

Abstract
The open reading frame 3 of the severe acute respiratory syndrome coronavirus (SARS‐CoV) genome encodes a predicted protein 3a, consisting of 274 amino acids, that lacks any significant similarities to any known protein. We generated specific antibodies against SARS protein 3a by using a synthetic peptide (P2) corresponding to amino acids 261–274 of the putative protein. Anti‐P2 antibodies and the sera from SARS patients could specifically detect the recombinant SARS protein 3a expressed in Escherichia coli and in Vero E6 cells. Expression of SARS protein 3a was detected at 8–12 h after infection and reached a higher level after ∼24 h in SARS‐CoV‐infected Vero E6 cells. Protein 3a was also detected in the alveolar lining pneumocytes and some intra‐alveolar cells of a SARS‐CoV‐infected patient's lung specimen. Recombinant protein 3a expressed in Vero E6 cells and protein 3a in the SARS‐CoV‐infected cells was distributed over the cytoplasm in a fine punctate pattern with partly concentrated staining in the Golgi apparatus. Our study demonstrates that SARS‐CoV indeed expresses a novel protein 3a, which is present only in SARS‐CoV and not in other known CoVs.
Keywords: SARS, severe acute respiratory syndrome; CoV, coronavirus; SARS; Nuclocapsid; Vero E6 cell; Atypical pneumonia; Golgi
1. Introduction
An epidemic of severe acute respiratory syndrome (SARS) has been associated with an outbreak of atypical pneumonia in many countries [1, 2]. There is now clear evidence that a novel coronavirus (CoV) is associated with this outbreak [3, 4]. Genome sequences of SARS‐CoV isolates obtained from clinical index patients have been published and are available in the GenBank (http://www.ncbi.nlm.nih.gov/).
Coronaviruses are exceptionally large RNA viruses. The genome of SARS‐CoV is 29.7 kb in length and has the same frame of sequence structure as do other CoVs such as the spike glycoprotein (S), envelope protein (E), membrane protein (M), and nucleocapsid protein (N), which have been identified in SARS‐CoV [5]. However, 14 putative open reading frames (ORFs) were identified within the SARS‐CoV genome, encoding six known and eight unknown proteins [6, 7]. Theoretically, all of these proteins play a unique and important role as SARS‐CoV infection progresses and could serve as targets for addressing the mechanisms and therapeutic developments for this disease. For example, the four structural proteins: the S protein, M protein, N protein, and small E protein have been broadly studied for their important roles in receptor binding, virion budding, and viral life cycle during general processes of viral infection [8, 9]. The gene of ORF 3a (CDS: 25268–26092) is located between the gene of the S glycoprotein (CDS: 21492–25259) and E protein (CDS: 26117–26347) [6, 7]. The minimal transcription regulatory sequence (5′‐ACGAAC‐3′) is also located upstream of ORF 3a (CDS: 25260–25265) [7]. Due to its novelty, we investigated ORF 3a that encodes the putative protein 3a, designated as protein X1 by Rota et al. [4].
In this study, we demonstrated that protein 3a was expressed in SARS‐CoV‐infected cells and a SARS‐CoV‐infected patient's lung specimen. Protein 3a was a membrane associated protein that was distributed over the cytoplasm in a fine punctate pattern with partly concentrated staining in the Golgi apparatus. In addition, specific antibodies against protein 3a were also present in sera from SARS‐CoV‐infected patients. Thus, the identification of this novel protein 3a could be the first step toward exploring its role in the SARS infection process and the potential utilization of protein 3a in clinical diagnosis or therapeutic development.
2. Materials and methods
2.1. Cloning and expression of protein 3a
SARS‐CoV total RNA was isolated from SARS‐CoV (TW1 strain)‐infected Vero E6 cells by a single‐step extraction method as described previously [10, 11]. Oligonucleotide primers used for amplification and sequencing of the protein 3a gene were based on the SARS‐CoV‐TW1 sequence (Accession No. AY291451). Two oligonucleotides based on the N‐ and C‐terminal sequences of the protein 3a gene were synthesized. The sense primer used was 5′‐ATGGATTTGTTTATGAGATTTTTTACT‐3′, encoding the eight N‐terminal amino acids (MDLFMRFFT), whereas the antisense primer was 5′‐CAAAGGCACGCTAGTAGTCGTCGT‐3′, encoding the C‐terminus (TTTTSVPL). The coding sequence of the protein 3a gene was then amplified by PCR, and the amplified product was analyzed by electrophoresis, subcloned into the pSTBlue vector (Novagen, Madison, WI), then transformed into Escherichia coli strain DH5. After transformation, plasmids from positive clones were subjected to sequence analysis by use of an ABI 3100 sequencer (Applied Biosystems, Foster City, CA) and the dye terminator cycle sequencing FS Ready reaction. For expression of the recombinant protein 3a, the SARS virus protein 3a gene from the pSTBlue construct was inserted into vector pGEX 4T‐2 (Amersham Biosciences Inc., Sweden) in frame with the EcoRI restriction enzyme site for expression as a glutathione S‐transferase (GST) fusion protein. After being transformed into E. coli strain BL21 (DE3) (Novagen, Madison, WI), the fusion protein was expressed by induction with 0.5 mM isopropyl‐β‐d‐thiogalactopyranoside, for 3 h at 37 °C. For obtaining the pure recombinant protein 3a, the GST‐fusion protein was affinity purified by glutathione–Sepharose 4B chromatography (Amersham Biosciences Inc.) according to the manufacturer's instructions. The recombinant protein 3a was then released from the Sepharose‐bound GST moiety by specific digestion with thrombin protease and collected. Briefly, the induced cells were suspended in phosphate‐buffered saline (PBS) buffer with 1 mM phenylmethanesulfonyl fluoride and then sonicated in an ice bath for 1 min. The lysed cells were centrifuged at 20 000×g at 4 °C for 15 min and the pellets were discarded. The supernatant was applied to a glutathione–Sepharose 4B column equilibrated with PBS buffer and the column was washed with 10× the bed volume of the same buffer. After the GST‐fusion SARS protein 3a was bound, the column was incubated with PBS containing 50 units of thrombin at 25 °C for 2 h. The released recombinant protein 3a lacking the GST carrier was eluted with PBS buffer and collected. The protein purity was determined by 12.5% SDS–PAGE electrophoresis and visualized with Coomassie blue.
For transient expression of the recombinant protein 3a in Vero E6 cells, the coding region of protein 3a was subcloned into the EcoRI site of the pcDNA3.1/myc‐His vector (Invitrogen, San Diego, CA). Vero E6 (ATCC CRL‐1586) cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco‐BRL) supplemented with 2 mM l‐glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum. The pcDNA3.1/Myc‐His/protein 3a plasmid was mixed with Lipofectin reagent (Invitrogen, San Diego, CA) according to the manufacturer's instructions, and approximately 15 μg of DNA was transfected into 1 × 106 Vero E6 cells that had been infected 2 h earlier with 1 × 105 infectious units of a vaccinia virus/bacteriophage T7 RNA polymerase hybrid [12, 13, 14]. After 48 h, cells were harvested, washed twice with PBS, and then the expression of protein 3a was verified by Western blotting.
2.2. Polyclonal antibody preparation
New Zealand White rabbits were immunized with keyhole limpet hemocyanin‐conjugated synthetic peptide CPIYDEPTTTTSVPL (peptide P2), corresponding to residues 261–274 of protein 3a, or SDNGPQSNQRSAC (peptide N1), corresponding to residues 1–12 of the nucleocapsid protein. The conjugated‐peptides in 0.5 ml of PBS were emulsified with an equal volume of Titer Max adjuvant (CytRx Inc., Norcross, GA) and 3‐month‐old rabbits were immunized. After 3 weeks, boosters of each conjugated‐peptide emulsified in Freund's incomplete adjuvant were given by intradermal injection; this was followed by another injection of peptide in another 3 weeks. Anti‐sera were collected at 12‐day intervals starting from the last boost and the IgG specificity was monitored by Western blotting assay.
2.3. Immunodetection assays
Protein samples were separated by SDS–PAGE on a 12.5% separation gel. For immunodetection of the antigenicity of protein 3a, the separated proteins were electroblotted onto a PVDF membrane, which was then blocked with skimmed milk and incubated for 16 h at 4 °C with anti‐P2 rabbit anti‐serum diluted 1:2000–3000 or a human serum pool diluted 1:100. Bound IgG antibodies were detected by use of horseradish peroxidase‐labeled donkey anti‐rabbit IgG antibodies (Amersham Biosciences Inc.) or sheep anti‐human IgG antibodies (Amersham Biosciences Inc.). The target proteins were revealed by an enhanced chemiluminescence (ECL, Amersham Biosciences Inc.) development system.
2.4. Immunohistochemistry assay
A 73‐year‐old man was admitted to National Taiwan University Hospital because of recurrent chest pain and dyspnea on April 23, 2003. He had an episode of myocardial infarction two years before. After admission, a low‐grade fever developed and serial follow‐up chest X‐rays showed progressive lung infiltration. SARS‐CoV was isolated from his throat swab and RT‐PCR for SARS‐CoV further confirmed the presence of viral genome in the specimen of his throat swab. He died of acute myocardial infarction at the 7th day after admission. A restricted autopsy was performed immediately, random sampling of the lungs, heart, kidneys, liver, small intestine, and spleen was taken. All the tissue specimens were fixed in 10% neutral formalin and paraffin‐embedded blocks and serial sections were prepared in a conventional manner. The hematoxylin and eosin staining of the lung tissue from this SARS patient showed congestion and focal edema. Another three lung biopsy specimens without SARS‐CoV infection were also analyzed as negative controls. For immunohistochemical staining, antigen retrieval was carried out with the steam heat method for 10 min using the Trilogy retrieval buffer. (Cell Marque Cooperation, Austin, TX). Endogenous peroxidase activity was quenched with 0.3% hydrogen peroxide for 20 min. For SARS‐CoV detection anti‐P2 antibody diluted in 1:500 was used. Pre‐immune rabbit serum was used as a negative control. The previously heat‐treated tissue sections were incubated with the primary antibodies and non‐immune serum at 4 °C overnight. The slides were then sequentially stained on a Nexis autostainer (Ventana, Tucson, AZ) using the Ventana Basic Alkaline Phosphatase Red Detection Kit (Ventana) and counterstained with hematoxylin.
2.5. Immunofluorescence microscopy
About 6 × 104 Vero E6 cells were grown on coverslips within 12‐well plates and then infected /transfected with 1 μg pcDNA3.1/myc‐His protein 3a plasmid by Lipofectin reagent (Invitrogen, San Diego, CA) as described above. After 48 h, cells were washed with PBS twice followed by fixation with 4% paraformaldehyde in PBS for 5 min at room temperature and permeabilized in methanol for 5 min on ice. Indirect immunofluorescence staining was performed as described previously [15]. The cells were blocked with 0.2% bovine serum albumin, 0.1% saponin in PBS, followed by incubation with anti‐P2 peptide‐specific rabbit anti‐serum or anti‐SARS M protein rabbit anti‐serum for 30 min. Mouse monoclonal antibodies to Golgi 58K protein (Sigma), mouse monoclonal antibodies to β‐COP protein (Sigma), mouse monoclonal antibodies to calnexin protein (Affinity BioReagents), mouse monoclonal antibodies to lysosomal‐associated membrane proteins‐2 (LAPM‐2) (BD Biosciences) or mouse monoclonal antibodies to cytochrome c oxidase subunit 1 (COX 1) (Molecular Probes) were diluted in 1:100 and co‐incubated with anti‐P2 antibody, respectively. The optimal anti‐P2 anti‐serum dilution (1:600) for the use of SARS‐protein 3a was determined by titration experiments on non‐infected cells. After three washes with PBS, cells were incubated with secondary antibody, Alexa 488‐conjugated anti‐rabbit IgG antibody (Molecular Probes), Alexa 594‐conjugated anti‐mouse IgG antibody, and Hoechst 33258 (DNA‐binding dye). Finally, cells were washed 3× with PBS, mounted on Mowiol, and examined with a Zeiss Axiophot microscope equipped for epifluorescence and a Bio‐Rad Radiance 2100 confocal microscope for confocal fluorescence.
2.6. Subcellular fractionation
Cytosol (C) and membrane (M) fractions of Vero E6 cells were prepared using a CNM compartment protein extraction kit (BioChain Institute Inc., Hayward, CA) according to the manufacturer's instructions. Briefly, infected–transfected Vero E6 cells (1 × 106 cells) were harvested, washed with PBS, and then centrifuged at 1000×g for 5 min. The cell pellet was homogenized in 100 μl of buffer C plus a mixture of protease inhibitors, rotated at 4 °C for 20 min followed by centrifugation at 4 °C, 12 000×g for 20 min to generate soluble cytosolic (C) fraction. The insoluble pellet was washed with 200 μl of buffer W and resuspended in 50 μl of buffer N at 4 °C for 20 min. The nuclear proteins (N) were extracted and recovered in the supernatant after centrifugation at 4 °C, 12 000×g for 20 min. Finally, to obtain the membrane proteins, the cell pellet containing cell debris was extracted with 50 μl of buffer M and then rotated at 4 °C for 20 min. The supernatant was centrifuged at 4 °C, 12 000×g for 20 min to generate soluble membrane (M) fraction. All of the fractionated protein solutions were stored at −70 °C until analyzed.
3. Results and discussion
3.1. Analysis of the novel SARS‐CoV protein 3a
The ORF 3a of the SARS‐CoV genome encodes a predicted protein 3a containing 274 amino acids with a theoretical molecular mass of approximately 30.9 kDa (Swiss‐Prot: P59632). Using BLAST program analysis [16], we showed that the N‐terminal domain of SARS protein 3a has ∼29% identity with the putative cytochrome B‐561 transmembrane protein from bacteria (Ralstonia solanacearum) and 24% identity with the opsin fragments from the hawkmoth (Manduca sexta) and the rhodopsin from the butterfly (Popilio glaucus), as indicated in Fig. 1 . The C‐terminus of protein 3a has moderate similarity to the calcium‐transporting ATPase (calcium pump) from a parasite (Plasmodium falciparum) and the outer‐membrane porin from a bacterium (Shewanella oneidensis), 41% and 27% identity, respectively. However, overall the putative full‐length sequence of protein 3a lacks significant similarities to any known proteins. When we used hydrophobicity and polarity analysis programs [17, 18], there were three regions with low polarity and high hydrophobicity in the N‐terminus of the protein 3a sequence. Membrane‐spanning regions, predicted by the Dense Alignment Surface method [19], were located in three sequence segments, amino acids 43–56, 79–97, and 106–116, suggesting that protein 3a may play a role in SARS‐CoV interaction with the cellular membrane. Based on bioinformation of this novel protein, we further demonstrate that protein 3a is expressed both in SARS‐CoV‐infected cells and a SARS‐CoV‐infected patient's tissue, which is a first step in exploring its role in the SARS‐CoV infection process.
Figure 1.
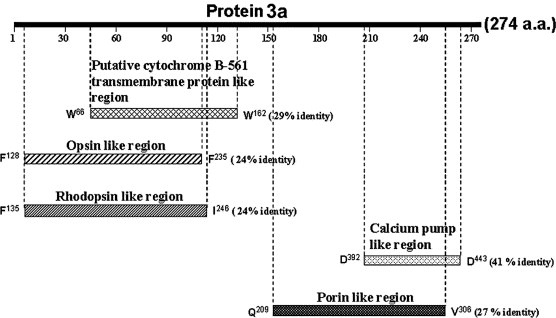
Sequence analysis of putative SARS‐CoV protein 3a. Schematic representation of protein 3a aligned with the putative cytochrome B‐561 transmembrane protein from bacteria (R. solanacearum) (TrEMBL: http://Q8XVV8), opsin from hawkmoth (M. sexta) (TrEMBL: http://O02465), rodopsin from butterfly (P. glaucus) (TrEMBL: http://Q9UAM8), calcium pump from parasite (P. falciparum) (Swiss Prot: Q08853), and outer‐membrane porin from bacteria (S. oneidensis) (TrEMBL: http://Q8EAK6). The numbering system is based on the individual sequences.
3.2. Preparation of anti‐serum to recombinant protein 3a
The cDNA of SARS protein 3a was obtained from the SARS‐CoV‐TW1 strain by reverse‐transcription PCR and was subcloned to a pGEX 4T‐2 vector. The protein 3a/GST‐fusion protein was expressed in E. coli and purified with glutathione chromatography. To explore the possibility of immunologic detection of SARS protein 3a, we prepared rabbit antibodies to a synthetic peptide (P2), corresponding to amino acids 261–274 of the putative SARS protein 3a. Western blot analysis showed that partially purified recombinant protein 3a from E. coli can be recognized by anti‐P2 antibody (Fig. 2A , lane 3) and pooled sera from SARS‐CoV‐infected patients (Fig. 2A, lane 4). This protein was not detected by preimmune serum (data not shown).
Figure 2.
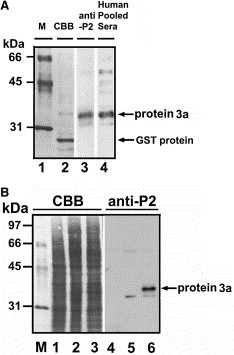
Expression and immunodetection of recombinant SARS‐CoV protein 3a in E. coli or Vero E6 cells. (A) Recombinant SARS protein 3a expressed and partially purified from E. coli cells was separated by 12.5% SDS–PAGE and transferred to PVDF membrane. Purified protein was stained with Coomassie brilliant blue (lane 2) or probed with anti‐P2 antibody (lane 3) or pooled sera from SARS‐CoV‐infected patients (lane 4). Molecular weight markers were in lane 1. Recombinant protein 3a (∼33–34 kDa) containing additional 14 amino acids from pGEX 4T‐2 vector was indicated. GST protein degraded from GST‐protein 3a was also indicated. (B) A pcDNA3.1/myc‐His/protein 3a plasmid was transfected into vaccinia virus‐infected Vero E6 cells. After 48 h, non‐infected–transfected cell lysates (lanes 1 and 4), vaccinia virus‐infected Vero E6 cell lysates (lanes 2 and 5), and infected–transfected cell lysates (lanes 3 and 6) were prepared for Coomassie blue staining (lanes 1–3) and Western blot analysis with anti‐P2 antibody (lanes 4–6). This recombinant myc‐His tagged protein 3a (∼37 kDa) contains additional 38 amino acids from pcDNA3.1/myc‐His vector.
3.3. Expression of recombinant SARS protein 3a in Vero E6 cells
To determine whether recombinant protein 3a can be expressed in mammalian cells, the cDNA of protein 3a was subcloned into the pcDNA3.1/myc‐His vector and then transfected into Vero E6 cells. However, expression of protein 3a was not detected in the Vero E6 cells by Western blot analysis (data not shown). We also attempted to express SARS protein 3a in several cell lines, but all attempts were unsuccessful, suggesting that protein 3a was selectively expressed in SARS‐CoV‐infected cells. We next used an infected–transfection transient expression system to enhance the expressed level of protein 3a in Vero E6 cells [11, 12, 13]. Vero E6 cells were infected with a vaccinia virus expressing T7 RNA polymerase for 2 h, then transfected with the pcDNA3.1/Myc‐His/protein 3a construct. Forty eight hours after infection‐transfection, cell lysates were separated and analyzed by Western blotting. Fig. 2B shows that a myc‐His tagged protein 3a with a molecular mass of ∼37 kDa in the infected–transfected cell lysates was recognized by the anti‐P2 antibody. No signal was detected in the non‐infected–transfected or vaccinia virus‐infected cell lysates. Our data demonstrated that the putative SARS protein 3a can be expressed in E. coli and Vero E6 cells, although the expression level is relatively low. We also showed that SARS‐CoV‐infected patients' sera appeared to have specific antibodies against the recombinant protein 3a. Moreover, the P2 peptide from the C‐terminus of protein 3a appeared to be a good epitope for generating antibodies against protein 3a that will be useful for further studies of SARS‐CoV.
3.4. Detection of protein 3a in SARS‐CoV‐infected cells
To determine whether protein 3a can be detected in SARS‐CoV‐infected cells, we infected Vero E6 cells with SARS‐CoV TW1 and harvested cells at the indicated time after infection. Cell lysates were analyzed by Western blotting with anti‐P2 or anti‐nucleocapsid (anti‐N1) antibodies. Fig. 3 shows that expression of SARS protein 3a (panel B), like that of the viral nucleocapsid protein (panel C), was detected at 8–12 h after infection and reached a higher level after ∼24 h in the SARS‐CoV‐infected Vero E6 cells. Anti‐P2 antibody pre‐incubated with the P2 peptide, but not the N1 peptide, prevented binding of anti‐P2 antibodies to the protein 3a, indicating that protein 3a was specifically detected (Fig. 3B, lane 6; and data not shown).
Figure 3.
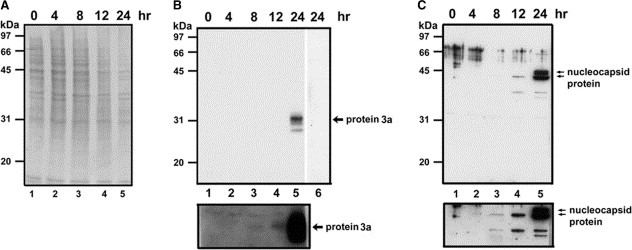
Detection of protein 3a in SARS‐CoV‐infected cells. (A) Vero E6 cells uninfected (lane 1) or infected with SARS‐CoV TW1 for 4, 8, 12, or 24 h (lanes 2–5) were harvested, treated with SDS‐sample buffer, and separated by 12.5% SDS–PAGE, transferred to PVDF membrane, and stained with Coomassie blue. (B) Protein samples in panel A were analyzed by Western blotting using an anti‐P2 antibody (lanes 1–5) or anti‐P2 antibody pre‐incubated with 5 μM of P2 peptide (lane 6). (C) Protein samples in panel A were analyzed by Western blotting using an anti‐nucleocapsid (anti‐N1) antibody. Lower panels in B and C show longer exposure time for lanes 1–5.
3.5. Detection of protein 3a in the lung tissue of a SARS‐CoV‐infected patient
We next determined whether protein 3a could be detected in a SARS‐CoV‐infected patient by immunohistochemistry (described in Section 2). Our data showed that protein 3a was detected only in the lung section (Fig. 4 ), but not in small intestine, heart, liver, spleen, kidney, and lymph node of the SARS patient (data not shown). No staining was seen in the lung tissue sections using rabbit pre‐immune serum (data not shown). The positive staining cells were mostly located in the alveolar lining pneumocytes and some intra‐alveolar cells. The hyaline membrane along the alveolar space was occasionally positive. The positive staining area tended to be found in the periphery of the lung with a patch‐like distribution. The bronchial epithelium and the endothelium of pulmonary vessels were negative. Protein 3a could not be detected in any of the tissue sections from non‐SARS patients. Our data clearly showed that protein 3a was expressed in the SARS‐CoV‐infected lung tissue. The function of this molecule within the alveolar lining pneumocytes was unclear, but its presence highlights the importance of further characterization of this novel protein and identification of its interacting cellular proteins in future studies.
Figure 4.
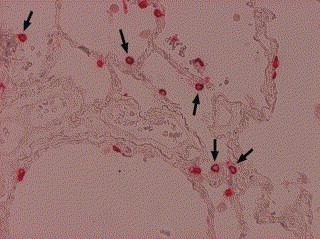
Immunohistochemical detection of protein 3a in lung section from an SARS‐CoV‐infected patient. The expression of protein 3a was detected using anti‐P2 antibody diluted 1:500 as described in Section 2. Arrowheads indicate protein 3a in the cytoplasm of some pneumocytes (100×).
3.6. Subcellular localization of SARS protein 3a
We next determined the subcellular localization of the SARS protein 3a in Vero E6 cells by confocal microscopy and fractionation. Fig. 5A shows that recombinant protein 3a expressed in Vero E6 cells as well as protein 3a in the SARS‐CoV‐infected cells was distributed over the cytoplasm in a fine punctate pattern with partly concentrated staining in the Golgi apparatus. Similar immunofluorescence staining of protein 3a was also observed in Vero cells after 6 h of infection with SARS‐CoV, however the expression level was lower than that of 24 h (data not shown). Furthermore, by using commercially prepared slides with SARS‐CoV‐infected cells (Euroimmun Inc., Germany), we showed that the expression of protein 3a was also detected in a punctate pattern with partly concentrated staining in the Golgi apparatus (Fig. 5B, a–f), but not co‐localized with endoplasmic reticulum (ER), lysosome, or mitochondria (Fig. 5B, g–o). The distribution pattern of protein 3a as well as the SARS‐CoV M protein was co‐localized with the β‐COP Golgi apparatus marker protein shown in Fig. 5B, d–f and p–r, respectively. To assess the subcellular distribution of recombinant protein 3a expressed in Vero E6 cells, homogenates were fractionated by differential centrifugation. Membrane (M) and cytosol (C) fractions were separated and protein 3a, calnexin (ER membrane marker), and α‐tubulin (cytoplasmic marker) in subcellular fractions were identified by Western blot analysis. Protein 3a was detected in the membrane fraction (Fig. 5C). Our data demonstrate that SARS‐CoV indeed expressed the putative membrane‐associated protein 3a in vivo. It is known that the viral membrane proteins, including the major proteins S (spike) and M (membrane), are inserted into the ER–Golgi intermediate compartment, whereas full‐length replicated RNA (+ strands) assembles with the N (nucleocapsid) protein. This RNA protein complex then associates with the M protein embedded in the membranes of the ER and virus particles form as the nucleocapsid complex buds into the ER. The virus then migrates through the Golgi complex and eventually exits from the cell, probably by exocytosis [9, 20, 21, 22]. It is of interest to note that protein 3a was partially co‐localized with Golgi marker protein p58 and that the punctate pattern may be the result of protein 3a being a membrane‐associated molecule. Whether protein 3a can, like the S and M proteins, play a role in SARS‐CoV assembly in the ER–Golgi exocytosis pathway needs to be investigated further.
Figure 5.
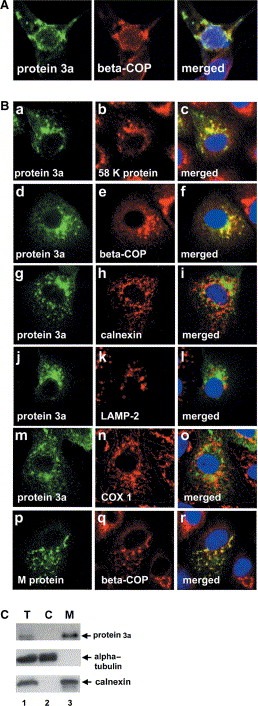
Subcellular localization of protein 3a in SARS‐CoV‐infected cells. (A) Vaccinia virus‐infected Vero E6 cells were transfected with the pcDNA3.1/myc‐His/protein 3a plasmid and grown on glass coverslips. After 48 h, cells were fixed with formaldehyde and incubated with anti‐P2 or anti‐β‐COP antibody (Golgi marker). (B) Commercial slides with SARS‐CoV‐infected cells were examined by indirect immunofluorescence staining and confocal microscopy. The left panels show protein 3a or membrane (M) protein fluorescence in green, the middle panels show marker fluorescence including 58 K protein (Golgi marker), β‐COP (Golgi marker), calnexin (ER marker), LAMP‐2 (lysosome marker), and cytochrome c oxidase subunit 1 (COX 1) (mitochondria marker) in red, and the right panels show overlays of fluorescence images of protein 3a or M protein and the subcellular marker proteins where yellow indicates co‐localization of green (protein 3a or M protein) and red (marker proteins) signals. Hoechst 33258 nuclear DNA staining is shown in blue. The confocal fluorescence was visualized by Bio‐Rad Radiance 2100 confocal microscope (60×). (C) Subcellular distribution of protein 3a. Membrane (M), and cytosolic (C) fractions of recombinant protein 3a expressed in Vero E6 were prepared as described in Section 2. Equivalent amounts (from total homogenate, T) of each fraction were analyzed by electrophoresis and immunoblotting using specific antibodies against the P2 peptide of protein 3a, α‐tubulin (cytosolic marker), or calnexin (ER membrane marker).
In conclusion, we have identified and characterized a novel SARS‐CoV protein, protein 3a, that lacks significant identity with any known proteins. Recently, based on molecular epidemiology and genome evolution studies of SARS‐CoV, Yeh et al. [23] reported that two of the 11 protein‐coding regions in the SARS‐CoV genome, spike and orf3 (protein 3a), showed a sign of positive selection during virus evolution. Although the function(s) of protein 3a is yet to be known, the spike protein is thought to be of particular importance in the infectious process, based on the studies of other CoVs [24, 25, 26]. The high‐frequency mutations or evolution adaptive property of protein 3a implied that its function may be, like spike protein, very important in the virus life cycle and/or disease development. Our findings provide an opportunity to explore protein 3a's function in the SARS‐CoV infection process. The SARS‐CoV protein 3a may provide insight into the mechanisms of infection of the virus as well as serve as a target for therapeutic developments for this disease, allowing for rapid and rational development and testing of candidate vaccines and therapeutics against this important human pathogen.
Acknowledgements
We thank Yi‐Hen Kou for generating ORF3a cDNA and Kuan‐Yu Chou for operating Confocal microscope. We also thank Drs. S.‐M. Hsu and Randy Haun for critical reading of the manuscript. This work was supported by grants partly from the National Science Council, R.O.C. (NSC 92‐3112‐B‐002‐029) to M.‐F. Chang, (NSC 92‐3112‐B‐002‐031) to L.‐M. Huang, (NSC 92‐2751‐B‐002‐014‐Y) to H.‐N. Ho, and by use of Yung‐Shin Biomedical Research Fund to F.‐J.S. Lee, and by an internal fund of the National Taiwan University Hospital (NTUH‐92A10‐2, ‐93A17).
Yu Chia-Jung,Chen Yee-Chun,Hsiao Cheng-Hsiang,Kuo Tse-Chun,Chang Shin C.,Lu Chun-Yi,Wei Wen-Chin,Lee Chia-Huei,Huang Li-Min,Chang Ming-Fu,Ho Hong-Nerng and Lee Fang-Jen S.(2004), Identification of a novel protein 3a from severe acute respiratory syndrome coronavirus, FEBS Letters, 565, doi: 10.1016/j.febslet.2004.03.086
References
- 1. Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., N. Engl. J. Med, 348, (2003), 1953– 1966. [DOI] [PubMed] [Google Scholar]
- 2. Kuiken T., Fouchier R.A., Schutten M., Rimmelzwaan G.F., van Amerongen G., van Riel D., Laman J.D., de Jong T., van Doornum G., Lim W., Lancet, 362, (2003), 263– 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holmes K.V., Enjuanes L., Science, 300, (2003), 1377– 1378. [DOI] [PubMed] [Google Scholar]
- 4. Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Science, 300, (2003), 1394– 1399. [DOI] [PubMed] [Google Scholar]
- 5. Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., N. Engl. J. Med, 348, (2003), 1967– 1976. [DOI] [PubMed] [Google Scholar]
- 6. Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E., J. Mol. Biol, 331, (2003), 991– 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J., J. Gen. Virol, 84, (2003), 2305– 2315. [DOI] [PubMed] [Google Scholar]
- 8. Yu X.J., Luo C., Lin J.C., Hao P., He Y.Y., Guo Z.M., Qin L., Su J., Liu B.S., Huang Y., Acta. Pharmacol. Sin, 24, (2003), 481– 488. [PubMed] [Google Scholar]
- 9. Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Science, 300, (2003), 1399– 1404. [DOI] [PubMed] [Google Scholar]
- 10. Hsueh, P.R., Hsiao, C.H., Yeh, S.H., Wang, W.K., Chen, P.J., Wang, J.T., Chang, S.C., Kao, C.L., Yang, P.C. and SARS Research Group of National Taiwan University College of National Taiwan University College of Medicine and National Taiwan University Hospital (2003) Emerg. Infect. Dis. 9, 1163–1167
- 11. Chomczynski P., Sacchi N., Anal. Biochem, 162, (1987), 156– 159. [DOI] [PubMed] [Google Scholar]
- 12. Wyatt L.S., Moss B., Rozenblatt S., Virology, 210, (1995), 202– 205. [DOI] [PubMed] [Google Scholar]
- 13. Fuerst T.R., Niles E.G., Studier F.W., Moss B., Proc. Natl. Acad. Sci. USA, 83, (1986), 8122– 8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lewis T.L., Matsui S.M., J. Virol, 70, (1996), 2869– 2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin C.Y., Huang P.H., Liao W.L., Cheng H.J., Huang C.F., Kuo J.C., Patton W.A., Massenburg D., Moss J., Lee F.J., J. Biol. Chem, 275, (2000), 37815– 37823. [DOI] [PubMed] [Google Scholar]
- 16. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J., Nucleic Acids Res, 25, (1997), 3389– 3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hopp T.P., Woods K.R., Proc. Natl. Acad. Sci. USA, 78, (1981), 3824– 3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grantham R., Science, 185, (1974), 862– 864. [DOI] [PubMed] [Google Scholar]
- 19. Cserzo M., Wallin E., Simon I., von Heijne G., Elofsson A., Prot. Eng, 10, (1997), 673– 676. [DOI] [PubMed] [Google Scholar]
- 20. Lai M.M., Holmes K.V., Knipe D.M. Howley P.M. Griffin D.E. Martin M.A. Lamb R.A. Roizman B. Straus S.E. Fields Virology 4 (2001), Lippincott Williams & Wilkins ; Philadelphia: 1163– 1185. [Google Scholar]
- 21. Nguyen V.P., Hogue B.G., J. Virol, 71, (1997), 9278– 9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krijnse-Locker J., Ericsson M., Rottier P.J., Griffiths G., J. Cell Biol, 124, (1994), 55– 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yeh S.H., Wang H.Y., Tsai C.Y., Kao C.L., Yang J.Y., Liu H.W., Su I.J., Tsai S.F., Chen D.S., Chen P.J., Proc. Natl. Acad. Sci. USA, 101, (2004), 2542– 2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Collins A.R., Knobler R.L., Powell H., Buchmeier M.J., Virology, 119, (1982), 358– 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Groot R.J., Van Leen R.W., Dalderup M.J., Vennema H., Horzinek M.C., Spaan W.J., Virology, 171, (1989), 493– 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jimenez G., Correa I., Melgosa M.P., Bullido M.J., Enjuanes L., J. Virol, 60, (1986), 131– 139. [DOI] [PMC free article] [PubMed] [Google Scholar]