pore. Under physiological conditions, a supercoiled pDNA has a diameter of 10 nm or larger if in a relaxed conformation, thus suggesting that passive diffusion of pDNA through the NPC is highly unlikely [72]. Therefore, targeting of pDNA to nuclear ‘shuttle’ proteins has become part of the design of GTAs developed to transfer non-dividing cells, as in CF gene therapy.
Several approaches have been taken to improve nuclear entry of pDNA, including the electrostatic binding of pDNA to NLS-containing proteins, such as HMG-1, and the covalent attachment of NLS-motifs to double-stranded DNA. With regard to the latest approach, most of the studies published so far have used the NLS of the simian virus 40 (SV40) large T antigen because its trafficking to the nucleus is well characterised and has also shown to mediate nuclear import of non-karyophilic proteins. By using this ‘piggyback’ approach, Sebestyen et al. demonstrated nuclear accumulation in digitonin-permeabilised cells, but failed to show uptake of the modified DNA in nuclei of intact cells. This suggests that covalent modification of pDNA with a signal peptide may alter its behaviour and interaction with other cellular factors [73]. By capping a luciferase gene with a single NLS peptide, Zanta et al. showed a 10–1000-fold increase in gene transfer efficiency [74]. However, the major drawback of this technique is the relatively low amount of NLS-pDNA that can be produced.
The development of peptide nucleic acids (PNAs), oligonucleotide analogues in which the sugar phosphate backbone of nucleic acid has been replaced by a synthetic peptide backbone, has led to further developments in this field. PNA is capable of sequence-specific recognition of DNA and RNA following the Watson–Crick hydrogen-bonding scheme and can be used to link peptides, such as NLS-motifs, to plasmid DNA. The advantage over the other strategies is that the NLS peptide can be linked to very specific regions of plasmid DNA, with no effects on the transcription of genes located elsewhere on the plasmid. Furthermore, a precise number of NLS-motifs can be attached and large quantities of the modified pDNA obtained [75]. However, up to now, none of the strategies described has been used to enhance nuclear import and gene transfer expression in airway epithelial cells in vivo. In addition to this, there are reports suggesting that some polycations, such as PEI [71] and lactosylated poly-l-lysine [76], may facilitate the nuclear uptake of pDNA and that, unlike lipoplexes, the complexes remain intact during nuclear translocation. This might suggest the existence of nuclear import pathways distinct from the conventional NLS one.
Unlike plasmid-based approaches, viral vectors have evolved efficient ways to enter the nucleus. The cytoplasmic movement of viral DNA towards the nucleus is facilitated by the interaction of viral proteins, such as polymerase or capsid proteins, with the microtubular network [77]. It has recently been shown that adenovirus type 2 docks at the CAN/Nup214 protein of the nuclear pore, then hijacks histone H1 and specific H1-import receptors to effect a targeted uncoating of its nucleocapsid at the nuclear pore. Consequently, the viral DNA is liberated near the opening of the pore and positioned for translocation into the nucleus [78].
For most of the viral vectors, but not retroviruses, nuclear import does not depend on the mitotic status of the cell. It has recently been reported that a polypurine tract (cPPT) present in the HIV-1 genome leads to the formation of a triple-stranded DNA structure, the HIV-1 flap, which is recognised by the nuclear import machinery of the host cell. This ultimately results in the transport of the HIV-1 genome into the nucleus across the nuclear pore complex [79]. Introduction of the cPPT sequence in recombinant lentiviral vectors has been shown to enhance nuclear import and, therefore, transgene expression [80]. For other viruses, such as Sendai virus, nuclear import is not a rate-limiting step since replication and transcription both occur in the cytoplasm.
5. Host immune responses
As transgene expression is transient, and unless lung-resident stem cells are targeted, the treatment of CF by gene therapy will require repeated administrations throughout the lifetime of the patient. The outcome of repeated administrations is, of course, dependent on the nature of the GTA.
5.1. Virus delivery
For virus-based approaches, many hurdles have to be overcome in order to re-administer the vector. These include: (i) an antigen-nonspecific, cytokine-dependent response resulting in acute inflammation; (ii) a cytotoxic (CD8+) T-lymphocyte (CTL)-dependent response directed at cells expressing viral or transgene proteins, resulting in chronic inflammation and lack of persistent transgene expression; and (iii) a helper (CD4+) T lymphocyte-dependent response directed at adenoviral capsid proteins present during vector delivery, resulting in the production of neutralising antibodies (NAB) that limit repeated vector administration.
Several approaches have been taken to reduce expression-limiting immune responses in recipients of virus-based gene therapy.
(i) Immunosuppressant drugs such as cyclophosphamide, cyclosporine and FK506 have been reported to prolong transgene expression and facilitate repeated gene transfer in the lung [81]. However, the potential to induce severe systemic side effects may limit the clinical application of these drugs. Furthermore, prolonged general immunosuppression of CF patients, where lungs are colonised by pathogenic bacteria, could be injurious.
(ii) Topical corticosteroids, such as budesonide, have allowed successful re-administration of adenovirus, at least twice. Compared to saline-treated animals, budesonide treatment resulted in significantly higher transgene expression and lower amounts of NAB both in BALF and serum. However, these differences disappeared after five previous exposures to the virus [82].
(iii) Co-administration of interferon-γ (IFN-γ) or interleukin-12 (IL-12) has been shown to diminish the activity of TH2 cells and formation of NAB, allowing efficient re-administration (at least once) of recombinant virus [83]. A potential drawback of this approach is that TH2 cells are inhibited at the expense of increased TH1 activation. Thus, both IFN-γ and IL-12, while capable of inhibiting humoral immunity, might enhance the elimination of adenovirus-transduced cells by CTLs.
(iv) Both CTL and B cell responses require activation of CD4+ T cells of the TH1 and TH2 subsets, respectively. The central role of CD4+ T cells in the activation of both arms of the immune response suggests that a transient blockade of CD4+ T cells at the time of virus administration might prevent the activation of humoral and cellular immune response to viruses, thus causing prolonged transgene expression and efficient re-administration. Several strategies have been adopted including the use of non-depleting monoclonal antibodies to the CD4 molecule [84] and the blockade of either CD40-CD40 ligand [85] or CD28-B7 (with CTLA4-Ig) [86], co-stimulatory signals necessary for complete T-cell activation. These treatments have resulted in suppression of cellular and humoral responses, prolonged transgene expression and, in some cases, vector re-administration up to four times [84]. However, in non-human primates, treatment with an anti-CD40 ligand monoclonal antibody did not prevent the elicitation of a virus-specific antibody response upon secondary challenge with vector [87]. It is possible that a combination of blocking agents may provide a more complete abrogation of T- and B-cell immune responses.
(v) Antibody neutralisation of the virus can be reduced by coating the vector particle with polyethylene glycol (PEG) [88], GL67–DOPE-PEG [89] and poly-l-lysine or DEAE-dextran [59]. These treatments have the added advantage of providing a means to retarget the vector through ligands coupled to the coating polymer. Another potential strategy for effective repeated delivery is ‘serotype switching’ where gene therapy is initiated with one virus serotype, then switched to a virus derived from a second serotype for subsequent administration, thereby avoiding neutralising antibodies induced by the first serotype. However, transgene expression may be limited by cross-reactive CTLs that can also target cells infected by the second virus serotype [90].
If the use of these strategies were to be combined with adenovirus vectors that are devoid of all viral sequences (‘gutless’ or helper-dependent adenoviral vectors), thereby avoiding a cell-mediated immune response, it might be possible to repeatedly deliver the virus. The use of these ‘stealth’ adenoviruses could eliminate the requirement for systemic immunosuppression with repeated administration [91].
Recombinant AAV (rAAV) vectors are generally less immunogenic in the airways and Beck et al. have recently suggested that AAV can evade immunological surveillance and can be repeatedly delivered to rabbit airways, because they are unable to transduce antigen presenting cells (dendritic cells) [92]. However, this possibility has recently been ruled out unless different serotypes [93] or some form of immunosuppression [94], [95] is used. Furthermore, the small packaging size limit of the virion has restricted the use of genetic control elements that can drive CFTR expression compared to the relatively weak promoter activity of the native viral inverted terminal repeat (ITR) sequence. Recent progress to overcome this problem has included the use of a shortened version of the CFTR gene (‘mini-gene’) [96]. Another approach is to expand rAAV packaging capacity with trans-splicing or overlapping vectors. The first reconstitutes gene expression from two independent rAAV vectors, each encoding unique, non-overlapping halves of a transgene. The overlapping vector approach uses homologous recombination between overlapping regions in two independent vectors [97]. Preliminary results seem to indicate that the trans-splicing approach is 4–10-fold more efficient than the overlapping approach [98].
5.2. Plasmid-based delivery
Plasmid-based gene delivery strategies are generally regarded as safer and less immunogenic alternatives to viral vectors. Repeated administration of lipoplexes in mice [99] and CF patients [17] resulted in similar levels of transgene expression as observed after a single delivery, thus suggesting that lipoplexes can be re-administered without apparent loss of efficacy. However, the efficacy of repeated administrations is dependent on the dose used and the time interval between administrations. Lee et al. reported that very little or no loss in efficacy was observed provided the dose of lipoplexes was low or if the time interval between successive instillations was sufficiently long [100]. It appears that the inflammation elicited by the complexes may affect the efficacy of repeat administration.
Non-viral gene delivery agents can indeed have inflammatory and toxic effects in vivo. Scheule et al. observed a dose-dependent pulmonary inflammation characterised by infiltrates of neutrophils and, to a lesser extent, macrophages and lymphocytes when the cationic lipid GL-67 was administered to mouse lungs in vivo. Associated with this were elevated levels of the pro-inflammatory cytokines IL-6, TNF-α and IFN-γ that peaked at day 1–2 post-instillation, but resolved to normal limits by day 14 [101]. Histopathological analysis of lung sections from mice treated with the individual components of the lipoplex suggested that the cationic lipid was the major mediator of the observed inflammation. However, results of clinical studies in which CF patients were subjected to either aerosolised liposomes alone [102] or cationic lipid–pDNA complexes indicated that bacterially derived pDNA may also be inflammatory [16], [19]. Each of the cationic lipid–pDNA-treated patients, but not the liposome-treated controls, exhibited mild flu-like symptoms (including fever, myalgia and a reduction in FEV1 of approximately 15%) over a period of 24 h.
One possible explanation for this response may be related to the presence of unmethylated CpG dinucleotide sequences in bacterially derived pDNA. Compared with DNA of eukaryotic origin, bacterial genomic DNA contains a 20-fold higher frequency of the dinucleotide sequence CpG. Further, unlike eukaryotic DNA, in which approximately 80% of the cytosines are methylated, bacterial DNA is relatively unmethylated. Instillation of bacterial DNA or oligonucleotides containing immunostimulatory CpG motifs into mouse lungs resulted in inflammation of the lower respiratory tract [103]. Several strategies have been employed to decrease the immunostimulatory properties of pDNA, including (i) methylation of CpG sequences [104], (ii) reduction of the CpG frequency by eliminating non-essential regions or by site-directed mutagenesis [105] and (iii) the use of inhibitors of the CpG signalling pathway, such as chloroquine or quinacrine [105]. Independent of the strategy used, the CpG-reduced pDNAs were found to be less pro-inflammatory. However, methylation of the CpG motifs can severely reduce the expression of the transgene [104].
6. Expression of the therapeutic gene
One of the main obstacles to the development of gene therapy for the airways is the inability of current viral and nonviral gene transfer vectors to direct sustained expression of a therapeutic transgene. This may be due to several causes including loss of the vector (especially if present in an episomal form), transcriptional silencing of the transgene promoter, loss of the transfected cell through cell turnover, or the generation of an immune response to the transgene product or the transfected cell itself.
Several approaches have therefore been taken to achieve longer-term expression following each gene transfer treatment. As outlined in Section 5.1, immunosuppressant drugs have been reported to prolong transgene expression [106], because of their ability to block T cell-mediated response. In addition, Scaria et al. showed that by using an adenovirus co-expressing both human CFTR and ICP47 (a gene shown to block the transporter associated with MHC class I-mediated antigen presentation to CD8+T cells) a prolonged expression (up to 21 days) was observed in monkey lungs, even though natural killer cell activity was enhanced [107].
However, several lines of evidence suggest that attenuation of promoter function may be the most significant factor in the lack of persistence of transgene expression. The transcriptional activity of the widely used cytomegalovirus immediate early gene promoter (CMV) is highly robust, but prone to inactivation over time. Cytokines induced by adenovirus- or plasmid-mediated gene delivery have been shown to down-regulate CMV-driven expression. For this reason, many investigators have evaluated alternative promoters, with many showing increased persistence. These include the polyubiquitin C promoter (high-level transgene expression for up to 8 weeks and still detectable after 6 months), the elongation factor 1α promoter (expression up to 4 weeks) [108] and the CMV–ubiquitin B hybrid promoter (expression up to 3 months, with 50% of day 2 levels remaining at day 84) [109]. Similar prolonged transgene expression was obtained when the E4 region from adenovirus 2 or simply the open reading frame 3 (ORF3) of E4 were cloned upstream of the CMV promoter on a plasmid backbone [110]. However, a potential disadvantage of this approach is the immunogenicity of the E4 ORF3 product once expressed in the transfected cell, thereby limiting its usefulness in vivo. In addition, there is growing evidence that genomic sequences, either within or flanking the gene, might be essential to provide in vivo long-term expression [111].
An alternative approach to achieve prolonged transgene expression is to use artificial chromosome vectors or integrating viruses, since they are stable over time and will propagate to daughter cells should cell division occur. Huertas et al. have recently developed a circular yeast artificial chromosome (YAC) carrying the human CFTR sequence and the oriP and EBNA-1 genes from Epstein–Barr (EBV) virus [112]. Plasmids carrying these two EBV genes have been shown to allow long-term episomal maintenance of the DNA, being able to replicate and segregate in the daughter cells. However, unless lung-resident stem cells are targeted, these vectors are unlikely to have greater advantages over conventional plasmids, since the airway epithelium is mainly composed of non-dividing cells. Furthermore, their size makes in vivo delivery and subsequent nuclear trafficking quite difficult.
With regard to integrating viruses, AAV and, more recently, lentiviruses have been considered as good candidates for prolonged expression. Wild-type AAV persists by site-specific integration into human chromosome 19. However, recombinant AAVs persist predominantly in an episomal form and integrate randomly in the host genome at a much lower frequency than wild-type AAV, probably because of the lack of rep gene products [113]. Furthermore, results demonstrate that, at least in the liver extrachromosomal, not integrated genomes, are the primary source of rAAV-mediated gene expression [114].
Lentiviral vectors from different strains including human, feline and equine immunodeficiency virus have been used to transduce airway epithelial cells because of their potential to provide long-term expression through the integration of the provirus into the host cell genome. Kobinger et al. recently used an HIV-based vector pseudotyped with the envelope from the Ebola (EboZ) virus to efficiently transduce airway epithelia in vivo. Animals receiving EboZ-pseudotyped HIV vector demonstrated minimal expression at day 7, but strong expression in the airway epithelium, including submucosal glands, by day 28 that persisted at day 63. On average, 30% of the entire tracheal epithelium was transduced by the vector at day 28 and 24% at day 63 [58].
Despite these results, integrating vectors still have some problems. The first is that chromosomal position and structure can negatively affect transgene expression, thus leading to host shut-off of the transferred expression cassette, a problem that has plagued retrovirus gene transfer vectors. This effect, known as transcriptional silencing, can be mitigated through the use of chromatin insulators, which are protein-binding DNA elements that lack intrinsic promoter/enhancer activity, but shelter genes from transcriptional influence of surrounding chromatin. The second problem is that the majority of the epithelium is comprised of differentiated cells that are replaced every few months, so that a transgene integrated in the host genome will eventually be lost due to cell turnover.
An alternative would be to target lung resident stem cells so that the integrated transgene is continuously propagated to the daughter cells when cell division occurs. There has been a long-standing debate regarding the identity of airway epithelial stem cells and two theoretical models of cell lineage in the pseudostratified airway epithelium have been suggested. The ‘stem cell niche’ model suggests that airway epithelial stem cells are localised to distinct and potentially inaccessible compartments of the lung. However, there is evidence for great plasticity in growth and differentiation potential of airway epithelial cells and earlier studies showed that both basal and non-basal cells could regenerate a complete mucociliary epithelium in tracheal grafts. This observation led to an alternative ‘unlimited plasticity’ model where many non-terminal cells with ample progenitorial capacity are scattered throughout the epithelium [115]. A better understanding of this issue is also likely to benefit any gene therapy strategy using integrating vectors, considering the different accessibility of stem cells in the two models.
7. How many and which cells should be corrected to achieve clinical benefit?
A key issue is to distinguish between two concepts: ‘percent of cells corrected’ and ‘level of CFTR transduced/cell’.
7.1. Percent of cells corrected
An in vitro study has shown that approximately 6–10% of ‘corrected’ cells are needed to restore normal Cl− transport function [116]. The amplification of functional correction reflects the fact that Cl− can move via gap junctions from non-corrected cells into adjacent corrected cells for secretion. An in vivo study has shown that 5% of the normal level of Cftr gene expression can correct the chloride abnormality (50% of normal) and, importantly, the intestinal pathology seen in mice with CF [117]. However, different levels of correction may be required to restore the various functions of CFTR. The relationship between efficiency of gene transfer and normalisation of sodium transport is likely to be linear, reflecting the fact that CFTR directly regulates ENaC channels within individual cells. This suggests that virtually every affected cell (100%) should be corrected [116]. The level of transfection required for other functions of CFTR to be restored, e.g., sulphation/sialylation defects or transport of other molecules, remains unknown and appears to depend on the cell type transduced and the GTA used. Zhang et al. showed that cationic liposome-mediated CFTR transfer achieved very low transgene expression with insignificant correction of the chloride defect, but mucus sulphation was reduced to levels seen in non-CF airways. The converse was seen with adenovirus that, despite higher levels of expression, did not transduce goblet cells [118]. In addition, the route of administration itself seems to have a role with regard to the localisation of the delivered gene. Instillation of GTAs into the mouse lungs results predominantly in transfection of the alveolar and terminal bronchial cells, while, when they are aerosolised, a higher level of transfection is observed in the airway epithelium [119]. Aerosolisation is more likely to lead to a more even deposition of the GTAs throughout the lung than could be attained by instillation, which presumably primarily deposits the complexes in the parenchyma.
7.2. Level of CFTR expression per cell
A delivered gene would ideally be expressed in a manner similar to the normal pattern of the defective gene that it is replacing. To counterbalance the poor gene transfer efficiency with current GTAs, very strong non-specific viral promoters such as RSV and CMV have been used to drive transgene expression. However, because the number of CFTR molecules per respiratory epithelial cell is low (20–100 channel proteins/cell) and that the CFTR protein regulates other ionic channels implicated in water and salt secretion, it is possible that high levels of CFTR expression may perturb the function of other proteins or alter physiological properties of the cell. It has been reported that a high level of CFTR expression can cause growth arrest and increased cell volume [120], [121], thus suggesting that either regulatable expression cassettes [121] or epithelium-specific promoters, such as that for the human cytokeratin 18 gene, will have to be used to drive CFTR expression. To produce more physiological CFTR expression other approaches such as the use of (i) genomic context vectors in which CFTR expression is driven by its natural promoter and regulatory sequences and (ii) ‘gene targeting’ molecules, such as RNA–DNA chimeric oligos and small DNA fragments for homologous replacement may be needed. This latter strategy would allow the mutation within the CFTR gene to be ‘surgically’ modified without altering the promoter and the regulatory sequences of the CFTR gene.
8. Non-conventional approaches
Because of the hurdles encountered by GTAs in getting into airway epithelial cells, many less conventional strategies than those described above have been developed and will be reviewed here.
8.1. Oligonucleotide-mediated strategies
Because of their size, oligonucleotides have the potential to enter the cell and the nucleus much more easily than plasmid DNA. Goncz et al. have developed a new strategy based on gene targeting by small fragment homologous replacement (SFHR). Specific genomic sequences are targeted with small fragments of exogenous DNA (400–800 bp) that are homologous to the targeted endogenous DNA sequences except for the particular base pairs that encode the desired modification. Gene targeting is thought to have several advantages over classic gene complementation, including long-term and tissue specific expression of the functional gene, no introduction of foreign sequences and no immune response. For the first time, Goncz et al. showed the modification of specific genomic sequences in exon 10 of the mouse CFTR after small DNA fragments were delivered to the lungs of normal mice [122]. Recently, conversion of wild-type CFTR to the G551D mutation in primary rat hepatocytes has been reported by using a different molecule, RNA–DNA chimeraplasts [123].
Another strategy is to use oligonucleotides as antisense molecules. Friedman et al. used antisense oligoribonucleotides to correct the CFTR splicing mutation 3849+10 kb C→T in human and mouse epithelial cells [124]. In another report, antisense inhibition of B cell antigen receptor-associated protein (BAP) 31 increased expression of both wild-type CFTR and [ΔPhe508]CFTR and enabled cAMP-activated Cl− currents in [ΔPhe508]CFTR-expressing CHO cells [125]. Antisense strategy might suggest a new way to correct the defects present in CF, such as mucin production or sodium hyperabsorption.
8.2. Spliceosome-mediated RNA trans-splicing (SMaRT)
A very recent technology developed by Mitchell and collaborators takes advantage of the cell’s endogenous splicing machinery as a strategy for modifying pre-mRNA. The SMaRT process uses pre-therapeutic RNA molecules (PTMs) that are designed to base pair with the intron of a targeted pre-mRNA to suppress target cis-splicing while enhancing trans-splicing between the PTM and target. The aim is to repair mutant pre-mRNA molecules and generate full length repaired mRNA that is translated and processed into mature CFTR protein [126]. In an in vivo model of ΔF508 CF airway epithelia, Liu et al. showed that human CF bronchial xenografts infected with a recombinant adenovirus encoding a PTM targeted to CFTR intron 9 demonstrated partial correction of CFTR-mediated Cl− permeability to 22% of that seen in non-CF xenograft [127]. This strategy would allow a more physiological expression of CFTR, as previously discussed. Furthermore, as PTM expression cassettes can be much smaller than those encoding full-length cDNAs, SMaRT also allows for the use of smaller and less immunogenic vectors, such as rAAV with limited packaging capacity. However, the very high titres required to achieve correction and the potential of trans-splicing into non-CFTR mRNAs are disadvantages which should not be ignored.
8.3. Physical methods
Because of the inefficiency of currently available GTAs, newer ways of increasing gene transfer have to be developed. Physical methods such as magnetism, electroporation and ultrasound have been employed by several groups as a means to enhance gene uptake. The feasibility of these methods is very organ- and tissue-specific and for the lung represents a challenge with many unknown aspects. Gersting et al. recently reported that application of a magnetic field to human primary airway epithelial cells transfected with plasmid DNA mixed with superparamagnetic nanoparticles (magnetofection) resulted in more than 100-fold increase in gene transfer [128]. The challenge is now to see whether this very promising technique [129] can be applied to the airway epithelium in vivo.
8.4. In utero gene transfer
Because of the hurdles and barriers involved with conventional gene transfer, several groups have begun to look into in utero gene delivery as a new way potentially to increase the efficacy and duration of transgene expression. CF is a particularly inviting target disease for the development of in utero gene therapy because amniotic fluid circulation provides vector exposure to pulmonary, gastrointestinal and sinus epithelia, all primary sites of CF pathology. Viral vector introduced into amniotic fluid of mice, rats, sheep and rabbits results in reporter gene expression in both pulmonary and gastrointestinal epithelia. Transgene expression was also demonstrated in pulmonary epithelia after intratracheal instillation of vector in utero. Persistence of transgene expression in the lung ranged from 14 to 30 days post-infection. Most of these studies have been carried out with adenoviral vectors, and many have led to substantial inflammation and subsequent foetal loss. Fewer adverse events have been observed when AAV is used. Retroviral vector use for applications in utero has been limited because the amniotic fluid reduces infectivity (see Ref. [130] for an extended overview).
The potential advantage of in utero gene transfer over other approaches is that, as the host immune system is not fully developed, it may be possible to tolerise the organism to viral vectors. However, recent reports have ruled out the possibility of successful repeated administration of viral vectors during adult life despite previous in utero exposure [131]. In humans this approach would be even less successful since the immune system becomes responsive by midgestation.
Larson et al. have recently presented an interesting and controversial report, showing that treatment of primate foetuses with an adenovirus expressing the cftr gene resulted in accelerated differentiation of the lung [132]. Furthermore, after in utero gene transfer with an adenovirus containing the cftr gene, a permanent reversion of the lethal phenotype in CF knockout mice was observed [133]. This might suggest that CF is a developmental disease that could be prevented by transient in utero cftr gene expression at the proper time of lung and intestine differentiation.
9. Methodological issues
Clinical trials have shown that there is clearly a requirement for newer approaches to improve delivery and efficiency, increase duration of expression and permit repeated administration of GTAs. In addition there is an increasing need by the scientific community that all these new GTAs and technologies are tested on relevant airway test systems (i.e., only highly differentiated epithelial cells in vitro and a spectrum of in vivo systems) before entering clinical trials. CF mice have been a great source of information in understanding the basic defect in cystic fibrosis. However, the development of animal models that reproduce the lung pathology in CF would greatly help the development and testing of gene-based therapies. For this reason big efforts are being made to develop a CF ferret [134] and a CF sheep [135], their lung biology being more similar to humans.
In addition, improved methods to detect whether the relevant airway epithelial cells in vivo have been transduced are needed. Several techniques have been developed to determine the location, duration and magnitude of transgene expression in living animals, including positron emission tomography (PET) and magnetic resonance imaging (MRI). Rooney et al. have recently developed a laser-induced fluorescence bronchoscopy as a method for non-invasively detecting adenoviral-mediated expression of green fluorescent protein in airway epithelia of rhesus monkeys [136]. Although these techniques are currently not sensitive enough, they might become in the near future a feasible option to detect gene expression in a non-invasive way in human airways.
10. Conclusion
In conclusion, in the 13 years since the cloning of the CFTR gene and after about 20 clinical trials, some of the crucial barriers limiting gene transfer have become clearer. The combination of newly developed GTAs and technologies, better models to test gene-based therapies and new detection systems will hopefully allow these barriers to be overcome.
Acknowledgements
This work was supported by the Cystic Fibrosis Research Trust (SF) and a Wellcome Trust Senior Clinical Fellowship (EWFWA). The authors are members of the UK Cystic Fibrosis Gene Therapy Consortium (www.cfgenetherapy.org.uk).
References
- 1.Zabner J., Couture L.A., Gregory R.J., Graham S.M., Smith A.E., Welsh M.J. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelium of patients with cystic fibrosis. Cell. 1993;75:207–216. doi: 10.1016/0092-8674(93)80063-k. [DOI] [PubMed] [Google Scholar]
- 2.Crystal R.G., McElvaney N.G., Rosenfeld M.A., Chu C.S., Mastrangeli A., Hay J.G., Brody S.L., Jaffe H.A., Eissa N.T., Danel C. Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat. Genet. 1994;8:42–51. doi: 10.1038/ng0994-42. [DOI] [PubMed] [Google Scholar]
- 3.Hay J.G., McElvaney N.G., Herena J., Crystal R.G. Modification of nasal epithelial potential differences of individuals with cystic fibrosis consequent to local administration of a normal CFTR cDNA gene transfer vector. Hum. Gene Ther. 1995;6:1487–1496. doi: 10.1089/hum.1995.6.11-1487. [DOI] [PubMed] [Google Scholar]
- 4.Knowles M.R., Hohneker K., Zhou Z., Olsen J.C., Noah T.L., Hu P.C., Leigh M.W., Engelhardt J.F., Edwards L.J., Jones K.R., Grossman M., Wilson J.M., Johnson L.G., Boucher R.C. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. New Engl. J. Med. 1995;333:823–831. doi: 10.1056/NEJM199509283331302. [DOI] [PubMed] [Google Scholar]
- 5.Zabner J., Ramsey B.W., Meeker D.P., Aitken M.L., Balfour R.P., Gibson R.L., Launspach J., Moscicki R.A., Richards S.M., Standaert T.A., Williams-Warren J., Wadsworth S.C., Smith A.E., Welsh M.J. Repeat administration of an adenovirus vector encoding cystic fibrosis transmembrane conductance regulator to the nasal epithelium of patients with cystic fibrosis. J. Clin. Invest. 1996;97:1504–1511. doi: 10.1172/JCI118573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellon G., Michel-Calemard L., Thouvenot D., Jagneaux V., Poitevin F., Malcus C., Accart N., Layani M.P., Aymard M., Bernon H., Bienvenu J., Courtney M., Doring G., Gilly B., Gilly R., Lamy D., Levrey H., Morel Y., Paulin C., Perraud F., Rodillon L., Sené C., So S., Touraine-Moulin F., Schatz C., Pavirani A. Aerosol administration of a recombinant adenovirus expressing CFTR to cystic fibrosis patients: a phase I clinical trial. Hum. Gene Ther. 1997;8:15–25. doi: 10.1089/hum.1997.8.1-15. [DOI] [PubMed] [Google Scholar]
- 7.Harvey B.G., Leopold P.L., Hackett N.R., Grasso T.M., Williams P.M., Tucker A.L., Kaner R.J., Ferris B., Gonda I., Sweeney T.D., Ramalingam R., Kovesdi I., Shak S., Crystal R.G. Airway epithelial CFTR mRNA expression in cystic fibrosis patients after repetitive administration of a recombinant adenovirus. J. Clin. Invest. 1999;104:1245–1255. doi: 10.1172/JCI7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuckerman J.B., Robinson C.B., McCoy K.S., Shell R., Sferra T.J., Chirmule N., Magosin S.A., Propert K.J., Brown-Parr E.C., Hughes J.V., Tazelaar J., Baker C., Goldman M.J., Wilson J.M. A phase I study of adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator gene to a lung segment of individuals with cystic fibrosis. Hum. Gene Ther. 1999;10:2973–2985. doi: 10.1089/10430349950016384. [DOI] [PubMed] [Google Scholar]
- 9.Perricone M.A., Morris J.E., Pavelka K., Plog M.S., O’Sullivan B.P., Joseph P.M., Dorkin H., Lapey A., Balfour R., Meeker D.P., Smith A.E., Wadsworth S.C., St. George J.A. Aerosol and lobar administration of a recombinant adenovirus to individuals with cystic fibrosis. II. Transfection efficiency in airway epithelium. Hum. Gene Ther. 2001;12:1383–1394. doi: 10.1089/104303401750298544. [DOI] [PubMed] [Google Scholar]
- 10.Wagner J.A., Reynolds T., Moran M.L., Moss R.B., Wine J.J., Flotte T.R., Gardner P. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet. 1998;351:1702–1703. doi: 10.1016/S0140-6736(05)77740-0. [DOI] [PubMed] [Google Scholar]
- 11.Aitken M.L., Moss R.B., Waltz D.A., Dovey M.E., Tonelli M.R., McNamara S.C., Gibson R.L., Ramsey B.W., Carter B.J., Reynolds T.C. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum. Gene Ther. 2001;12:1907–1916. doi: 10.1089/104303401753153956. [DOI] [PubMed] [Google Scholar]
- 12.Caplen N.J., Alton E.W.F.W., Middleton P.G., Dorin J.R., Stevenson B.J., Gao X., Durham S.R., Jeffery P.K., Hodson M.E., Coutelle C., Huang L., Porteous D.J., Williamson R., Geddes D.M. Liposome-mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Nat. Med. 1995;1:39–46. doi: 10.1038/nm0195-39. [DOI] [PubMed] [Google Scholar]
- 13.Zabner J., Cheng S.H., Meeker D., Launspach J., Balfour R., Perricone M.A., Morris J.E., Marshall J., Fasbender A., Smith A.E., Welsh M.J. Comparison of DNA-lipid complexes and DNA alone for gene transfer to cystic fibrosis airway epithelia in vivo. J. Clin. Invest. 1997;100:1529–1537. doi: 10.1172/JCI119676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gill D.R., Southern K.W., Mofford K.A., Seddon T., Huang L., Sorgi F., Thomson A., MacVinish L.J., Ratcliff R., Bilton D., Lane D.J., Littlewood J.M., Webb A.K., Middleton P.G., Colledge W.H., Cuthbert A.W., Evans M.J., Higgins C.F., Hyde S.C. A placebo-controlled study of liposome-mediated gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 1997;4:199–209. doi: 10.1038/sj.gt.3300391. [DOI] [PubMed] [Google Scholar]
- 15.Porteous D.J., Dorin J.R., McLachlan G., Davidson-Smith H., Davidson H., Stevenson B.J., Carothers A.D., Wallace W.A.H., Moralee S., Hoenes C., Kallmeyer G., Michaelis U., Naujoks K., Ho L.P., Samways J.M., Imrie M., Greening A.P., Innes J.A. Evidence for safety and efficacy of DOTAP cationic liposome mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 1997;4:210–218. doi: 10.1038/sj.gt.3300390. [DOI] [PubMed] [Google Scholar]
- 16.Alton E.W.F.W., Stern M., Farley R., Jaffe A., Chadwick S.L., Phillips J., Davies J., Smith S.N., Browning J., Davies M.G., Hodson M.E., Durham S.R., Li D., Jeffery P.K., Scallan M., Balfour R., Eastman S.J., Cheng S.H., Smith A.E., Meeker D., Geddes D.M. Cationic lipid-mediated CFTR gene transfer to the lungs and nose of patients with cystic fibrosis: a double-blind placebo-controlled trial. Lancet. 1999;353:947–954. doi: 10.1016/s0140-6736(98)06532-5. [DOI] [PubMed] [Google Scholar]
- 17.Hyde S.C., Southern K.W., Gileadi U., Fitzjohn E.M., Mofford K.A., Waddell B.E., Gooi H.C., Goddard C.A., Hannavy K., Smyth S.E., Egan J.J., Sorgi F.L., Huang L., Cuthbert A.W., Evans M.J., Colledge W.H., Higgins C.F., Webb A.K., Gill D.R. Repeat administration of DNA/liposomes to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 2000;7:1156–1165. doi: 10.1038/sj.gt.3301212. [DOI] [PubMed] [Google Scholar]
- 18.Noone P.G., Hohneker K.W., Zhou Z., Johnson L.G., Foy C., Gipson C., Jones K., Noah T.L., Leigh M.W., Schwartzbach C., Efthimiou J., Pearlman R., Boucher R.C., Knowles M.R. Safety and biological efficacy of a lipid-CFTR complex for gene transfer in the nasal epithelium of adult patients with cystic fibrosis. Mol. Ther. 2000;1:105–114. doi: 10.1006/mthe.1999.0009. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz F.E., Clancy J.P., Perricone M.A., Bebok Z., Hong J.S., Cheng S.H., Meeker D.P., Young K.R., Schoumacher R.A., Weatherly M.R., Wing L., Morris J.E., Sindel L., Rosenberg M., Van Ginkel F.W., McGhee J.R., Kelly D., Lyrene R.K., Sorscher E.J. A clinical inflammatory syndrome attributable to aerosolised lipid-DNA administration in Cystic Fibrosis. Hum. Gene Ther. 2001;12:751–761. doi: 10.1089/104303401750148667. [DOI] [PubMed] [Google Scholar]
- 20.Sanders N.N., De Smedt S.C., Van Rompaey E., Simoens P., De Baets F., Demeester J. Cystic fibrosis sputum: a barrier to the transport of nanospheres. Am. J. Respir. Crit. Care Med. 2000;162:1905–1911. doi: 10.1164/ajrccm.162.5.9909009. [DOI] [PubMed] [Google Scholar]
- 21.Sanders N.N., Van Rompaey E., De Smedt S.C., Demeester J. Structural alterations of gene complexes by cystic fibrosis sputum. Am. J. Respir. Crit. Care Med. 2001;164:486–493. doi: 10.1164/ajrccm.164.3.2011041. [DOI] [PubMed] [Google Scholar]
- 22.Yonemitsu Y., Kitson C., Ferrari S., Farley R., Griesenbach U., Judd D., Steel R., Scheid P., Zhu J., Jeffery P.K., Kato A., Hasan M.K., Nagai Y., Masaki I., Fukumura M., Hasegawa M., Geddes D.M., Alton E.W. Efficient gene transfer to airway epithelium using recombinant Sendai virus. Nat. Biotechnol. 2000;18:970–973. doi: 10.1038/79463. [DOI] [PubMed] [Google Scholar]
- 23.Kitson C., Angel B., Judd D., Rothery S., Severs N.J., Dewar A., Huang L., Wadsworth S.C., Cheng S.H., Geddes D.M., Alton E.W.F.W. The extra- and intracellular barriers to lipid and adenovirus-mediated pulmonary gene transfer in native sheep airway epithelium. Gene Ther. 1999;6:534–546. doi: 10.1038/sj.gt.3300840. [DOI] [PubMed] [Google Scholar]
- 24.Ferrari S., Kitson C., Farley R., Steel R., Marriott C., Parkins D.A., Scarpa M., Wainwright B., Evans M.J., Colledge W.H., Geddes D.M., Alton E.W. Mucus altering agents as adjuncts for nonviral gene transfer to airway epithelium. Gene Ther. 2001;8:1380–1386. doi: 10.1038/sj.gt.3301525. [DOI] [PubMed] [Google Scholar]
- 25.Stern M., Caplen N.J., Browning J.E., Griesenbach U., Sorgi F., Huang L., Gruenert D.C., Marriot C., Crystal R.G., Geddes D.M., Alton E.W.F.W. The effect of mucolytic agents on gene transfer across a CF sputum barrier in vitro. Gene Ther. 1998;5:91–98. doi: 10.1038/sj.gt.3300556. [DOI] [PubMed] [Google Scholar]
- 26.Perricone M.A., Rees D.D., Sacks C.R., Smith K.A., Kaplan J.M., St. George J.A. Inhibitory effect of cystic fibrosis sputum on adenovirus-mediated gene transfer in cultured epithelial cells. Hum. Gene Ther. 2000;11:1997–2008. doi: 10.1089/10430340050143426. [DOI] [PubMed] [Google Scholar]
- 27.Virella-Lowell I., Poirier A., Chesnut K.A., Brantly M., Flotte T.R. Inhibition of recombinant adeno-associated virus (rAAV) transduction by bronchial secretions from cystic fibrosis patients. Gene Ther. 2000;7:1783–1789. doi: 10.1038/sj.gt.3301268. [DOI] [PubMed] [Google Scholar]
- 28.Ferrari S., Pettenazzo A., Garbati N., Zacchello F., Behr J.P., Scarpa M. Polyethylenimine shows properties of interest for cystic fibrosis gene therapy. Biochim. Biophys. Acta. 1999;1447:219–225. doi: 10.1016/s0167-4781(99)00153-0. [DOI] [PubMed] [Google Scholar]
- 29.Pickles R.J., Fahrner J.A., Petrella J.M., Boucher R.C., Bergelson J.M. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J. Virol. 2000;74:6050–6057. doi: 10.1128/jvi.74.13.6050-6057.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griesenbach U., Chonn A., Cassady R., Hannam V., Ackerley C., Post M., Tanswell A.K., Olek K., O’Brodovich H., Tsui L.C. Comparison between intratracheal and intravenous administration of liposome-DNA complexes for cystic fibrosis lung gene therapy. Gene Ther. 1998;5:181–188. doi: 10.1038/sj.gt.3300562. [DOI] [PubMed] [Google Scholar]
- 31.Zhu N., Liggitt D., Liu Y., Debs R. Systemic gene expression after intravenous DNA delivery into adult mice. Science. 1993;261:209–211. doi: 10.1126/science.7687073. [DOI] [PubMed] [Google Scholar]
- 32.Koehler D.R., Hannam V., Belcastro R., Steer B., Wen Y., Post M., Downey G., Tanswell A.K., Hu J. Targeting transgene expression for cystic fibrosis gene therapy. Mol. Ther. 2001;4:58–65. doi: 10.1006/mthe.2001.0412. [DOI] [PubMed] [Google Scholar]
- 33.Chirmule N., Propert K., Magosin S., Qian R., Wilson J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999;6:1574–1583. doi: 10.1038/sj.gt.3300994. [DOI] [PubMed] [Google Scholar]
- 34.Harvey B.G., Hackett N.R., El-Sawy T., Rosengart T.K., Hirschowitz E.A., Lieberman M.D., Lesser M.L., Crystal R.G. Variability of human systemic humoral immune responses to adenovirus gene transfer vectors administered to different organs. J. Virol. 1999;73:6729–6742. doi: 10.1128/jvi.73.8.6729-6742.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pickles R.J., Barker P.M., Ye H., Boucher R.C. Efficient adenovirus-mediated gene transfer to basal but not columnar cells of cartilaginous airway epithelia. Hum. Gene Ther. 1996;7:921–931. doi: 10.1089/hum.1996.7.8-921. [DOI] [PubMed] [Google Scholar]
- 36.Jiang C., O’Connor S.P., Fang S.L., Wang K.X., Marshall J., Williams J.L., Wilburn B., Echelard Y., Cheng S.H. Efficiency of cationic lipid-mediated transfection of polarized and differentiated airway epithelial cells in vitro and in vivo. Hum. Gene Ther. 1998;9:1531–1542. doi: 10.1089/hum.1998.9.11-1531. [DOI] [PubMed] [Google Scholar]
- 37.Matsui H., Johnson L.G., Randell S.H., Boucher R.C. Loss of binding and entry of liposome-DNA complexes decreases transfection efficiency in differentiated airway epithelial cells. J. Biol. Chem. 1997;272:1117–1126. doi: 10.1074/jbc.272.2.1117. [DOI] [PubMed] [Google Scholar]
- 38.Fasbender A., Zabner J., Zeiher B.G., Welsh M.J. A low rate of cell proliferation and reduced DNA uptake limit cationic lipid-mediated gene transfer to primary cultures of ciliated human airway epithelia. Gene Ther. 1997;4:1173–1180. doi: 10.1038/sj.gt.3300524. [DOI] [PubMed] [Google Scholar]
- 39.Pickles R.J., McCarty D., Matsui H., Hart P.J., Randell S.H., Boucher R.C. Limited entry of adenovirus vectors into well-differentiated airway epithelium is responsible for inefficient gene transfer. J. Virol. 1998;72:6014–6023. doi: 10.1128/jvi.72.7.6014-6023.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan D., Yue Y., Yan Z., McCray P.B., Jr., Engelhardt J.F. Polarity influences the efficiency of recombinant adenoassociated virus infection in differentiated airway epithelia. Hum. Gene Ther. 1998;9:2761–2776. doi: 10.1089/hum.1998.9.18-2761. [DOI] [PubMed] [Google Scholar]
- 41.Wang G., Zabner J., Deering C., Launspach J., Shao J., Bodner M., Jolly D.J., Davidson B.L., McCray P.B., Jr. Increasing epithelial junction permeability enhances gene transfer to airway epithelia in vivo. Am. J. Respir. Cell Mol. Biol. 2000;22:129–138. doi: 10.1165/ajrcmb.22.2.3938. [DOI] [PubMed] [Google Scholar]
- 42.Chu Q., St. George J.A., Lukason M., Cheng S.H., Scheule R.K., Eastman S.J. EGTA enhancement of adenovirus-mediated gene transfer to mouse tracheal epithelium in vivo. Hum. Gene Ther. 2001;12:455–467. doi: 10.1089/104303401300042348. [DOI] [PubMed] [Google Scholar]
- 43.Parsons D.W., Grubb B.R., Johnson L.G., Boucher R.C. Enhanced in vivo airway gene transfer via transient modification of host barrier properties with a surface-active agent. Hum. Gene Ther. 1998;9:2661–2672. doi: 10.1089/hum.1998.9.18-2661. [DOI] [PubMed] [Google Scholar]
- 44.Man Y., Hart V.J., Ring C.J.A., Sanjar S., West M.R. Loss of epithelial integrity resulting from E-cadherin dysfunction predisposes airway epithelial cells to adenoviral infection. Am. J. Respir. Cell Mol. Biol. 2000;23:610–617. doi: 10.1165/ajrcmb.23.5.4046. [DOI] [PubMed] [Google Scholar]
- 45.Coyne C.B., Kelly M.M., Boucher R.C., Johnson L.G. Enhanced epithelial gene transfer by modulation of tight junctions with sodium caprate. Am. J. Respir. Cell Mol. Biol. 2000;23:602–609. doi: 10.1165/ajrcmb.23.5.4164. [DOI] [PubMed] [Google Scholar]
- 46.Croyle M.A., Cheng X., Sandhu A., Wilson J.M. Development of novel formulations that enhance adenoviral-mediated gene expression in the lung in vitro and in vivo. Mol. Ther. 2001;4:22–28. doi: 10.1006/mthe.2001.0411. [DOI] [PubMed] [Google Scholar]
- 47.Weiss D.J., Strandjord T.P., Jackson J.C., Clark J.G., Liggitt D. Perfluorochemical liquid-enhanced adenoviral vector distribution and expression in lungs of spontaneously breathing rodents. Exp. Lung Res. 1999;25:317–333. doi: 10.1080/019021499270222. [DOI] [PubMed] [Google Scholar]
- 48.Weiss D.J., Bonneau L., Allen J.M., Miller A.D., Halbert C.L. Perfluorochemical liquid enhances adeno-associated virus-mediated transgene expression in lung. Mol. Ther. 2000;2:624–630. doi: 10.1006/mthe.2000.0207. [DOI] [PubMed] [Google Scholar]
- 49.Das A., Niven R. Use of perfluorocarbon (Fluorinert) to enhance reporter gene expression following intratracheal instillation into the lungs of Balb/c mice: implications for nebulized delivery of plasmids. J. Pharm. Sci. 2001;90:1336–1344. doi: 10.1002/jps.1086. [DOI] [PubMed] [Google Scholar]
- 50.Lee J.H., Baker T.J., Mahal L.K., Bertozzi C.R., Wiemer D.F., Welsh M.J. Engineering novel cell surface receptors for virus-mediated gene transfer. J. Biol. Chem. 1999;274:21878–21884. doi: 10.1074/jbc.274.31.21878. [DOI] [PubMed] [Google Scholar]
- 51.Kreda S.M., Pickles R.J., Lazarowski E.R., Boucher R.C. G-protein-coupled receptors as targets for gene transfer vectors using natural small-molecules ligands. Nat. Biotechnol. 2000;18:635–640. doi: 10.1038/76479. [DOI] [PubMed] [Google Scholar]
- 52.R.J. Pickles, A.J. Hirsh, R.J. Gerard, R.C. Boucher, Retargeting Ad5 fibers to bradykinin receptors expressed on the lumenal surface of human airway epithelium, Pediatr. Pulmonol. Suppl. 22 (2001) Abstr. 215.
- 53.Drapkin P.T., O’Riordan C.R., Yi S.M., Chiorini J.A., Cardella J., Zabner J., Welsh M.J. Targeting the urokinase plasminogen activator receptor enhances gene transfer to human airway epithelia. J. Clin. Invest. 2000;105:589–596. doi: 10.1172/JCI8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.A.G. Ziady, T. Kelley, P.B. Davis, Molecular conjugate mediated CFTR gene transfer in CF mice affects secondary CF defects, Pediatr. Pulmonol. Suppl. 22 (2001) Abstr. 236.
- 55.Jost P.J., Harbottle R.P., Knight A., Miller A.D., Coutelle C., Schneider H. A novel peptide, THALWHT, for the targeting of human airway epithelia. FEBS Lett. 2001;489:263–269. doi: 10.1016/s0014-5793(00)02236-5. [DOI] [PubMed] [Google Scholar]
- 56.Wang G., Deering C., Macke M., Shao J., Burns R., Blau D.M., Holmes K.V., Davidson B.L., Perlman S., McCray P.B., Jr. Human coronavirus 229E infects polarized airway epithelia from the apical surface. J. Virol. 2000;74:9234–9239. doi: 10.1128/jvi.74.19.9234-9239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Slepushkin V.A., Staber P.D., Wang G., McCray P.B., Jr., Davidson B.L. Infection of human airway epithelia with H1N1, H2N2, and H3N2 influenza A virus strains. Mol. Ther. 2001;3:395–402. doi: 10.1006/mthe.2001.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobinger G.P., Weiner D.J., Yu Q.C., Wilson J.M. Filovirus-pseudotyped lentiviral vector can efficiently and stably transduce airway epithelia in vivo. Nat. Biotechnol. 2001;19:225–230. doi: 10.1038/85664. [DOI] [PubMed] [Google Scholar]
- 59.Kaplan J.M., Pennington S.E., St. George J.A., Woodworth L.A., Fasbender A., Marshall J., Cheng S.H., Wadsworth S.C., Gregory R.J., Smith A.E. Potentiation of gene transfer to the mouse lung by complexes of adenovirus vector and polycations improves therapeutic potential. Hum. Gene Ther. 1998;9:1469–1479. doi: 10.1089/hum.1998.9.10-1469. [DOI] [PubMed] [Google Scholar]
- 60.Fasbender A., Lee J.H., Walters R.W., Moninger T.O., Zabner J., Welsh M.J. Incorporation of adenovirus in calcium phosphate precipitates enhances gene transfer to airway epithelia in vitro and in vivo. J. Clin. Invest. 1998;102:184–193. doi: 10.1172/JCI2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zabner J., Zeiher B.G., Friedman E., Welsh M.J. Adenovirus-mediated gene transfer to ciliated airway epithelia requires prolonged incubation time. J. Virol. 1996;70:6994–7003. doi: 10.1128/jvi.70.10.6994-7003.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walters R.W., Yi S.M., Keshavjee S., Brown K.E., Welsh M.J., Chiorini J.A., Zabner J. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J. Biol. Chem. 2001;276:20610–20616. doi: 10.1074/jbc.M101559200. [DOI] [PubMed] [Google Scholar]
- 63.L. Zhang, P.L. Collins, R.C. Boucher, R.J. Pickles, Respiratory syncytial virus (RSV) infects ciliated cells of airway epithelium via the lumenal membrane, Pediatr. Pulmonol. Suppl. 22 (2001) Abstr. 207.
- 64.Boussif O., Lezoualc’h F., Zanta M.A., Mergny M.D., Scherman D., Demeneix B., Behr J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Behr J.P. L’èponge à protons: un moyen d’entrer dans une cellule auquel les virus n’ont pas pensé. Mèd. Sci. 1996;12:56–59. [Google Scholar]
- 66.Hansen J., Qing K., Srivastava A. Adeno-associated virus type 2-mediated gene transfer: altered endocytic processing enhances transduction efficiency in murine fibroblasts. J. Virol. 2001;75:4080–4090. doi: 10.1128/JVI.75.9.4080-4090.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yonemitsu Y., Kaneda Y., Muraishi A., Yoshizumi T., Sugimachi K., Sueishi K. HVJ (Sendai virus)-cationic liposomes: a novel and potentially effective liposome-mediated technique for gene transfer to the airway epithelium. Gene Ther. 1997;4:631–638. doi: 10.1038/sj.gt.3300463. [DOI] [PubMed] [Google Scholar]
- 68.Lechardeur D., Sohn K.J., Haardt M., Joshi P.B., Monck M., Graham R.W., Beatty B., Squire J., O’Brodovich H., Lukacs G.L. Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer. Gene Ther. 1999;6:482–497. doi: 10.1038/sj.gt.3300867. [DOI] [PubMed] [Google Scholar]
- 69.Pollard H., Toumaniantz G., Amos J.L., Avet-Loiseau H., Guihard G., Behr J.P., Escande D. Ca2+-sensitive cytosolic nucleases prevent efficient delivery to the nucleus of injected plasmids. J. Gene Med. 2001;3:153–164. doi: 10.1002/jgm.160. [DOI] [PubMed] [Google Scholar]
- 70.Duan D., Yue Y., Yan Z., Yang J., Engelhardt J.F. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J. Clin. Invest. 2000;105:1573–1587. doi: 10.1172/JCI8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pollard H., Remy J.S., Loussouarn G., Demolombe S., Behr J.P., Escande D. Polyethylenimine but not cationic lipids promotes transgene delivery to the nucleus in mammalian cells. J. Biol. Chem. 1998;273:7507–7511. doi: 10.1074/jbc.273.13.7507. [DOI] [PubMed] [Google Scholar]
- 72.Tachibana R., Harashima H., Shinohara Y., Kiwada H. Quantitative studies on the nuclear transport of plasmid DNA and gene expression employing nonviral vectors. Adv. Drug Deliv. Rev. 2001;52:219–226. doi: 10.1016/s0169-409x(01)00211-3. [DOI] [PubMed] [Google Scholar]
- 73.Sebestyen M.G., Ludtke J.J., Bassik M.C., Zhang G., Budker V., Lukhtanov E.A., Hagstrom J.E., Wolff J.A. DNA vector chemistry: the covalent attachment of signal peptides to plasmid DNA. Nat. Biotechnol. 1998;16:80–85. doi: 10.1038/nbt0198-80. [DOI] [PubMed] [Google Scholar]
- 74.Zanta M.A., Belguise-Valladier P., Behr J.P. Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc. Natl. Acad. Sci. USA. 1999;96:91–96. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dean D.A. Peptide nucleic acids: versatile tools for gene therapy strategies. Adv. Drug Deliv. Rev. 2000;44:81–95. doi: 10.1016/s0169-409x(00)00087-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klink D.T., Chao S., Glick M.C., Scanlin T.F. Nuclear translocation of lactosylated poly-l-lysine/cDNA complex in cystic fibrosis airway epithelial cells. Mol. Ther. 2001;3:831–841. doi: 10.1006/mthe.2001.0332. [DOI] [PubMed] [Google Scholar]
- 77.Whittaker G.R., Helenius A. Nuclear import and export of viruses and virus genomes. Virology. 1998;246:1–23. doi: 10.1006/viro.1998.9165. [DOI] [PubMed] [Google Scholar]
- 78.Trotman L.C., Mosberger N., Fornerod M., Stidwill R.P., Greber U.F. Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 2001;3:1092–1100. doi: 10.1038/ncb1201-1092. [DOI] [PubMed] [Google Scholar]
- 79.Zennou V., Petit C., Guetard D., Nerhbass U., Montagnier L., Charneau P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 2000;101:173–185. doi: 10.1016/S0092-8674(00)80828-4. [DOI] [PubMed] [Google Scholar]
- 80.Follenzi A., Ailles L.E., Bakovic S., Geuna M., Naldini L. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat. Genet. 2000;25:217–222. doi: 10.1038/76095. [DOI] [PubMed] [Google Scholar]
- 81.Jooss K., Yang Y., Wilson J.M. Cyclophosphamide diminishes inflammation and prolongs transgene expression following delivery of adenoviral vectors to mouse liver and lung. Hum. Gene Ther. 1996;7:1555–1566. doi: 10.1089/hum.1996.7.13-1555. [DOI] [PubMed] [Google Scholar]
- 82.Kolb M., Inman M., Margetts P.J., Galt T., Gauldie J. Budesonide enhances repeated gene transfer and expression in the lung with adenoviral vectors. Am. J. Respir. Crit. Care Med. 2001;164:866–872. doi: 10.1164/ajrccm.164.5.2008066. [DOI] [PubMed] [Google Scholar]
- 83.Yang Y., Trinchieri G., Wilson J.M. Recombinant IL-12 prevents formation of blocking IgA antibodies to recombinant adenovirus and allows repeated gene therapy to mouse lungs. Nat. Med. 1995;1:890–893. doi: 10.1038/nm0995-890. [DOI] [PubMed] [Google Scholar]
- 84.Chirmule N., Truneh A., Haecker S.E., Tazelaar J., Gao G., Raper S.E., Hughes J.V., Wilson J.M. Repeated administration of adenoviral vectors in lungs of human CD4 transgenic mice treated with a nondepleting CD4 antibody. J. Immunol. 1999;163:448–455. [PubMed] [Google Scholar]
- 85.Scaria A., St. George J.A., Gregory R.J., Noelle R.J., Wadsworth S.C., Smith A.E., Kaplan J.M. Antibody to CD40 ligand inhibits both humoral and cellular immune responses to adenoviral vectors and facilitates repeated administration to mouse airway. Gene Ther. 1997;4:611–617. doi: 10.1038/sj.gt.3300431. [DOI] [PubMed] [Google Scholar]
- 86.Jooss K., Turka L.A., Wilson J.M. Blunting of immune responses to adenoviral vectors in mouse liver and lung with CTLA4Ig. Gene Ther. 1998;5:309–319. doi: 10.1038/sj.gt.3300595. [DOI] [PubMed] [Google Scholar]
- 87.Chirmule N., Raper S.E., Burkly L., Thomas D., Tazelaar J., Hughes J.V., Wilson J.M. Readministration of adenovirus vector in nonhuman primate lungs by blockade of CD40-CD40 ligand interactions. J. Virol. 2000;74:3345–3352. doi: 10.1128/jvi.74.7.3345-3352.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.O’Riordan C.R., Lachapelle A., Delgado C., Parkes V., Wadsworth S.C., Smith A.E., Francis G.E. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum. Gene Ther. 1999;10:1349–1358. doi: 10.1089/10430349950018021. [DOI] [PubMed] [Google Scholar]
- 89.Chillon M., Lee J.H., Fasbender A., Welsh M.J. Adenovirus complexed with polyethylene glycol and cationic lipid is shielded from neutralizing antibodies in vitro. Gene Ther. 1998;5:995–1002. doi: 10.1038/sj.gt.3300665. [DOI] [PubMed] [Google Scholar]
- 90.Mack C.A., Song W.R., Carpenter H., Wickham T.J., Kovesdi I., Harvey B.G., Magovern C.J., Isom O.W., Rosengart T., Falck-Pedersen E., Hackett N.R., Crystal R.G., Mastrangeli A. Circumvention of anti-adenovirus neutralizing immunity by administration of an adenoviral vector of an alternate serotype. Hum. Gene Ther. 1997;8:99–109. doi: 10.1089/hum.1997.8.1-99. [DOI] [PubMed] [Google Scholar]
- 91.Croyle M.A., Chirmule N., Zhang Y., Wilson J.M. ‘Stealth’ adenovirus blunt cell-mediated and humoral immune responses against the virus and allow for significant gene expression upon readministration in the lung. J. Virol. 2001;75:4792–4801. doi: 10.1128/JVI.75.10.4792-4801.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beck S.E., Jones L.A., Chesnut K., Walsh S.M., Reynolds T.C., Carter B.J., Askin F.B., Flotte T.R., Guggino W.B. Repeated delivery of adeno-associated virus vectors to the rabbit airway. J. Virol. 1999;73:9446–9455. doi: 10.1128/jvi.73.11.9446-9455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Halbert C.L., Rutledge E.A., Allen J.M., Russel D.W., Miller A.D. Repeat transduction in the mouse lung by using adeno-associated virus vectors with different serotypes. J. Virol. 2000;74:1524–1532. doi: 10.1128/jvi.74.3.1524-1532.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Manning W.C., Zhou S., Bland M.P., Escobedo J.A., Dwarki V. Transient immunosuppression allows transgene expression following readministration of adeno-associated viral vectors. Hum. Gene Ther. 1998;9:477–485. doi: 10.1089/hum.1998.9.4-477. [DOI] [PubMed] [Google Scholar]
- 95.Halbert C.L., Standaert T.A., Wilson C.B., Miller A.D. Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J. Virol. 1998;72:9795–9805. doi: 10.1128/jvi.72.12.9795-9805.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang L., Wang D., Fischer H., Fan P.D., Widdicombe J.H., Kan Y.W., Dong J.Y. Efficient expression of CFTR function with adeno-associated virus vectors that carry shortened CFTR genes. Proc. Natl. Acad. Sci. USA. 1998;95:10158–10163. doi: 10.1073/pnas.95.17.10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duan D., Yue Y., Engelhardt J.F. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol. Ther. 2001;4:383–391. doi: 10.1006/mthe.2001.0456. [DOI] [PubMed] [Google Scholar]
- 98.D. Duan, Y. Yue, J.F. Engelhardt, Expanding AAV packaging capacity with trans-splicing and overlapping vectors: a quantitative comparison, Pediatr. Pulmonol. Suppl. 22 (2001) Abstr. 221. [DOI] [PubMed]
- 99.Goddard C.A., Ratcliff R., Anderson J.R., Glenn E., Brown S., Gill D.R., Hyde S.C., MacVinish L.J., Huang L., Higgins C.F., Cuthbert A.W., Evans M.J., Colledge W.H. A second dose of a CFTR cDNA-liposome complex is as effective as the first dose in restoring cAMP-dependent chloride secretion to null CF mice trachea. Gene Ther. 1997;4:1231–1236. doi: 10.1038/sj.gt.3300515. [DOI] [PubMed] [Google Scholar]
- 100.Lee E.R., Marshall J., Siegel C.S., Jiang C., Yew N.S., Nichols M.R., Nietupski J.B., Ziegler R.J., Lane M.B., Wang K.X., Wan N.C., Scheule R.K., Harris D.J., Smith A.E., Cheng S.H. Detailed analysis of structures and formulations of cationic lipids for efficient gene transfer to the lung. Hum. Gene Ther. 1996;7:1701–1717. doi: 10.1089/hum.1996.7.14-1701. [DOI] [PubMed] [Google Scholar]
- 101.Scheule R.K., St. George J.A., Bagley R.G., Marshall J., Kaplan J.M., Akita G.Y., Wang K.X., Lee E.R., Harris D.J., Jiang C., Yew N.S., Smith A.E., Cheng S.H. Basis of pulmonary toxicity associated with cationic lipid-mediated gene transfer to the mammalian lung. Hum. Gene Ther. 1997;8:689–707. doi: 10.1089/hum.1997.8.6-689. [DOI] [PubMed] [Google Scholar]
- 102.Chadwick S.L., Kingston H.D., Stern M., Cook R.M., O’Connor B.J., Lukasson M., Balfour R.P., Rosenberg M., Cheng S.H., Smith A.E., Meeker D.P., Geddes D.M., Alton E.W. Safety of a single aerosol administration of escalating doses of the cationic lipid GL-67/DOPE/DMPE-PEG5000 formulation to the lungs of normal volunteers. Gene Ther. 1997;4:937–942. doi: 10.1038/sj.gt.3300481. [DOI] [PubMed] [Google Scholar]
- 103.Schwartz D.A., Quinn T.J., Thorne P.S., Sayeed S., Yi A.K., Krieg A.M. CpG motifs in bacterial DNA cause inflammation in the lower respiratory tract. J. Clin. Invest. 1997;100:68–73. doi: 10.1172/JCI119523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yew N.S., Wang K.X., Przybylska M., Bagley R.G., Stedman M., Marshall J., Scheule R.K., Cheng S.H. Contribution of plasmid DNA to inflammation in the lung after administration of cationic lipid:pDNA complexes. Hum. Gene Ther. 1999;10:223–234. doi: 10.1089/10430349950019011. [DOI] [PubMed] [Google Scholar]
- 105.Yew N.S., Zhao H., Wu I.H., Song A., Tousignant J.D., Przybylska M., Cheng S.H. Reduced inflammatory responses to plasmid DNA vectors by elimination and inhibition of immunostimulatory CpG motifs. Mol. Ther. 2000;1:255–262. doi: 10.1006/mthe.2000.0036. [DOI] [PubMed] [Google Scholar]
- 106.Cassivi S.D., Liu M., Boehler A., Pierre A., Tanswell A.K., O’Brodovich H., Mullen J.B.M., Slutsky A.S., Keshavjee S.H. Transplant immunosuppression increases and prolongs transgene expression following adenoviral-mediated transfection of rat lungs. J. Heart Lung Transplant. 2000;19:984–994. doi: 10.1016/s1053-2498(00)00166-2. [DOI] [PubMed] [Google Scholar]
- 107.Scaria A., Sullivan J.A., St. George J.A., Kaplan J.M., Lukason M.J., Morris J.E., Plog M., Nicolette C., Gregory R.J., Wadsworth S.C. Adenoviral vector expressing ICP47 inhibits adenovirus-specific cytotoxic T lymphocytes in nonhuman primates. Mol. Ther. 2000;2:505–514. doi: 10.1006/mthe.2000.0197. [DOI] [PubMed] [Google Scholar]
- 108.Gill D.R., Smyth S.E., Goddard C.A., Pringle I.A., Higgins C.F., Colledge W.H., Hyde S.C. Increased persistence of lung gene expression using plasmids containing the ubiquitin C or elongation factor 1α promoter. Gene Ther. 2001;8:1539–1546. doi: 10.1038/sj.gt.3301561. [DOI] [PubMed] [Google Scholar]
- 109.Yew N.S., Przybylska M., Ziegler R.J., Liu D., Cheng S.H. High and sustained transgene expression in vivo from plasmid vector containing a hybrid ubiquitin promoter. Mol. Ther. 2001;4:75–82. doi: 10.1006/mthe.2001.0415. [DOI] [PubMed] [Google Scholar]
- 110.Yew N.S., Marshall J., Przybylska M., Wysokenski D.M., Ziegler R.J., Rafter P.W., Li C., Armentano D., Cheng S.H. Increased duration of transgene expression in the lung with plasmid DNA vectors harbouring adenovirus E4 open reading frame 3. Hum. Gene Ther. 1999;10:1833–1843. doi: 10.1089/10430349950017518. [DOI] [PubMed] [Google Scholar]
- 111.Stoll S.M., Sclimenti C.R., Baba E.J., Meuse L., Kay M.A., Calos M.P. Epstein–Barr virus/human vector provides high-level, long-term expression of α1-antitrypsin in mice. Mol. Ther. 2001;4:122–129. doi: 10.1006/mthe.2001.0429. [DOI] [PubMed] [Google Scholar]
- 112.Huertas D., Howe S., McGuigan A., Huxley C. Expression of the human CFTR gene from episomal oriP-EBNA1-YACs in mouse cells. Hum. Mol. Genet. 2000;9:617–629. doi: 10.1093/hmg/9.4.617. [DOI] [PubMed] [Google Scholar]
- 113.Kearns W.G., Afione S.A., Fulmer S.B., Pang M.C., Erikson D., Egan M., Landrum M.J., Flotte T.R., Cutting G.R. Recombinant adeno-associated virus (AAV-CFTR) vectors do not integrate in a site-specific fashion in an immortalized epithelial cell line. Gene Ther. 1996;3:748–755. [PubMed] [Google Scholar]
- 114.Nakai H., Yant S.R., Storm T.A., Fuess S., Meuse L., Kay M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Borthwick D.W., Shahbazian M., Todd Krantz Q., Dorin J.R., Randell S.H. Evidence for stem-cell niches in the tracheal epithelium. Am. J. Respir. Cell Mol. Biol. 2001;24:662–670. doi: 10.1165/ajrcmb.24.6.4217. [DOI] [PubMed] [Google Scholar]
- 116.Johnson L.G., Olsen J.C., Sarkadi B., Moore K.L., Swanstrom R., Boucher R.C. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 117.Dorin J.R., Farley R., Webb S., Smith S.N., Farini E., Delaney S.J., Wainwright B.J., Alton E.W.F.W., Porteous D.J. A demonstration using mouse models that successful gene therapy for cystic fibrosis requires only partial gene correction. Gene Ther. 1996;3:797–801. [PubMed] [Google Scholar]
- 118.Zhang Y., Jiang Q., Dudus L., Yankaskas J.R., Engelhardt J.F. Vector-specific complementation profiles of two independent primary defects in cystic fibrosis airways. Hum. Gene Ther. 1998;9:635–648. doi: 10.1089/hum.1998.9.5-635. [DOI] [PubMed] [Google Scholar]
- 119.Gautam A., Densmore C.L., Golunski E., Xu B., Waldrep J.C. Transgene expression in mouse airway epithelium by aerosol gene therapy with PEI-DNA complexes. Mol. Ther. 2001;3:551–556. doi: 10.1006/mthe.2001.0300. [DOI] [PubMed] [Google Scholar]
- 120.Schiavi S.C., Abdelkader N., Reber S., Pennington S., Narayana R., McPherson J.M., Smith A.E., Hoppe H., IV, Cheng S.H. Biosynthetic and growth abnormalities are associated with high-level expression of CFTR in heterologous cells. Am. J. Physiol. 1996;270:C341–C351. doi: 10.1152/ajpcell.1996.270.1.C341. [DOI] [PubMed] [Google Scholar]
- 121.Ye L., Chan S., Chow Y.H., Tsui L.C., Hu J. Regulated expression of the human CFTR gene in epithelial cells. Mol. Ther. 2001;3:723–733. doi: 10.1006/mthe.2001.0314. [DOI] [PubMed] [Google Scholar]
- 122.Goncz K.K., Colosimo A., Dallapiccola B., Gagné L., Homg K., Novelli G., Papahadjopoulos D., Sawa T., Schreier H., Wiener-Kronish J., Xu Z., Gruenert D.C. Expression of ΔF508 CFTR in normal lung after site-specific modification of CFTR sequences by SFHR. Gene Ther. 2001;8:961–965. doi: 10.1038/sj.gt.3301476. [DOI] [PubMed] [Google Scholar]
- 123.U. Griesenbach, B.T. Kren, M. Stern, R.L. Cassady, S. Jueliger, E. Hillery, R. Farley, D.M. Geddes, C.J. Steer, E.W. Alton, Conversion of wild-type CFTR to the G551D mutation in primary rat hepatocytes using RNA/DNA oligonucleotides, Pediatr. Pulmonol. Suppl. 22 (2001) Abstr. 231.
- 124.Friedman K.J., Kole J., Cohn J.A., Knowles M.R., Silverman L.M., Kole R. Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J. Biol. Chem. 1999;274:36193–36199. doi: 10.1074/jbc.274.51.36193. [DOI] [PubMed] [Google Scholar]
- 125.Lambert G., Becker B., Schreiber R., Boucherot A., Reth M., Kunzelmann K. Control of cystic fibrosis transmembrane conductance regulator expression by BAP31. J. Biol. Chem. 2001;276:20340–20345. doi: 10.1074/jbc.M011209200. [DOI] [PubMed] [Google Scholar]
- 126.Mansfield S.G., Kole J., Puttaraju M., Yang C.C., Garcia-Blanco M.A., Cohn J.A., Mitchell L.G. Repair of CFTR mRNA by spliceosome-mediated RNA trans-splicing. Gene Ther. 2000;7:1885–1895. doi: 10.1038/sj.gt.3301307. [DOI] [PubMed] [Google Scholar]
- 127.Liu X., Jiang Q., Mansfield S.G., Puttaraju M., Zhang Y., Zhou W., Cohn J.A., Garcia-Blanco M.A., Mitchell L.G., Engelhardt J.F. Partial correction of endogenous ΔF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat. Biotechnol. 2002;20:47–52. doi: 10.1038/nbt0102-47. [DOI] [PubMed] [Google Scholar]
- 128.S. Gersting, C. Rudolph, P. Nicklaus, C. Plank, J. Rosenecker, Magnetofection of permanent and primary human airway epithelial cells, Mol. Ther. 3 (No. 5, Part 2 of 2 Parts) (2001) Abstr. 1017.
- 129.Scherer F., Anton M., Schillinger U., Henke J., Bergemann C., Kruger A., Gansbacher B., Plank C. Magnetofection: enhancing and targeting gene delivery by magnetic force in vitro and in vivo. Gene Ther. 2002;9:102–109. doi: 10.1038/sj.gt.3301624. [DOI] [PubMed] [Google Scholar]
- 130.Boyle M.P., Enke R.A., Adams R.J., Guggino W.B., Zeitlin P.L. In utero AAV-mediated gene transfer to rabbit pulmonary epithelium. Mol. Ther. 2001;4:115–121. doi: 10.1006/mthe.2001.0428. [DOI] [PubMed] [Google Scholar]
- 131.Lipshutz G.S., Flebbe-Rehwaldt L., Gaensler K.M.L. Reexpression following readministration of an adenoviral vector in adult mice after initial in utero adenoviral administration. Mol. Ther. 2000;2:374–380. doi: 10.1006/mthe.2000.0136. [DOI] [PubMed] [Google Scholar]
- 132.Larson J.E., Morrow S.L., Delcarpio J.B., Bohm R.P., Ratterree M.S., Blanchard J.L., Cohen J.C. Gene transfer into the fetal primate: evidence for the secretion of transgene product. Mol. Ther. 2000;2:631–639. doi: 10.1006/mthe.2000.0209. [DOI] [PubMed] [Google Scholar]
- 133.Larson J.E., Delcarpio J.B., Farberman M.M., Morrow S.L., Cohen J.C. CFTR modulates lung secretory cell proliferation and differentiation. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;279:L333–L341. doi: 10.1152/ajplung.2000.279.2.L333. [DOI] [PubMed] [Google Scholar]
- 134.Q. Jiang, Z. Li, Y. Zhang, J.F. Engelhardt, Development of a ferret model of the cystic fibrosis, Pediatr. Pulmonol. Suppl. 20 (2000) Abstr. 164.
- 135.Harris A. Towards an ovine model of cystic fibrosis. Hum. Mol. Genet. 1997;6:2191–2194. doi: 10.1093/hmg/6.13.2191. [DOI] [PubMed] [Google Scholar]
- 136.C. Rooney, M. Suter, M. Donnelly, J. Reinhardt, A. Delsing, E.A. Hoffman, G. McLennan, J. Zabner, Laser induced fluorescence bronchoscopy for the detection of gene transfer, Mol. Ther. 3 (No. 5, Part 2 of 2 Parts) (2001) Abstr. 1152.