

Graphical abstract
Novel potent inhibitors of 3C-like protease (3CLpro) of severe acute respiratory syndrome associated coronavirus (SARS-CoV) were identified through structure-based virtual screening and validated the inhibitory activity in vitro.
Keywords: 3CL Protease, Severe acute respiratory syndrome, SARS, Coronavirus, FRET-based assays, Autodock, Virtual screening, Grid
Abstract
The 3C-like protease (3CLpro) of severe acute respiratory syndrome associated coronavirus (SARS-CoV) is vital for SARS-CoV replication and is a promising drug target. Structure based virtual screening of 308 307 chemical compounds was performed using the computation tool Autodock 3.0.5 on a WISDOM Production Environment. The top 1468 ranked compounds with free binding energy ranging from −14.0 to −17.09 kcal mol−1 were selected to check the hydrogen bond interaction with amino acid residues in the active site of 3CLpro. Fifty-three compounds from 35 main groups were tested in an in vitro assay for inhibition of 3CLpro expressed by Escherichia coli. Seven of the 53 compounds were selected; their IC50 ranged from 38.57 ± 2.41 to 101.38 ± 3.27 μM. Two strong 3CLpro inhibitors were further identified as competitive inhibitors of 3CLpro with Ki values of 9.11 ± 1.6 and 9.93 ± 0.44 μM. Hydrophobic and hydrogen bond interactions of compound with amino acid residues in the active site of 3CLpro were also identified.
Severe acute respiratory syndrome (SARS) is a severe febrile respiratory illness caused by SARS-associated coronavirus (SARS-CoV).1, 2 A global outbreak of SARS between March 2003 and July 2003 caused over 8 000 probable or confirmed cases and 774 deaths.3 A mortality rate as high as 10% has been estimated by the World Health Organization.3, 4 The possibility of the re-emergence of SARS is a serious threat, since efficient therapy and a vaccine are not currently available.5 The SARS-CoV genome contains 14 functional open reading frames (ORFs).6 Two large 5′-terminal ORFs, designated 1a and 1b, encode two overlapping polyproteins, respectively designated pp1a and pp1b, which have to be cleaved extensively to produce proteins necessary for viral RNA synthesis and genome replication.6, 7 SARS-CoV 3C-like protease (3CLpro) plays a major role in the processing of the viral polyproteins and control of the activity of the replicase complex.8 The enzymatic activity of 3CLpro is essential for the viral life cycle and, therefore, represents an attractive target for the development of antiviral drugs directed against SARS-CoV and other coronavirus infections.9
Over the past decade, high-throughput virtual screening (VS) has emerged; especially structure based virtual screening (SBVS), as a reliable, cost-effective and time-saving technique for the discovery of lead compounds as an alternative to high-throughput screening.10 VS applied to the discovery of new enzyme inhibitors involves docking, computational fitting of structures of compounds to the active site of an enzyme, and scoring and ranking of each compound.11
In this study, we identified novel 3CLpro inhibitors among 308,307 compounds by SBVS with docking calculations on a WISDOM (Wide In Silico Docking On Malaria) production environment (WPE).12 Additionally, the inhibition activity and enzyme kinetics were characterized by in vitro enzyme assay. A 3-Dimensional coordinate in the X-ray crystal structure of 3CLpro (PDB accession code 2ZU5)13 obtained from the Protein Data Bank (PDB; http://www.pdb.org)14 was used as the receptor model in the SBVS with docking simulations. After removing the water molecules, the polar hydrogen (H) atoms were added to the macromolecule, histidine residues were made neutral and Kollman charges were assigned for all atoms.15 The docking library for 3CLpro was comprised of 308 307 compounds from the Chembridge database (http://www.chembridge.com). All compounds were filtered to remove unsuitable components that would not reach and pass clinical trials due to undesired and toxic properties. The three dimensional atomic coordinates of the compounds included in the docking library were generated by the Corina program (Molecular Networks GmbH, Erlangen, Germany). AutoDock version 3.0.5 was used for the computational molecular docking simulation of flexible small molecules to rigid proteins with ligand and rigid proteins.16 Large scale computations were conducted between 2ZU5 and 308 307 compounds using the KISTI grid infrastructure.17 In the first Postdocking filtering strategy based on the free binding energy of the lowest energy conformation, 1468 top ranked compounds having a free binding energy ranging from −14.0 to −17.09 kcal mol−1 were selected. To further narrow drug candidate selection, the Chimera software 1.4.1 program (University of California, San Francisco)18 was used to identify potential H-bonds between residues in the active site pocket of 3CLpro. Selected compounds were analyzed for their hydrophobic and H-bond interactions using the Ligplot program.19 Among 1468 compounds, 22 compounds (1.5% of the top scoring compounds) exhibited no potential hydrogen bond (H-bond) interactions and 381 compounds (25.95% of the top scoring compounds) showed weak H-bond interactions with amino acid residues in the active site pocket of 3CLpro. According to their free binding energy and H-bond interactions with key residues, 214 compounds were selected and classified into 35 groups by library MCS 0.7 (ChemAxon, San Francisco, CA, USA). Fifty-three compounds were selected from the 35 main clusters for in vitro inhibitory activity against 3CLpro. The compounds provided by the vendor were each >90% pure and were used without further treatment.
The gene encoding 3CLpro from SARS-CoV polyprotein (amino acid residues 3241–3546, GenBank accession no. AY274119) was constructed by a custom gene synthesis service (GenScript, Piscataway, NJ, USA). The 3CLpro enzyme was expressed in E. coli BL21 (DE3) and purified using a Ni-Sepharose resin (GE Healthcare, Buckinghamshire, UK). The K m value of 3CLpro calculated from the double reciprocal Lineweaver–Burk plot by fluorogenic peptide Dabcyl-KTSAVLQSGFRKME-Edans using a SpectraMax Gemini XPS apparatus (Molecular Devices, Eugene, OR, USA) with excitation and fluorescence emission wavelengths of 355 and 538 nm, respectively. Each compound from VS was tested in duplicate at a concentration of 100 μM for its ability to inhibit 3CLpro activity. Seven compounds showed optimal inhibition 3CLpro at 100 μM (Table 1 ). The chemical structure of each compound is depicted in Figure 1 , with physicochemical and chemical names of each compound is supplied in the Supplementary data. The seven compounds displayed 3CLpro inhibitory activities with IC50 values ranging between 38.57 ± 2.41 and 101.38 ± 3.27 μM (Table 1). These compounds can be considered as a new inhibitor scaffold for further development by structure activity relationship or de novo design studies. Compounds 6 and 7, which were similar in structure and which displayed more than 60% inhibition activity at 100 μM, were subjected to kinetic characterization. To elucidate the inhibitory mechanism of both compounds, inhibition kinetic experiments were performed at different constant inhibitor concentrations (0–60 μM) and different substrate concentrations. For analysis of the modes of inhibition of these compounds, both Lineweaver–Burk and Dixon plots were used. A K i value was calculated from the Dixon plots using the SigmaPlot program (SPSS, San Diego, CA, USA). Both compounds were competitive inhibitors. The inhibition of 3CLpro by compound 7 is illustrated in Figure 2 . The Lineweaver–Burk plot of 1/v versus 1/[S] resulted in a family of straight lines with the same y-axis intercept reflecting competitive inhibition toward 3CLpro (Fig. 2A). This activity suggests that compound 7 potently impairs the catalytic activity of 3CLpro by binding in the enzyme’s catalytic site. The K i value of compounds 6 and 7 were determined to be 9.11 ± 1.61 and 9.93 ± 0.44 μM, respectively, from the common x-axis intercept of lines on the corresponding Dixon plot (Fig. 2B). Compound 7 was analyzed by molecular docking as a potent binder to the active site pocket of 3CLpro (Fig. 3 A). The predicted free binding energy between compound 7 and 3CLpro is shown in Table 1. Figure 3B provides the details of the specific interactions between compound 7 and 3CLpro; carbon atoms of compound 7 formed hydrophobic interactions with His41, Leu141, Phe140, Cys145, Glu166, His163, Gly170, and His172 of 3CLpro. The O2 atom of methylbenzamide group of compound 7 accepted H-bonding with the side chain carboxamide of Asn142 with a distance of 3.15 Å (Fig. 3B). The N2 atom of the methacrylamide group of compound 7 donated 2.66 and 2.73 Å H-bond to the carboxyl group of Phe140 and the side chain carbonyl group of Glu166. The O4 atom of the nitrophenyl group of compound 7 formed 2.65 Å H-bonds with the backbone N atom of Gly143. The O1 atom of the nitrophenyl group of compound 7 has two H-bonds: one H-bond with side chain of Cys145 with distance 3.23 Å and another one with the N atom of the main chain of Cys145 with a distance 3.23 Å. Viewing the H-bond between compound 7 and the amino acid residues in the active site of 3CLpro revealed that compound 7 bound to the S1 site (the substrate binding pocket) of SARS-CoV through H-bonds with Phe140, Gly143, Cy145, and Glu166. S1 of SARS-CoV 3CLpro confers absolute specificity for the Gln-P1 substrate residue.20 The nitrophenyl group of compound 7 is very likely crucial in the 3CLpro inhibition activity, given its H-bond formation with Cys145 and Gly143, as well as hydrophobic interactions with His41, Cys145 and Cys145. Hence, one of the catalytic dyad residues of 3CLpro is vital for the binding of compound 7 to 3CLpro.
Table 1.
Inhibitory activities of the identified compounds against recombinant 3CLpro
| Compounds | Chembridge ID | Free binding energy (kcal mol−1) | Inhibitiona (%) | IC50 (μM) |
|---|---|---|---|---|
| 1 | 7007610 | −14.5 | 58.23 | 58.35 ± 1.41 |
| 2 | 6939388 | −15.09 | 61.36 | 62.79 ± 3.19 |
| 3 | 7742065 | −15.17 | 49.14 | 101.38 ± 3.27 |
| 4 | 5623347 | −15.20 | 56.11 | 77.09 ± 1.94 |
| 5 | 7935169 | −15.75 | 53.27 | 90.72 ± 5.54 |
| 6 | 5874274 | −15.02 | 82.59 | 38.57 ± 2.41 |
| 7 | 7112399 | −15.13 | 81.43 | 41.39 ± 1.17 |
Inhibition by 100 μM.
Figure 1.
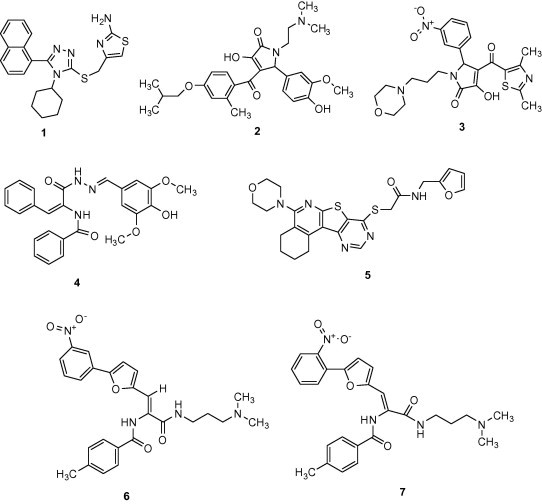
Chemical structure of the novel 3CLpro inhibitors.
Figure 2.
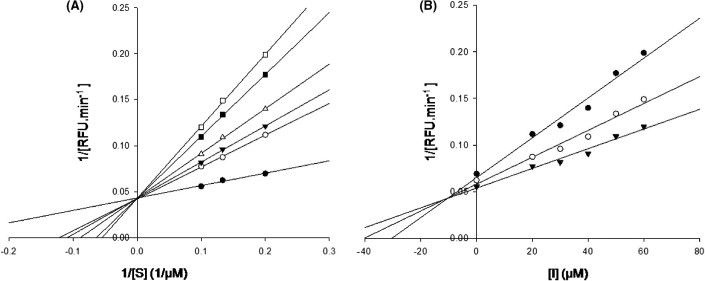
Lineweaver–Burk plot (A) and Dixon plot (B) analyses for the inhibition of 3CLpro by compound 7. The kinetic constants, Km and Ki, were calculated using linear regression analysis. (A) Compound 7 concentration of 0 μM (●), 20 μM (○), 30 μM (▾), 40 μM (Δ), 50 μM (■), and 60 μM (□). (B) FRET substrate concentration of 5 μM (●), 7.5 μM (○), and 10 μM (▾).
Figure 3.
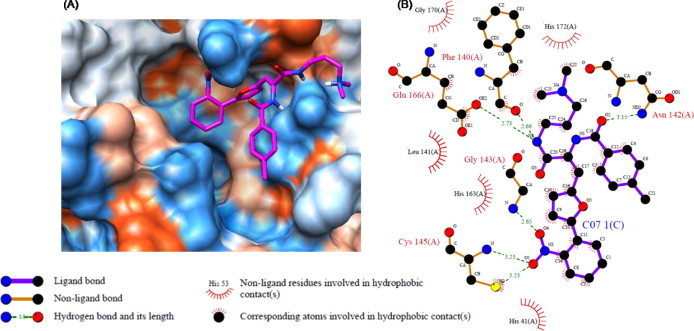
Computational docking, hydrophobic and H-bond interactions of compound 7 with residues in the active site pocket of 3CLpro. (A) Comparison of binding modes of compound 7 (magenta) in the active site pocket of 3CLpro. (B) Hydrophobic and H-bond interactions of compound 7 with amino acid residues in the active site of 3CLpro. H-Bond interactions are represented by green dashed lines.
In summary, VS of 308,307 compounds was done to identify novel 3CLpro inhibitors. Fifty-three compounds were tested for their inhibitory activity towards 3CLpro expressed from E. coli. The IC50 of seven especially potent compounds ranged from 38.57 ± 2.41 to 101.38 ± 3.27 μM, and two of these compounds (6 and 7) were competitive inhibitors of 3CLpro with K i values of 9.11 ± 1.61 and 9.93 ± 0.44 μM. Detailed docking simulation binding mode analyses showed that the inhibitors could be stabilized by the formation of H-bonds with catalytic residues and the establishment of hydrophobic contacts at the opposite regions of the active site. More detailed inhibition kinetics and molecular modeling studies are underway to elucidate the inhibitory mechanism of compounds 6 and 7.
Acknowledgement
This work was partially supported by the Regional Technology Innovation Program of the Ministry of Knowledge Economy (MKE), South Korea. Authors acknowledge support from FKPPL Internal Associated Laboratory for grid computing resources.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2011.03.034.
Supplementary data
References and notes
- 1.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Eng. J. Med. 2003;348:1967. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S.X., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J., Grp S.W. N. Eng. J. Med. 2003;348:1953. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization (2004) Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003.
- 4.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemistry. 2004;43:4906. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 5.Xu T., Ooi A., Lee H.C., Wilmouth R., Liu D.X., Lescar J. Acta Crystallogr., Sect. F. 2005;61:964. doi: 10.1107/S1744309105033257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan Y.J., Lim S.G., Hong W. Antiviral Res. 2005;65(2):69. doi: 10.1016/j.antiviral.2004.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee P., Desai P., Ross L., White E.L., Avery M.A. Bioorg. Med. Chem. 2008;16:4138. doi: 10.1016/j.bmc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Virus Res. 2008;133:63. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou T.J., Xu X.J. Curr. Pharm. Des. 2004;10:1011. doi: 10.2174/1381612043452721. [DOI] [PubMed] [Google Scholar]
- 11.Guido R.V.C., Oliva G., Andricopulo A.D. Curr. Med. Chem. 2008;15:37. doi: 10.2174/092986708783330683. [DOI] [PubMed] [Google Scholar]
- 12.Kasam V., Zimmermann M., Maass A., Schwichtenberg H., Wolf A., Jacq N., Breton V., Hofmann-Apitius M. J. Chem. Inf. Model. 2007;47:1818. doi: 10.1021/ci600451t. [DOI] [PubMed] [Google Scholar]
- 13.Lee C.C., Kuo C.J., Ko T.P., Hsu M.F., Tsui Y.C., Chang S.C., Yang S., Chen S.J., Chen H.C., Hsu M.C., Shih S.R., Liang P.H., Wang A.H. J. Biol. Chem. 2009;284:7646. doi: 10.1074/jbc.M807947200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanner M.F., Olson A.J., Spehner J.C. Biopolymers. 1996;38:305. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 16.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. J. Comput. Chem. 1998;19:1639. [Google Scholar]
- 17.Jacq N., Salzemann J., Jacq F., Legre Y., Medernach E., Montagnat J., Maass A., Reichstadt M., Schwichtenberg H., Sridhar M., Kasam V., Zimmermann M., Hofmann M., Breton V. J. Grid Computing. 2008;6:29. [Google Scholar]
- 18.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. J. Comput. Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 19.Wallace A.C., Laskowski R.A., Thornton J.M. Protein Eng. 1995;8:127. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 20.Yang H.T., Yang M.J., Ding Y., Liu Y.W., Lou Z.Y., Zhou Z., Sun L., Mo L.J., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z.H.P. Natl. Acad. Sci. U.S.A. 2003;100:13190. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.