

Abstract
The innate immune system is comprised of a sophisticated network of recognition and effector molecules that act together to protect the host in the first minutes or hours of exposure to an infectious challenge. The mannose-binding lectin (MBL) is an evolutionary conserved circulating host defense protein that acts as a broad-spectrum recognition molecule against a wide variety of infectious agents. Target binding triggers the MBL pathway of complement activation. MBL can be considered conceptually as an ‘ante-antibody’ because it has a role in mammals during the lag period that is required to develop an antibody response against infectious agents. Additionally, there are MBL-like homologues in animals that lack adaptive immunity that activate a primitive complement system, and under these circumstances these MBL-like molecules play an analogous role to antibodies in higher animals. These molecules might be considered to be functional antecedents of antibodies. Recent work also indicates that MBL recognizes altered self-antigens, and as such MBL has a role that extends beyond a traditional role in first line host defense as it appears to play a role as a modulator of inflammation.
Introduction
The innate immune system is considered to be the first barrier to infection. The skin, the mucociliary escalator in the lung, antimicrobial peptides, bacterial permeability increasing protein, natural antibodies, the complement system, lipopolysaccharide-binding protein, certain classes of lymphocytes, NK cells, and tissue macrophages conspire to limit an infectious challenge. This repertoire of molecules, together with other molecules and cells and their many biologically active products, undoubtedly act in concert and represent a network of first-line host defense.
Intrinsic to innate immunity is the ability to recognize infectious agents from self. One basic tenant of innate immunity is the evolution of classes of molecules that are termed pattern recognition receptors and molecules. These molecules recognize the patterns that adorn the so-called pathogen associated molecular patterns (PAMPs) of infectious agents [1]. PAMPs represent invariant exposed structures such as lipopolysaccharide, peptidoglycans, mannans, double-stranded RNA and methylated bacterial DNA. Specified pattern recognition receptors either directly recognize and or sense infectious agents. These include Toll-like receptors, peptidoglycan recognition proteins, scavenger receptors and lectin-like molecules such as the mannose receptor and the mannose-binding lectin (MBL). Recent work indicates that the role of MBL extends beyond pathogen recognition to include recognition of cryptic self antigens that are exposed by reperfusion injury or are modified by disease states such as diabetes [2, 3]. In this context it is noteworthy that the expression of Mbl2 gene is regulated, so that certain MBL haplotypes are associated with a dramatic decrease in circulating MBL levels. The prevalence of these low secretor MBL haplotypes vary between ethic populations, ranging from 5% in Caucasians to 30% in certain African populations [4]. Low MBL levels are associated with susceptibility to infection in the context of a compromised host, which is in keeping with the role of MBL as an ante-antibody. By contrast, high MBL levels might exacerbate the renal complications of diabetes and the inflammatory consequences of reperfusion injury. Therefore, MBL might represent a paradigm of the multifunctional roles of molecules of the innate immune system that extend beyond their traditional roles in host defense.
In this review, we attempt to provide an overview of the role of MBL in health and disease and illustrate that this molecule represents many nuances that are emerging as a paradigm for molecules involved in innate immunity.
Molecular characterization of MBL
An important preface to this section is that the MBL acts in a context of many circulating pattern recognition molecules that contribute to limiting the initial spread of an infectious disease. Some of these molecules are known, but it is likely that many are yet to be discovered. The liver is the major source of MBL synthesis, although other sites such as the intestine have been proposed [5, 6, 7]. MBL exists in serum as a multimer of homotrimers. The regular collagen repeats of Gly–Xaa–Yaa region have an interruption of a single Gly–Xaa–Gly that forms a kink in this domain that follows the amino-terminal cysteine-rich domain of MBL.
In the human MBL protein, a neck region arranges the carbohydrate recognition domains (CRDs) so that they are 45 Å apart and is followed by the collagen helix (Figure 1a) [1]. MBL belongs to the collectin family. This includes four other proteins that have calcium-dependent CRDs and collagen stalks [8]. MBL recognizes specific carbohydrates such as d-mannose, l-fucose and N-acetylglucosamine that are represented on the surface of a wide variety of infectious agents (Figure 1b) [1]. Pathogens targeted by MBL include certain Gram-positive and Gram-negative bacteria, yeast, parasites and viruses [9]. Recently it has become apparent that MBL (like two other collectins — surfactant proteins A [SP-A] and D [SP-D]) is also able to recognize apoptotic cells, free DNA and a variety of altered-self antigens, among other targets (Table 1 ). The ability of MBL to recognize either altered-self or self molecules that are not normally accessible relates directly to the role of this protein that extends beyond the first-line host defense and suggests a broader function as a general modulator of inflammation.
Figure 1.
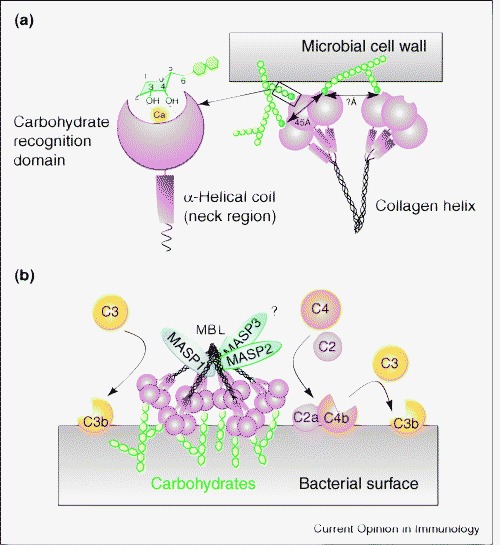
(a) A schematic diagram of proposed binding of MBL to hexose sugars that have hydroxyl groups in the equatorial position and are thereby accommodated in the binding pocket that is stabilized by calcium ions. Also depicted is a model of MBL binding to complex ligands that require interactions of multiple binding pockets with ligand, which results in high affinity binding. (b) A model of the interactions of MBL with MASPs. Engagement of ligand by MBL activates MASP2, which then cleaves the C2C4 convertase and results in the cleavage of C3 and the generation of C3b. It has also been proposed that MASP1 can directly cleave C3; what is not known is whether this requires an MBL interaction with ligand. Our model proposes that this might be a prerequisite for this reaction to occur and that this in turn would activate the alternative complement pathway. Adapted from [1].
Table 1.
The endogenous ligands of MBL.
| Endogenous ligands of MBL | References |
|---|---|
| Dying cells | |
| Apoptosis | [53•, 55] |
| Necrosis | [56] |
| Ischemic tissues | |
| Myocardial reperfusion injury | [51•] |
| Renal reperfusion injury | [50, 57] |
| Gastrointestinal reperfusion injury | [49] |
| Anoxic endothelia cells | |
| Endothelial oxidative stress | [2, 58] |
| Transformed cells | |
| Colon adenocarcinoma | [59, 60] |
| Colorectal carcinoma | |
| Immunogloublins | |
| Agalactosyl IgG | [61, 62, 63] |
| Dimeric/polymeric IgA | [21•] |
| IgM | |
| Nucleic acids | |
| DNA | [64, 65] |
| RNA | |
| Phospholipids | |
| Phosphatidylserine | [66, 67] |
| Phosphatidylinositol | |
| Phosphatidylcholine | |
| Zinc metalloproteases | |
| Meprin α and β | [68] |
MBL and the complement cascades
There are three pathways in the complement cascade: the classical, alternative and lectin pathways [10•]. Initial antigen recognition of the classical pathway depends on antibodies. In the alternative pathway recognition occurs directly by cleaved third complement component C3b, and in the lectin pathway recognition is by way of either MBL or ficolins. For the classical pathway, recognition depends upon IgM or certain isotypes of antibody [11] that interact with the first component of complement C1q, which is structurally similar to MBL. Binding of C1q results in a conformation change that activates two homologous serine proteases — C1r and C1s [12]. These proteases cleave complement components C4 and C2 to form C4bC2b convertase, which in turn cleaves the third complement component C3. Cleaved C3 (C3b) either opsonizes the target for uptake by phagocytic cells or leads to the assembly of the membrane attack complex that permeabilizes the target membrane. The effector ability of MBL in host defense is facilitated by activation of the lectin pathway through a specific interaction between MBL and MBL-associated serine protease (MASP) (Figure 1).
Recent work indicates that the minimum functional unit required to activate MBL:MASP2 complement activation is a MASP2 dimer bound to two MBL trimeric units [13]. However, there are three known MASPs that have been termed MASP-1, -2 and -3 [14, 15]. MASP1 and MASP2 are encoded by distinct genes whereas MASP3 represents an alternative splice form of the MASP1 gene that lacks a serine protease domain. MASP2 is the functionally relevant enzyme in initiation of the lectin complement pathway. MASP2 is made up of six protein domains that include two CUB domains, a calcium-binding EGF-like domain, two complement consensus repeat domains and a terminal serine protease domain [15]. Recent structural work confirms that the amino-terminal domain of CUB-1:EGF:CUB-2 mediates homodimerization of the molecule as well binding to MBL [15]. The MASP2 dimer appears to nestle in a distinct region of the collagen domain on the carboxy-terminal side of the Gly–X–Gly kink in the collagen region and ends proximal to the neck region of the molecule. Ligand binding induces a conformational change in the MASP2 that activates the terminal serine protease domain. It appears that MASP1 and MASP3 bind to distinct but overlapping areas of MBL schematically represented in Figure 1. Their respective roles in the lectin complement pathway require further clarification.
A key question is regarding the relative roles of the classical, alternative and lectin pathways. L- and H-ficolins, like MBL, are circulating proteins that participate in the lectin pathway by binding MASPs [16]. L-ficolin contains a collagen-like domain and a fibrinogen-like domain that appears to bind N-acetylglucosamine and other acetylated compounds [17]. Compared with MBL, L-ficolin might have a more selective recognition repertoire. From an evolutionary perspective it appears that the lectin pathway is the most ancient pathway of complement activation. A glucose-binding lectin isolated from the solitary ascidian Halocynthia roretzi is structurally homologous to MBL [18]. This animal also has a MASPa, a MASP1 homologue and an ancestral form of C3 that is cleaved by MASPa [19]. In mammals, MASP1 can cleave C3 directly [14]. Therefore, MASPa appears to be the primordial form of human MASPs and this system seems to represent the ancestral complement pathway that preceded adaptive immunity.
In mammals it has been suggested that there is cross-talk between the three complement pathways that goes beyond the obvious convergence on cleavage of C3. It has been demonstrated under certain circumstances, such as ischemia-reperfusion injury complement-mediated damage is initiated by monoclonal IgM [20]. One might speculate that target-bound IgM undergoes conformational change, binds MBL and subsequently activates MASP2 to generate C3 convertase without requirement of the C1 complex. Serving as a cautionary note to this attractive idea is a recent in vitro study that indicated that only 20% of IgM binds to immobilized MBL [21•]. However, once IgM binds antigen, IgM is unable to bind MBL because the target glycans on IgM become inaccessible owing to a conformational change in its structure [21•]. These authors speculate that MBL might bind and clear IgM complexes [21•].
Another issue that is not fully understood is the relationship between the alternative and the lectin pathways. The alternative pathway relies on the cleaved C3b to activate the alternative pathway convertase that catalyses the reaction. It seems that the initially cleaved C3 product might be generated by MASP1 that circulates in a complex with MBL and/or ficolins. Accordingly, the initial recognition event for the alternative pathway is hypothetically mediated by the lectin binding. Therefore, these ideas raise the question of whether the alternative pathway acts as an amplification pathway for the lectin and classical pathways or whether it exists as a truly independent physiological cascade.
MBL as a disease susceptibility gene
Charles Janeway [22] proposed that molecules that participate in the innate immune response were invariant because variations in their gene expression would be detrimental to the host and consequently they would be lost by natural selection. It now appears that single nucleotide polymorphisms (SNPs) in a variety of genes in which the gene products play a key role in first-line host defense can determine an individual's relative resistance or susceptibility to an infectious challenge. Although classical Mendelian inheritable forms of immunodeficiency are important [23], it is clear that there are many genes that participate in first-line host defense and that their inheritance patterns do not follow the rules of Mendelian genetics. It appears reasonable that the innate immune system of each individual is specified by their innate immune genotype, which is likely to reflect a diversification in several genes that play a key role in host defense [24•]. Variations in the MBL gene, Mbl2, are amongst the most intensely studied and as such represent a paradigm that will apply to other genes in which the products play a key role in first-line host defense.
There is a rapidly expanding body of literature that focuses on the impact of variations in MBL haplotypes or serum concentrations on human diseases. At least ten distinct MBL haplotypes have been described in humans, four of which (LYPB, LYQC, HYPD and LXPA) dictate low serum MBL concentrations [4]. Human populations from diverse geographic locations and ethnic genetic backgrounds have a high rate of haplotype variation, with a range of heterozygosity from 15% in white populations to 30% in certain African populations [25]. Importantly, there is no consensus about the range of MBL concentrations that constitutes normal serum values. In this context, contradictory findings in the literature reflect a combination of intrinsic genetic differences, lack of methodological standardization and discrepant interpretation of data. Nevertheless, the available evidence validates correlations between certain MBL genotypes and/or MBL serum concentrations on the one hand and infectious, autoimmune, inflammatory, vascular and metabolic diseases, as well as ischemia-reperfusion injury, on the other hand [25, 26].
To establish standardized and reproducible interpretative criteria, we propose that it would be helpful to always correlate low levels of MBL with a functional measurement of the MBL:MASP pathway. This assumption would correlate with structural variants of MBL that are unable to bind MASP2 because of disruption of the MASP2-binding site in the MBL collagen stalk, consequently reducing lectin pathway activity [27].
Low levels of MBL are associated with increased susceptibility to infection
One area of intensive research during the past few decades has been the impact of MBL on human infections. A key question is whether low MBL levels per se contribute to susceptibility to infection. There are examples in apparently immunocompetent individuals whereby low MBL levels contribute to severity of illness in infections of pathogenic bacteria such as Neisseria meningitidis, Staphylococcus aureus and Streptococcus pneumoniae [28, 29, 30]. However, a more generalizable paradigm is that low levels of MBL modify susceptibility to infection in an already immunocompromised host. Examples of underlying diseases in which low MBL concentrations contribute to disease severity include cystic fibrosis [31] and a broad category of immunosuppression that is often secondary to cancer chemotherapy. In this way, an individual's MBL haplotype might represent that person's premorbid innate immune genotype, which contributes to their susceptibility or resistance to infection in the face of neutropenia.
These observations have led to the idea that recombinant human MBL (rhMBL) therapy might be used effectively as prophylaxis against infection in the context of cancer chemotherapy in those individuals that have low-producing MBL haplotypes. The concept of patient pre-selection for therapeutic intervention based on their molecular phenotype has set a precedent in the use of specific anticancer agents that target mutations found in a subset of patients with diseases such as chronic myeloid leukemia and non-small-cell lung cancers [32]. In addition, infants have a period of vulnerability to infection between the ages of 6 and 18 months of age; this is associated with delayed maturation of MBL synthesis and waning maternally-derived immunoglobulin concentrations when the adaptive immune system is still immature [33, 34, 35].
Healthy individuals that have low-producing MBL variants also might have increased susceptibility to certain viruses. However, it is possible that implicated viruses transiently suppress adaptive immunity and thereby exacerbate disease expression in individuals that have MBL deficiency. There is ample evidence in the literature of interactions between MBL and certain viral pathogens such as human immunodeficiency virus, influenza viruses, severe acute respiratory syndrome coronavirus (SARS-CoV) and hepatitis B virus, among others. MBL binds specifically to the human immunodeficiency virus gp120 envelope proteins, efficiently opsonizes the virus and prevents infection of T lymphocytes [36]. MBL mediates hemagglutination and neutralization of influenza virus [37] and complement-associated lysis of infected cells [38, 39]. More recently, epidemiologic correlation studies have revealed associations between MBL deficiency and certain viral infections. In infections caused by SARS-CoV, MBL appears to play a primary role in host defense. Certain mutant MBL alleles are susceptibility factors for acquisition of coronavirus infection [40, 41]. Furthermore, severity, chronicity and bacterial complications of hepatitis B virus infection and its sequelae such as cirrhosis and hepatocellular carcinoma are associated with MBL genotypes that dictate low serum concentrations, whereas high MBL levels are associated with containment of the infection [42, 43].
Disease models of MBL deficiency
Bacterial infections
The human association studies provide useful insights into the role of MBL in health and disease, but to evaluate a causal role of MBL in host susceptibility we set out to create a mouse model of MBL deficiency. Humans and New World monkeys have a single MBL gene, whereas rodents have two genes — mbl1 located on chromosome 14 and mbl2 on chromosome 19 that encode for MBL-A and MBL-C, respectively [25•]. These two forms of rodent MBL bear 50% homology to one another, have distinct and overlapping ligand-binding specificity and are both able to activate the MBL:MASP pathway [7]. We created MBL-null mice that lack both forms of MBL and that were devoid of MBL:MASP pathway activity [44••]. We then challenged these mice with various infectious agents under certain defined conditions.
First, we subjected MBL-null and wild-type mice to an intravenous inoculation of S. aureus [44••]. We selected this organism because it causes significant worldwide morbidity and mortality, and it is associated with development of high-level resistance to currently available antibiotics. In addition, it appears that MBL plays a key role in mobilizing the complement cascades in response to S. aureus infection. This raises the idea that the presence of low-producing mbl2 gene haplotypes might be a genetic factor that predisposes certain humans to poor outcomes as a result of S. aureus infection. We found that MBL-null mice are indeed highly susceptible to S. aureus systemic infection, because all MBL-null mice died by two days after infection compared with a survival rate of 55% in wild-type mice. Importantly, pre-treatment of MBL-null mice with rhMBL restored the MBL:MASP pathway in vivo and reversed the phenotype. Compared with wild-type mice, there was a 10–100-fold increase in bacterial titers in the blood, liver, spleen, kidneys and lungs of MBL-null mice. The bacterial challenge evoked a muted cytokine response in the MBL-null mice and the organism was able to proliferate in these animals; this was a marked contrast to wild-type or MBL-null mice that were reconstituted with rhMBL.
In summary, this study indicated that MBL acts in the same way as an opsonin, but also is important in stimulating an appropriate pro-inflammatory response. Recent unpublished work from our laboratory indicates that MBL probably regulates TNF-α secretion by co-opting known pathways within the cell (WKE Ip, unpublished). This work also provides credence to the idea that MBL is an important component of first-line host defense against certain pathogens. However, under most circumstances a lack of MBL does not predispose the immunocompetent host to bacterial infection. It is in the setting of relative immune insufficiency that low-producing MBL haplotypes are associated with enhanced susceptibility to infection, as is observed in the setting of cancer chemotherapy.
We found that intraperitoneal inoculation of a dose of S. aureus that was lower than that used for intravenous experiments did not cause infectious complications in MBL-null mice unless they were rendered neutropenic [44••]. This was in contrast to neutropenic wild-type mice that were able to contain and eliminate the infection. These results confirm that, under certain circumstances, MBL plays a non-redundant role in first-line host defense against certain microorganisms.
Burn models and infection
One problem with these types of mouse models of infection is that the initial inoculum of organisms is unrealistically high. Therefore, we sought to establish a more physiological model for the role of MBL in limiting infections. We elected a burn injury model. Burn injury disrupts the mechanical and biological barrier that the skin presents against bacterial pathogens. A frequent cause of morbidity and mortality in burn victims is infection with the Gram-negative bacterial symbiont Pseudomonas aeruginosa, among others. The skin utilizes a combination of antimicrobial peptides and resident effector cells such as Langerhans cells, NKT cells, γδ T cells and tissue macrophages to combat the infectious challenge following burn injuries [45]. Disruption of skin integrity induces rather chaotic inflammatory responses that include influx of phagocytes and serum factors such as complement and MBL.
We examined whether MBL deficiency increases the risk of P. aeruginosa infection in a burned host. We found that both wild-type and MBL-null mice were resistant to a 5% total body surface area dry-burn. However, when mice were burned and then inoculated with P. aeruginosa (1 x 104 cfu) at the burn site, all of the MBL-null mice died by 42 h as a result of septicemia, whereas only one-third of wild-type mice succumbed (p = 0.0005) [46•]. Importantly, in the skin at the inoculation site, there was no difference in growth rates of the bacteria between the MBL-null mice and the wild-type mice. However, we found that the bacterial titer in the blood of MBL-null mice was significantly higher than that of wild-type mice. Elevated bacterial titers were also observed in kidneys and livers of MBL-null mice. These results indicate that MBL does not play a role in local host defense in the skin but rather plays a key role in containing and preventing the systemic spread of P. aeruginosa infection. These observations suggest that low MBL haplotypes might contribute to the infectious complications in burn patients, a subject hitherto that has not been addressed.
Viral infections
In addition to bacterial infection models, a viral infection model has been also explored. MBL-null mice demonstrated increased susceptibility to systemic infection with herpes simplex virus-2 (HSV-2) [47]. These results provide in vivo evidence that MBL plays an important role in first-line host defense against infection with S. aureus, P. aeruginosa and HSV-2 and support the clinical associations between MBL deficiency and increased susceptibility to certain infections.
Ischemia-reperfusion injury
Animal models of ischemia-reperfusion injury have clearly indicated a role for complement components as key mediators of reperfusion damage [20]. The relative role of the three complement pathways has been a source of much investigation. Recent work has indicated that inhibition of the MBL:MASP pathway in a rat myocardial reperfusion model caused protection against subsequent reperfusion injury [48]. Three sets of studies in MBL-null mice examined the role of the MBL:MASP pathway in reperfusion injuries of the gut, kidney and heart [49, 50, 51•]. All three studies revealed that lack of the MBL:MASP pathway was protective, which indicates that this pathway did have a role in mediating reperfusion injury. One note of caution is that it appears that the effects depend on the length of time of ischemic injury. It is clear that factors other than the MBL:MASP pathway contribute to the reperfusion injury. Important next steps in this crucial area of investigation are to define the precise antigens that are recognized by MBL and to establish the relative role of MBL as initiators of the reperfusion injury.
Autoimmunity
MBL also recognizes altered-self in the guise of apoptotic cells and therefore joins the collectins, SP-A and SP-D, and C1q in this function. C1q deficiency sets up a predisposition in man and mice for systemic lupus erythematosus [52]. It has been suggested that the failure of C1q apoptotic cell clearance leads to the presence of apoptotic cells that are available as self-antigens that then trigger autoimmunity. Like C1q-null mice, MBL-null mice also demonstrated a defect in apoptotic cell clearance but they did not develop spontaneous autoimmune diseases, lymphoproliferation or germinal center expansion, although the mice did have expanded numbers of peritoneal B1 cells [53•]. These results indicate that failure of apoptotic cell clearance alone can be dissociated from the development of autoimmunity in this model and on a mixed genetic background. Clearly, propensity to autoimmunity is a multi-step process that involves several genes. Our animal experiments together with the human association studies suggest that MBL is, at best, a weak modifier of the autoimmune phenotype.
Conclusions
A significant body of work now indicates that MBL fulfils the criteria of a molecule that plays an important role in the host's initial response to infection. MBL is able to co-opt a unique pathway of complement activation as well as to mediate MBL-dependent phagocytosis by circulating and sessile leukocytes. What is not clear is the relative role of complement receptors versus MBL or collectin receptors in mediating MBL-dependent clearance of bound particles. In this regard, it appears that MBL-mediated clearance of infectious agents provokes a pro-inflammatory response, whereas MBL-dependant clearance of apoptotic cells promotes an anti-inflammatory response. The precise signals that mediate these different outcomes have not been defined but clearly are of great interest.
Another mystery is why the MBL locus displays such variation. One can speculate that dysfunctional MBL disables the MASP pathway of complement activation, thereby predisposing the host to infection under immunocompromising conditions or in the setting of certain viral infections; by contrast, this same phenotype reduces the complications of ischemia-injuries. Noteworthy in this regard is a recent report that examined mutations in the human-expressed MBL pseudogene. The authors speculate that the same mechanisms that are at play in silencing the pseudogene over time also impact the selection for low-producing MBL haplotypes [54]. However, the evolutionary pressure for this proposed drift to low-producing haplotypes is not clear. Therefore, in many respects MBL represents a paradigm for a molecule that has a broad role in innate immunity and inflammation.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
The authors wish to thank members of Laboratory of Developmental Immunology for their helpful comments. Furthermore, we were unable to cover the entire field in this review and so we acknowledge the work that we had to omit. This work was supported by grants from National Institute of Health to RABE. Conflict of interest: RABE is on the Board of and has a financial interest in Natimmune, a privately held biotechnology company that plans to explore the therapeutic potential of MBL.
References
- 1.Hoffmann J.A., Kafatos F.C., Janeway C.A., Ezekowitz R.A. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- 2.Collard C.D., Vakeva A., Morrissey M.A., Agah A., Rollins S.A., Reenstra W.R., Buras J.A., Meri S., Stahl G.L. Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol. 2000;156:1549–1556. doi: 10.1016/S0002-9440(10)65026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen T.K. Mannose-binding lectin (MBL) and vascular complications in diabetes. Horm Metab Res. 2005;37:95–102. doi: 10.1055/s-2005-861372. [DOI] [PubMed] [Google Scholar]
- 4.Madsen H.O., Satz M.L., Hogh B., Svejgaard A., Garred P. Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J Immunol. 1998;161:3169–3175. [PubMed] [Google Scholar]
- 5.Uemura K., Saka M., Nakagawa T., Kawasaki N., Thiel S., Jensenius J.C., Kawasaki T. L-MBP is expressed in epithelial cells of mouse small intestine. J Immunol. 2002;169:6945–6950. doi: 10.4049/jimmunol.169.12.6945. [DOI] [PubMed] [Google Scholar]
- 6.Sheriff S., Chang C.Y., Ezekowitz R.A. Human mannose-binding protein carbohydrate recognition domain trimerizes through a triple alpha-helical coiled-coil. Nat Struct Biol. 1994;1:789–794. doi: 10.1038/nsb1194-789. [DOI] [PubMed] [Google Scholar]
- 7.Liu H., Jensen L., Hansen S., Petersen S.V., Takahashi K., Ezekowitz A.B., Hansen F.D., Jensenius J.C., Thiel S. Characterization and quantification of mouse mannan-binding lectins (MBL-A and MBL-C) and study of acute phase responses. Scand J Immunol. 2001;53:489–497. doi: 10.1046/j.1365-3083.2001.00908.x. [DOI] [PubMed] [Google Scholar]
- 8.Sastry K.N., Ezekowitz R.A.B. Collectins and Innate Immunity. In: Sastry K.N., Ezekowitz R.A.B., Reid K.B.M., editors. Collectins and Innate Immunity. RG Lnasders Company; 1996. pp. 1–7. [Google Scholar]
- 9.Fraser I.P., Koziel H., Ezekowitz R.A. The serum mannose-binding protein and the macrophage mannose receptor are pattern recognition molecules that link innate and adaptive immunity. Semin Immunol. 1998;10:363–372. doi: 10.1006/smim.1998.0141. [DOI] [PubMed] [Google Scholar]
- 10•.Fujita T., Matsushita M., Endo Y. The lectin-complement pathway – its role in innate immunity and evolution. Immunol Rev. 2004;198:185–202. doi: 10.1111/j.0105-2896.2004.0123.x. [DOI] [PubMed] [Google Scholar]; A recent update on the lectin-complement pathway and its molecular evolution. In this study, a lectin-based complement system, similar to that in the lectin pathway triggered by MBL and ficolins, is shown to be present in ascidians — our closest invertebrate relatives. This suggests that the complement system played a pivotal role in innate immunity before the evolution of an acquired immune system.
- 11.Miletic V.D., Frank M.M. Complement–immunoglobulin interactions. Curr Opin Immunol. 1995;7:41–47. doi: 10.1016/0952-7915(95)80027-1. [DOI] [PubMed] [Google Scholar]
- 12.Arlaud G.J., Gabortaud C., Thielens N.M., Budayova-Spano M., Rossi V., Fontecilla-Camps J.C. Structural biology of the C1 complex of complement unveils the mechanism of activations of its activation and proteolytic activity. Mol Immunol. 2002;39:383–394. doi: 10.1016/s0161-5890(02)00143-8. [DOI] [PubMed] [Google Scholar]
- 13.Chen C.B., Wallis R. Two mechanisms for mannose-binding protein modulation of the activity of its associated serine proteases. J Biol Chem. 2004;279:26058–26065. doi: 10.1074/jbc.M401318200. [DOI] [PubMed] [Google Scholar]
- 14.Dahl M.R., Thiel S., Matsushita M., Fujita T., Willis A.C., Christensen T., Vorup-Jensen T., Jensenius J.C. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001;15:127–135. doi: 10.1016/s1074-7613(01)00161-3. [DOI] [PubMed] [Google Scholar]
- 15.Feinberg H., Uitdehaag J.C.M., Davies J.M., Wallis R., Drickamer K., Weis W.I. Crystal structure of the CUB1-EGF-CUB2 region of mannose-binding protein associated serine protease-2. EMBO J. 2003;22:2348–2359. doi: 10.1093/emboj/cdg236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsushita M., Fujita T. The role of ficolins in innate immunity. Immunobiology. 2002;205:490–497. doi: 10.1078/0171-2985-00149. [DOI] [PubMed] [Google Scholar]
- 17.Cseh S., Vera L., Matsushita M., Fujita T., Arlaud G.J., Thielens N.M. Characterization of the interaction between L-ficolin/p35 and mannan-binding lectin-associated serine proteases-1 and -2. J Immunol. 2002;169:5735–5743. doi: 10.4049/jimmunol.169.10.5735. [DOI] [PubMed] [Google Scholar]
- 18.Sekine H., Kenjo A., Azumi K., Ohi G., Takahashi M., Kasukawa R., Ichikawa N., Nakata M., Mizuochi T., Matsushita M. An ancient lectin-dependent complement system in an ascidian: novel lectin isolated from the plasma of the solitary ascidian, Halocynthia roretzi. J Immunol. 2001;167:4504–4510. doi: 10.4049/jimmunol.167.8.4504. [DOI] [PubMed] [Google Scholar]
- 19.Matsushita M., Endo Y., Fujita T. MASP1 (MBL-associated serine protease 1) Immunobiology. 1998;199:340–347. doi: 10.1016/S0171-2985(98)80038-7. [DOI] [PubMed] [Google Scholar]
- 20.Chan R.D., Ibrahim S.I., Vena N., Carroll M., Moore F.D., Jr., Hechtman H.B. Ischaemia-reperfusion is an event triggered by immune complexes and complement. Br J Surg. 2003;12:1470–1478. doi: 10.1002/bjs.4408. [DOI] [PubMed] [Google Scholar]
- 21•.Arnold J.N., Wormald M.R., Suter D.M., Radcliffe C.M., Harvey D.J., Dwek R.A., Rudd P.M., Sim R.B. Human serum IgM glycosylation: identification of glycoforms that can bind to mannan-binding lectin. J Biol Chem. 2005;280:29080–29087. doi: 10.1074/jbc.M504528200. [DOI] [PubMed] [Google Scholar]; These authors constructed a glycosylated model of pentameric IgM and identified that non-antigen-bound IgM can bind to MBL through a complex of glycans, which becomes inaccessible once IgM binds antigen.
- 22.Janeway C.A., Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13:11–16. doi: 10.1016/0167-5699(92)90198-G. [DOI] [PubMed] [Google Scholar]
- 23.Casanova J.L., Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 24•.Hughes A.L., Parker B., Welch R., Chanock S.J., Yeager M. High level of functional polymorphism indicates a unique role of natural selection at human immune system loci. Immunogenetics. 2005;29:1–7. doi: 10.1007/s00251-005-0052-7. [DOI] [PubMed] [Google Scholar]; This work addresses the question of whether there is a selection pressure on genes involved immunity and supports the idea that innate immune system is not hard-wired in that there are distinct differences between individuals rather than all these being invariant.
- 25.Takahashi K., Ezekowitz R.A. The role of the mannose-binding lectin in innate immunity. Clin Infect Dis. 2005;41:S440–S444. doi: 10.1086/431987. [DOI] [PubMed] [Google Scholar]
- 26.Kilpatrick D.C. Mannan-binding lectin: clinical significance and applications. Biochim Biophys Acta. 2002;1572:401–413. doi: 10.1016/s0304-4165(02)00321-5. [DOI] [PubMed] [Google Scholar]
- 27.Petersen S.V., Thiel S., Jensen L., Steffensen R., Jensenius J.C. An assay for the mannan-binding lectin pathway of complement activation. J Immunol Methods. 2001;257:107–116. doi: 10.1016/s0022-1759(01)00453-7. [DOI] [PubMed] [Google Scholar]
- 28.Hibberd M.L., Sumiya M., Summerfield J.A., Booy R., Levin M. Association of variants of the gene for mannose-binding lectin with susceptibility to meningococcal disease. Meningococcal Research Group. Lancet. 1999;353:1049–1053. doi: 10.1016/s0140-6736(98)08350-0. [DOI] [PubMed] [Google Scholar]
- 29.Kars M., van Dijk H., Salimans M.M., Bartelink A.K., van de Wiel A. Association of furunculosis and familial deficiency of mannose-binding lectin. Eur J Clin Invest. 2005;35:531–534. doi: 10.1111/j.1365-2362.2005.01521.x. [DOI] [PubMed] [Google Scholar]
- 30.Roy S., Knox K., Segal S., Griffiths D., Moore C.E., Welsh K.I., Smarason A., Day N.P., McPheat W.L., Crook D.W. MBL genotype and risk of invasive pneumococcal disease: a case-control study. Lancet. 2002;359:1569–1573. doi: 10.1016/S0140-6736(02)08516-1. [DOI] [PubMed] [Google Scholar]
- 31.Garred P., Pressler T., Madsen H.O., Frederiksen B., Svejgaard A., Hoiby N., Schwartz M., Koch C. Association of mannose-binding lectin gene heterogeneity with severity of lung disease and survival in cystic fibrosis. J Clin Invest. 1999;104:431–437. doi: 10.1172/JCI6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 33.Thorarinsdottir H.K., Ludviksson B.R., Vikingsdottir T., Leopoldsdottir M.O., Ardal B., Jonsson T., Valdimarsson H., Arason G.J. Childhood levels of immunoglobulins and mannan-binding lectin in relation to infections and allergy. Scand J Immunol. 2005;61:466–474. doi: 10.1111/j.1365-3083.2005.01588.x. [DOI] [PubMed] [Google Scholar]
- 34.Kielgast S., Thiel S., Henriksen T.B., Bjerke T., Olsen J., Jensenius J.C. Umbilical cord mannan-binding lectin and infections in early childhood. Scand J Immunol. 2003;57:167–172. doi: 10.1046/j.1365-3083.2003.01202.x. [DOI] [PubMed] [Google Scholar]
- 35.Koch A., Melbye M., Sorensen P., Homoe P., Madsen H.O., Molbak K., Hansen C.H., Andersen L.H., Hahn G.W., Garred P. Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA. 2001;285:1316–1321. doi: 10.1001/jama.285.10.1316. [DOI] [PubMed] [Google Scholar]
- 36.Ezekowitz R.A., Kuhlman M., Groopman J.E., Byrn R.A. A human serum mannose-binding protein inhibits in vitro infection by the human immunodeficiency virus. J Exp Med. 1989;169:185–196. doi: 10.1084/jem.169.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kase T., Suzuki Y., Kawai T., Sakamoto T., Ohtani K., Eda S., Maeda A., Okuno Y., Kurimura T., Wakamiya N. Human mannan-binding lectin inhibits the infection of influenza A virus without complement. Immunology. 1999;97:385–392. doi: 10.1046/j.1365-2567.1999.00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anders E.M., Hartley C.A., Reading P.C., Ezekowitz R.A. Complement-dependent neutralization of influenza virus by a serum mannose-binding lectin. J Gen Virol. 1994;75:615–622. doi: 10.1099/0022-1317-75-3-615. [DOI] [PubMed] [Google Scholar]
- 39.Reading P.C., Hartley C.A., Ezekowitz R.A., Anders E.M. A serum mannose-binding lectin mediates complement-dependent lysis of influenza virus-infected cells. Biochem Biophys Res Commun. 1995;217:1128–1136. doi: 10.1006/bbrc.1995.2886. [DOI] [PubMed] [Google Scholar]
- 40.Ip W.K., Chan K.H., Law H.K., Tso G.H., Kong E.K., Wong W.H., To Y.F., Yung R.W., Chow E.Y., Au K.L. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis. 2005;191:1697–1704. doi: 10.1086/429631. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first study that provides evidence from both case-control and in vitro experiments to suggest MBL plays an important role played in the first-line host defense against SARS-CoV infection, a newly emerged infectious disease. This also shows that MBL deficiency is a susceptibility factor for acquisition of SARS.
- 41.Zhang H., Zhou G., Zhi L., Yang H., Zhai Y., Dong X., Zhang X., Gao X., Zhu Y., He F. Association between mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect Dis. 2005;192:1355–1361. doi: 10.1086/491479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chong W.P., To Y.F., Ip W.K., Yuen M.F., Poon T.P., Wong W.H., Lai C.L., Lau Y.L. Mannose-binding lectin in chronic hepatitis B virus infection. Hepatology. 2005;42:1037–1045. doi: 10.1002/hep.20891. [DOI] [PubMed] [Google Scholar]
- 43.Hakozaki Y., Yoshiba M., Sekiyama K., Seike E., Iwamoto J., Mitani K., Mine M., Morizane T., Ohtani K., Suzuki Y. Mannose-binding lectin and the prognosis of fulminant hepatic failure caused by HBV infection. Liver. 2002;22:29–34. doi: 10.1046/j.0106-9543.2001.01516.x. [DOI] [PubMed] [Google Scholar]
- 44••.Shi L., Takahashi K., Dundee J., Shahroor-Karni S., Thiel S., Jensenius J.C., Gad F., Hamblin M.R., Sastry K.N., Ezekowitz R.A. Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J Exp Med. 2004;199:1379–1390. doi: 10.1084/jem.20032207. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence from an MBL-null mouse infection model that MBL is an important first-line host defense molecule that initiates opsonophagocytosis and stimulates proinflammatory cytokines in response to challenge with intravenous S. aureus.
- 45.Allgower M., Schoenenberger G.A., Sparkes B.G. Burning the largest immune organ. Burns. 1995;21:S7–S47. doi: 10.1016/0305-4179(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 46•.Møller-Kristensen M, Shi L, Ip WKE, Gowda LG, Hamblin M, Thiel S, Jensenius JC, Ezekowitz RAB, Takahashi K: Burn injury in the context of deficiency of mannose-binding lectin, greatly increases susceptibility to Pseudomonas aeruginosa infection. J Immunology, in press. [DOI] [PMC free article] [PubMed]; This work describes that MBL is indeed an opsonin in vivo.
- 47.Gadjeva M., Paludan S.R., Thiel S., Slavov V., Ruseva M., Eriksson K., Lowhagen G.B., Shi L., Takahashi K., Ezekowitz A. Mannan-binding lectin modulates the response to HSV-2 infection. Clin Exp Immunol. 2004;138:304–311. doi: 10.1111/j.1365-2249.2004.02616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jordan J.E., Montalto M.C., Stahl G.L. Inhibition of mannose-binding lectin reduces postischemic myocardial reperfusion injury. Circulation. 2001;104:1413–1418. doi: 10.1161/hc3601.095578. [DOI] [PubMed] [Google Scholar]
- 49.Hart M.L., Ceonzo K.A., Shaffer L.A., Takahashi K., Rother R.P., Reenstra W.R., Buras J.A., Stahl G.L. Gastrointestinal ischemia-reperfusion injury is lectin complement pathway dependent without involving C1q. J Immunol. 2005;174:6373–6380. doi: 10.4049/jimmunol.174.10.6373. [DOI] [PubMed] [Google Scholar]
- 50.Moller-Kristensen M., Wang W., Ruseva M., Thiel S., Nielsen S., Takahashi K., Shi L., Ezekowitz A., Jensenius J.C., Gadjeva M. Mannan-binding lectin recognizes structures on ischaemic reperfused mouse kidneys and is implicated in tissue injury. Scand J Immunol. 2005;61:426–434. doi: 10.1111/j.1365-3083.2005.01591.x. [DOI] [PubMed] [Google Scholar]
- 51•.Walsh M.C., Bourcier T., Takahashi K., Shi L., Busche M.N., Rother R.P., Solomon S.D., Ezekowitz R.A., Stahl G.L. Mannose-binding lectin is a regulator of inflammation that accompanies myocardial ischemia and reperfusion injury. J Immunol. 2005;175:541–546. doi: 10.4049/jimmunol.175.1.541. [DOI] [PubMed] [Google Scholar]; MBL-deficient mice were shown to be protected from cardiac ischemia-reperfusion injury, which confirms the key regulatory role of the lectin complement pathway. This study supports the contention that MBL has a dual role in modifying inflammatory responses to sterile and infectious injuries.
- 52.Cortes-Hernandez J., Fossati-Jimack L., Petry F., Loos M., Izui S., Walport M.J., Cook H.T., Botto M. Restoration of C1q levels by bone marrow transplantation attenuates autoimmune disease associated with C1q deficiency in mice. Eur J Immunol. 2004;34:3713–3722. doi: 10.1002/eji.200425616. [DOI] [PubMed] [Google Scholar]
- 53•.Stuart L.M., Takahashi K., Shi L., Savill J., Ezekowitz R.A. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol. 2005;174:3220–3226. doi: 10.4049/jimmunol.174.6.3220. [DOI] [PubMed] [Google Scholar]; These data indicate that MBL-null mice are similar to C1q-deficient mice to the extent that they have delayed removal of apoptotic cells. However, unlike C1q-deficient animals, MBL-null mice do not spontaneously develop autoimmunity or lymphoproliferation.
- 54.Seyfarth J., Garred P., Madsen H.O. The ‘involution’ of mannose-binding lectin. Hum Mol Genet. 2005;14:2859–2869. doi: 10.1093/hmg/ddi318. [DOI] [PubMed] [Google Scholar]
- 55.Ogden C.A., deCathelineau A., Hoffmann P.R., Bratton D., Ghebrehiwet B., Fadok V.A., Henson P.M. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194:781–795. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nauta A.J., Raaschou-Jensen N., Roos A., Daha M.R., Madsen H.O., Borrias-Essers M.C., Ryder L.P., Koch C., Garred P. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol. 2003;33:2853–2863. doi: 10.1002/eji.200323888. [DOI] [PubMed] [Google Scholar]
- 57.de Vries B., Walter S.J., Peutz-Kootstra C.J., Wolfs T.G., van Heurn L.W., Buurman W.A. The mannose-binding lectin-pathway is involved in complement activation in the course of renal ischemia-reperfusion injury. Am J Pathol. 2004;165:1677–1688. doi: 10.1016/S0002-9440(10)63424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collard C.D., Montalto M.C., Reenstra W.R., Buras J.A., Stahl G.L. Endothelial oxidative stress activates the lectin complement pathway: role of cytokeratin 1. Am J Pathol. 2001;159:1045–1054. doi: 10.1016/S0002-9440(10)61779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muto S., Sakuma K., Taniguchi A., Matsumoto K. Human mannose-binding lectin preferentially binds to human colon adenocarcinoma cell lines expressing high amount of Lewis A and Lewis B antigens. Biol Pharm Bull. 1999;22:347–352. doi: 10.1248/bpb.22.347. [DOI] [PubMed] [Google Scholar]
- 60.Ma Y., Uemura K., Oka S., Kozutsumi Y., Kawasaki N., Kawasaki T. Antitumor activity of mannan-binding protein in vivo as revealed by a virus expression system: mannan-binding proteindependent cell-mediated cytotoxicity. Proc Natl Acad Sci USA. 1999;96:371–375. doi: 10.1073/pnas.96.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malhotra R., Wormald M.R., Rudd P.M., Fischer P.B., Dwek R.A., Sim R.B. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med. 1995;1:237–243. doi: 10.1038/nm0395-237. [DOI] [PubMed] [Google Scholar]
- 62.Roos A., Bouwman L.H., van Gijlswijk-Janssen D.J., Faber-Krol M.C., Stahl G.L., Daha M.R. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol. 2001;167:2861–2868. doi: 10.4049/jimmunol.167.5.2861. [DOI] [PubMed] [Google Scholar]
- 63.Hisano S., Matsushita M., Fujita T., Endo Y., Takebayashi S. Mesangial IgA2 deposits and lectin pathway-mediated complement activation in IgA glomerulonephritis. Am J Kidney Dis. 2001;38:1082–1088. doi: 10.1053/ajkd.2001.28611. [DOI] [PubMed] [Google Scholar]
- 64.Palaniyar N., Nadesalingam J., Reid K.B. Innate immune collectins bind nucleic acids and enhance DNA clearance in vitro. Ann N Y Acad Sci. 2003;1010:467–470. doi: 10.1196/annals.1299.084. [DOI] [PubMed] [Google Scholar]
- 65.Palaniyar N., Nadesalingam J., Clark H., Shih M.J., Dodds A.W., Reid K.B. Nucleic acid is a novel ligand for innate, immune pattern recognition collectins surfactant proteins A and D and mannose-binding lectin. J Biol Chem. 2004;279:32728–32736. doi: 10.1074/jbc.M403763200. [DOI] [PubMed] [Google Scholar]
- 66.Honma T., Kuroki Y., Tsunezawa W., Ogasawara Y., Sohma H., Voelker D.R., Akino T. The mannose-binding protein A region of glutamic acid185-alanine221 can functionally replace the surfactant protein A region of glutamic acid195-phenylalanine228 without loss of interaction with lipids and alveolar type II cells. Biochemistry. 1997;36:7176–7184. doi: 10.1021/bi962967e. [DOI] [PubMed] [Google Scholar]
- 67.Kilpatrick D.C. Phospholipid-binding activity of human mannan-binding lectin. Immunol Lett. 1998;61:191–195. doi: 10.1016/s0165-2478(98)00031-5. [DOI] [PubMed] [Google Scholar]
- 68.Hirano M., Ma B.Y., Kawasaki N., Okimura K., Baba M., Nakagawa T., Miwa K., Oka S., Kawasaki T. Mannan-binding protein blocks the activation of metalloproteases meprin alpha and beta. J Immunol. 2005;175:3177–3185. doi: 10.4049/jimmunol.175.5.3177. [DOI] [PubMed] [Google Scholar]