

Abstract
Vascular endothelial activation, marked by de novo expression of E-selectin, is an early and essential event in the process of leukocyte extravasation and inflammation. Evidence suggests that hepatocyte growth factor (HGF) ameliorates inflammation in animal models of renal disease, implying that HGF might inhibit specific components of the inflammatory response. This study examined the effect of HGF on endothelial E-selectin expression in acute inflammation induced by tumor necrosis factor (TNF)-α. In vitro, HGF suppressed TNF-α-induced cell surface expression of E-selectin in human umbilical vein endothelial cells (HUVEC) and inhibited E-selectin mediated monocytic adhesion to endothelial monolayers. HGF activated phosphatidylinositol 3-kinase (PI3K)–Akt that in turn inhibited its downstream transducer glycogen synthase kinase (GSK)3. Blockade of the PI3K–Akt pathway with specific inhibitors abrogated HGF induced inhibitory phosphorylation of GSK3 and suppression of E-selectin. In addition, selective inhibition of GSK3 activity by lithium suppressed TNF-α-induced E-selectin expression and monocytic adhesion, reminiscent of the action of HGF. Moreover, ectopic expression of an uninhibitable mutant GSK3β, in which the regulatory serine-9 is replaced by alanine, abolished HGF's suppressive effect on endothelial E-selectin. In vivo, administration of exogenous HGF reduced endothelial expression of E-selectin induced by bolus injection of TNF-α. This was associated with less sequestration of circulating fluorescence-labeled macrophages in the kidney. These findings suggest that HGF ameliorates acute renal inflammation in part by downregulating E-selectin mediated macrophage adhesion to the inflamed endothelium.
KEYWORDS: hepatocyte growth factor, inflammation, monocyte to endothelial adhesion, glycogen synthase kinase 3
Inflammation, characterized by tissue infiltration by leukocytes, is a basic biological reaction and part of the innate defense to injuries induced by various pathogenic factors.1 An inflammatory response of appropriate magnitude and timing is crucial to tissue repair and homeostasis.1., 2. Most inflammatory responses are acute and self-limiting; however, an excessive inflammatory reaction may result in critical and fatal conditions as systemic inflammatory response syndrome, severe acute respiratory syndrome, and acute renal failure. In addition, if the inflammatory response is prolonged or frequently relapsing chronic persistent inflammation develops, which may promote fibrosis and loss of organ function.3 Immunosuppressants including glucocorticoids are widely used to treat patients with excessive or chronic inflammation and reduce the likelihood of these complications, despite an increased risk of opportunistic infections.1., 2., 3.
Vascular endothelial activation and dysfunction play a critical role in the inflammatory response.4., 5., 6., 7., 8. Normally, leukocytes continuously patrol the vasculature, alert for signals of inflammation. Proinflammatory substances released by pathogens (e.g. lipopolysaccharide) or by damaged tissue (e.g. tumor necrosis factor (TNF)-α) upregulate the expression of adhesion molecules on the endothelium and initiate the migration of leukocytes to the inflamed area. Leukocyte migration from blood to tissues involves several steps: rolling, sticking, diapedesis, and chemotaxis.9 Among these processes, rolling is the earliest and indispensable event initiating leukocyte extravasation and inflammation. Rolling is mediated by the selectin family of adhesion molecules, endothelial E-selectin, platelet P-selectin, and leukocyte L-selectin.10 Both E- and P-selectins are expressed by endothelial cells; L-selectin is found only on leukocytes. P- and L-selectins are constitutively expressed whereas E-selectin is elicited by proinflammatory stimulate and is considered essential for leukocyte trafficking.10 The importance of E-selectin in initiating inflammation is demonstrated by the potent anti-inflammatory effect of E-selectin blockade.11., 12., 13., 14., 15., 16.
Hepatocyte growth factor (HGF) is a mesenchymal-derived, pleiotropic multifunctional growth factor.17 Upon binding to its receptor, c-Met, HGF triggers several signal transduction pathways, including phosphatidylinositol 3-kinase (PI3K)–Akt, Ras–Mek–Erk, and Stat3 pathway, and modulates diverse cell processes including mitogenesis, motogenesis, morphogenesis, and antiapoptosis/survival in epithelial and endothelial cells.17., 18. Evidence suggests that HGF ameliorates both acute and chronic injury in various organs including kidney,19 liver,20 lung,21 and intestine.22 Of note, inflammation is an invariable finding in both acute and chronic disease. Inflammation subsides in response to HGF treatment; however, the potential beneficial effects of HGF on inflammation have been largely overlooked. Recently, we reported that HGF treatment substantially attenuated inflammation in the rat remnant kidney model of chronic renal failure;23 however, the mechanisms responsible for this action remain uncertain. In this study, we show that HGF abrogates monocyte to endothelial adhesion and ameliorates acute renal inflammation by suppressing endothelial E-selectin expression. These findings suggest that the beneficial effects of HGF in both acute and chronic disease may be partially ascribed to its systemic anti-inflammatory action on the endothelium.
RESULTS
HGF suppresses TNF-α induced endothelial E-selectin expression in vitro
As presented in Figure 1a , TNF-α strongly stimulated E-selectin expression at low doses without significantly reducing the human umbilical vein endothelial cells (HUVEC) viability. Western immunoblot of whole-cell lysates showed that HGF treatment suppressed TNF-α induced total E-selectin expression in a time-dependent manner. E-selectin is found both on the cell membrane and in the intracellular reservoir in activated endothelial cells.24 Only membrane E-selectin is accessible to leukocytes and functionally active in mediating the endothelial–leukocyte adhesion. To examine whether HGF treatment modulated cell surface expression of E-selectin, flow cytometry (Figure 1b) and fluorescent immunocytochemistry (Figure 1c–f) without cell membrane permeabilization were employed. As shown in Figure 1c–f, control and HGF alone treated HUVEC cells are negative for E-selectin. TNF-α markedly induced E-selectin expression with a typical surface distribution pattern, and HGF pretreatment significantly decreased the surface E-selectin staining. Flow cytometry analysis corroborated the immunocytochemistry findings. HGF prevented TNF-α induced surface expression of E-selectin in HUVEC cells.
Figure 1.
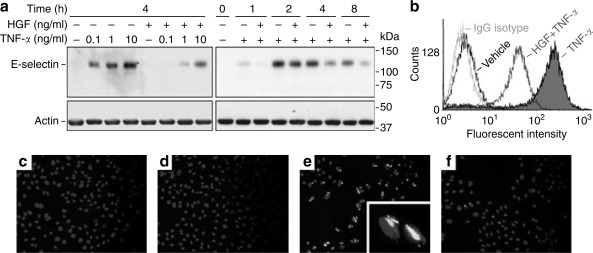
HGF suppresses TNF-α induced endothelial E-selectin expression in HUVEC cells. (a) HUVEC cells were pretreated with HGF (100 ng/ml) or vehicle for 30 min before stimulation of TNF-α (0.1 ng/ml or otherwise as indicated). Cell lysates were harvested at different time points after TNF-α stimulation and analyzed for E-selectin by Western immunoblot. Actin served as a standard molecule for normalization. (b) Flow cytometric analysis of cell surface E-selectin in HUVEC cells after 4 h of TNF-α (0.1 ng/ml) stimulation with or without HGF (100 ng/ml) pretreatment. (c–f) Representative micrographs of fluorescent immunocytochemistry depicted cell surface E-selectin expression on HUVEC cells pretreated with vehicles (c, e), or 100 ng/ml GF (d, f) before stimulation of 0.1 ng/ml TNF-α (e, f) or vehicle (c, d) for 4 h. Original magnification: (c–f) × 200 and (inset in e) × 400.
HGF blunts TNF-α elicited monocyte to endothelial adhesion in vitro
To determine whether HGF suppression of endothelial E-selectin expression reduces leukocyte adhesion to endothelium, we employed the monocytic static adhesion assay.25 Few fluorescent THP-1 cells were found adherent to vehicle (Figure 2a ) or HGF (Figure 2b) treated HUVEC monolayers. TNF-α promoted monocyte adhesion (Figure 2c), and HGF strikingly prevented it (Figure 2d). To quantify monocytes adherent to HUVEC monolayers, cells were lysed and subjected to fluorometric analysis (Figure 2f), which was in agreement with the microscopic findings. Of note, addition of a specific rabbit anti-E-selectin antibody blocked monocyte adhesion, suggesting that E-selectin mediates endothelial to monocyte adhesion and that suppression of endothelial expression of E-selectin by HGF accounts for the reduction in monocytic adhesion.
Figure 2.
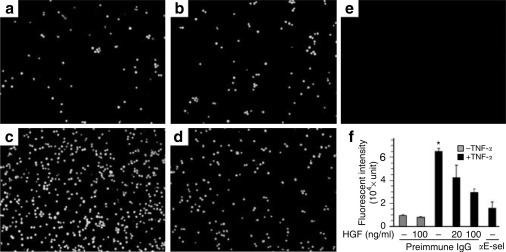
HGF functionally attenuates TNF-α elicited monocyte adhesion to HUVEC monolayers. Representative fluorescent micrographs show human monocyte adhesion to HUVEC monolayers. HUVEC cells were pretreated with (a, c) vehicle or (b, d) 100 ng/ml HGF before addition of (c, d) 0.1 ng/ml TNF-α or (a, b) vehicle. After 4 h, Calcein-AM-labeled (green fluorescence) THP-1 cells were applied. Prior to TNF-α stimulation a rabbit anti-E-selectin antibody (2 μg/ml) or preimmune IgG (2 μg/ml) was given to demonstrate the role of E-selectin in monocyte adhesion. (e) Nonlabeled THP-1 cells were applied to TNF-α treated HUVEC cells and served as negative controls. (f) Aliquots of cell lysates were subjected to fluorometric analysis to quantify the amount of adherent monocytes. αE-sel, anti-E-selectin blocking antibody. *P<0.05 vs other treatments. Original magnification: (a–e) × 100.
The PI3K–Akt pathway is required for HGF suppression of E-selectin
After binding to its cognate receptor, c-Met, HGF triggers multiple signaling pathways including the PI3K–Akt pathway, Ras–Mek–Erk pathway, and Stat3 pathway.18 HGF activated all three pathways in HUVEC cells, while TNF-α had only a minor effect (Figure 3a ). To determine which signaling pathway mediates HGF suppression of E-selectin, we pretreated HUVEC with various inhibitors specific for each pathway. As shown in Figure 3b, the suppressive effect of HGF on TNF-α -induced E-selectin was blocked by two different inhibitors specific for the PI3K–Akt pathway, wortmannin and LY294002. In contrast, U0126, the selective inhibitor for the Ras–Mek–Erk pathway and PpYLKTK-mts, the Stat3 inhibitor, failed to abolish the HGF's inhibitory action (Figure 3c). These data suggest that the PI3K–Akt pathway mediates HGF's suppression of E-selectin in endothelial cells.
Figure 3.
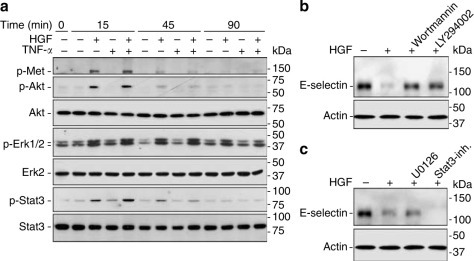
HGF activates c-Met and triggers multiple signaling pathways in endothelial cells, including PI3K–Akt, which is required for suppression of E-selectin. (a) HUVEC cells were pretreated with HGF (100 ng/ml) for 30 min before TNF-α (0.1 ng/ml) stimulation. At different time points after TNF-α stimulation, cell lysates were analyzed by immunoblotting for different molecules. (b) Pretreatment for 30 min with wortmannin (50 nM) and LY294002 (20 μM), specific chemical inhibitors for PI3K, diminished HGF's suppressive effect on E-selectin expression in HUVEC cells. (c) Pretreatment with a Stat3 inhibitor (160 μM PpYLKTK-mts, Calbiochem, San Diego, CA, USA), or with U0126 (10 μM) to block the Mek–Erk pathway, failed to override HGF suppression of E-selectin.
HGF inhibits glycogen synthase kinase (GSK) 3 via PI3K–Akt mediated phosphorylation in endothelial cells
GSK3 is an important downstream transducer of the PI3K–Akt signaling pathway. GSK3 is inactivated in response to PI3K signaling, as a result of Akt-mediated phosphorylation of an N-terminal serine, serine-9 in GSK3β and Ser-21 in GSK3α.26 Because phosphorylation of GSK3 at these two residues denotes GSK3 inactivation, we probed using a specific antibody against phosphorylated GSK3α (S21) and GSK3β (S9). In HUVEC cells, HGF treatment immediately elicited inhibitory phosphorylation of GSK3β and, to a lesser extent, GSK3α (Figure 4a ). This effect persisted for at least 90 min in the presence or absence of TNF-α, while TNF-α alone had only a minor effect. In addition, HGF-induced inhibitory phosphorylation of GSK3 was abolished by wortmannin (Figure 4b), implying that activation of PI3K–Akt pathway is required for this action.
Figure 4.
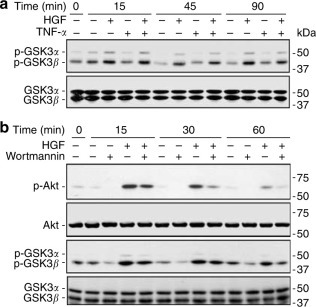
HGF induces GSK3 inhibition through PI3K–Akt-dependent phosphorylation in endothelial cells. (a) HUVEC cells were pretreated with HGF (100 ng/ml) for 30 min, and/or then stimulated with TNF-α (0.1 ng/ml) for different intervals before cell lysates were harvested for immunoblot analysis of different molecules. (b) Pretreatment for 30 min with wortmannin (50 nM) diminished HGF activation of PI3K–Akt–GSK3 pathway assessed by phosphorylation of Akt and GSK3.
Inhibition of GSK3 mimics HGF suppression of TNF-α induced E-selectin expression in endothelial cells
To avoid cell loss due to induction of apoptosis, the general apoptosis inhibitor BOC-Asp-CH2F was added to the culture.27 At the optimal concentration not associated with significant apoptosis, lithium (20 mM), a selective inhibitor for GSK3,28 induced inhibitory phosphorylation of GSK3 (Figure 5a ), and attenuated basal and TNF-α-induced E-selectin in HUVEC (Figure 5b), reminiscent of the action of HGF. Sodium, an osmolality control, had no effect.
Figure 5.
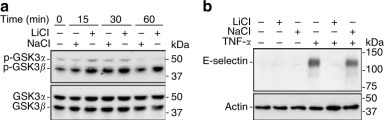
Specific inhibition of GSK3 by lithium (LiCl) mimics HGF suppression of TNF-α induced E-selectin expression in endothelial cells. (a) LiCl (20 mM) induced inhibitory phosphorylation in HUVEC cells. As an osmolality control, sodium chloride (20 mM) had little or no effect. (b) Lithium (20 mM), but not sodium (20 mM), abolished HGF suppression of E-selectin in HUVEC cells.
Blockade of PI3K–Akt–GSK3 pathway by specific chemical inhibitors functionally inhibits endothelial to monocyte adhesion
The finding that activation of PI3K–Akt and subsequent inhibition of GSK3 mediates HGF suppression of E-selectin prompted us to investigate whether the PI3K–Akt–GSK3 cascade regulates endothelial to monocyte adhesion. HUVEC monolayers were activated with TNF-α and subjected to static adhesion assay after pretreatment with HGF or different selective chemical inhibitors of the PI3K–Akt–GSK3 pathway. Fluorometric analysis of cell lysates demonstrated that inhibition of GSK3 by lithium attenuates TNF-α-induced endothelial to monocyte adhesion, similar to the effect of HGF. Sodium, the osmolality control, had no effect. Blockade of PI3K activation by wortmannin also blocked the inhibitory action of HGF on monocytic adhesion from 4 h on (Figure 6 ).
Figure 6.
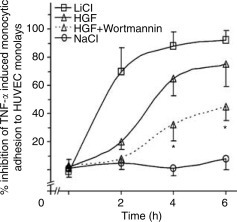
Blockade of the PI3K–Akt–GSK3 pathway by specific chemical inhibitors functionally impedes endothelial to monocyte adhesion. HUVEC monolayers were pretreated with LiCl (20 mM), NaCl (20 mM), HGF (100 ng/ml), wortmannin (50 nM), or vehicle before addition of TNF-α (0.1 ng/ml). At different time intervals, Calcein-AM labeled THP-1 cells were applied for monocyte adhesion assay as described in the Materials and Methods. Aliquots of cell lysates were subjected to fluorometric analysis to measure the quantity of adherent monocytes. Values are expressed as mean relative inhibition over HUVEC monolayers pretreated with vehicle±s.d. from three separate experiments. *P<0.05 vs HGF treatment at the same time points.
Ectopic expression of uninhibitable mutant GSK3β abolishes HGF inhibition of endothelial E-selectin in vitro
GSK3 consists of two distinct isoforms, GSK3α and GSK3β. Previous studies suggested that GSK3β, but not GSK3α, is essential for TNF-α or IL-1β-induced inflammatory responses.29 To further examine the role of inhibitory phosphorylation of GSK3β in HGF inhibition of E-selectin, we studied the effect of forced expression of GSK3β on E-selectin in HUVEC cells. Vectors encoding the hemagglutin (HA) tagged wild type (WT) GSK3β or uninhibitable mutant GSK3β,30 in which the regulatory serine-9 residue was replaced by alanine (S9A-GSK3β), were transfected into HUVEC cells. As a control, pcDNA3 was used in transfection. To evaluate the levels of expression, whole-cell lysates were analyzed by immunoblotting for HA or HA-GSK3β (Figure 7a ). Immunofluorescent detection using an antibody against the HA epitope revealed that over 50% of the cells expressed the HA-tagged constructs 24 h after transfection. As shown in Figure 7a–c, HGF inhibition of TNF-α induced E-selectin expression was evident in HUVEC cells transfected with pcDNA3 or WT-GSK3β. In contrast, ectopic expression of S9A-GSK3β abolished the suppressive action of HGF on E-selectin expression. Collectively, these findings suggest that inhibitory phosphorylation of GSK3β at serine-9 is required for HGF inhibition of E-selectin in HUVEC cells.
Figure 7.
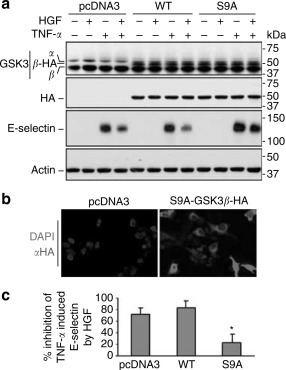
Forced expression of the uninhabitable mutant GSK3β abolishes HGF inhibition of endothelial E-selectin in HUVEC cells. HUVEC were transfected by electroporation with the empty vector pcDNA3 or vectors encoding the HA-tagged wild type (WT) or the uninhibitable mutant (S9A). (a) At 24 h after transfection, cells were stimulated with TNF-α (0.1 ng/ml) after HGF (100 ng/ml) pretreatment. Cell lysates were subjected to Western immunoblot anslysis. (b) Fluorescent immunocytochemistry staining shows expression of HA tag in the transfected cells (Original magnification: 200). (c) Relative inhibition of E-selectin by HGF was estimated by densitometric analysis of the immunoblot bands as shown in (a). *P<0.01 vs cells transfected with pcDNA3 or WT (n=3).
HGF suppresses TNF-α provoked endothelial E-selectin expression in vivo
The behavior of endothelial cells in vivo may differ from HUVEC cells in culture. To examine the effect of HGF on E-selectin expression in acute inflammation in vivo, rats were given a bolus intra-arterial injection of TNF-α. After 4 h, fluorescent immunohistochemistry revealed abundant staining for E-selectin in the kidney (Figure 8c ) that was distributed in the pattern of the interstitial microvasculature (Figure 8e). HGF treatment reduced E-selectin staining (Figure 8d). The quantitative fluorescence intensity of E-selectin (Figure 8f) was significantly weakened in kidneys from HGF treated group. Western immunoblot analysis of total kidney homogenates confirmed that HGF prevented TNF-α induced E-selectin expression (Figure 8g).
Figure 8.
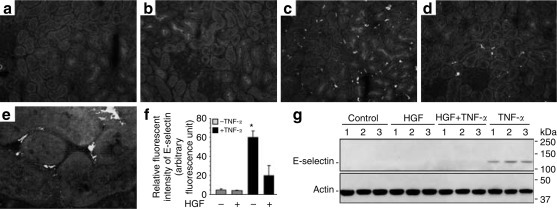
HGF suppresses TNF-α provoked endothelial E-selectin expression in rats. (a–e) Representative microphotographs of fluorecent immunohistochemistry show the expression of E-selectin in rats receiving an arterial injection of (c–e) TNF-α (2 μg/kg) or (a, b) vehicle after pretreatment of (b, d) HGF (100 μg/kg) or (a, c, e) vehicle for 30 min. After 4 h, kidney sections were snap frozen and processed for immunofluorescent staining of E-selectin. Tissues were counterstained with 4′,6-diamidino-2-phenylindole and Evans' blue. Original magnification (a–d) × 200 and (e) × 400. (f) Relative fluorescent intensity of E-selectin as estimated by quantitative immunofluorescence analysis; *P<0.01 vs other groups (n=3). (g) Representative pictures of Western immunoblot analysis of E-selectin expression in kidney homogenates.
HGF protects against acute renal inflammation via suppression of E-selectin
To confirm the functional significance of HGF suppression of E-selectin in the kidney, fluorescent viable rat alveolar macrophages (RAM) were infused into the carotid artery after administration of HGF and/or TNF-α as described in Materials and Methods. Tissues were processed for fluorescent histological analysis after 30 min. As shown in Figure 9c , many fluorescent RAM cells were found in the kidneys in TNF-α treated animals, most prominently in the interstitium of juxtamedullary cortex. An anti-E-selectin blocking antibody (figure not shown) or HGF (Figure 9d) markedly decreased the number of RAM cells. Quantification of RAM cells by fluorometric analysis of kidney homogenates (Figure 9e) was in agreement with the morphologic findings.
Figure 9.
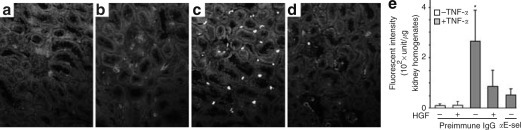
HGF decreases renal sequestration of macrophages, predominantly located in the interstitium of juxtamedullary cortex, after TNF-α stimulation. (a–d) Representative fluorescent micrographs depict renal sequestration of Calcein-AM labeled (green fluorescent) RAM cells in rats receiving an arterial injection of (c, d) TNF-α (2 μg/kg) or (a, b) vehicle after pretreatment of (b, d) HGF (100 μg/kg) or (a, c) vehicle for 30 min. After 4 h, Calcein-AM labeled RAM cells were infused. Kidneys were harvested after 30 min and processed for fluorescent histological examination. Prior to TNF-α injection, a rabbit anti-E-selectin antibody or preimmune IgG was given to demonstrate the role of E-selectin in inflammation. Tissues were double counterstained with 4′,6-diamidino-2-phenylindole and Evans' blue. (a–d) Original magnification × 200. (e) Aliquots of kidney homogenates from differently treated groups were subjected to fluorometry to quantify the sequestrated RAM cells. αE-sel, anti-E-selectin blocking antibody. *P<0.05 vs other groups (n=4).
DISCUSSION
Inflammation is a common finding in both acute and chronic injuries of diverse etiologies. Leukocyte to endothelium adhesion mediated by the selectin family of adhesion molecules, in particular E-selectin, is an indispensable event initiating the inflammatory reaction.6., 7., 8., 9., 10. In the present study, we found that HGF suppresses monocyte to endothelial adhesion and attenuates acute renal inflammation via inhibition of endothelial E-selectin expression. HGF exerts this action via activation of PI3K–Akt–GSK3β pathway in endothelial cells. This finding complements our recent report demonstrating that HGF attenuates renal inflammation in the rat remnant kidney model of chronic renal disease.23 HGF has also recently been reported to be anti-inflammatory in non-renal diseases.21., 31., 32., 33., 34. In a murine model of inflammatory bowl disease (IBD), HGF gene transfection diminished inflammatory infiltrates in the intestinal epithelium.31 Similarly, Arthur et al. 32., 33. reported that direct intravenous infusion of exogenous HGF to rats with IBD significantly ameliorated gross and microscopic bowl inflammation. In a murine model of airway hyperresponsiveness, airway inflammation was reduced by administration of recombinant HGF.34 In the present study, exogenous HGF significantly attenuated TNF-α induced macrophage infiltration and acute renal inflammation in rats. The finding that HGF has systemic anti-inflammatory effects suggests that HGF may intercept common processes in the general inflammatory reaction rather than organ-specific mechanisms.
Leukocyte adhesion to an activated endothelium is a prerequisite for generating an inflammatory infiltrate and is found in virtually all-inflammatory diseases.8., 9. Increased expression of particular adhesion molecules is an important early marker of endothelial activation. E-selectin is of particular interest because it is only found on the inflamed endothelium in contrast to other adhesion molecules, which have a wide constitutive tissue distribution.9 Previous studies demonstrated that inhibition of endothelial activation by glucocorticoids35 or statins36 attenuates inflammation by decreasing endothelial expression of adhesion molecules, including E-selectin. Similarly, blocking the interaction between the endothelium and leukocytes with E-selectin antibodies as well as selectin antagonists also ameliorates various inflammatory diseases.11., 12., 13., 14., 15., 16. For instance, anti-E-selectin blocking antibody or E-selectin gene disruption protected mice from ischemia–reperfusion induced acute renal failure.12 Similarly, antibody blockade of E-selectin decreased adventitial inflammation and attenuated intimal hyperplasia in rat carotid arteries after balloon injury.13 Moreover, transgenic mice producing soluble E-selectin that can competitively inhibit the binding of inflammatory cells to E-selectins on the endothelium are resistant to bleomycin induced chronic pulmonary inflammation and lung fibrosis.14 All these studies suggest the therapeutic potential of E-selectin blockade in inflammatory diseases. In our study, HGF was found to suppress endothelial E-selectin expression in cultured HUVEC cells and in vivo in rats injected with TNF-α. Suppression of E-selectin in turn was responsible, at least in part, for HGF-induced suppression of monocyte to endothelial adhesion and inflammation in the kidney.
The mechanism by which HGF regulates endothelial E-selectin expression is not well studied. The PI3K–Akt pathway is a major signaling cascade triggered by the binding of HGF to its cognate receptor c-Met on endothelial cells.17., 18., 19. In the present study, HGF activated PI3K–Akt in endothelial cells and specific blockade of PI3K–Akt overrode HGF suppression of E-selectin expression. These data are consistent with another study in a murine model of endotoxemia,37 in which blocking the PI3K–Akt pathway with wortmannin or LY294002 enhanced LPS-induced E-selectin levels and exacerbated macrophage infiltration in liver and kidney. GSK3 is an important downstream substrate of the PI3K–Akt signaling pathway that has been shown to regulate the inflammatory response.29., 38. A ubiquitously expressed serine-threonine kinase, GSK3 exists in two isoforms, GSK3α and GSK3β and is a unique signal transducer in that it is constitutively active under normal conditions. GSK3 is inactivated in response to PI3K signaling, as a result of Akt-mediated inhibitory phosphorylation.39., 40. We found that HGF induced inhibitory phosphorylation of both GSK3α and GSK3β in endothelial cells. Selective inhibition of GSK3 by lithium also inhibited phosphorylation of GSK3 and suppressed TNF-α-induced E-selectin expression in HUVEC cells, reminiscent of the action of HGF. Furthermore, ectopic expression of a mutant construct encoding the uninhibitable GSK3β blunted HGF-induced suppression of E-selectin, again suggesting that HGF inhibits E-selectin via inhibition of GSK3β. A regulatory role for GSK3β in E-selectin gene expression was also reported by Hoeflich et al. 29 In that study, E-selectin-luciferase transcription induced by TNF-α or IL-1β was reduced in GSK3β -/- cells and could be restored by the expression of exogenous GSK-3.
The mechanism by which GSK3β inactivation modulates E-selectin is unclear. Recent data suggest that GSK3β is an essential element for NF-κB activation.41., 42., 43. Sequence analysis reveals the presence of multiple putative κB elements in the promoter region of the E-selectin gene.44 In addition, interaction between NF-κB and κB elements in the E-selectin promoter is required for E-selectin expression.44., 45. Therefore, it is possible that HGF inhibition of endothelial E-selectin and inflammation might be due to suppression of NF-κB via GSK3β inactivation. GSK3β has been recently suggested to be a key regulator of multiple cellular processes implicated in the pathogenesis of diabetes and chronic inflammatory diseases.26., 38. Due to its regulatory effect on GSK3β, HGF might represent a novel potential strategy for the treatment of inflammatory diseases.
In summary, HGF suppresses E-selectin expression in the activated endothelium and thereby attenuates monocyte to endothelial adhesion and alleviates acute inflammation in the kidney. Our findings suggest that HGF might exert its beneficial effects in various disease models at least in part through its potent anti-inflammatory action on vascular endothelium.
MATERIALS AND METHODS
Cell culture
HUVEC were purchased from VEC Technologies (Rensselaer, NY, USA) and maintained in MCDB-131 complete media. HUVEC cells (2–8 passages) were seeded on gelatin (1.5%) coated cultures at approximately 80% confluence. After growth for 24 h in complete media, cells underwent serum starvation for 6 h in Medium 199. Human recombinant HGF (Genentech, South San Francisco, CA, USA) and human recombinant TNF-α (R&D systems, Minneapolis, MN, USA) were added to the culture with fresh serum-free medium at a final concentration of 100 and 0.1 ng/ml respectively, or as otherwise indicated. In experiments with GSK3 blockade, the general apoptosis inhibitor BOC-Asp-CH2F (Enzyme Systems Products, Dublin, CA, USA) was added to the culture at the final concentration of 50 μ M to reduce cell loss due to induction of apoptosis.27 Cell viability was assessed by Trypan blue exclusion. Human monocytes (THP-1) and RAM were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in suspension respectively in RPMI supplemented with 10% fetal bovine serum and Ham's F12K containing 15% fetal bovine serum. For fluorescent viable labeling, THP-1 and RAM cells (1 × 107) were incubated in their respective media containing 5 μg/ml Calcein-AM (Molecular Probes, Eugene, OR, USA) at 37°C for 30 min. Excess dye was removed by washing three times with phosphate-buffered saline.
Monocytic adhesion assay
Adhesion studies were performed with the THP-1 cells under static conditions.25 Briefly, HUVEC monolayers with equal cell numbers in 12-well plates were treated with HGF and/or TNF-α for 4 h. After addition of 1 ml fluorescence-labeled THP-1 cells (1 × 106 cells/ml) per well, the plates were incubated for 30 min at 37°C. Monolayers were gently washed three times with phosphate-buffered saline after incubation. Adherent monocytes were lysed with RIPA buffer23 and fluorescent intensity was measured in a Spectramax GEMINI EM fluorescence plate reader (Molecular Devices, Sunnyvale, CA, USA) at an excitation wavelength of 485 nm and emission at 530 nm. HUVEC monolayers adhered with non-labeled THP-1 cells served as negative controls.
Transient transfection
The expression vectors encoding the HA-tagged wild-type (WT-GSK3β-HA/pcDNA3) and uninhibitable mutant GSK3β (S9A-GSK3β-HA/pcDNA3) were kindly and respectively provided by Dr Jim Woodgett (University of Toronto, Totonto, Ontario, Canada)46 and Dr Gail VW Johnson (University of Alabama at Birmingham, Birmingham, AL, USA).30 HUVEC cells were transfected by electroporation using the Amaxa HUVEC Nucleofector kit (Amaxa GmbH, Koeln, Germany). After transfection with equal amounts of expression plasmid or empty vector pcDNA3 (Invitrogen, Carlsbad, CA, USA), HUVECs were treated as indicated.
Flow cytometry
Flow cytometric analysis of E-selectin expression on HUVEC cells was performed as previously described24 using the FACS flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Primary anti-E-selectin (CTB202) mouse mAb (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and secondary mAbs were used at saturating concentrations. Isotype-matched primary mAb were used as negative controls. The mean fluorescence intensity in negative controls was consistently <10 fluorescence units.
Fluorescent immunocytochemistry
Indirect immunofluorescence staining was performed as before.47 Briefly, cells cultured on chamber slides were fixed with 4% paraformadehyde. Following donkey serum blocking for 30 min, cells were incubated with the specific primary and then secondary antibodies. Finally, cells were stained with 4′,6-diamidino-2-phenylindole to visualize the nuclei. Stained cells were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) and the extent of stained assessed using a fluorescence microscope.
Animal studies
Male Sprague–Dawley rats (Harlan Sprague–Dawley, Indianapolis, IN, USA) with initial weights of 200–250 g were housed in an approved animal care facility and fed standard chow. On the day of study, rats were anesthetized, placed on a heated table to keep constant body temperature, and maintained euvolemic state as described before.23 A polyethylene catheter was inserted in the left carotid artery as an access for drug administration. After a 45-min equilibration period, HGF (100 μg/kg wt) or an equal volume of vehicle was administrated as a bolus injection into the left carotid artery. After 30 min, a bolus injection of rat TNF-α (2 μg/kg wt) (R&D system) or vehicle was given to stimulate the systemic inflammation. To demonstrate the pro-inflammatory role of E-selectin, a rabbit anti-E-selectin antibody (Santa Cruz Biotechnology) or preimmune IgG was given prior to TNF-α injection. At 4 h after TNF-α injection, fluorescent-labeled RAM cells (1 × 104) resuspended in normal saline were infused via the carotid artery. Rats were killed before or 30 min after RAM cell infusion and various organs harvested for further investigation. One portion of the kidney was immediately frozen for cryostat sectioning. To quantify the fluorescent RAM cells sequestrated in the kidney, kidney homogenates with equal amount of protein (100 μg) were subjected to fluorometric analysis in a fluorescence plate reader as described above.
Morphologic studies
Indirect immunofluoresent staining of E-selectin was carried out on methanol/acetone (1:1) fixed frozen cryostat sections using rabbit polyclonal anti-E-selectin antibody (Santa Cruz Biotechnology). The Alexa Fluor goat anti-rabbit (Molecular probes) was used as secondary antibody. As a negative control, the primary antibody was replaced by nonimmune serum from the same species; no staining occurred. Frozen sections were double stained with 4′,6-diamidino-2-phenylindole, counterstained with Evan's blue and mounted with Vectashield mounting medium. To visualize fluorescent macrophages sequestrated in the tissue, cryostat sections were directly fixed and then subjected to counterstaining.
In quantitative immunofluorescence studies, all sections were stained and analyzed at the same time to exclude artifacts due to variable decay of the fluorochrome. Sections were examined at × 400 magnification with a Nikon Microphot-FX fluorescence microscope equipped with a Spot II digital camera. Captured images were analyzed with NIH Image (v1.62). The mean fluorescence intensity was calculated using the arbitrary fluorescence units obtained in 20 random fields per rat in three rats per group.
Western immunoblot analysis
Rat kidneys were homogenized and HUVEC monolayers were lysed in RIPA buffer. Protein concentrations were determined using a bicinchoninic acid protein assay kit (Sigma, St Louis, MO, USA). Samples with equal amounts of total protein (40–80 μg/ml) were fractionated by 7.5–10% SDS-polyacrylamide gels under reducing condition and analyzed by Western immunoblot as described previously.48 The antibodies against E-selection, Akt, Erk2, p-Stat3, Stat3, GSK3, and actin were purchased from Sata Cruz Biotechnology and those for p-Akt, p-Erk1/2, p-GSK3, and HA were purchased from Cell Signaling Technology (Beverly, MA, USA).
Statistics
For immunoblot analysis, bands were scanned and the integrated pixel density was determined using a densitometer and the NIH image analysis program. All data are expressed as mean±s.d. Statistical analysis of the data from multiple groups was performed by ANOVA followed by Student–Newman–Kuels tests. Data from two groups were compared by Student's t-test.
ACKNOWLEDGMENTS
This work was supported by the Young Investigator Research Fund from Rhode Island Foundation for Health (R Gong), National Institutes of Health Grant RO1-DK52314 (LD Dworkin) and AT001465-01A2 (A Rifai).
published online 22 Februry 2006
Footnotes
This paper is dedicated to Professor Lei-Shi Li, Founder of contemporary nephrology in China and Chair of our renal sister center at Nanjing University, on the occasion of his 80th birthday.
REFERENCES
- 1.Ward P.A., Marks R.M. The acute inflammatory reaction. Curr Opin Immunol. 1989;2:5–9. doi: 10.1016/0952-7915(89)90090-3. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 3.Levy J.H. The human inflammatory response. J Cardiovasc Pharmacol. 1996;27(Suppl 1):S31–S37. doi: 10.1097/00005344-199600001-00008. [DOI] [PubMed] [Google Scholar]
- 4.Hack C.E., Zeerleder S. The endothelium in sepsis: source of and a target for inflammation. Crit Care Med. 2001;29(Suppl):S21–S27. doi: 10.1097/00003246-200107001-00011. [DOI] [PubMed] [Google Scholar]
- 5.Ali H., Haribabu B., Richardson R.M., Snyderman R. Mechanisms of inflammation and leukocyte activation. Med Clin N Am. 1997;81:1–28. doi: 10.1016/s0025-7125(05)70503-4. [DOI] [PubMed] [Google Scholar]
- 6.Kevil C.G. Endothelial cell activation in inflammation: lessons from mutant mouse models. Pathophysiology. 2003;9:63–74. doi: 10.1016/s0928468002000834. [DOI] [PubMed] [Google Scholar]
- 7.Luscinskas F.W., Gimbrone M.A., Jr Endothelial-dependent mechanisms in chronic inflammatory leukocyte recruitment. Annu Rev Med. 1996;47:413–421. doi: 10.1146/annurev.med.47.1.413. [DOI] [PubMed] [Google Scholar]
- 8.Bevilacqua M.P., Nelson R.M., Mannori G., Cecconi O. Endothelial–leukocyte adhesion molecules in human disease. Annu Rev Med. 1994;45:361–378. doi: 10.1146/annurev.med.45.1.361. [DOI] [PubMed] [Google Scholar]
- 9.Carlos T.M., Harlan J.M. Leukocyte–endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 10.Ley K. The role of selectins in inflammation and disease. Trends Mol Med. 2003;9:263–268. doi: 10.1016/s1471-4914(03)00071-6. [DOI] [PubMed] [Google Scholar]
- 11.Welply J.K., Keene J.L., Schmuke J.J., Howard S.C. Selectins as potential targets of therapeutic intervention in inflammatory diseases. Biochim Biophys Acta. 1994;1197:215–226. doi: 10.1016/0304-4157(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 12.Singbartl K., Ley K. Protection from ischemia–reperfusion induced severe acute renal failure by blocking E-selectin. Crit Care Med. 2000;28:2507–2514. doi: 10.1097/00003246-200007000-00053. [DOI] [PubMed] [Google Scholar]
- 13.Gotoh R., Suzuki J., Kosuge H. E-selectin blockade decreases adventitial inflammation and attenuates intimal hyperplasia in rat carotid arteries after balloon injury. Arterioscler Thromb Vasc Biol. 2004;24:2063–2068. doi: 10.1161/01.ATV.0000145942.31404.20. [DOI] [PubMed] [Google Scholar]
- 14.Azuma A., Takahashi S., Nose M. Role of E-selectin in bleomycin induced lung fibrosis in mice. Thorax. 2000;55:147–152. doi: 10.1136/thorax.55.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman G., Jankowski S., Shahla M. Administration of an antibody to E-selectin in patients with septic shock. Crit Care Med. 1996;24:229–233. doi: 10.1097/00003246-199602000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Huang J., Choudhri T.F., Winfree C.J. Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke. 2000;31:3047–3053. [PubMed] [Google Scholar]
- 17.Boros P., Miller C.M. Hepatocyte growth factor: a multifunctional cytokine. Lancet. 1995;345:293–295. doi: 10.1016/s0140-6736(95)90279-1. [DOI] [PubMed] [Google Scholar]
- 18.Birchmeier C., Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 1998;8:404–410. doi: 10.1016/s0962-8924(98)01359-2. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y. Hepatocyte growth factor and the kidney. Curr Opin Nephrol Hypertens. 2002;11:23–30. doi: 10.1097/00041552-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 20.Matsumoto K., Nakamura T. Hepatocyte growth factor: molecular structure and implications for a central role in liver regeneration. J Gastroenterol Hepatol. 1991;6:509–519. doi: 10.1111/j.1440-1746.1991.tb00897.x. [DOI] [PubMed] [Google Scholar]
- 21.Ware L.B., Matthay M.A. Keratinocyte and hepatocyte growth factors in the lung: roles in lung development, inflammation, and repair. Am J Physiol Lung Cell Mol Physiol. 2002;282:L924–L940. doi: 10.1152/ajplung.00439.2001. [DOI] [PubMed] [Google Scholar]
- 22.Dignass A.U., Sturm A. Peptide growth factors in the intestine. Eur J Gastroenterol Hepatol. 2001;13:763–770. doi: 10.1097/00042737-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Gong R., Rifai A., Tolbert E.M. Hepatocyte growth factor ameliorates renal interstitial inflammation in rat remnant kidney by modulating tubular expression of MCP-1 and RANTES. J Am Soc Nephrol. 2004;15:2868–2881. doi: 10.1097/01.ASN.0000141962.44300.3A. [DOI] [PubMed] [Google Scholar]
- 24.Grabner R., Till U., Heller R. Flow cytometric determination of E-selectin, vascular cell adhesion molecule-1, and intercellular cell adhesion molecule-1 in formaldehyde-fixed endothelial cell monolayers. Cytometry. 2000;40:238–244. doi: 10.1002/1097-0320(20000701)40:3<238::aid-cyto9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Ludwig A., Lorenz M., Grimbo N. The tea flavonoid epigallocatechin-3-gallate reduces cytokine-induced VCAM-1 expression and monocyte adhesion to endothelial cells. Biochem Biophys Res Commun. 2004;316:659–665. doi: 10.1016/j.bbrc.2004.02.099. [DOI] [PubMed] [Google Scholar]
- 26.Jope R.S., Johnson G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez J.F., Sniderhan L.F., Williamson A.L. Glycogen synthase kinase 3β-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor κB signaling. Mol Cell Biol. 2003;23:4649–4662. doi: 10.1128/MCB.23.13.4649-4662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen P., Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 29.Hoeflich K.P., Luo J., Rubie E.A. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 30.Cho J.H., Johnson G.V. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating tau's ability to bind and stabilize microtubules. J Neurochem. 2004;88:349–358. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- 31.Oh K., Iimuro Y., Takeuchi M. Ameliorating effect of hepatocyte growth factor on inflammatory bowel disease in a murine model. Am J Physiol Gastrointest Liver Physiol. 2005;288:G729–G735. doi: 10.1152/ajpgi.00438.2004. [DOI] [PubMed] [Google Scholar]
- 32.Arthur L.G., Schwartz M.Z., Kuenzler K.A., Birbe R. Hepatocyte growth factor treatment ameliorates diarrhea and bowel inflammation in a rat model of inflammatory bowel disease. J Pediatr Surg. 2004;39:139–143. doi: 10.1016/j.jpedsurg.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Arthur L.G., Kuenzler K.A., Schwartz M.Z. Hepatocyte growth factor ameliorates inflammatory bowel disease in a rat model. J Gastrointest Surg. 2003;7:1062–1068. doi: 10.1016/j.gassur.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 34.Ito W., Kanehiro A., Matsumoto K. Hepatocyte growth factor attenuates airway hyperresponsiveness, inflammation, and remodeling. Am J Respir Cell Mol Biol. 2005;32:268–280. doi: 10.1165/rcmb.2004-0058OC. [DOI] [PubMed] [Google Scholar]
- 35.Cronstein B.N., Kimmel S.C., Levin R.I. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial–leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1992;89:9991–9995. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Omi H., Okayama N., Shimizu M. Statins inhibit high glucose-mediated neutrophil–endothelial cell adhesion through decreasing surface expression of endothelial adhesion molecules by stimulating production of endothelial nitric oxide. Microvasc Res. 2003;65:118–124. doi: 10.1016/s0026-2862(02)00033-x. [DOI] [PubMed] [Google Scholar]
- 37.Schabbauer G., Tencati M., Pedersen B. PI3K–Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24:1963–1969. doi: 10.1161/01.ATV.0000143096.15099.ce. [DOI] [PubMed] [Google Scholar]
- 38.Martinez A., Castro A., Dorronsoro I., Alonso M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med Res Rev. 2002;22:373–384. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- 39.Cohen P., Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 40.Ali A., Hoeflich K.P., Woodgett J.R. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101:2527–2540. doi: 10.1021/cr000110o. [DOI] [PubMed] [Google Scholar]
- 41.Demarchi F., Bertoli C., Sandy P., Schneider C. Glycogen synthase kinase-3β regulates NF-κB1/p105 stability. J Biol Chem. 2003;278:39583–39590. doi: 10.1074/jbc.M305676200. [DOI] [PubMed] [Google Scholar]
- 42.Haefner B. A model for NF-κB regulation by GSK-3β. Drug Discov Today. 2003;8:1062–1063. doi: 10.1016/s1359-6446(03)02898-8. [DOI] [PubMed] [Google Scholar]
- 43.Pomerantz J.L., Baltimore D. Signal transduction. A cellular rescue team. Nature. 2000;406:26–27. doi: 10.1038/35017673. [DOI] [PubMed] [Google Scholar]
- 44.Schindler U., Baichwal V.R. Three NF-κB binding sites in the human E-selectin gene required for maximal tumor necrosis factor alpha-induced expression. Mol Cell Biol. 1994;14:5820–5831. doi: 10.1128/mcb.14.9.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyle E.M., Jr, Sato T.T., Noel R.F., Jr Transcriptional arrest of the human E-selectin gene. J Surg Res. 1999;82:194–200. doi: 10.1006/jsre.1998.5536. [DOI] [PubMed] [Google Scholar]
- 46.Xavier I.J., Mercier P.A., McLoughlin C.M. Glycogen synthase kinase 3β negatively regulates both DNA-binding and transcriptional activities of heat shock factor 1. J Biol Chem. 2000;275:29147–29152. doi: 10.1074/jbc.M002169200. [DOI] [PubMed] [Google Scholar]
- 47.Gong R., Rifai A., Dworkin L.D. Activation of PI3K–Akt–GSK3β pathway mediates hepatocyte growth factor inhibition of RANTES expression in renal tubular epithelial cells. Biochem Biophys Res Commun. 2005;330:27–33. doi: 10.1016/j.bbrc.2005.02.122. [DOI] [PubMed] [Google Scholar]
- 48.Gong R., Rifai A., Tolbert E.M. Hepatocyte growth factor modulates matrix metalloproteinases and plasminogen activator/plasmin proteolytic pathways in progressive renal interstitial fibrosis. J Am Soc Nephrol. 2003;14:3047–3060. doi: 10.1097/01.asn.0000098686.72971.db. [DOI] [PubMed] [Google Scholar]