

Abstract
In this work, a gold complex is used as electroactive label for monitoring hybridization assays on glassy carbon electrodes.
Ionic gold is bound to a 30-mer sequence of the SARS (severe acute respiratory syndrome) virus, responsible for the atypical pneumonia, using sodium aurothiomalate.
In order to label this single strand, a mixture of sodium aurothiomalate and the strand is prepared. Then, it is incubated for 24 h at 37 °C and, finally, free gold is separated from the labeled strand by a dialysis against a 0.15 M NaCl solution (pH 7.5).
The DNA hybridization sensor is designed immobilizing the complementary probe on the pre-treated electrode surface and, then, the hybridization reaction takes place with the gold labeled strand. The electrochemical determination is based on the catalytic effect of electrodeposited gold on the reduction of silver ions.
In non-stringent experimental conditions, a limit of detection of 15 fmol (30 μL) is obtained, and discrimination between a complementary oligonucleotide and a three-based mismatch complementary oligonucleotide is achieved. For the discrimination of a single-base mismatch, is needed to use stringent conditions (50% of formamide in the hybridization buffer).
Keywords: Genosensor, Glassy carbon electrode, Electrocatalytic deposition of silver, Electroactive label, Sodium aurothiomalate
1. Introduction
DNA diagnostic has become an important area of molecular biology and biotechnology studies. The detection of specific base sequences in human, viral and bacterial nucleic acid is becoming increasingly important in several areas, with applications ranging from the detection of disease-causing and food-contaminating organisms to forensic and environmental research.
Conventional methods for the analysis of specific gene sequences are based on either direct sequencing or DNA hybridization methods. Although the DNA sequencing has undergone very big progress in the last decade (Kling, 2003, Kartalov and Quake, 2004, Aborn et al., 2005), these traditional methods, based on the coupling of electrophoretic separations and radioisotopic (32P) detection, are generally labour intensive and time consuming. Therefore, the second option is more commonly used in diagnostic laboratories.
Genosensors (or DNA hybridization biosensors) offer a promising alternative to carry out the specific gene sequence identification (De los Santos-Álvarez et al., 2004, Wang, 2002, Paleček et al., 1998). These biosensors commonly rely on the immobilization of a single-stranded (ss) oligonucleotide probe onto a transducer surface to recognize – by hybridization – its complementary target sequence.
Among the different kind of transducers found in the literature – chemiluminescent (Nguyen and Heffelfinger, 1995), quartz crystal microbalance (Hashimoto et al., 1994, Steel et al., 1998), fiber optical (Piunno et al., 1995), evanescent wave (Watts et al., 1995) or an-acoustic wave (Su et al., 1996) – those based on electrochemical transduction, due to their high sensitivity, small dimensions, low cost and compatibility with micro-fabrication technology provide a very interesting alternative.
These electrochemical biosensing devices can monitor sequence-specific hybridization processes: (i) directly, based on the intrinsic DNA electroactivity (Kerman et al., 2005, Erdem et al., 2004, Ozkan et al., 2002, Lucarelli et al., 2002) or (ii) indirectly by measuring changes in the electrical properties of the electrode–solution interface – electrochemical properties of conducting polymers (Thompson et al., 2003, Cha et al., 2003, Wang et al., 1999), capacitance (Berney et al., 2000) or impedance (Souteyrand et al., 1997, Alfonta et al., 2001) – or using different covalently and non-covalently bound markers.
Non-covalent markers (hybridization indicators) are electroactive compounds – metal complexes (Wang et al., 1997, Zhao and Ju, 2004, Nojima et al., 2003, Kara et al., 2002, Aoki and Umezawa, 2003) or organic compounds (Zhu et al., 2005, Teh et al., 2005, Marrazza et al., 1999, Hashimoto et al., 1998) – that interact in a different way with single strands (ss-) and double strands (ds-) of DNA.
Covalently bound markers can be subdivided in two groups, electroactive and non-electroactive. Ferrocene and its derivatives (Nakayama et al., 2002, Xu et al., 2000, Xu et al., 2001), Pt(II) complex (Hernández-Santos et al., 2005), colloidal gold (Ozsoz et al., 2003, Cai et al., 2002), or tris(2,2′-bipyridyl)cobalt(III)-doped silica nanoparticles (Zhu et al., 2003) are used as electroactive markers, while as non-electroactive marker, the use of enzymes, linked directly to the DNA strand (Zhang et al., 2003, Zhang et al., 2004) or indirectly through biotin–avidin (Abad-Valle et al., 2005, Azek et al., 2000) or fluorescein–antifluorescein bridges (Hernández-Santos et al., 2004), has been extensively reported.
As can be seen, metal complexes and metal nanoparticles have an important role in the development of electrochemical genosensors. In addition to the works cited above, where metal complexes are used as hybridization indicators or electroactive labels, examples of transition metal complexes, e.g. Ru(bpy)3 2+, Os(bpy)3 2+, mediating the electro-oxidation of nucleobases, have also been found in literature (Yang et al., 2002, Gore et al., 2003).
In this work, it was used for first time sodium aurothiomalate as electroactive label in a DNA hybridization biosensor. This gold(I) complex has been previously used in our laboratory with success as electroactive label for immunosensor devices using glassy carbon electrodes (GCEs) as electrochemical transducers (de la Escosura-Muñiz et al., 2004a). The electrochemical detection in these immunosensors is based on the catalytic effect of ionic gold on silver electrodeposition (de la Escosura-Muñiz et al., 2004b).
GCEs have been widely used in the construction of electrochemical DNA hybridization biosensors in combination with different oligonucleotides immobilization methods, such as (i) film entrapment using polymers (Wang et al., 1999, Xu et al., 2001, Cai et al., 2002, Zhu et al., 2003), (ii) covalent attachment on functionalized glassy carbon electrodes—polymers, carbon nanotubes, etc. (Zhao and Ju, 2004, Kara et al., 2002, Zhu et al., 2005, Teh et al., 2005, Piro et al., 2005, Cai et al., 2003) or (iii) by electrostatic adsorption on gold colloid modified surfaces (Lin et al., 2002).
Direct adsorption of oligonucleotides onto pre-oxidized glassy carbon electrodes is used in this work, in combination with the electroactive label sodium aurothiomalate for the electrochemical detection of a SARS virus sequence, using a silver catalytic electrodeposition method.
2. Experimental
2.1. Apparatus and electrodes
Cyclic voltammetric experiments were performed with an ECO CHEMIE Autolab PGSTAT 10 potentiostat interfaced to a Pentium 120 computer system and controlled by an Autolab GPES Version 4.6 for Windows 98.
All measurements were carried out at room temperature in a 20 mL cell (protected from light) with a three-electrode configuration. Working glassy carbon electrodes (GCEs) were home made, using 3 mm diameter glassy carbon rods (Goodfellow, Spain), sealed into Teflon holders with a spurr resin for electron microscopy purchased from Sigma (Spain). Electrical contact was a brass rod welded to glassy carbon with a silver loaded conductive epoxy resin purchased from Circuit Works (USA). The renewal of the glassy carbon surface was achieved by polishing with 1.0 and 0.3 μm alpha-alumina on a microcloth polishing sheet of 8 in, followed by washing in an ultrasonic Selecta bath for 5 min.
A platinum wire as counter electrode and an Ag/AgCl reference electrode were used.
A Metrohm AG Herisau magnetic stirrer was used for the electrochemical pre-treatment of the electrode surface and the gold electrodeposition and oxidation.
2.2. Reagents and solutions
Synthetic 30-mer oligonucleotides were obtained from Eurogentec (Spain). The target sequence employed corresponds to a portion of the severe acute respiratory syndrome (SARS) virus, precisely the bases comprised between 29218 and 29247, both included. For selectivity studies, single-base and three-base-mismatch strands were also purchased. Mismatches are located in bases number 12 and 5, 15 and 26, respectively.
Target (50 nmol): 5′-ACA-GAG-CCT-AAA-AAG-GAC-AAA-AAG-AAA-AAG-3′;
Thiolated target (50 nmol): 5′-ACA-GAG-CCT-AAA-AAG-GAC-AAA-AAG-AAA-AAG-SH-3′;
Single-base-mismatch target (49 nmol): 5′-ACA-GAG-CCT-AAC-AAG-GAC-AAA-AAG-AAA-AAG-3′;
Three-base-mismatch target (184 nmol): 5′-ACA-GCG-CCT-AAA-AAC-GAC-AAA-AAG-AGA-AAG-3′;
The probe is a complementary strand of the target that was also obtained from Eurogentec. Probe (50 nmol): 5′-CTT-TTT-CTT-TTT-GTC-CTT-TTT-AGG-CTC-TGT-3′.
Oligonucleotide solutions were prepared in TE buffer, pH 8 (10 mM Tris–HCl buffer solution, 1 mM in EDTA). Aliquots were prepared and maintained at −20 °C.
Working solutions of the oligonucleotide probe were made in 0.1 M Tris, pH 7.2 buffer, while all gold labeled oligonucleotide target strands were diluted in a 2 × SSC buffer (300 mM sodium chloride/30 mM sodium citrate), pH 7.2. These solutions were conserved at 4 °C. For selectivity studies, oligonucleotide strands were diluted in 2 × SCC buffer or in this buffer containing 50% of formamide. For the labeling procedure, target strands were diluted in an aqueous solution of 0.15 M NaCl with pH adjusted to 7.5 with 0.1 M NaOH.
Sodium aurothiomalate was obtained from Aldrich (Spain). It was reconstituted in 0.15 M NaCl solution and protected from light. Dilutions of this stock solution were prepared daily in an unbuffered aqueous solution of 0.15 M NaCl, with pH adjusted to 7.5 with 0.1 M NaOH.
Tris(hydroxymethyl)aminomethane (Tris) and formamide were purchased from Sigma (Spain). EDTA was obtained from Fluka (Spain).
Silver nitrate was obtained from Sigma (Spain). Solutions were prepared using ultra-pure water and were protected from light.
Analytical grade (Merck, Spain) NaCl, HCl, H2SO4, NH3, KCN, NaOH and sodium citrate were used. Their solutions were prepared with Millipore Milli-Q system, excepting KCN solutions, which were prepared with a 0.1 M NaOH solution.
Ultra-pure water obtained with a Milli-Q plus 185 from Millipore Ibérica S.A. (Spain) was used for all solutions.
Alumina powders of 1.0 and 0.3 μm grain sizes and microcloth polishing sheet of 8 in were purchased from Buehler (Germany).
Dialysis procedures were carried out with Slide-A-Lizer dialysis cassettes, 3500 MWCO from Pierce (USA).
2.3. Methods
Fig. 1 shows a scheme of the analytical procedure.
Fig. 1.
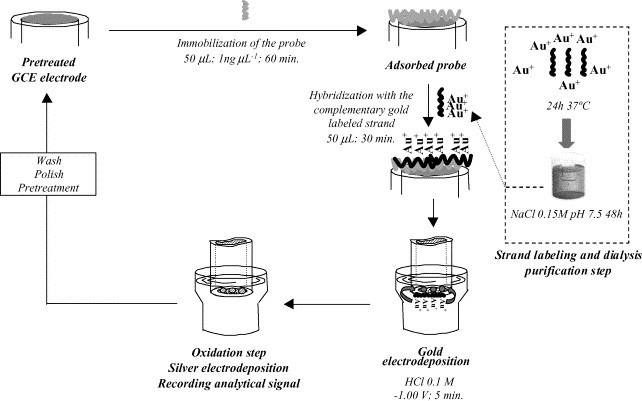
Scheme of the DNA hybridization biosensor and the analytical signal recording.
2.3.1. Labeling of the target strand with sodium aurothiomalate
The conjugation of aurothiomalate to single strands was carried out according to the procedure described in a previous work to label immunoglobulins (de la Escosura-Muñiz et al., 2004a). An aliquot of 50 μL of a 200 ng μL−1 target strand solution was mixed with an aliquot of 450 μL of a 390 ng μL−1 sodium aurothiomalate solution. The reaction was carried out at 37 °C for 24 h. After that, the conjugate (strand-Au) was purified by dialysis against 200 mL of 0.15 M NaCl, pH 7.5, unbuffered solution, for 48 h at room temperature.
Thiolated and non-thiolated targets, single-base and three-base-mismatch complementary strands and a non-complementary strand were labeled following this procedure.
2.3.2. Electrode pre-treatment
The smoothed glassy carbon surfaces were pre-treated before each assay by an electrochemical pre-treatment in 0.1 M HCl. Working electrodes were immersed in a 0.1 M HCl stirred solution and a potential of +1.40 V was applied for 2 min. After that, the electrode surfaces were washed with 0.1 M Tris–HCl, pH 7.2, buffer.
2.3.3. Immobilization of the probe
The probe immobilization was performed by physical adsorption, onto the inverted electrode, at room temperature. Five GCEs were used simultaneously and the adsorption was carried out depositing a 50 μL droplet of a 1 ng μL−1 oligonucleotide probe solution on each electrode surface, and left there for 60 min. Then, a washing step was performed with the hybridization buffer solution.
2.3.4. Hybridization reaction
After the probe immobilization and the washing step, the hybridization reaction took place for 30 min with a 30 μL droplet of the complementary gold labeled strand, 2 × SCC buffer pH 7.2 diluted, at room temperature. After that, the electrode surfaces were washed with ultra-pure water.
The same procedure was performed to obtain the background signals, using a gold labeled non-complementary strand, and for selectivity studies with gold labeled single-base and three-base-mismatch complementary strands. For the selectivity studies, 2 × SCC buffer, pH 7.2, and this buffer containing 50% of formamide were used.
2.3.5. Recording the analytical signal: catalytic effect of gold on the silver electrodeposition
The electrodes were immersed in a stirred 0.1 M HCl solution, and the gold, which was bound to the target strand, was electrodeposited on the electrode surfaces by applying a potential of −1.00 V for 5 min. After a washing step with ultra-pure water, the electrodes were immersed in a stirred 0.1 M H2SO4 solution and an oxidation step was carried out applying a potential of +1.40 V for 60 s. After that, the electrodes were rinsed with ultra-pure water and introduced in an unstirred 1.0 M NH3 solution containing silver nitrate at a fixed concentration (2.0 × 10−4 M) and held at a deposition potential of −0.14 V for 60 s. Then, cyclic voltammograms were scanned from deposition potential to +0.30 V at a scan rate of 50 mV s−1, obtaining the analytical signal. Finally, in order to remove the gold from the electrode surfaces, the GCEs were immersed after each measurement in another cell containing a 0.1 M KCN stirred solution for 2 min in open circuit.
This analytical procedure, based on the catalytic effect of electrodeposited gold on the electroreduction of silver ions (de la Escosura-Muñiz et al., 2004b) has been previously optimized, using bovine serum albumin (BSA) and a rabbit immunoglobulin G (RIgG) labeled with aurothiomalate (de la Escosura-Muñiz et al., 2004a). Briefly, this detection protocol involves a reduction step at −1.00 V in HCl 0.1 M for the gold label electrodeposition on the electrode surface, followed by an oxidation step in 0.1 M H2SO4 necessary to remove the hydrogen generated in the gold deposition step and to oxidize this gold. Then, by selecting an adequate potential (−0.14 V), the silver contained in the solution of AgNO3 2 × 10−4 M–NH3 1.0 M is reduced to metallic silver only in presence of gold deposited on the electrode surface. Finally, an anodic scan is performed and the reduced silver is oxidized at +0.08 V, which constitutes the analytical signal.
As the amount of silver electrodeposited at this potential is proportional to the gold deposited on the electrode surface, the silver stripping allows the determination of the gold labeled target that has hybridized with the probe. When a non-complementary gold labeled strand is assayed no hybridization takes place, so no gold is deposited on the electrode surface. Thus, no silver is reduced at the chosen deposition potential, and no signal is obtained.
3. Results and discussion
3.1. Single strand DNA labeling
It is well known that sodium aurothiomalate binds to the albumin molecules mainly through the thiol group of the cysteine 34, by exchanging a proton (Moller-Pedersen, 1987). Based on this interaction between gold and thiol, our group has successfully used sodium autothiomalate for labeling different biological molecules, such as BSA and immunoglobulins, in order to design electrochemical metal immunoassays and immunosensors (de la Escosura-Muñiz et al., 2004a).
Thus, this high affinity between gold and thiol group was applied in order to label a thiolated oligonucleotide. The product of the dialysis step was then evaluated by direct adsorption on the electrode surface, previously pre-treated at +1.40 V (versus Ag/AgCl) for 2 min in 0.1 M HCl stirred solution. A 50 μL droplet of a 1 ng μL−1 solution of the thiolated oligonucleotide, after the labeling process, was deposited on the electrode surface and left there for 30 min, before recording the analytical signal.
To verify the gold(I)–oligonucleotide binding and to know if this interaction is through the thiol group, a solution of sodium aurothiomalate 390 ng μL−1 that was not mixed with any oligonucleotide, and a non-thiolated oligonucleotide with identical sequence, were subjected to the same procedure (reaction + dialysis + adsorption).
The results obtained are showed in Fig. 2 . As it can be seen in this figure, very similar analytical signals were obtained for both, the thiolated (Fig. 2a) and the non-thiolated oligonucleotide (Fig. 2b), while, as it could be expected, no analytical signal was obtained for the aurothiomalate solution assayed (Fig. 2c), because, as it is known by our previous works, sodium aurothiomalate is totally dialysed and anyway, it is not adsorbed on the electrode surface under these experimental conditions. The reproducibility of the signals showed in Fig. 2a and b was checked, using five different GCEs, obtaining a R.S.D. of 8% (n = 5, Ip average = 6.6 μA) and 7% (n = 5, Ip average = 6.4 μA), respectively.
Fig. 2.
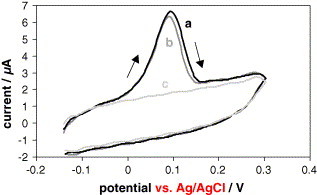
Cyclic voltammograms recorded in aqueous 1.0 M NH3 −2.0 × 10−4 M AgNO3 from −0.14 to +0.30 V for 50 μL of: (a) 1 ng μL−1 thiolated oligonucleotide solution, (b) 1 ng μL−1 non-thiolated oligonucleotide solution and (c) 39 ng μL−1 dialysed sodium aurothiomalate solution, adsorbed during 30 min on the GCE surface. Gold deposition potential: −1.00 V; gold deposition time: 5 min; oxidation step in 0.1 M H2SO4 for 60 s after the gold deposition; silver deposition time: 60 s; silver deposition potential: −0.14 V; scan rate: 50 mV s−1.
These results indicate that oligonucleotide is adsorbed on the GCE surface and it has been successfully labeled with sodium aurothiomalate.
The DNA binding properties of sodium aurothiomalate and other gold(I) complexes have been studied by Blank and Dabrowiak (1984) using absorption and circular dichroism spectroscopies. This study concludes that sodium aurothiomalate does not bind to calf thymus DNA, while others gold(I) complexes which have an easily displaced ligand, e.g. Cl− or Br− are capable of interacting in a non-denaturing fashion with the guanine and cytosine residues of calf thymus DNA.
To the best of our knowledge, this is the unique study of gold(I)–DNA complexes that includes sodium aurothiomalate. So, we only have evidence of its not interaction with calf thymus DNA, but we have no information about its binding to ssDNA or to DNA nucleosides.
Therefore, the aurothiomalate–oligonucleotide binding mechanism is not clear, since not only the thiolated strand was labeled, being the interaction not through the thiol group, at least in its totality, as it could be expect. Besides, this fact allows us to label unmodified oligonucleotides which is economically advantageous.
3.2. Immobilization of the probe
Physical adsorption is used in order to immobilize the oligonucleotide on the pre-oxidized GCE surface at room temperature. The influence of the probe strand concentration and adsorption time on the analytical signal were evaluated following the analytical procedure previously described, using in both cases 30 min of hybridization and 0.1 ng μL−1 gold labeled target solution.
Fig. 3 shows the influence of the probe strand concentration on the analytical signal, using an adsorption time of 60 min. The peak current increases with the probe concentration up to 1 ng μL−1, where it reaches a plateau. So, 1 ng μL−1 was chosen for further studies.
Fig. 3.
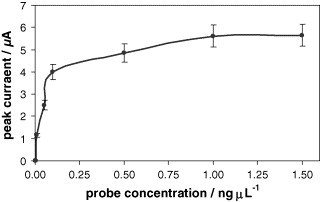
Influence of the probe strand concentration on the analytical signal. Probe adsorption time: 60 min; labeled target concentration: 0.1 ng μL−1; hybridization time: 30 min. The rest of experimental conditions are as in Fig. 2. Data are given as average ± S.D. (n = 3).
A very important fact obtained from this optimization assay is that non-specific adsorptions of the gold labeled target on the electrode surface are not observed for the concentration assayed (0.1 ng μL−1), since when no probe is immobilized, no analytical signal is recorded. The absence of non-specific adsorptions is a very important fact in the development of DNA hybridization sensors, since a blocking step is not needed, which simplifies the procedure, and the sensitivity of the assay is notably improved.
Two factors could be responsible for this fact. One of them is that the gold labeled target concentration is not higher enough to adsorb on the electrode surface in 30 min. The other one is that, although the strand is adsorbed, the low amount of gold deposited on the electrode does not catalyze the silver reduction.
Once the probe concentration was optimized, the effect of the probe adsorption time (from 5 to 90 min) on the analytical signal was evaluated. The obtained results (data not shown) show that the peak current increases upon raising the adsorption time up to 60 min. Beyond this point, the electrode response changes slightly with the adsorption time. Thus, an adsorption time of 60 min is chosen for further studies. Besides of the peak current increase observed with the adsorption time, a better reproducibility of the analytical signal is also obtained when the adsorption time is increased.
Electrostatic adsorption of the oligonucleotide probe was also tested using different times (1–10 min) and potentials (+0.3 to +1.0 V) without success.
3.3. Hybridization reaction
After the optimization of the genosensor sensing phase, the hybridization reaction time (between 5 and 60 min) was studied using a 30 μL droplet of a 0.1 ng μL−1 solution of gold labeled target strand in 2 × SCC, pH 7.2, buffer solution. The analytical signal recorded increases with the reaction time, reaching a plateau for a hybridization time of 30 min (data not shown). So, a hybridization time of 30 min is chosen for further studies.
The background signal was also evaluated using a 0.1 ng μL−1 solution of the gold labeled non-complementary strand. When this strand is assayed, no hybridization reaction is expected, so this gold labeled strand will not be captured by the probe immobilized on the electrode surface. As the analytical signal is based on the catalytic effect of electrodeposited gold on the silver electroreduction, when no gold is deposited on the electrode surface, no silver is reduced at the chosen deposition potential, and no signal is obtained.
As can be seen in Fig. 4 , no hybridization reaction takes place working with the gold labeled non-complementary strand, and a perfect discrimination between complementary (Fig. 4a) and non-complementary (Fig. 4b) strand is obtained under the experimental conditions optimized.
Fig. 4.
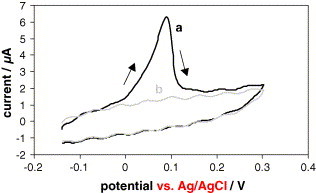
Cyclic voltammograms recorded in aqueous 1.0 M NH3 −2.0 × 10−4 M AgNO3 from −0.14 to +0.30 V for the DNA hybridization sensor with the probe immobilized from a solution of 1 ng μL−1 that is left to react for 30 min with 30 μL of: (a) 0.1 ng μL−1 gold labeled target solution and (b) 0.1 ng μL−1 gold labeled non-complementary strand solution (background). The rest of experimental conditions are as in Fig. 2.
3.4. Analytical characteristics of the DNA hybridization sensor
Following the analytical procedure previously described, the higher the concentration of labeled target in solution, the higher is the amount of gold that is electrodeposited, and subsequently, an increase in the analytical signal is obtained.
The peak current showed a good linear relationship with the concentration of gold labeled target prepared in 2 × SCC, pH 7.2, buffer solution, in the range from 10 to 200 pg μL−1, with a correlation coefficient of 0.9984, according to the following equation:
The limit of detection (calculated as the concentration corresponding to three times the standard deviation of the estimate) of the target strand was 5.0 pg μL−1 (15 fmol in 30 μL).
Background signals, using the gold labeled non-complementary strand, and non-specific adsorption of the gold labeled target were also evaluated. As it was expected in both cases, no analytical signals were obtained working with concentrations between 10 and 200 pg μL−1.
The reproducibility of the analytical signal was checked. Using five different GCEs, a R.S.D. of 10% (n = 5, Ip average = 2.1 μA) was obtained for a 50 pg μL−1 solution of the complementary strand.
Finally, it was also evaluated the selectivity of the DNA hybridization sensor, using different concentrations (10, 25, 50, 100 and 200 pg μL−1) of both single-base and three-base-mismatch gold labeled complementary strands prepared in 2 × SCC, pH 7.2, buffer solution. As it is shown in Fig. 5 , the sensor allows us to discriminate between the complementary strand and the three-base-mismatch complementary strand. However, with the single-base mismatch complementary strand, the analytical signals recorded are similar than those obtained for the complementary strand.
Fig. 5.
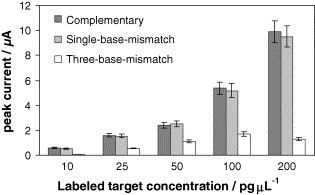
Selectivity of the DNA hybridization sensor for the gold labeled strands: complementary, single-base-mismatch and three-base-mismatch in non-stringent experimental conditions. Data are given as average ± S.D. (n = 3).
In order to discriminate a single-base-mismatch, other saline concentrations of the hybridization buffer were tested, but the discrimination between single-base-mismatch complementary strand and the target strand was not improved.
More stringent experimental conditions were also tested, adding different concentrations of formamide to the 2 × SCC, pH 7.2, hybridization buffer. It is well known that this molecule makes the hybridization reaction more difficult by decreasing the melting point of DNA.
A concentration of 50% of formamide was necessary to discriminate between single-base-mismatch complementary strand and the target strand. The results obtained for 10, 25, 50, 100 and 200 pg μL−1 strand concentrations are shown in Fig. 6 . Working with this formamide concentration, the analytical signals obtained for the target strand decrease, while the analytical signals obtained for the three-base-mismatch complementary strand were approaching zero.
Fig. 6.
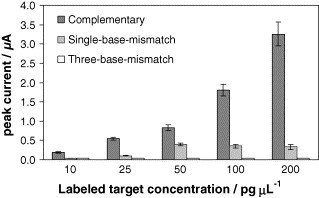
Selectivity of the DNA hybridization sensor for the gold labeled strands: complementary, single-base-mismatch and three-base-mismatch in stringent experimental conditions. Data are given as average ± S.D. (n = 3).
Under these more stringent conditions, a new calibration curve was obtained. A linear relationship between peak current and concentration of oligonucleotide target was obtained for concentrations between 10 and 200 pg μL−1, with a correlation coefficient of 0.997, according to the following equation:
The limit of detection calculated as described above, was 10 pg μL−1 (30 fmol in 30 μL).
As can be seen, the sensitivity (slope) of the assay decreases under these more stringent conditions, due to the minor peak current recorded for all concentrations of gold labeled target assayed. However, the same linear range from 10 to 200 pg μL−1 were obtained working with both non-stringent and stringent experimental conditions.
The R.S.D. (50 pg μL−1 target strand) for five parallel experiments under these experimental conditions was 8.9% with a mean peak current of 0.84 μA.
4. Conclusions
This work describes, for the first time, the use of sodium aurothiomalate as electroactive label in a DNA hybridization biosensor.
About the interaction between this gold(I)complex and oligonucleotides, we only can conclude that the presence of a thiol group on the strand, which acts as a binding site for gold in other biological molecules, is not necessary to carry out an effective interaction.
However, the hybridization of gold labeled target strand onto the surface of a probe modified GCE is performed successfully, demonstrating that DNA bases of the target are available for the hybridization after its labeling with the gold(I) complex.
Although the sensitivity of the sensor is lower than that obtained with an enzymatic method on gold band electrodes (Abad-Valle et al., 2005), the analysis time is considerably shorter, since in the enzymatic method, a blocking step, a biotin–streptavidin interaction to link the enzyme to the duplex and an enzymatic reaction are needed before to obtain the analytical signal.
Besides, the sensitivity of this genosensor is competitive with others reported that use Pt(II) complex – 20-mer oligonucleotide (Hernández-Santos et al., 2005) – or tris(2,2′-bipyridil)cobalt(III)-doped silica nanoparticle – 24-mer oligonucleotide (Zhu et al., 2003) – electroactive label; and with others gold nanoparticle-based hybridization assays – 19-mer oligonucleotide (Wang et al., 2001) – that use electrochemical detection.
The reported genosensor is simple, economical and it is able to discriminate single-base mismatch on the target.
Work is in progress trying to elucidate the binding mechanism between aurothiomalate and oligonucleotides and adapting the system to another carbon electrodes in order to improve the sensitivity.
Acknowledgment
This work has been supported by Spanish project BIO2003-06008-C03-01.
References
- Abad-Valle P., Fernández-Abedul M.T., Costa-García A. Biosens. Bioelectron. 2005;20:2251–2260. doi: 10.1016/j.bios.2004.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aborn J.H., El-Difrawy S.A., Novotny M., Gismondi E.A., Lam R., Matsudaira P., Mckenna B.K., O’Neil T., Streechon P., Ehrlich D.J. Lab. Chip. 2005;6:669–674. doi: 10.1039/b501104c. [DOI] [PubMed] [Google Scholar]
- Alfonta L., Singh A.K., Willner I. Anal. Chem. 2001;73:91–102. doi: 10.1021/ac000819v. [DOI] [PubMed] [Google Scholar]
- Aoki H., Umezawa Y. Analyst. 2003;128:681–685. doi: 10.1039/b300465a. [DOI] [PubMed] [Google Scholar]
- Azek F., Grossiord C., Joannes M., Limoges B., Brossier P. Anal. Biochem. 2000;284:107–113. doi: 10.1006/abio.2000.4692. [DOI] [PubMed] [Google Scholar]
- Berney H., West J., Haefele E., Alderman J., Lane W., Collins J.K. Sens. Actuators B. 2000;68:100–108. [Google Scholar]
- Blank C.E., Dabrowiak J.C. J. Inorg. Biochem. 1984;21:21–29. doi: 10.1016/0162-0134(84)85036-9. [DOI] [PubMed] [Google Scholar]
- Cai H., Cao X., Jiang Y., He P., Fang Y. Anal. Bioanal. Chem. 2003;375:287–293. doi: 10.1007/s00216-002-1652-9. [DOI] [PubMed] [Google Scholar]
- Cai H., Wang Y., He P., Fang Y. Anal. Chim. Acta. 2002;469:165–172. [Google Scholar]
- Cha J., Ham J.I., Choi Y., Yoon D.S., Oh K.W., Lim G. Biosens. Bioelectron. 2003;18:1241–1247. doi: 10.1016/s0956-5663(03)00088-5. [DOI] [PubMed] [Google Scholar]
- de la Escosura-Muñiz A., González-García M.B., Costa-García A. Anal. Chim. Acta. 2004;524:355–363. [Google Scholar]
- de la Escosura-Muñiz A., González-García M.B., Costa-García A. Electroanalysis. 2004;16:1561–1568. [Google Scholar]
- De los Santos-Álvarez P., Lobo-Castañón M.J., Miranda-Ordieres A.J., Tuñón-Blanco P. Anal. Bioanal. Chem. 2004;378:104–118. doi: 10.1007/s00216-003-2369-0. [DOI] [PubMed] [Google Scholar]
- Erdem A., Prividori M.I., del Valle M., Alegret S. J. Electroanal. Chem. 2004;567:29–37. [Google Scholar]
- Gore M.R., Szalai V.A., Ropp P.A., Yang I.V., Silverman J.S., Thorp H.H. Anal. Chem. 2003;75:6586–6592. doi: 10.1021/ac034918v. [DOI] [PubMed] [Google Scholar]
- Hashimoto K., Ito K., Ishimori Y. Sens. Actuators B. 1998;46:220–225. [Google Scholar]
- Hashimoto K., Ito K., Ishimori Y. Anal. Chem. 1994;66:3830–3833. doi: 10.1021/ac00093a045. [DOI] [PubMed] [Google Scholar]
- Hernández-Santos D., González-García M.B., Costa-García A. Anal. Chem. 2005;77:2868–2874. doi: 10.1021/ac048091w. [DOI] [PubMed] [Google Scholar]
- Hernández-Santos D., Díaz-González M., González-García M.B., Costa-García A. Anal. Chem. 2004;76:6887–6893. doi: 10.1021/ac048892z. [DOI] [PubMed] [Google Scholar]
- Kara P., Ozkan D., Kerman K., Meric B., Erdem A., Ozsoz M. Anal. Bioanal. Chem. 2002;373:710–716. doi: 10.1007/s00216-002-1301-3. [DOI] [PubMed] [Google Scholar]
- Kartalov E.P., Quake S.R. Nucleic Acids Res. 2004;32:2873–2879. doi: 10.1093/nar/gkh613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerman K., Morita Y., Takamura Y., Tamiya E. Anal. Bioanal. Chem. 2005;381:1114–1121. doi: 10.1007/s00216-004-3007-1. [DOI] [PubMed] [Google Scholar]
- Kling J. Nat. Biotechnol. 2003;21:1425–1427. doi: 10.1038/nbt1203-1425. [DOI] [PubMed] [Google Scholar]
- Lin X., Zheng S., Miao Q., Jin B. Anal. Lett. 2002;35:1373–1385. [Google Scholar]
- Lucarelli F., Marrazza G., Palchetti I., Cesaretti S., Mascini M. Anal. Chim. Acta. 2002;469:93–99. doi: 10.1016/j.aca.2008.03.027. [DOI] [PubMed] [Google Scholar]
- Marrazza G., Chianella I., Mascini M. Biosens. Bioelectron. 1999;14:43–51. doi: 10.1016/s0956-5663(98)00102-x. [DOI] [PubMed] [Google Scholar]
- Moller-Pedersen S. Biochem. Pharm. 1987;36:2661–2666. doi: 10.1016/0006-2952(87)90548-x. [DOI] [PubMed] [Google Scholar]
- Nakayama M., Ihara T., Nakano K., Maeda M. Talanta. 2002;56:857–866. doi: 10.1016/s0039-9140(01)00659-2. [DOI] [PubMed] [Google Scholar]
- Nguyen Q., Heffelfinger D.M. Anal. Biochem. 1995;226:59–67. doi: 10.1006/abio.1995.1191. [DOI] [PubMed] [Google Scholar]
- Nojima T., Yamashita K., Takagi A., Takagi M., Ikeda Y., Kondo H., Takenaka S. Anal. Sci. 2003;19:79–83. doi: 10.2116/analsci.19.79. [DOI] [PubMed] [Google Scholar]
- Ozkan D., Erdem A., Kara P., Kerman K., Meric B., Hassmann J., Ozsoz M. Anal. Chem. 2002;74:5931–5936. doi: 10.1021/ac0257905. [DOI] [PubMed] [Google Scholar]
- Ozsoz M., Erdem A., Kerman K., Ozkan D., Tugrul B., Topcuoglu N., Ekren H., Taylan M. Anal. Chem. 2003;75:2181–2187. doi: 10.1021/ac026212r. [DOI] [PubMed] [Google Scholar]
- Paleček E., Fojta M., Tomschik M., Wang J. Biosens. Bioelectron. 1998;13:621–628. doi: 10.1016/s0956-5663(98)00017-7. [DOI] [PubMed] [Google Scholar]
- Piro B., Haccoun J., Pham M.C., Tran L.D., Rubin A., Perrot H., Gabrielli C.J. Electroanal. Chem. 2005;577:155–165. [Google Scholar]
- Piunno P.A.E., Krull U.J., Hudson R.H.E., Damha M.J. Anal. Chem. 1995;67:2635–2643. doi: 10.1021/ac00111a022. [DOI] [PubMed] [Google Scholar]
- Souteyrand E., Cloarec J.P., Martin J.R., Wilson C., Lawrence I., Mikkelsen S., Lawrence M.F. J. Phys. Chem. B. 1997;101:2980–2985. [Google Scholar]
- Steel A.B., Herne T.M., Tarlov M.J. Anal. Chem. 1998;70:4670–4677. doi: 10.1021/ac980037q. [DOI] [PubMed] [Google Scholar]
- Su H., Chong S., Thompson M. Langmuir. 1996;12:2247–2255. [Google Scholar]
- Teh H.F., Gong H., Dong X.-D., Zeng X., Tan A.L.K., Yang X., Tan S.N. Anal. Chim. Acta. 2005;551:23–29. [Google Scholar]
- Thompson L.A., Kowalik J., Josowicz M., Janata J. J. Am. Chem. Soc. 2003;125:324–325. doi: 10.1021/ja027929z. [DOI] [PubMed] [Google Scholar]
- Wang J. Anal. Chim. Acta. 2002;469:63–71. [Google Scholar]
- Wang J., Xu D., Kawde A.-N., Polsky R. Anal. Chem. 2001;73:5576–5581. doi: 10.1021/ac0107148. [DOI] [PubMed] [Google Scholar]
- Wang J., Jiang M., Fortes A., Mukherjee B. Anal. Chim. Acta. 1999;402:7–12. [Google Scholar]
- Wang J., Rivas G., Cai X., Dontha N., Shiraishi H., Luo D., Valera F.S. Anal. Chim. Acta. 1997;337:41–48. [Google Scholar]
- Watts H.J., Yeung D., Parkes H. Anal. Chem. 1995;67:4283–4289. doi: 10.1021/ac00119a013. [DOI] [PubMed] [Google Scholar]
- Xu C., Cai H., He P., Fang Y. Analyst. 2001;126:62–65. doi: 10.1039/b005847p. [DOI] [PubMed] [Google Scholar]
- Xu C., He P., Fang Y. Anal. Chim. Acta. 2000;411:31–36. [Google Scholar]
- Yang I.V., Ropp P.A., Thorp H.H. Anal. Chem. 2002;74:347–354. doi: 10.1021/ac0109063. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Pothukuchy A., Shin W., Kim Y., Heller A. Anal. Chem. 2004;76:4093–4097. doi: 10.1021/ac0495034. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Kim H.-H., Heller A. Anal. Chem. 2003;75:3267–3269. doi: 10.1021/ac034445s. [DOI] [PubMed] [Google Scholar]
- Zhao H., Ju H. Electroanalysis. 2004;16:1642–4646. [Google Scholar]
- Zhu N., Chang Z., He P., Fang Y. Anal. Chim. Acta. 2005;545:21–26. [Google Scholar]
- Zhu N., Cai H., He P., Fang Y. Anal. Chim. Acta. 2003;481:181–189. [Google Scholar]