

Graphical abstract

Keywords: SARS 3CL protease, Inhibitor, Decahydroisoquinolin, Non-prime site, Thioacetal
Abstract
A non-prime site substituent and warheads combined with a decahydroisoquinolin scaffold was evaluated as a novel inhibitor for severe acute respiratory syndrome (SARS) chymotrypsin-like protease (3CLpro). The decahydroisoquinolin scaffold has been demonstrated to be an effective hydrophobic center to interact with S2 site of SARS 3CLpro, but the lack of interactions at S3 to S4 site is thought to be a major reason for the moderate inhibitory activity. In this study, the effects of an additional non-prime site substituent on the scaffold as well as effects of several warheads are evaluated. For the introduction of a desired non-prime site substituent, amino functionality was introduced on the decahydroisoquinolin scaffold, and the scaffold was constructed by Pd(II) catalyzed diastereoselective ring formation. The synthesized decahydroisoquinolin inhibitors showed about 2.4 times potent inhibitory activities for SARS 3CLpro when combined with a non-prime site substituent. The present results indicated not only the expected additional interactions with the SARS 3CLpro but also the possibility of new inhibitors containing a fused-ring system as a hydrophobic scaffold and a new warhead such as thioacetal.
1. Introduction
In 2003, a life-threatening atypical pneumonia called severe acute respiratory syndrome (SARS) spread worldwide from its likely origin in southern China and affected about 8500 patients with more than 800 fatalities.1, 2, 3 The causative virus is a new beta coronavirus (CoV) containing 29.7 kb positive-strand RNA genome which encodes two large replicative polyproteins: pp1a (486 kDa) and pp1ab (790 kDa).4, 5, 6 The polyprotein contains a 3C-like cysteine protease (SARS 3CLpro) which can process the polyprotein to provide a variety of proteins necessary to re-construct the virus particle. Thus, the SARS 3CLpro is indispensable for viral replication but not found in the host cell, which makes the SARS 3CLpro an ideal target for antiviral agents. Numerous studies on substrate-peptide-based and non-peptide inhibitors for SARS 3CLpro have been reported,7, 8 but no therapeutic agents are available at present.
During our studies on SARS 3CLpro inhibitors,9 a substrate-based peptide aldehyde 1 was found to show potent inhibitory activity (IC50 = 98 nM) for R188I SARS 3CLpro.10 Based on X-ray crystal structure analyses of 1 complexed with R188I SARS 3CLpro, the following structures regarding interactions with the protease are clarified: (i) C-terminal aldehyde of 1 is located at hydrogen-bond distance from the active center of the protease forming no covalent bond, (ii) P1 site imidazole is inserted tightly into S1 pocket of the protease, (iii) P2 site cyclohexyl ring interacts with the protease at S2 pocket by hydrophobic interaction, and (iv) distance of o-position carbon of cyclohexyl-ring from α-nitrogen of cyclohexylalanine (Cha) is 3.48 Å (Fig. 1 ). These results suggest that the cyclohexyl ring might be connected to the peptide chain by a methylene linker, yielding a new non-peptide inhibitor scaffold containing a hydrophobic decahydroisoquinolin ring. The resulting decahydroisoquinolin-type inhibitor 2 was synthesized and confirmed to be an inhibitor as expected, but the potency was moderate (IC50 = 63 μM).11 The X-ray crystal structure analyses of inhibitor 2 complexed with R188I SARS 3CLpro revealed the lack of interactions covering the P3 to P4 sites of substrate-based inhibitor 1, which suggest that these missing interactions at non-prime sites might be a major reason of the moderate inhibitory activity of 2.
Fig. 1.

Design of a decahydroisoquinolin inhibitor 2.
Based on the above evaluations, in this study, a non-prime site substituent was introduced to the decahydroisoqunolin scaffold to improve its inhibitory activity. From the overlay of the structures of 1 and 2, both complexed with R188I SARS 3CLpro, the distance between a 4-position carbon of decahydroisoquinolin ring of 2 and a nitrogen of α-amine of P2-site Cha in 1 is estimated to be 1.63 Å, nearly the same distance as single C—N covalent bond. This estimation suggests the 4-position carbon of decahydroisoqunolin is a suitable point to introduce a non-prime site substituent to yield a possible non-peptide-based inhibitor 3 (Fig. 2 ). As a non-prime site structure, dipeptide, Ac-Thr-Gly-OH, instead of an original Ac-Thr-Val sequence in 1 was selected since an isopropyl side chain of the Val is directed to an outward of SARS 3CLpro and no interactions with SARS 3CLpro at the Val site is detected (Fig. 1).
Fig. 2.

Design of a decahydroisoquinolin inhibitor 3 containing a non-prime site substituent. The overlay of X-ray crystal structures of inhibitors 1 (yellow) and 2 (green) into the active site of SARS 3CLpro (PDB codes 3ATW and 4TWW).
2. Results and discussion
2.1. Chemistry
Retrosynthetic route for 3 is shown in Scheme 1 . The non-prime site substituent is introduced via an amino group at 4-position of decahydroisoquinolin ring. With regard to the configuration of the amino group at the 4-position, a preliminary overlay of inhibitor 1 and 2 (Fig. 2) did not eliminate either diastereomer as a non-interacting inhibitor compared with the other, although the 4-S configuration was expected to be preferable. Thus, a synthetic route applicable for the construction of both configurations at the 4-position was employed. P1 site His residue is condensed by reductive-amination via an aldehyde introduced by oxidative cleavage of a terminal olefin at 3-position of decahydroisoquinolin ring of 4. The key intermediate 4 is synthesized from a precursor 5 by Pd(II) catalyzed diastereoselective cyclization.12, 13 The selectivity and efficiency have been well established by our previous syntheses of decahydroisoquinolin scaffold11 and natural products containing piperidine14 and pyrrolidine15 scaffolds. As an acyl substituent of an amino group, the 4-bromobenzoyl group was selected owing to the expected additional interactions with SARS 3CLpro, as inhibitors containing 4-halogenated benzoyl substituents exhibited better inhibitory activities than those containing 4-phenyl substituted benzoyl substituents in our previous study.11 Precursor 5 can be synthesized by conventional homologation and acylation reactions from 6. Amino functional group of 6 is stereoselectively introduced by Sharpless asymmetric dihydroxylation16 followed by Mitsunobu reaction,17 using a commercially available diol compound 7.
Scheme 1.

Retrosynthetic route for a decahydroisoquinolin inhibitor 3.
Precursor compound 13 for the Pd(II) catalyzed cyclization was synthesized according to the route shown in Scheme 2 . One alcohol group of the starting diol compound 7 was protected with a benzyl group, and the other alcohol was oxidized by tetrapropylammonium perruthenate (TPAP)/N-methylmorpholine-N-oxide (NMO) to an aldehyde, which was then homologated by Wittig reaction to give compound 8. The terminal olefin of 8 was converted to a 1, 2-diol by Sharpless asymmetric dihydroxylation and the resulting primary alcohol was selectively protected with TBDMS group to give an alcohol 9 as 4:1 mixture with an epimer alcohol. After purification, the configuration of a secondary hydroxyl group in the major product was confirmed to be S by Mosher procedure (Fig. S-1).18 To proceed with the synthesis, without further purification, the hydroxyl group of the mixture of diastereomers was converted to Boc-protected amine 10 by Mitsunobu reaction and following catalytic hydrogenation in the presence of (Boc)2O. After purification, compound 10 having an amino functional group for introduction of a non-prime site substituent was obtained as a single diastereomer. Similar combination of Mitsunobu reaction and reduction converted an alcohol of 10 to an amine, which was then acylated with p-bromobenzoic acid to give an amide 11. The primary hydroxyl group was oxidized by TPAP/NMO, and the resulting aldehyde was homologated by the Wittig reaction to give an ester 12. Reduction of 12 with DIBALH yielded the precursor compound 13.
Scheme 2.

Synthetic route for a precursor compound 13.
Next, epi-13 containing an opposite amine-configuration at the carbon atom connecting the non-prime site substituent was synthesized according to the route shown in Scheme 3 . The terminal olefin of above synthesized 8 was converted to an epi-1, 2-diol by Sharpless asymmetric dihydroxylation using a different ligand from the one used for 9. A primary alcohol of the product was protected with TBDMS group to yield epi-9 as 8:1 diastereomixture. Without further purification, the mixture was treated with diphenyl phosphorazidate under Mitsunobu reaction followed by catalytic hydrogenation in the presence of (Boc)2O. During the reactions, in addition to the desired epi-10 as diastereomixtures, TBDMS protecting group was removed and a diol 14 was obtained as a single diastereomer. To obtain epi-13 as a single diastereomer, the diol 14 was used for further synthesis. One primary alcohol of 14 was protected as an acetonide and the other alcohol was converted to an amine as above by Mitsunobu reaction and following catalytic hydrogenation. The resulting amine was acylated with p-bromobenzoic acid as above to give compound 16. Oxidation with TPAP and following homologation and reduction yielded a desired epi-13 as a single diastereomer.
Scheme 3.

Synthetic route for a precursor compound epi-13.
Pd(II)-catalyzed diastereoselective cyclization was then examined (Scheme 4 ). Treatment of a precursor 13 with bis-acetonitrile palladium chloride gave a desired decahydroisoquinolin compound 17 with 78% yield as well as an unreacted precursor 13 with 17% recovery. The recovered 13 could be reused for the cyclization to yield 17. In contrast, the same cyclization reaction for epi-13 gave neither a desired cyclized product nor starting precursor. Although the product was a complex of multi-products, one of the side products was estimated as compound 19 based on the NMR spectrum. The side-product 19 might be derived from partially produced initial cyclization product 18, in which a distance between the terminal olefin and nitrogen atom at 4-position of decahydroisoquinolin ring is estimated by MD calculation to be 2.54 Å, a distance capable for recyclization to yield byproduct 19. In contrast, the corresponding distance in 17 was estimated to be 4.29 Å, which is too far to have any interaction. Thus, the cyclized product 17 was used to introduce a substituent at a non-prime site.
Scheme 4.

Pd(II) catalyzed cyclization of 13 and epi-13. Models of Compound 17 and 18 were constructed by using the MMFF94s force field in the Molecular Operating Environment (MOE) software package (Chemical Computing Group Inc.: Montreal, Quebec, Canada, 2007). These models were constructed based on the estimated maximum stabilization structure.
The P1 site fragment and non-prime site substituent, covering P3 to P4 site, to be introduced on the decahydroisoquinolin scaffold were prepared as shown in Scheme 5 . For preparation of P1 site fragment, a commercially available Fmoc-His(Trt)-OH was converted to Weinreb amide,19 Fmoc-His(Trt)-N(OMe)Me, and the amide was reduced to an aldehyde by the treatment with DIBALH. The resulting Fmoc-His(Trt)-al 20 was converted to α,β-unsaturated ester or thioacetal followed by Fmoc-deprotection with 20% Et2NH to yield 21a or 21b. P3 to P4 fragment 22 was prepared by EDC20/DMAP mediated coupling of Ac-Thr-OH and H-Gly-OBn and following debenzylation by catalytic hydrogenation.
Scheme 5.

Syntheses of P1 site fragment and P3 to P4 site fragment.
Finally, the above synthesized P1 site fragment and P3 to P4 site fragment were condensed with a cyclized product 17 (Scheme 6 ). The Boc group of 17 was removed by HCl and the resulting amine was coupled with P1 fragment 22 using BOP21 as a coupling reagent. A terminal olefin of the product 23 was oxidized with O3 and an aldehyde of the product was reductively coupled with P1 fragment 21b to give 24b. Removal of the Trt group at the His-imidazole of 24b was conducted by the treatment with TFA:TIS:CH2Cl2:H2O = 10:1:10:1 to yield 25b quantitatively. To evaluate the effect of warheads on the inhibitory activity, P1 fragment 21a and H-His(Trt)-N(OMe)Me were similarly coupled with 23 to yield 25a and 25c, respectively. Brief treatment of 25b with NBS22 converted the thioacetal to an aldehyde and the product was purified by preparative HPLC to give 26 in high homogeneity (Fig. S-2).
Scheme 6.

Introduction of P1 site and P3 to P4 site substituent.
2.2. Inhibitory activity
The inhibitory activities of 26 as well as 25a–c were evaluated based on IC50 values calculated from the decrease in the substrate digested by R188I SARS 3CLpro according to the published procedure.9 The results are summarized in Table 1 and a typical sigmoidal curve used for estimation of the IC50 value is shown in Figure S-3. The IC50 value of 26 (26 μM) was about 2.4 times that of the parent compound 2 (63 μM), and the value of 26 was identical with that of a substrate-based tetrapeptide aldehyde inhibitor (26 μM of Ac-Thr-Ser-Ala-Val-Leu-NHCH(CH2CH2CON(CH3)2)-CHO).9 The result strongly suggests the additional interactions with R188I SARS 3CLpro are caused by the introduction of a non-prime site substituent, although the interactions would not be sufficient as in substrate-based inhibitor 1.
Table 1.
IC50 values of 26 and 25a–c.
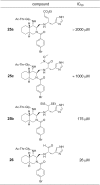 |
The inhibitory activities of 25a–c suggest a possible warhead combined with the decahydroisoquinolin scaffold. The results showed that a well-known Michael-type warhead in 25a was ineffective and a Weinreb amide warhead in 25c was also rather weak one when combined with a decahydroisopuinolin scaffold. In contrast, 25b containing a thioacetal as a warhead showed clear inhibitory activity although the IC50 value (175 μM) is still moderate but comparable with a substrate-based tetrapeptide aldehyde inhibitor9 (155 μM of Ac-Ala-Val-Leu-NHCH(CH2CH2CON(CH3)2)-CHO). These results indicate the possibility of the thioacetal functional group as a new warhead when combined with a decahydroisopuinolin or related fused ring scaffold. Structure optimization of the thioacetal functional group combined with the non-prime site substituents is now underway.
The interactions of 26 with R188I SARS 3CLpro were suggested from a docking model constructed using the Molecular Operating Environment (MOE) software. Template-guided docking by MOE estimated two interaction modes with almost the same high score. The overlay of each estimated interaction mode with that of the substrate-based inhibitor 1 is shown in Fig. 3 -i and 3-ii. In both interaction modes, the newly introduced non-prime site substituent of 26 directed to S3 to S4 pocket of SARS 3CLpro, as expected, as shown in Fig. 2. In addition, hydrogen bond interactions at the non-prime site substituent were estimated (Fig. 3-ii) and the modes were almost the same as those of 1 10 complexed with SARS 3CLpro. To confirm the estimated interactions, X-ray crystallography analyses of 26 complexed with R188I SARS 3CLpro are now underway.
Fig. 3.

Docking model of 26 with R188I SARS 3CLpro: (i) interaction mode with GBVI/WSA dG score, −11.0665 kcal/mol: (ii) interaction mode with GBVI/WSA dG score, −10.9356 kcal/mol. Each docking model of 26 with R188I SARS 3CLpro was constructed by using an X-ray crystal structure of a complex of SARS 3CLpro and inhibitor 2 (PDB 4TWW) as a template. The possible binding mode was obtained by a docking simulation of inhibitor 26 and SARS 3CLpro using an automated template-guided docking protocol with the Amber10:EHT force field in the Molecular Operating Environment (MOE) 2018.01 software package (Chemical Computing Group Inc., Montreal, Quebec, Canada).
3. Conclusion
As a novel non-peptide inhibitor for SARS 3CLpro, a non-prime site substituent was introduced to a 4-position carbon of the decahydroisoquinolin scaffold of a previously synthesized fused-ring inhibitor. The substitution position was selected by overlay of a substrate-based inhibitor and the fused-ring inhibitor. The inhibitor 26 was synthesized using Pd(II) catalyzed diastereoselective cyclization as a key reaction. Synthesized inhibitor 26 showed a clear but moderate inhibitory activity for R188I SARS 3CLpro. The results strongly suggested increased interactions of inhibitor 26 with SARS 3CLpro at S3 to S4 pocket, although the interactions were expected to be insufficient. Thus, detailed analyses of the interactions based on X-ray crystal analyses of 26 complexed with R188I SARS 3CLpro are the next objects. In addition, it was demonstrated that a thioacetal warhead would be an alternative warhead to aldehyde, which has rather high reactivity causing a possibility of non-selective interactions with proteins. The present data suggested that a combination of thioacetal functional group with decahydroisoquinolin or related fused-ring scaffold would be another approach to design novel inhibitors for SARS 3CL protease, and studies on this line are now underway.
4. Experimental
4.1. General
Silica gel 70PF 254 Plate-Wako was used for TLC (thin layer chromatography) and column chromatography was performed on Wakogel®, 60 N, (particle size, 63–212 μm) or Wakogel®, C-300E, (particle size, 45–75 μm). Melting points were obtained on a Yanaco micro melting point apparatus and are uncorrected. Low-resolution mass spectra (LRMS) were recorded on Shimadzu LCMS-2010EV (ESI) and high-resolution mass spectra (HRMS) were recorded on Shimadzu LCMS-IT-TOF (ESI) or JEOL GC mate II (EI). 1H NMR spectra were recorded using Bruker AV 300 spectrometer, chemical shifts (δ) are quoted in parts per million (ppm) referenced to tetramethylsilane (0 ppm) or the residual solvent peak. 13C NMR spectra were recorded on the same spectrometer at 75 MHz, using the residual solvent peak as the internal reference. Optical rotations were recorded using a HORIBA SEPA-300 polarimeter.
4.2. (1S, 2S)-2-[(benzyloxy)methyl]cyclohexanemethanol
Ag2O (17.4 g, 75 mmol) was added to a solution of (1S, 2S)-1, 2-cyclohexanedimethanol 7 (7.2 g, 50 mmol) and benzyl bromide (6.5 mL, 55 mmol) in CH2Cl2 (250 mL), and the mixture was stirred for 24 h at room temperature. The reaction mixture was filtered and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 5:1) to give a title alcohol (10.5 g, 95%) as a colorless oil. [α]D 25−9.3 (c 1.2, CHCl3); 1H NMR (300 MHz): δ = 7.34–7.28 (m, 5H), 4.50 (s, 2H), 3.60 (dd, J = 11.4, 3.6 Hz, 1H), 3.50–3.43 (m, 2H), 3.39 (dd, J = 9.6, 3.6 Hz, 1H), 3.22 (br s, 1H), 1.74–1.62 (m, 4H), 1.58–1.47 (m, 1H), 1.33–0.76 (m, 5H); 13C NMR (75 MHz): δ = 137.7, 128.4, 127.69, 127.66, 75.6, 73.3, 66.9, 45.0, 40.2, 30.1, 29.9, 26.0; HRMS (ESI) calcd for C15H22O2Na [M+Na]+: 257.1512. Found: 257.1515.
4.3. Benzyl (1S, 2R)-2-vinylcyclohexylmethyl ether, 8
TPAP (tetra-n-propyl ammonium perrutenate, 316 mg, 0.9 mmol) was added to a solution of above alcohol (10.5 g, 47.5 mmol) and NMO (N-methylmorphline-N-oxide, 21.1 g, 180 mmol) in CH2Cl2 (180 mL) at 0 °C. The temperature was gradually raised to 25 °C. After being stirred for 30 min, the reaction mixture was filtered through a silica gel layer and the filtrate was concentrated. Resulting residue was immediately used for the next step without purification. t-BuOK (15.1 g, 135 mmol) was added to a solution of methyltriphenylphosphonium bromide (48.2 g, 135 mmol) in THF (225 mL) at 0 °C under an argon gas atmosphere and the mixture was stirred for 1 h. Above residue obtained by TPAP oxidation was added dropwise at 0 °C and the mixture was warmed to 25 °C and stirred for 12 h. The reaction was quenched with saturated aqueous NH4Cl and was extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 30:1) to give 8 (6.9 g, 63%, 2 steps) as a colorless oil. [α]D 25+50 (c 1.49, CHCl3); 1H NMR (300 MHz): δ = 7.33–7.26 (m, 5H), 5.62 (ddd, J = 17.2, 8.6, 1.4, 1H), 4.96 (dd, J = 16.7, 1.4 Hz, 1H), 4.93 (dd, J = 8.1, 2.1 Hz, 1H), 4.49 (d, J = 12.0 Hz, 1H), 4.41 (d, J = 12.3 Hz, 1H), 3.48 (dd, J = 9.2, 3.5 Hz, 1H), 3.23 (dd, J = 9.2, 7.4 Hz, 1H), 2.03–1.94 (m, 1H), 1.89–1.78 (m, 1H), 1.75–1.61 (m, 3H), 1.45–1.33 (m, 1H), 1.32–1.05 (m, 4H); 13C NMR (75 MHz): δ = 143.2, 138.9, 127.4, 127.4, 127.3, 113.7, 74.0, 73.0, 45.0, 42.1, 33.5, 29.9, 25.9, 25.8.
4.4. (R)-1-{(1S, 2S)-2-[(benzyloxy)methyl]cyclohexyl}ethane-1, 2-diol
To a stirred solution of H2O (60 mL) and t-BuOH (60 mL) under an argon gas atmosphere were sequentially added K2CO3 (12.4 g, 90 mmol), K3Fe(CN)6 (29.6 g, 90 mmol), (DHQD)2AQN (hydroquinidine anthraquinone-1,4-diyl diether, 255 mg, 0.3 mmol) and K2OsO2(OH)2 (43.7 mg, 0.12 mmol) at 25 °C. The mixture was cooled to 0 °C and stirred for 30 min, and then olefin 8 (6.9 g, 30 mmol) in H2O/t-BuOH (1:1, 30 mL) was added. After stirring for 24 h at 0 °C, the reaction was quenched with aqueous Na2SO3 (20 mL) and the mixture was stirred for an additional 10 min. The mixture was extracted with EtOAc and the organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 2:1) to give title alcohol (7.73 g, 98%, dr = 4:1) as a colorless oil. 1H NMR (300 MHz): δ = 7.31–7.24 (m, 5H), 4.50 (d, J = 12.0 Hz, 1H), 4.45 (d, J = 11.7 Hz, 1H), 4.12 (br s, 0.2H), 3.86 (br s, 0.8H), 3.71–3.69 (m, 0.2H), 3.63 (t, J = 9.8 Hz, 1H), 3.48–3.41 (m, 3.8H), 3.10 (m, 0.8H), 2.66–2.60 (m, 0.2H), 1.79–1.67 (m, 4H), 1.52–1.38 (m, 1H), 1.34–0.99 (m, 5H); 13C NMR (75 MHz): δ = 137.8, 137.9, 128.3, 127.6, 127.5, 74.8, 73.8, 73.2, 73.0, 72.8, 65.0, 63.9, 44.7, 44.4, 40.4, 38.9, 30.4, 26.9, 25.9, 25.7, 25.6, 25.5; HRMS (EI) calcd for C16H24O3 [M]+: 264.1726. Found: 264.1717.
4.5. (R)-1-{(1S, 2S)-2-[(benzyloxy)methyl]cyclohexyl}-2-[(tert-butyldimethylsilyl) oxy]ethan-1-ol, 9
TBDMS-Cl (tert-butyldiphenylsilyl chloride, 4.86 g, 32.2 mmol) was added to a solution of the above alcohol (7.73 g, 29.3 mmol) and Et3N (6.1 mL, 44 mmol) in CH2Cl2 (120 mL). After DMAP (4-(dimethylamino)pyridine, 0.36 g, 2.9 mmol) was added to the mixture, the mixture was stirred for 16 h at 25 °C. The reaction was quenched with saturated aqueous NH4Cl and was extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 20:1) to give 9 (10.5 g, 95%) as a colorless oil. 1H NMR (300 MHz): δ = 7.36–7.25 (m, 5H), 4.52 (d, J = 12.0 Hz, 1H), 4.46 (d, J = 12.0 Hz, 1H), 3.90–3.86 (m, 0.8H), 3.81–3.78 (m, 0.2H), 3.71–3.51 (m, 2.2H), 3.47–3.46 (d, J = 4.8 Hz, 1.6H), 3.40–3.35 (m, 0.2H), 2.04 (br s, 1H), 1.78–1.68 (m, 4H), 1.58–1.55 (d, J = 9.0 Hz, 1H), 1.37–1.22 (m, 5H), 0.91 (s, 7.2H), 0.90 (s, 1.8H), 0.07 (s, 2.4H), 0.06 (s, 2.4H), 0.05 (s, 0.6H), 0.04 (s, 0.6H); 13C NMR (75 MHz): δ = 138.4, 128.31, 128.28, 127.6, 127.5, 127.4, 74.3, 74.1, 73.2, 73.0, 65.4, 64.1, 43.3, 42.7, 39.7, 38.9, 30.6, 30.3, 26.5, 26.0, 25.9, 25.7, 25.1, 18.3, −5.3, −5.37, −5.42; HRMS (EI) calcd for C22H38O3Si [M]+: 378.2590 Found: 378.2596.
4.6. 2-{(S)-1- [N-(tert-butoxycarbonyl)amino]-2-[(tert-butyldimethylsilyl)oxy]ethyl} [(1S,2S)-cyclohexyl]methanol, 10
DIAD (diisopropyl azodicarboxylate, 21.9 mL, 111 mmol) and DPPA (diphenylphosphoryl azide, 24.1 mL, 111 mmol) were added to a solution of the above alcohol (10.5 g, 27.8 mmol) and PPh3 (triphenylphosphine, 29.2 g, 111 mmol) in THF (500 mL) under an argon atmosphere at 0 °C. The mixture was warmed to at 25 °C and stirred for 14 h. Then the reaction mixture was concentrated. The residue was passed through a silica gel column chromatography (hexane/EtOAc = 30:1). The product was immediately used for the next step without further purification. The product was dissolved in EtOAc (10 mL) and added to (Boc)2O (9.6 mL, 41.7 mmol) and Pd(OH)2-C (1.2 g, 10 wt%). The mixture was stirred for 16 h under hydrogen gas and the mixture was filtered and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 5:1) to give 10 (5.38 g, single diastereomer, 50%, 2 steps) as a colorless oil. [α]D 25+59 (c 1.2, CHCl3); 1H NMR (300 MHz): δ = 4.96 (br d, J = 7.5 Hz, 1H), 3.83–3.81 (m, 2H), 3.65–3.55 (m, 2H), 3.48–3.44 (m, 1H), 3.29 (br s, 1H), 1.66–1.57 (m, 4H), 1.39 (s, 9H), 1.39–1.34 (m, 2H), 1.23–1.05 (m, 4H), 0.85 (s, 9H), 0.01 (s, 6H); 13C NMR (75 MHz): δ = 156.1, 79.3, 77.2, 65.0, 61.8, 52.0, 41.2, 41.0, 29.7, 28.3, 26.6, 25.9, 25.7, 18.1, −5.55, −5.58; HRMS (ESI) calcd for C20H41NO4SiNa [M+Na]+: 410.2697. Found: 410.2699.
4.7. 4-Bromo-N-[(2-{(S)-1- [N-(tert-butoxycarbonyl)amino]-2-hydroxyethyl} (1S,2S)-cyclohexyl)methyl]benzamide, 11
DIAD (11.0 mL, 55.6 mmol) and DPPA (12.1 mL, 55.6 mmol) were added to a solution of the above alcohol 10 (5.38 g, 13.9 mmol) and PPh3 (14.6 g, 55.6 mmol) in THF (140 mL) under an argon atmosphere at 0 °C. The mixture was warmed to 25 °C, stirred for 14 h, and concentrated. The residue was passed through silica gel column chromatography (hexane/EtOAc = 30:1). Without further purification, the crude product was dissolved in THF (30 mL) and added to LiAlH4 (1.2 g, 30.6 mmol) in THF (30 mL) under an argon atmosphere at 0 °C. After stirring for 1 h, the reaction was quenched with H2O and 1 M NaOH aq. The mixture was filtered through celite® and silica gel and the filtrate was concentrated. Resulting residue in MeOH (20 mL) was added to a solution of NMM (2.3 mL, 20.9 mmol), DMT (4-(4, 6-Dimethoxy-1, 3, 5-triazin-2-yl-4-methylmorpholinium chloride)-MM (4.2 g, 15.3 mmol), and 4-bromobenzoic acid (3.1 g, 15.3 mmol) in MeOH (40 mL) at 0 °C. The mixture was stirred for 12 h at 25 °C. The reaction was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 2:1) to afford 11 (3.16 g, 50%, 3 steps) as a white solid. Mp 193–194 °C; [α]D 25+51.8 (c 0.3, CHCl3); 1H NMR (300 MHz): δ = 8.03 (br d, J = 7.2 Hz, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.7 Hz, 2H), 5.34 (d, J = 6.6 Hz, 1H), 4.07–3.99 (m, 2H), 3.75 (dd, J = 11.3, 3.5 Hz, 1H), 3.58 (dd, J = 11.3, 8.6, 1H), 3.37–3.33 (m, 1H), 3.10 (br s, 0.8H), 2.16 (br s, 0.2H), 1.76–1.56 (m, 6H), 1.47 (s, 9H), 1.26–0.95 (m, 4H); 13C NMR (75 MHz): δ = 167.1, 156.8, 133.1, 131.3, 129.3, 125.9, 79.9, 60.7, 53.0, 42.2, 41.5, 39.3, 30.9, 28.4, 26.2, 26.0, 25.9; HRMS (EI) calcd for C21H31BrN2O4 [M]+: 454.1451. Found: 454.1467.
4.8. Ethyl (R, E)-4-{[(1S, 2S)-2-(4-bromobenzamido)methyl]cyclohexyl}-4-[N-(tert-butoxycarbonyl)amino]but-2-enoate, 12
TPAP (48.9 mg, 0.14 mmol) was added to a solution of the before mentioned alcohol 11 (3.16 g, 6.96 mmol), NMO (3.26 g, 27.8 mmol), (carbethoxymethylene) triphenylphosphorane (4.85 g, 13.9 mmol), and MS 4 Å in CH2Cl2 (70 mL) under an argon atmosphere at 0 °C and stirred for 16 h. The mixture was filtered through a silica gel layer, and the filtrate was concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 3:1) to afford 12 (2.33 g, 67%) as a colorless oil. [α]D 24 + 275 (c 0.3, CHCl3); 1H NMR (300 MHz): δ = 7.91 (d, J = 8.4 Hz, 2H), 7.69 (br d, J = 6.9 Hz, 1H), 7.53 (d, J = 8.7 Hz, 2H), 6.91 (dd, J = 15.8, 5.6 Hz, 1H), 5.98 (dd, J = 15.9, 1.8 Hz, 1H), 5.24 (d, J = 8.1 Hz, 1H), 4.73–4.68 (m, 1H), 4.22 (q, J = 7.2 Hz , 2H), 4.11 (dd, J = 13.8, 9.6 Hz, 1H), 3.33–3.28 (m, 1H), 1.85–1.58 (m, 7H), 1.50 (s, 9H), 1.31 (t, J = 7.2 Hz, 3H), 1.25–1.18 (m, 3H); 13C NMR (75 MHz): δ = 166.7, 165.8, 155.9, 143.9, 133.2, 131.4, 129.2, 125.8, 121.9, 80.5, 60.7, 52.1, 44.1, 41.3, 39.1, 30.7, 28.4, 26.5, 25.8, 25.6, 14.2; HRMS (ESI) calcd for C25H35BrN2O5Na [M+Na]+: 545.1605. Found: 545.1606.
4.9. 4-Bromo-N-[(2-{(R, E)-1-[N-(tert-butoxycarbonyl)amino]-4-hydroxybut-2- enyl})(1S,2S)-cyclohexyl]methylbenzamide, 13
To a solution of 12 (2.33 g, 4.45 mmol) in CH2Cl2 (18 mL), DIBAL-H (diisobutylaluminum hydride, 13.4 mL, 13.4 mmol, 1.0 M solution in hexane) was added at −78 °C. After being stirred for 20 min at the same temperature, the reaction was quenched with MeOH (14 mL). The mixture was warmed to 25 °C and filtered through Celite® and a silica gel layer. The filtrate was concentrated, and the residue was purified by silica gel column chromatography (hexane/EtOAc = 1:1) to give a title alcohol (1.75 g, 82%) as a white solid. Mp 101–102 °C; [α]D 26 + 283 (c 0.2, CHCl3); 1H NMR (300 MHz): δ = 7.91 (d, J = 8.4 Hz, 2H), 7.80 (br d, J = 8.1 Hz, 1H), 7.52 (d, J = 8.4 Hz, 2H), 5.85 (dt, J = 15.4, 4.9 Hz, 1H), 5.66 (dd, J = 15.6, 6.6 Hz, 1H), 5.08 (br d, J = 6.0 Hz, 1H), 4.49 (m, 1H), 4.17 (d, J = 4.8 Hz, 2H), 4.10–4.02 (m, 1H), 3.29–3.24 (m, 1H), 2.54 (br s, 1H), 1.69–1.41 (m, 6H), 1.49 (s, 9H), 1.27–1.03 (m, 4H); 13C NMR (75 MHz): δ = 166.8, 155.9, 133.2, 131.9, 131.3, 129.2, 126.4, 125.8, 80.0, 77.2, 62.7, 52.5, 43.6, 41.5, 38.9, 30.6, 28.4, 25.8; HRMS (ESI) calcd for C23H33BrN2O4Na [M+Na]+: 503.1516. Found: 503.1507.
4.10. tert-Butyl ((R)-2-hydroxy-1-((1S, 2S)-2-(hydroxymethyl)cyclohexyl) ethyl)carbamate, 14
A title compound was similarly prepared from 7 as above to yield a colorless oil; yield 5.0% (7 steps): [α]D 25+6.1 (c 1.05, MeOH); 1H NMR (300 MHz): δ = 4.92 (br d, J = 8.7 Hz, 1H), 3.99 (m, 1H), 3.74 (dd, J = 11.1, 3.6 Hz, 1H), 3.67–3.56 (m, 3H), 3.17–3.16 (m, 1H), 3.00–2.90 (m, 1H), 1.92 (br s, 1H), 1.74–1.53 (m, 5H), 1.44 (s, 9H), 1.24–1.03 (m, 4H); 13C NMR (75 MHz): δ = 158.7, 80.2, 79.6, 65.8, 64.0, 53.4, 41.9, 40.4, 31.1, 29.1, 27.2, 27.1, 26.5; HRMS (ESI) calcd for C14H27BrNO4Na [M+Na]+: 296.1832. Found: 296.1837.
4.11. tert-Butyl(R)-4-((1S, 2S)-2-((4-bromobenzamido)methyl)cyclohexyl)-2,2- dimethyloxazolidine-3-carboxylate, 16
BF3·Et2O (18.9 μL, 0.15 mmol) was added to a solution of above an alcohol 14 (819 mg, 3.0 mmol), and 2, 2-dimethoxypropane (3.68 mL, 30 mmol) in acetone. The mixture was stirred at room temperature for 10 min. The reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed with NaHCO3, brine, dried over Na2SO4, and filtered. The filtrate was concentrated and resulting residue was purified by silica gel column chromatography (hexane/EtOAc = 10:1) to afford 15 (640 mg, 50%). Without further purification, DIAD (1.2 mL, 6.0 mmol) and DPPA (1.3 mL, 6.0 mmol) were added to a solution of 15 (640 mg, 1.5 mmol) and PPh3 (1.57 g, 6.0 mmol) in THF (15 mL) under an argon atmosphere at 0 °C. The mixture was warmed to 25 °C and stirred for 16 h. The mixture was concentrated, and the residue was roughly purified by silica gel column chromatography (hexane/EtOAc = 30:1). This compound was immediately used for the next step without further purification. To a solution of the product in CHCl3 (0.5 mL) was added Pd-C (80 mg, 10 wt%) and MeOH (5.5 mL). The mixture was stirred under hydrogen gas for 8 h. The mixture was filtered and concentrated. The residue was used without purification. The crude product in MeOH (4 mL) was added to a solution of NMM (0.25 mL, 2.3 mmol), DMT-MM (456 mg, 1.7 mmol), and 4-bromobenzoic acid (332 mg, 1.7 mmol) in MeOH (4 mL) at 0 °C. The mixture was stirred for 12 h at 25 °C. The reaction was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 1:1) to afford 16 (1.04 g, 42%, 3 steps) as a white wax. [α]D 24+3.3 (c 2.82, CHCl3); 1H NMR (300 MHz): δ = 8.47 (br s, 1H), 7.81 (br d, J = 8.1 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H), 4.27–4.26 (m, 1H), 4.08 (dd, J = 9.0, 7.2 Hz, 1H), 3.72 (dd, J = 9.0, 2.1 Hz, 1H), 3.47 (ddd, J = 14.0, 4.7, 3.6 Hz, 1H), 3.37 (m, 1H), 2.11 (br s, 1H), 1.86–1.69 (m, 4H), 1.60 (s, 3H), 1.53 (s, 9H), 1.46 (s, 3H), 1.41–1.07 (m, 5H); 13C NMR (75 MHz): δ = 165.9, 154.4, 133.5, 131.3, 128.9, 125.6, 94.7, 80.6, 69.3, 58.0, 55.3, 53.4, 49.8, 46.6, 39.5, 33.4, 28.3, 27.6, 25.9, 25.8, 24.2; HRMS (ESI) calcd for C24H35BrN2O4Na [M+Na]+: 517.1672. Found: 517.1678.
4.12. 4-Bromo-N-[(2-{(S, E)-1-[N-(tert-butoxycarbonyl)amino]-4-hydroxybut-2-enyl})(1S,2S)-cyclohexyl]methylbenzamide, epi-13
(CH3CN)2PdCl2 (76.5 mg, 0.3 mmol) was added to 16 in CH2Cl2 (3 mL) and the mixture was stirred for 30 h at 25 °C. The mixture was filtered and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 2:1) to afford alcohol epi-11 (157 mg, 55%) as a colorless oil. The product was similarly converted to the title compound epi-13 using the same procedure for 13 described above to yield a colorless oil; yield 21% (2 steps); [α]D 24 + 19.1 (c 1.0, CHCl3); 1H NMR (300 MHz): δ = 7.75 (br d, J = 8.1 Hz, 2H), 7.66 (br s, 1H), 7.52 (d, J = 8.7 Hz, 2H), 5.76 (td, J = 10.5, 1.2 Hz, 1H), 5.70 (dd, J = 11.1, 3.9 Hz, 1H), 4.72 (br d, J = 9.3 Hz, 1H), 4.55 (br d, J = 9.3 Hz, 1H), 4.15 (br d, J = 3.6 Hz, 1H), 3.59 (ddd, J = 14.0, 7.1, 4.7 Hz, 1H), 3.33–3.28 (m, 1H), 2.44 (br s, 0.7H), 2.15 (br s, 0.3H), 1.90 (br d, J = 10.2 Hz, 1H), 1.72–1.62 (m, 3H), 1.41 (s, 9H), 1.37–1.11 (m, 5H), 1.07–0.95 (m, 2H); 13C NMR (75 MHz): δ = 166.7, 156.2, 133.5, 131.6, 131.4, 129.6, 129.0, 125.7, 80.0, 62.8, 52.3, 46.9, 45.3, 39.2, 32.2, 28.3, 25.8, 25.5; HRMS (ESI) calcd for C23H33BrN2O4Na [M+Na]+: 503.1516. Found: 503.1525.
4.13. (3R, 4S, 4aS, 8aS)-[N-(4-Bromophenylcarbonyl)]-4-[N-(tert-butoxycarbonyl) amino]-3-vinyldecahydroisoquinolin, 17
To a solution of 13 (1.75 g, 3.65 mmol) in dry CH2Cl2 (37 mL), (CH3CN)2PdCl2 (189 mg, 0.73 mmol) was added at 0 °C under an argon gas atmosphere. The temperature was gradually raised up to 25 °C. After being stirred for 4 h, the reaction mixture was filtered and concentrated. The residue was purified by silica gel column chromatography (hexane/EtOAc = 3:1) to give 17 (1.32 g, 78%, 9:1 conformational isomers) as a colorless oil. [α]D 25 −36.9 (c 1.6, CHCl3); 1H NMR (300 MHz): δ = 7.56 (br d, J = 7.5 Hz, 0.2H), 7.47 (d, J = 8.1 Hz, 1.8H), 7.33–7.29 (m, 0.2H), 7.25 (d, J = 8.4 Hz, 1.8H), 5.89–5.87 (m, 0.1H), 5.79 (ddd, J = 17.3, 10.7, 3.5 Hz, 0.9H), 5.44 (m, 0.1H), 5.37 (dd, J = 10.8, 1.5 Hz, 0.9H), 5.25 (d, J = 17.4 Hz, 1H), 4.77 (d, J = 8.4 Hz, 0.9H), 4.62 (d, J = 8.1 Hz, 0.1H), 4.47 (d, J = 4.2 Hz, 0.2H), 4.43 (d, J = 3.3 Hz, 1.8H), 3.95 (d, J = 8.1 Hz, 0.1H), 3.62 (d, J = 8.4 Hz, 0.9H), 2.84 (t, J = 12.2 Hz, 0.1H), 2.61 (t, J = 12.5 Hz, 0.9H), 1.76–1.72 (m, 3H), 1.59–1.49 (m, 2H), 1.47 (s, 0.9H), 1.38 (s, 8.1H), 1.132–1.15 (m, 4H), 1.02–0.83 (m, 1H); 13C NMR (75 MHz): δ = 171.5, 170.8, 155.2, 134.6, 133.6, 131.6, 128.8, 128.1, 124.0, 117.9, 79.7, 62.7, 53.5, 43.0, 39.0, 35.0, 30.1, 28.4, 28.3, 28.0, 25.8, 25.3; HRMS (EI) calcd for C23H31BrN2O3 [M]+: 462.1518. Found: 462.1530.
Compound 19; A title compound was similarly prepared from epi-13 as above to yield a mixture with inseparable impurities; 1H NMR (300 MHz): δ = 7.56–7.52 (m, 2H), 7.37–7.22 (m, 2H), 5.95–5.89 (m, 1H), 5.45–5.33 (m, 1H), 4.46–4.42 (m, 1H), 4.23–4.20 (m, 1H), 4.16–4.11 (m, 1H), 3.68–3.54 (m, 1H), 2.80 (dd, J = 14.4, 9.6 Hz, 1H), 2.64–2.56 (m, 1H), 2.38–2.35 (m, 1H), 2.02–1.55 (m, 3H), 1.48 (s, 9H), 1.45–1.36 (m, 4H), 1.28–0.86 (m, 2H).
4.14. Fmoc-His(Trt)-CH CH-COOEt
To a solution of Fmoc-His(Trt)-N(OMe)Me (165 mg, 0.25 mmol) in CH2Cl2 (2 mL), DIBAL-H (0.5 mL, 0.5 mmol, 1.0 M solution in hexane) was added at −78 °C. After being stirred for 20 min at the same temperature, the reaction was quenched with MeOH (1.0 mL). The mixture was warmed to 25 °C and filtered through celite® and a silica gel layer. The filtrate was concentrated. The crude aldehyde was used without purification. (Carbethoxymethylene)triphenylphosphorane (175 mg, 0.5 mmol) was added to a solution of above crude aldehyde in CH2Cl2 under an argon atmosphere at 0 °C and the mixture was stirred for 16 h. The mixture was purified by silica gel column chromatography (hexane/EtOAc = 1:1) to afford a title product (160 mg, 95%, 2 steps) as a yellow oil. [α]D 23 −0.25 (c 0.75, CHCl3); 1H NMR (300 MHz): δ = 7.75 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.53–7.30 (m, 14H), 7.11–7.09 (m, 6H), 6.86 (dd, J = 15.6, 5.1 Hz, 1H), 6.59 (s, 1H), 6.64 (d, J = 7.8 Hz, 1H), 5.88 (br d, J = 15.6 Hz, 1H), 4.67 (m, 1H), 4.36–4.32 (m, 2H), 4.24–4.18 (m, 1H), 4.19 (q, J = 7.2 Hz, 2H), 3.00 (dd, J = 14.6, 4.7 Hz, 1H), 2.79 (dd, J = 14.6, 5.6 Hz, 1H), 1.28 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz): δ = 166.1, 155.7, 147.8, 144.0, 143.9, 142.2, 141.2, 138.7, 136.6, 129.7, 128.0, 127.6, 125.5, 121.1, 119.9, 119.8, 77.2, 66.9, 60.3, 51.9, 47.2, 31.9, 14.2; HRMS (ESI) calcd for C44H39N3O4Na [M + Na]+: 696.2833. Found: 696.2835.
4.15. H-His(Trt)–CH CH-COOEt, 21a
20% Et2NH in CH3CN solution was added to above Fmoc-His(Trt)-CH CH-COOEt (160 mg, 0.24 mmol). The mixture was stirred at 25 °C for 2 h. The mixture was concentrated under reduced pressure. The residue was roughly purified by silica gel column chromatography (CHCl3/MeOH = 20:1) and the product was used without further purification.
4.16. Fmoc-His(Trt)-CH(SEt)2
To a solution of Fmoc-His(Trt)-N(OMe)Me (165 mg, 0.25 mmol) in CH2Cl2 (2 mL), DIBAL-H (0.5 mL, 0.5 mmol, 1.0 M solution in hexane) was added at −78 °C. After being stirred for 20 min at the same temperature, the reaction was quenched with CH3OH (1.0 mL). The mixture was warmed to 25 °C and filtered through Celite® and a silica gel layer. The filtrate was concentrated. The crude aldehyde was used without purification. To a stirred solution of crude aldehyde, and EtSH (100 μL, 1.38 mmol) in AcOH (1 mL) was added BF3·Et2O (100 μL), and the mixture was stirred at 25 °C for 30 min·H2O (10 mL) was added, and the organic phase was washed with H2O, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 20:1) to afford a title product, Fmoc-His(Trt)-CH(SEt)2 (85.1 mg, 49%, 2 steps) as a white amorphous powder. [α]D 20 −2.54 (c 4.78, CHCl3); 1H NMR (300 MHz): δ = 7.79–7.39 (m, 2H), 7.63–7.59 (m, 2H), 7.42–7.35 (m, 4H),7.30–7.28 (m, 10H), 7.11–7.08 (m, 6H), 6.68 (s, 1H), 6.08 (br d, J = 8.4 Hz, 1H), 4.42–4.11 (m, 5H), 4.03 (d, J = 4.8 Hz, 1H), 3.08 (dd, J = 14.7, 4.8 Hz, 1H), 2.93 (dd, J = 14.6, 8.4 Hz, 1H), 2.74–2.62 (m, 5H), 1.28–1.19 (m, 6H); 13C NMR (75 MHz): δ = 156.0, 144.0, 143.9, 142.2, 141.1, 138.2, 137.4, 129.6, 127.9, 127.5, 126.9, 125.3, 119.8, 119.5, 75.2, 66.8, 55.2, 55.1, 47.1, 29.3, 26.1, 14.5; HRMS (ESI) calcd for C45H45N3O2S2 [M+Na]+: 710.2869. Found: 710.2865.
4.17. H-His(Trt)-CH(SEt)2, 21b
20% Et2NH in CH3CN solution was added to above Fmoc-His(Trt)-CH(SEt)2 (85 mg, 0.12 mmol). The mixture was stirred at 25 °C for 2 h and was concentrated. The residue was roughly purified by silica gel column chromatography (CHCl3/MeOH = 1:1) and the product was used without further purification.
4.18. Ac-Thr-Gly-OH, 22
H-Gly-OBn·HCl (1.2 g, 6.0 mmol) was added to a stirred solution of Ac-Thr-OH (645 mg, 4.0 mmol), Et3N (1.7 mL, 12.0 mmol), EDCI·HCl (844 mg, 4.4 mmol) and HOBt·H2O (735 mg, 4.8 mmol) in DMF (16 mL) at 0 °C. After DMAP (4.9 mg, 0.04 mmol) was added to mixture, the mixture was stirred for 8 h at 25 °C. The mixture was concentrated and quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 10:1) to afford Ac-Thr-Gly-OBn 22 (0.96 g, 78%) as a white solid. Mp.109–111 °C; [α]D 25 −8.0 (c 3.95, CHCl3); 1H NMR (300 MHz): δ = 7.41–7.32 (m, 5H), 5.25 (s, 2H), 4.68 (s, 0.5H), 4.44 (d, J = 4.2 Hz, 1H), 4.27–4.19 (m, 1H), 4.09 (d, J = 10.8 Hz, 1H), 4.05 (dd, J = 13.5, 4.2 Hz, 1H), 3.80 (s, 0.5H), 2.12 (s, 3H), 1.26 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz): δ = 173.9, 173.6, 171.4, 137.5, 129.9, 129.6, 128.6, 128.3, 68.6, 68.2, 60.5, 42.5, 42.2, 22.8, 20.2; HRMS (ESI) calcd for C15H20N2O5Na [M+Na]+: 331.1264. Found: 331.1266.
To a solution of above Ac-Thr-Gly-OBn in CHCl3 (1.0 mL) was added Pd-C (100 mg, 10 wt%) and MeOH (10 mL). The mixture was stirred under hydrogen gas for 8 h. The mixture was filtered and concentrated. The residue was used in the next step without purification.
4.19. Compound 23
To a solution of 17 (1.32 g, 2.85 mmol) in MeOH (6 mL), 12 M HCl (6 mL) was added at 0 °C. The mixture was warmed to 25 °C and stirred for 30 min. The mixture was concentrated, and the residue was used in the next step without purification. The crude product in DMF (6 mL) was added to a solution of BOP (1.89 g, 4.27 mmol), NMM (2.19 mL, 20.0 mmol), and Ac-Thr-Gly-OH (680 mg, 3.13 mmol) in DMF (6 mL) at 0 °C. The mixture was stirred for 16 h at 25 °C. The mixture was concentrated and quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 20:1) to afford 23 (1.19 g, 74%, 2 steps) as a colorless oil. [α]D 25 −20.3 (c 9.9, CHCl3); 1H NMR (300 MHz): δ = 7.86 (br t, J = 5.6 Hz, 0.45H), 7.66 (br t, J = 5.3 Hz, 0.55H), 7.57 (d, J = 8.4 Hz, 0.9H), 7.53 (br d, J = 3.0 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1.1H), 7.42 (d, J = 8.4 Hz, 0.9H), 7.24 (d, J = 8.1 Hz, 1.1H), 7.14 (br d, J = 7.5 Hz, 1H), 5.84 (ddd, J = 17.3, 10.5, 3.9 Hz, 0.45H), 5.77 (ddd, J = 17.3, 10.7, 3.8 Hz, 0.55H), 5.36 (dd, J = 10.5, 2.1 Hz, 1.1H), 5.20 (d, J = 17.4 Hz, 0.9H), 4.64 (d, J = 4.5 Hz, 0.45H), 4.52 (dd, J = 7.8, 2.7 Hz, 0.55H), 4.41–4.22 (m, 3H), 4.15 (dd, J = 15.3, 6.3 Hz, 0.45H), 4.04 (dd, J = 17.0, 5.9 Hz, 0.55H), 3.96 (br d, J = 8.4 Hz, 0.55H), 3.84 (dd, J = 17.0, 5.0 Hz, 0.55H), 3.57 (dd, J = 15.3, 5.7 Hz, 0.45H), 3.37 (br d, J = 12,6 Hz, 0.45H), 2.84 (br t, J = 11.7 Hz, 0.45H), 2.73 (br s, 1H), 2.59 (br t, J = 12.0 Hz, 0.55H), 2.06 (s, 1.65H), 2.03 (s, 1.35H), 1.71–1.40 (m, 6H), 1.26–1.07 (m, 3H), 1.17 (d, J = 6.3 Hz, 1.65H), 1.07 (d, J = 6.3 Hz, 1.35H), 0.99–0.82 (m, 1H); 13C NMR (75 MHz): δ = 172.6, 171.64, 171.58, 171.3, 169.5, 169.0, 134.6, 134.4, 133.5, 133.2, 131.8, 128.7, 127.9, 124.2, 124.0, 118.0, 117.9, 67.1, 66.2, 62.1, 58.5, 58.0, 56.4, 52.5, 51.3, 49.3, 44.4, 43.1, 42.8, 39.9, 39.1, 35.7, 34.3, 30.1, 29.7, 28.2, 25.8, 25.7, 25.1, 25.0, 23.0, 22.9, 19.4, 18.8; HRMS (ESI) calcd for C26H35N4O5Na [M+Na]+:585.1683. Found: 585.1688.
4.20. Compound 24a
A solution of 23 (56.2 mg, 0.1 mmol) in EtOAc (4 mL) at –78 °C was treated with ozone gas until the solution changed from colorless to blue. After that, Me2S (60 μL, 0.8 mmol) was added, and the mixture was warmed to 25 °C and was stirred for 10 min. The mixture was extracted with EtOAc. The organic layer was washed with H2O and brine, dried over Na2SO4, filtered, and concentrated. The residue was roughly purified by silica gel column chromatography (CHCl3/MeOH = 10:1). The product was immediately used without further purification. Amine 21a (54.1 mg, 0.12 mmol) was added to a solution of above aldehyde in CH2Cl2 (1 mL) at 0 °C. The mixture was stirred at 25 °C for 2 h and then NaBH3CN (22 mg, 0.35 mmol) was added. The mixture was stirred for 30 min. The reaction was quenched with 1 M HCl and the whole was extracted with EtOAc. The organic layer was washed with saturated aqueous NH4Cl and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (CHCl3/MeOH = 20:1) to give 24a (50 mg, 50%, 2 steps) as a colorless oil. [α]D 25 −18.8 (c 0.23, CHCl3); 1H NMR (300 MHz): δ = 7.61–7.52 (m, 1.2H), 7.48–7.37 (m, 4.8H), 7.33–7.31 (m, 10H), 7.14–7.03 (m, 6.67H), 6.98–6.93 (m, 0.33H), 6.87 (dd, J = 15.8, 7.7 Hz, 0.67H), 6.62 (dd, J = 15.8, 7.4 Hz, 0.33H), 6.62 (s, 0.33H), 6.53 (s, 0.67H), 5.84 (d, J = 15.6 Hz, 0.67H), 5.73 (d, J = 15.6 Hz, 0.33H), 4.78 (br t, J = 7.2 Hz, 1H), 4.44 (dd, J = 7.2, 2.1 Hz, 0.67H), 4.32–4.30 (m, 1.67H), 4.25–4.11 (m, 2.33H), 4.19 (qd, J = 7.2, 1.4 Hz, 2H), 3.94–3.93 (m, 1H), 3.84–3.79 (m, 0.33H), 3.58–3.51 (m, 0.66H), 3.47–3.40 (m, 0.67H), 3.34–3.28 (m, 0.67H), 2.86–2.57 (m, 4H), 2.43–2.34 (m, 1H), 2.04 (s, 1H), 2.02 (s, 2H), 1.71–1.40 (m, 5H), 1.31–1.19 (m, 3H), 1.28 (t, J = 7.1 Hz, 3H), 1.16 (d, J = 6.3 Hz, 1H), 1.07 (d, J = 6.3 Hz, 2H), 0.92–0.80 (m, 1H); 13C NMR (75 MHz): δ = 172.8, 171.8, 171.6, 171.5, 171.2, 169.3, 168.7, 166.2, 166.1, 150.2, 150.0, 142.4, 142.3, 140.1, 136.6, 138.4, 137.4, 137.0, 134.8, 131.6, 129.7, 129.5, 129.1, 129.0, 128.7, 128.6, 128.0, 124.0, 122.1, 120.1, 119.5, 75.2, 66.7, 65.8, 60.3, 60.2, 59.9, 59.7, 58.6, 58.1, 54.9, 49.9, 49.1, 46.2, 45.7, 44.5, 42.9, 42.7, 40.2, 39.1, 35.9, 34.7, 33.5, 30.3, 29.9, 29.7, 28.2, 25.8, 25.1, 23.0, 19.6, 19.2, 14.2; HRMS (ESI) calcd for C54H62BrN7O7Na [M+Na]+: 1022.3786. Found: 1022.3783.
4.21. Compound 24b
A solution of 23 (56.2 mg, 0.1 mmol) in EtOAc (4 mL) at −78 °C was treated with ozone gas as above. To a solution of the resulting aldehyde in CH2Cl2 (1 mL), amine 21b (58.4 mg, 0.12 mmol) was added at 0 °C. The mixture was stirred at 25 °C for 2 h and then NaBH3CN (22 mg, 0.35 mmol) was added. The resultant mixture was stirred for 30 min. The reaction was quenched with 1 M HCl and was extracted with EtOAc. The organic layer was washed with saturated aqueous NH4Cl and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (CHCl3/MeOH = 20:1) to give 24b (34.0 mg, 34%, 2 steps) as a yellow solid. [α]D 23 −10 (c 0.17, CHCl3); Mp 133–135 °C; 1H NMR (300 MHz): δ = 7.84–7.81 (m, 0.36H), 7.61–7.49 (m, 3.64H), 7.45–7.37 (m, 5.78H), 7.34–7.32 (m, 6.42H), 7.14–7.09 (m, 6.44H), 6.96–6.93 (m, 0.36H), 6.77 (m, 0.36H), 6.66 (m, 0.42H), 6.65 (m, 0.22H), 5.36–5.33 (m, 0.22H), 4.97–4.94 (m, 0.42H), 4.88–4.83 (m, 0.36H), 4.64–4.60 (m, 0.22H), 4.45–4.42 (m, 0.36H), 4.38–4.17 (m, 2.78H), 4.08–3.97 (m, 0.42H), 3.92–3.85 (m, 0.42H), 3.80–3.70 (m, 0.44H), 3.68–3.64(m, 0.36H), 3.62–3.43 (m, 0.58H), 3.37–3.08 (m, 2.42H), 3.06–2.87 (m, 1.64H), 2.83–2.75 (m, 1.78H), 2.66–2.28 (m, 7.58H), 2.08 (s, 0.78H), 2.04 (s, 0.42H), 2.03 (s, 1.8H), 1.73–1.33 (m, 6H), 1.26–0.99 (m, 12H), 0.92–0.80 (m, 1H); 13C NMR (75 MHz): δ = 172.5, 171.8, 171.6, 171.3, 169.4, 142.3, 140.3, 138.5, 137.8, 134.9, 131.5, 129.6, 129.5, 129.4, 128.9, 128.7, 128.6, 128.1, 123.9, 119.6, 119.5, 75.2, 66.0, 58.7, 58.6, 55.1, 49.3, 48.9, 45.2, 44.3, 40.4, 36.1, 30.0, 29.6, 28.2, 26.1, 26.0, 25.2, 23.0, 14.4, 14.2; HRMS (ESI) calcd for C54H67BrN7O5S2 [M+H]+: 1036.3823. Found: 1036.3820.
4.22. Compound 24c
A solution of 23 (56.2 mg, 0.1 mmol) in EtOAc (4 mL) at −78 °C was treated with ozone gas as above and the product aldehyde was reductively aminated by H-His(Trt)-N(OMe)Me (52.8 mg, 0.12 mmol) using the same procedure described above. The product was purified by flash column chromatography (CHCl3/MeOH = 20:1) to give 24c (25.7 mg, 26%, 2 steps) as a white wax. [α]D 25 −18.3 (c 0.43, CHCl3); 1H NMR (300 MHz): δ = 7.50 (d, J = 8.4 Hz, 1.8H), 7.43 (br d, J = 8.4 Hz, 1.4H), 7.39 (d, J = 8.4 Hz, 1.8H), 7.32–7.30 (m, 10H), 7.12–7.09 (m, 6.1H), 6.93 (br d, J = 8.1 Hz, 0.65H), 6.78 (d, J = 7.5 Hz, 0.25H), 6.61 (m, 0.1H), 6.65 (m, 0.65H), 6.51 (s, 0.25H), 5.36–5.34 (m, 0.25H), 5.12–5.07 (m, 0.1H), 4.82–4.80 (m, 0.1H), 4.72 (br t, J = 8.0 Hz, 0.65H), 4.44 (dd, J = 3.6, 2.4 Hz, 0.25H), 4.34–4.20 (m, 3.25H), 4.09–3.97 (m, 1.7H), 3.75–3.63 (m, 1H), 3.54 (s, 1.95H), 3.45–3.38 (m, 1.4H), 3.15 (s, 0.3H), 3.08 (s, 1.95H), 3.03 (s, 0.75H), 2.83–2.65 (m, 4H), 2.48–2.41 (m, 1H), 2.34–2.25 (m, 1H), 2.07 (s, 1.95H), 2.05 (s, 1.05H), 2.04–2.01 (m, 0.35H), 1.72–1.37 (m, 5H), 1.26–1.12 (m, 5H), 1.06 (d, J = 6.3 Hz, 3H), 0.99–0.83 (m, 2H); 13C NMR (75 MHz): δ = 172.9, 171.8, 171.6, 169.1, 142.4, 138.2, 134.9, 131.7, 129.7, 128.6, 128.0, 123.9, 119.3, 75.1, 65.4, 58.4, 57.4, 54.5, 49.0, 48.6, 45.9, 45.0, 40.3, 36.0, 32.3, 29.9, 29.7, 28.3, 25.8, 25.2, 23.1, 19.7, 14.2, 14.1; HRMS (ESI) calcd for C52H61BrN8O7Na [M+Na]+: 1011.3739. Found: 1011.3730.
4.23. Compound 25a
TFA (trifluoroacetic acid)/CH2Cl2/TIS (trisisopropylsilane)/H2O (10:10:1.0:1.0, 2.2 mL) was added to 24a (50 mg, 0.05 mmol) and the mixture was stirred at 25 °C for 30 min. The mixture was concentrated, and the product was isolated by ether precipitation to afford detritylated product 25a (37.8 mg, quant.) as a white amorphous. [α]D 24 −45.4 (c 1.19, MeOH); 1H NMR (300 MHz): δ = 8.87 (br s, 0.8H), 8.76 (br s, 0.2H), 8.45 (m, 0.4H), 7.93 (br d, J = 7.5 Hz, 1H), 7.66 (d, J = 8.1 Hz, 1.6H), 7.59 (br d, J = 7.8 Hz, 1H), 7.52–7.43 (m, 1H), 7.37–7.28 (m, 0.8H), 7.25–7.17 (m, 0.2H), 6.92–6.81 (m, 0.8H), 6.70 (dd, J = 15.6, 8.6 Hz, 0.2H), 6.19 (d, J = 15.3 Hz, 0.8H), 5.78 (d, J = 15.9 Hz, 0.2H), 5.04 (m, 1H), 4.39 (m, 1.2H), 4.28–4.15 (m, 4.8H), 4.11–3.78 (m, 3H), 3.69–3.39 (m, 3.2H), 3.32–2.77 (m, 2.8H), 2.12–2.03 (m, 0.2H), 2.08 (s, 2.4H), 2.03 (s, 0.6H), 1.81–1.66 (m, 4H), 1.55–1.52 (m, 1.8H), 1.42–1.20 (m, 3H), 1.32 (t, J = 7.1 Hz, 3H), 1.24 (d, J = 6.0 Hz, 3H), 1.00–0.97 (m, 1H); 13C NMR (75 MHz): δ = 174.7, 174.1, 174.0, 173.7, 172.5, 166.5, 163.1, 140.1, 136.4, 136.0, 135.8, 135.4, 133.4, 133.0, 131.1, 130.8, 130.1, 129.7, 128.2, 125.9, 125.1, 120.2, 119.7, 119.6, 118.7, 68.6, 68.3, 62.5, 62.3, 61.4, 61.0, 59.8, 54.0, 52.5, 51.8, 51.0, 50.6, 46.1, 44.7, 43.9, 43.6, 40.6, 39.6, 36.0, 35.6, 31.5, 31.0, 29.4, 28.6, 27.3, 26.7, 26.4, 22.9, 20.4, 14.7; HRMS (ESI) calcd for C35H49BrN7O7 [M+H]+: 758.2871. Found: 758.2875.
4.24. Compound 25b and 25c
Compound 24b and 24c were similarly treated TFA/CH2Cl2/TIS/H2O (10:10:1.0:1.0, 2.2 mL) as above to yield the corresponding de-tritylated products 25b and 25c.
Compound 25b; [α]D 24 −42.8 (c 1.57, MeOH); 1H NMR (300 MHz): δ = 8.90–8.74 (m, 1H), 7.97–7.90 (m, 0.25H), 7.73–7.63 (m, 2.5H), 7.58–7.40 (m, 3.75H), 7.37–7.22 (m, 2.5H), 4.76–4.72 (m, 0.5H), 4.43–4.27 (m, 4.5H), 4.12–3.91 (m, 2H), 4.04 (d, J = 13.5 Hz, 2H), 3.73–3.64 (m, 1H), 3.54–3.47 (m, 1H), 3.34–3.09 (m, 2H), 3.04–2.65 (m, 6.5H), 2.57–2.49 (m, 0.5H), 2.17 (s, 1.5H), 2.14–2.06 (m, 1H), 2.12 (s, 1.5H), 1.85–1.72 (m, 4H), 1.57–1.41 (m, 3H), 1.39–1.28 (m, 8H), 1.22–0.95 (m, 1H); 13C NMR (75 MHz): δ = 174.1, 173.8, 173.5, 172.0, 136.8, 134.9, 133.4, 133.3, 133.1, 131.1, 130.6, 130.2, 128.6, 125.7, 118.4, 118.0, 68.5, 68.3, 63.2, 62.2, 61.5, 61.1, 56.4, 51.0, 45.9, 40.5, 39.8, 36.2, 35.7, 29.6, 27.7, 27.5, 27.4, 27.2, 26.7, 23.0, 22.9, 20.4, 20.3, 15.4, 15.2; HRMS (ESI) calcd for C35H52BrN7O5S2Na [M + Na]+: 816.2547. Found: 816.2540.
Compound 25c; [α]D 24 −34.6 (c 0.57, MeOH); 1H NMR (300 MHz): δ = 8.88–8.83 (m, 1.5H), 7.92 (br d, J = 8.1 Hz, 0.5H), 7.71 (d, J = 8.7 Hz, 2H), 7.67 (s, 0.5H), 7.51–7.49 (m, 0.5H), 7.50 (d, J = 8.4 Hz, 2H), 7.35 (s, 0.5H), 7.32 (s, 0.5H), 4.88–4.73 (m, 1H), 4.50–4.25 (m, 4H), 4.11–3.94 (m, 2H), 3.78 (s, 1.5H), 3.84–3.61 (m, 2H), 3.65 (s, 1.5H), 3.52–3.43 (m, 1H), 3.33–3.04 (m, 3H), 3.32 (s, 1.5H), 3.27 (s, 1.5H), 2.98–2.71 (m, 2H), 2.63–2.53 (m, 1H), 2.17 (s, 1.5H), 2.12 (s, 1.5H), 1.95–1.70 (m, 4H), 1.60–1.28 (m, 7H), 1.21–0.95 (m, 1H); 13C NMR (75 MHz): δ = 174.1, 174.0, 173.7, 173.5, 172.1, 136.8, 136.2, 135.6, 135.1, 133.4, 133.1, 130.6, 130.1, 125.7, 124.9, 118.8, 118.4, 68.6, 68.3, 63.0, 62.8, 58.4, 58.1, 51.0, 51.7, 46.8, 44.7, 43.9, 43.6, 40.3, 39.5, 36.2, 35.6, 32.8, 31.6, 31.1, 29.5, 28.9, 28.1, 27.5, 27.3, 26.7, 26.6, 23.0, 22.9, 20.4, 20.2; HRMS (ESI) calcd for C33H47BrN8O7Na [M+Na]+: 769.2643. Found: 769.2651.
4.25. Compound 26
NBS (0.4 mL, 40 μmol, 0.1 M solution in THF) was added to a dithioacetal 25b (5 mg, 6.3 μmol). After being stirred at 25 °C for 5 min, the mixture was purified by HPLC [linear gradient of B, 5% to 45% in 40 min (B: 0.5% TFA in CH3CN, A: 0.5% TFA in H2O)] to give aldehyde 26 (0.7 mg, 16%) as a white solid. The product showed a single peak on an analytical HPLC (Figure S-2). 1H NMR (300 MHz): δ = 8.48 (m, 0.67H), 8.09 (m, 1.33H), 7.70 (d, J = 8.1 Hz, 2H), 7.59 (m, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.32–7.30 (m, 1.33H), 7.23 (br s, 0.67H), 4.37–4.36 (m, 1.67H), 4.28–4.18 (m, 3.33H), 4.03–3.97 (m, 3H), 3.12–3.11 (m, 1H), 2.92–2.68 (m, 6H), 2.13 (s, 1.2H), 2.07 (s, 1.8H), 1.81--1.26 (m, 7H), 0.94 (m, 1H); HRMS (ESI) calcd for C31H42BrN7O6Na [M+Na]+: 710.2272. Found: 710.2264.
4.26. Estimation of IC50 values
The peptide substrate [H-Thr-Ser-Ala-Val-Leu-Gln-Ser-Gly-Phe-Arg-Lys-NH2]9 (111 μM) in reaction solution (25 μL of 20 mM Tris-HCl buffer pH 7.5 containing 7 mM DTT) was incubated with the R188I SARS 3CLpro (56 nM)9 at 37 °C for 90 min in the presence of various concentrations of inhibitor. The mixture was injected into an analytical HPLC column [Cosmosil 5C18 (4.6 × 150 mm)]. A linear gradient of CH3CN (10%–20%) in an aqueous solution of 0.1% TFA over 30 min was used for elution, and the cleavage rates were calculated from the reduction in the substrate peak area. Each IC50 value was obtained from the sigmoidal dose–response curve (Figure S-3 for a typical sigmoidal curve). Each experiment was repeated three times and the results were averaged.
Acknowledgements
This work was supported, in part, by a Grant-in-aid for Scientific Research 16H05104 to KA and a Grant-in-aid for Scientific Research 16J10921 to KO from the Japan Society for the Promotion of Science.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2018.12.019.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Lee N., Hui D., Wu A., et al. N Engl J Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 2.Drosten C., Günther S., Preiser W., et al. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., et al. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Rota P.A., Oberste M.S., Monroe S.S., et al. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 5.Marra M.A., Jones S.J., Astell C.R., et al. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 6.Thiel V., Ivanov K.A., Putics Á., et al. J General Viol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 7.Pillaiyar T., Manickam M., Namasivayam V., et al. J Med Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akaji K. Amino Acids Pept Proteins. 2018;42:228–279. and references cited therein. [Google Scholar]
- 9.Akaji K., Konno H., Onozuka M., et al. Bioorg Med Chem. 2008;16:9400–9408. doi: 10.1016/j.bmc.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akaji K., Konno H., Mitsui H., et al. J Med Chem. 2011;54:7962–7973. doi: 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- 11.Shimamoto Y., Hattori Y., Kobayashi K., et al. Bioorg Med Chem. 2015;23:876–890. doi: 10.1016/j.bmc.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makabe H., Kong L.K., Hirota M. Org Lett. 2003;5:27–29. doi: 10.1021/ol0201916. [DOI] [PubMed] [Google Scholar]
- 13.Kurogome Y., Kogiso M., Looi K.K., et al. Tetrahedron. 2013;69:8349–8352. [Google Scholar]
- 14.Katsuyama M., Furuta M., Kobayashi K., et al. Heterocycles. 2015;91:959–969. [Google Scholar]
- 15.Ohnishi K., Sakurai H., Kobayashi K., et al. Heterocycles. 2015;91:573–582. [Google Scholar]
- 16.Hentges S.G., Sharpless K.B. J Am Chem Soc. 1980;102:4263–4265. [Google Scholar]
- 17.Mitsunobu O., Yamada M., Mukaiyama T. Bull Chem Soc Jpn. 1967;40:935–939. [Google Scholar]
- 18.Dale J.A., Mosher H.S. J Am Chem Soc. 1973;95:512–519. [Google Scholar]
- 19.Basha A., Lipton J.L., Weinreb S.M. Tetrahedron Lett. 1977;18:4171–4172. [Google Scholar]
- 20.Sheehan J., Cruickshank P., Boshart G. J Org Chem. 1961;26:2525–2528. [Google Scholar]
- 21.Castro B., Dormoy J.R., Evin G., et al. Tetrahedron Lett. 1975;16:1219–1222. [Google Scholar]
- 22.Konno H., Sema Y., Ishii M., et al. Tetrahedron Lett. 2013;54:4848–4850. doi: 10.1016/j.tetlet.2013.06.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.