

Abstract
A series of thiosemicarbazones and their platinum(II) and palladium(II) complexes have been synthesized. The chemical structures of ligands and their complexes were characterized by UV–Vis, IR, 1H NMR, 13C NMR, MS spectra, elemental analysis and TGA. The antiviral and cytotoxic activities of all compounds have been tested. Results of broad antiviral evaluation showed that none of the compounds evaluated endowed with anti-DNA or -RNA virus activity at subtoxic concentrations except for the palladium complex 1b. This compound exhibited slightly selective inhibition against cytomegalovirus. The platinum complex 4a exhibited the best cytostatic activities against human cervix carcinoma. Ligands 2, 4 and 5 showed cytostatic potential. The palladium complexes were in general less cytostatic than the corresponding platinum complexes or unliganded congeners.
Keywords: Thiosemicarbazones, Platinum(II) complexes, Palladium(II) complexes, Cytotoxic activity, Antiviral activity
Graphical abstract
Compound 1b showed slightly inhibitory activity against HCMV at 2.9–15.3 μM. The cytostatic potency of compound 4a (IC50: 1.7 μM) was significant against HeLa cell culture.
Highlights
▸ Some thiosemicarbazones and their Pt(II) and Pd(II) complexes were synthesized. ▸ All compounds were tested their in vitro antiviral and cytostatic/toxic activity. ▸ 1b exhibited some selective inhibition against HCMV. ▸ 4a showed significant cytostatic potency against HeLa cell culture.
1. Introduction
Since the discovery of the importance of metal containing compounds used in cancer treatment, reports on the use of metals are increasing [1], [2], [3], [4], [5]. In recent years new metal complexes have been identified as a very promising class of anticancer active compounds [6], [7], [8]. Metals bound to atoms such as N, O and S can form a chelate ring that binds the metal more tightly when compared to the non-chelate form. Large biological molecules (proteins, enzymes, DNA etc.) are electron-rich but metal ions are electron-deficient. Therefore, interactions occur between metal ions and many important biological molecules. This event has led to the use of metals or metal-containing agents to modulate biological systems [9]. Organometallic compounds have gained importance as enzyme inhibitors due to the capability of binding large biological molecules more strongly than metal-free organic compounds [10].
Metal complexes can inhibit metalloenzymes by chelate substitution and also inhibit non-metalloenzymes by coordinating to their active site. The other property of metals is to catalyze the formation of reactive oxygen species [11]. The anticancer potential of metal containing agents, from main group elements to early transition metals, have been evaluated [12], [13], [14]. Especially organo-platinum compounds such as cisplatin, carboplatin and oxaliplatin are metal-based drugs that are among the most active and widely used clinical drugs in cancer chemotherapy [15], [16], [17]. These platinum complexes react in vivo, binding to and causing crosslinking of DNA which finally activates programmed cell death [18]. However, the clinical utility of these drugs is limited to a relatively narrow range of tumors (sarcomas, small cell lung cancer, ovarian cancer, lymphomas and germ cell tumors) because of primary resistance and the development of resistance secondary to the initial treatment [19], [20]. Therefore, unconventional platinum complexes, that could be used in cisplatin-resistant tumors are made by different research groups [21], [22], [23], [24].
Lipophilicity that controls the rate of entry into the cell is altered by metal-coordination and some side-effects can be reduced by complexation. Moreover, metal complexes can exhibit biological activities more than free ligand [6]. Recently, thiosemicarbazones and their metal complexes have achieved importance due to their application in pharmaceutical chemistry and proved to be chemotherapeutic agents potentially useful for inhibiting the activities of cancer cells [25], [26]. For example, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine®; Vion Pharmaceuticals Inc. New Haven, CT) inhibit the biosynthesis of DNA in leukemia L1210 cells by blocking activity of ribonucleotide reductase [27]. Many heterocyclic thiosemicarbazone derivatives and their platinum and palladium complexes have a wide range of pharmacological activities, such as antituberculosis [28], antibacterial [29], antitumor [30], antiprotozoal [31], antimalarial [32], antimicrobial [33], antiviral [34], antifungal [35], anticonvulsant [36] and anti-trypanosomal [37] activities.
In our present work, the synthesis and characterization of thiosemicarbazones derived from 5-substitutedthiophene-2-carboxaldehydes and their platinum(II) and palladium(II) complexes are reported. The results of in vitro antiviral and cytostatic/toxic activities of ligands and their platinum and palladium complexes have been evaluated.
2. Chemistry
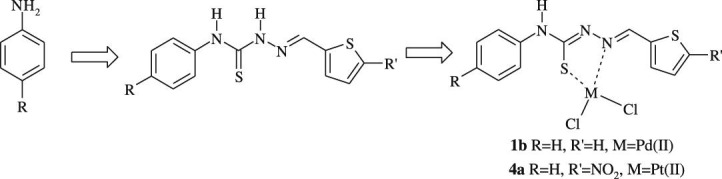
The precursors phenyl isothiocyanate and 4-nitrophenyl isothiocyanate were synthesized from aniline and 4-nitroaniline according to the method described in the literature [38]. N-phenylhydrazinecarbothioamide and N-(4-nitrophenyl)hydrazinecarbothioamide were prepared by stirring isothiocyanates with hydrazine monohydrate in diethyl ether at room temperature according to Ref. [39]. The ligands 1–5 used in this work were obtained by refluxing in methanol (20–30 mL) an equimolar amount of 5-substituted-2-thiophene carboxaldehydes with thiosemicarbazides according to Refs. [35], [40], [41], [42]. The chemical structures of the ligands are given in Scheme 1 . All thiosemicarbazone derivatives synthesized were characterized by UV–Vis, IR, 1H NMR, 13C NMR, MS spectral data and elemental analysis.
Scheme 1.

General synthesis of thiosemicarbazones (1–5).
The platinum(II) and palladium(II) complexes of thiosemicarbazone derivatives 1a–5a and 1b–5b were prepared by exposing a solution of the K2PtCl4 or K2PdCl4 in water to a solution of the appropriate ligand in ethanol in a 1:1 M:L molar ratio [43]. The structures of Pt(II) and Pd(II) complexes were confirmed by elemental analysis, UV–Vis, IR, 1H NMR, MS spectroscopies, elemental analyses and TGA thermogram and are given in Fig. 1 . The elemental analysis data of the ligands and their Pt(II) and Pd(II) complexes are presented in Table 1 .
Fig. 1.

The structures of Pt(II) and Pd(II) complexes (1a–5a and 1b–5b).
Table 1.
Physical data of thiosemicarbazone derivatives and their Pt(II) and Pd(II) complexes.
| Comp. no | Molecular formulas | Colour | Yield (%) | Mp/Dec. temp. (°C) | Found (calcd.) |
|||
|---|---|---|---|---|---|---|---|---|
| C | H | N | S | |||||
| 1 | C12H11N3S2 | Yellow | 85 | 188–190 | 55.14 (55.29) |
4.24 (4.28) |
16.08 (16.16) |
24.54 (24.32) |
| 1a | C12H10N3S2Cl2Pt | Orange | 69 | 212 | 27.38 (27.43) |
1.91 (1.98) |
7.98 (7.82) |
12.18 (12.67) |
| 1b | C12H10N3S2Cl2Pd | Brown | 58 | 272 | 32.93 (32.27) |
2.30 (2.35) |
9.60 (9.53) |
14.65 (14.45) |
| 2 | C12H10N3S2Cl | Yellow | 67 | 130–132 | 47.86 (47.72) |
3.21 (3.41) |
12.69 (12.21) |
22.62 (22.58) |
| 2a | C12H9N3S2Cl3Pt | Dark yellow | 53 | 243 | 25.65 (25.43) |
1.79 (1.58) |
7.48 (7.82) |
11.41 (11.67) |
| 2b | C12H9N3S2Cl3Pd | Brown | 62 | 250 | 30.53 (30.28) |
1.92 (1.81) |
8.90 (8.62) |
13.58 (13.85) |
| 3 | C18H15N3S2 | Yellow | 83 | 182–184 | 64.06 (64.72) |
4.48 (4.41) |
12.45 (12.21) |
19.00 (19.28) |
| 3a | C18H14N3S2Cl2Pt | Orange | 56 | 256 | 35.89 (35.62) |
2.34 (2.02) |
6.97 (6.73) |
10.64 (10.48) |
| 3b | C18H14N3S2Cl2Pd | Brown | 59 | 307 | 42.08 (42.19) |
2.75 (2.82) |
8.18 (8.33) |
12.48 (12.82) |
| 4 | C12H10N4O2S2 | Yellow | 82 | 190–192 | 47.04 (47.22) |
3.29 (3.34) |
18.29 (18.28) |
20.93 (20.79) |
| 4a | C12H9N4O2S2Cl2Pt | Dark brown | 52 | 220 | 25.23 (25.43) |
1.59 (1.19) |
9.81 (9.66) |
11.22 (11.83) |
| 4b | C12H9N4O2S2Cl2Pd | Dark brown | 54 | 302 | 29.86 (29.43) |
1.88 (1.89) |
11.61 (11.66) |
13.29 (13.72) |
| 5 | C12H10N4O2S2 | Yellow | 83 | 188–190 | 47.04 (47.16) |
3.29 (3.25) |
18.29 (18.32) |
20.93 (20.91) |
| 5a | C12H9N4O2S2Cl2Pt | Orange | 62 | 225 | 25.23 (25.61) |
1.59 (1.65) |
9.81 (9.96) |
11.22 (11.43) |
| 5b | C12H9N4O2S2Cl2Pd | Brown | 61 | 296 | 29.86 (29.67) |
1.88 (1.75) |
11.61 (11.84) |
13.29 (13.34) |
3. Results and discussion
3.1. Synthesis
All of the newly synthesized metal complexes 1a-5a and 1b-5b are solids and decomposed above ca. 200 °C. They are insoluble in organic solvents such as acetone, chloroform and methanol but soluble in DMF and DMSO. The elemental analyses data of the thiosemicarbazones and their platinum and palladium complexes (Table 1) were compatible with the structures of the ligands shown in Scheme 1. and the formulas of the complexes are shown in Fig. 1.
3.2. Spectral studies
3.2.1. Electronic spectral studies
The study of the electronic spectra in the ultraviolet and visible (UV–Vis) ranges for the ligand and metal complexes was carried out in DMF. Solid-state electronic spectra of all thiosemicarbazone derivatives reveal similar patterns, exhibiting two bands in the 433–344 nm and 28–258 nm regions. An intense band at ca. 433–344 nm is attributed to the n → π* transitions of C S group, C N group and thiophene ring, which are overlapped. The π → π* transitions of the thiophene ring and thiosemicarbazone imine function are rather weak, and observed at ca. 281–258 nm. These two bands are shifted to lower energies (bathochromic shift) after complexation (467–371 nm, 344–264 nm). Such observations have also been reported earlier in other platinum(II) and palladium(II) complexes of similar ligand systems [44].
3.2.2. IR spectral studies
IR spectral data of all thiosemicarbazones and their Pt(II) and Pd(II) complexes showed a holding the 5-substitutedthiophene-2-carboxaldehyde thiosemicarbazone moiety in all complexes. IR spectra were compared with free ligands and their Pt(II) and Pd(II) complexes and given in Table 2 .
Table 2.
Main characteristic IR vibrational bands of thiosemicarbazones and their platinum(II) and palladium(II) complexes.
| Compound |
vmax (cm−1) |
|||||
|---|---|---|---|---|---|---|
| N–H | C N | N–N | C S | M–N | M–S | |
| 1 | 3295 | 1588 | 1042 | 783 | – | – |
| 1a | 3272 | 1510 | 1095 | 748 | 521 | 400 |
| 1b | 3242 | 1527 | 1073 | 717 | 518 | 403 |
| 2 | 3316 | 1591 | 1064 | 787 | – | – |
| 2a | 3298 | 1524 | 1088 | 747 | 513 | 402 |
| 2b | 3205 | 1533 | 1105 | 748 | 511 | 405 |
| 3 | 3304 | 1592 | 1028 | 804 | – | – |
| 3a | 3288 | 1528 | 1049 | 791 | 507 | 418 |
| 3b | 3241 | 1527 | 1072 | 749 | 502 | 412 |
| 4 | 3342 | 1595 | 1110 | 825 | – | – |
| 4a | 3320 | 1565 | 1046 | 807 | 518 | 411 |
| 4b | 3255 | 1538 | 1099 | 778 | 510 | 408 |
| 5 | 3360 | 1582 | 1108 | 800 | – | – |
| 5a | 3289 | 1533 | 1172 | 776 | 516 | 409 |
| 5b | 3262 | 1547 | 1181 | 769 | 509 | 410 |
Although thiosemicarbazone derivatives are capable of interacting with metals in different ways, they usually behave as neutral NS donor bidentate ligands. The thiosemicarbazones can exhibit thione-thiol tautomerism in solution because of the thioamide (NH–C S) function. The ν(SH) band at ca. The 2500–2600 cm−1 region is absent from the IR spectra of the ligands but the v(NH) band at 3115–3140 cm−1 and the strong band due to v(C S) at 783–825 cm−1 are present, suggesting that in the solid phase the thiosemicarbazones remain as the thione form [45]. These bands are shifted to lower wavenumbers (13–66 cm−1) indicating the coordination of metal through thione/thiol sulfur in all complexes. This coordination is approved by the presence of two new bands at the 502–521 and 400–418 cm−1 region due to v(Pt–N, Pd–N and Pt–S, Pd–S), respectively [46]. The band due to v(C–S–C) of the thiophene ring remains unaltered in complexes (1a–5a and 1b–5b), indicating non-participation of the thiophene ring in coordination. The azomethine stretching vibrations ν(C N), characteristic of a Schiff base are observed at 1582–1595 cm−1 [7]. The negative shift (30–78 cm−1) of the C N band was observed in all complexes which exhibit the involvement of azomethine nitrogen upon complexation. On the other hand, the shift to higher wavenumbers of the v(N–N) band, observed for the platinum and palladium complexes, have also been related to the electronic delocalization and occurred as a consequence of coordination through the azomethine nitrogen atom and deprotonation of thiosemicarbazones [47]. Finally, the ligands 1–5 coordinate to platinum(II) and palladium(II) as NS bidentate.
3.2.3. NMR spectral analysis
The chemical structures of all thiosemicarbazones and their complexes were characterized by 1H NMR spectroscopy and given in Table 3 . Platinum(II) and palladium(II) complexes exhibited similar 1H NMR spectra and behavior.
Table 3.
1H NMR chemical shift values of (δ) ligands and their platinum(II) and palladium(II) complexes.
| Compound |
δ (ppm) |
|||
|---|---|---|---|---|
| CSNH | PhNH | N CH | Ar–H | |
| 1 | 11.83 | 9.81 | 8.36 | 7.70–7.15 |
| 1a | – | 9.94 | 8.48 | 8.00–6.92 |
| 1b | – | 9.83 | 8.66 | 8.02–7.09 |
| 2 | 11.89 | 9.86 | 8.24 | 7.54–7.16 |
| 2a | – | 9.85 | 8.62 | 7.92–6.93 |
| 2b | – | 9.80 | 8.55 | 7.68–7.18 |
| 3 | 11.90 | 9.84 | 8.33 | 7.73–7.21 |
| 3a | – | 9.81 | 8.62 | 7.78–6.98 |
| 3b | – | 9.79 | 8.64 | 8.00–7.33 |
| 4 | 12.19 | 10.19 | 8.31 | 8.11–7.24 |
| 4a | – | 10.12 | 8.63 | 8.18–7.02 |
| 4b | – | 10.27 | 8.36 | 8.31–7.20 |
| 5 | 11.91 | 10.23 | 8.40 | 8.25–7.17 |
| 5a | – | 10.69 | 9.09 | 8.12–7.64 |
| 5b | – | 10.26 | 8.91 | 8.38–7.22 |
Results of the 1H NMR integrations and signal multiplicities were in agreement with the proposed structures and other spectral data. 1H NMR spectra of all thiosemicarbazones (1–5) recorded in DMSO-d 6 exhibited a sharp singlet peak at δ 9.81–10.23 ppm due to the PhNH proton and at δ 11.83–12.19 ppm due to the = N–NH proton, which indicate that even in a polar solvent they remain in the thione form. A singlet peak at δ 8.24–8.40 ppm due to the HC N group was in accordance with the literature [48], [49], [50]. The = N–NH proton signals disappeared in the 1H NMR spectra of the platinum(II) and palladium(II) complexes indicating the deprotonation of the = N–NH group, while the signals of the other NH (PhNH) protons were observed at the same field (δ 9.80–10.69 ppm). The signals of the HC N protons shift to downfield in all complexes and appeared at 8.48–9.09 ppm. This information indicates the coordination of the metal center to the azomethine nitrogen and the thioamide sulfur and the PhNH group is not coordinated to the metal ion [51]. All aromatic protons were observed at the expected region (δ 7.15–8.25 ppm) in the NMR spectrum of the thiosemicarbazone ligands. These signals do not afford relevant changes in the chemical shifts for the platinum(II) and palladium(II) complexes.
Furthermore, the 13C NMR spectra of the ligands were recorded in DMSO-d 6 and gave the spectral signals in good agreement with the probable chemical structure. All ligands exhibited two important signals at δ 174.22–176.21 and δ 137.28–140.65 ppm assigned to the thioamide (C S) and imine (C N) carbon atoms, respectively [52]. The signals at δ 123.60–156.22 ppm were observed in the spectra due to the aromatic carbons.
3.2.4. Mass analysis
The MS spectra of the free ligands (1–5) and their complexes (1a–5a and 1b–5b) were recorded by electro spray impact (ESI) in positive ion mode. The mass fragmentation routes of compound 2 is given in Scheme 2 .
Scheme 2.

The mass fragmentation routes of compound 2.
The mass spectrum of compound 2 confirms the proposed molecular structure by showing a peak at m/z 296 corresponding to the molecular ion [M + H]+. The other thiosemicarbazone derivatives also showed the similar fragmentation pathway as given in Fig. 2 .
Fig. 2.

Proposed mass fragmentation pathway for ligands.
A general splitting pathway with the characteristic peaks was observed by the platinum(II) and palladium(II) complexes 1a–5a and 1b–5b. The spectra of all complexes showed several informative fragment ions confirming their molecular weights. The molecular ion peaks were conformed to [M]+ or [M + H]+ and the major fragmentation pathway involved the leaving of PtCl2 or PdCl2 [53].
3.2.5. Thermal decomposition
TGA thermograms of complexes (1a–5a and 1b–5b) were recorded under nitrogen with a heating rate of 10 °C/min between 30 °C and 900 °C. All complexes did not lose weight up to 230 °C. Further increment of the temperature causes decomposition of these complexes in two steps [54]. The first step being 250–415 °C for the palladium(II) and platinum(II) complexes where losses of mixed fragments are observed. The second step starts after the first step and continues until complete decomposition of the ligands and formation of MS [where, M = Pd(II) and Pt(II)]. The total weight loss of all complexes depends on the loss of the respective ligand, after one sulfur atom moving to the metal ion and residues conversion to the palladium sulfide and platinum sulfide.
3.3. Biological activity studies
The biological activity of the newly synthesized complexes were tested in terms of cytotoxic and antiviral activity compared with the free ligands.
3.3.1. Cytostatic/toxic activity
The all compounds were examined for their cytostatic activity against human lymphocyte (CEM), human cervix carcinoma (HeLa), Crandell feline kidney (CRFK), Madin Darby canine kidney (MDCK) and human lung fibroblast (HEL) cells and their activity results were given Table 4 .
Table 4.
Cytotoxic and cytostatic activity of the ligands and their platinum(II) and palladium(II) complexes.
| Compound | MICa (μM) |
IC50b (μM) |
|||||
|---|---|---|---|---|---|---|---|
| HEL | HeLa | MDCK | HEL | CrFK | HeLa | CEM | |
| 1a | ≥20 | 100 | 10.8 | 6.0 | 6.7 | 37 | 55 |
| 1b | 100 | >100 | >100 | 13.3 | 91.9 | 22 | 34 |
| 2 | ≥100 | 100 | 57.8 | 10.4 | 10.1 | 7.1 | 10 |
| 2a | 60 | 100 | 12.6 | 26.2 | 15.6 | 37 | 36 |
| 2b | 100 | >100 | >100 | 42.8 | >100 | 180 | 60 |
| 3 | ≥100 | ≥20 | >100 | 32.1 | >100 | 43 | 163 |
| 3a | 100 | 100 | 60.7 | 52.7 | 65.0 | 105 | 53 |
| 3b | ≥100 | 100 | 100 | 100 | >100 | 254 | 168 |
| 4 | ≥20 | ≥20 | 10.9 | 52.1 | 3.8 | 3.6 | 6.4 |
| 4a | 20 | 20 | 3.4 | 3.1 | 7.5 | 1.7 | 8.1 |
| 4b | ≥100 | 100 | 55.3 | 14.6 | 39.1 | 21 | 30 |
| 5 | ≥20 | 20 | >100 | 3.2 | 2.4 | 12 | 27 |
| 5a | ≥100 | >100 | >100 | 34 | >100 | 46 | 26 |
| 5b | >100 | >100 | >100 | 51.7 | >100 | 385 | 152 |
Minimal inhibitory concentration or compound concentration required to alter microscopically visible morphology of the confluent cell cultures.
50% Inhibitory concentration or compound concentration requred to inhibit MDCK (Madin Darby canine kidney), HEL (human lung fibroblast), CrFK (Cranell feline kidney), HeLa (human cervic carcinoma) or CEM (human lymphocyte) cells by 50%.
Among all ligands and their platinum and palladium complexes evaluated the platinum complex of 5-nitrothiophene-2-carboxaldeyde-N(4)phenyl thiosemicarbazone 4a showed the highest cytostatic activity against HeLa (IC50: 1.7 μM IC50: 0.22 μM for cisplatinum). However this compound was cytostatic against proliferating HEL fibroblasts (IC50: 3.1 μM), CRFK cells (IC50: 7.5 μM), MDCK cells and (IC50: 3.4 μM) CEM cells (IC50: 8.1 μM IC50: 2.2 μM for cisplatinum). Furthermore, the ligands 2, 4 and 5 showed moderately cytostatic activity against in cell cultures (IC50: 7.1–57.8 μM for compound 2, IC50: 3.6–52.1 μM for compound 4 and IC50: 2.4->100 μM for compound 5). Other ligands and complexes did not show appreciable cytostatic action against the evaluated confluent monolayer cell cultures (Table 4).
Regarding toxicity, it is striking that the ligands 2–5 (IC50: 2.4–163 μM) and their platinum complexes 1a–5a (IC50: 1.7–105 μM) were consistently more cytostatic than palladium complexes 1b–5b (IC50: 13.3–385 μM). Also, these compounds are clearly cytostatic (inhibition of cell proliferation) rather than cytotoxic (microscopically visible alteration of cell morphology).
3.3.2. Antiviral activity
The compounds 2–5, platinum(II) complexes 1a–5a and palladium (II) complexes 1b–5b were also evaluated for their antiviral activity against a wide variety of DNA and RNA viruses, including herpes simplex virus type 1 (HSV-1) (strain KOS), HSV-2 (strain G), vaccinia virus, vesicular stomatitis virus (VSV), varicella-zoster virus (VZV) strains OKA and 07/1 and human cytomegalovirus (HCMV) strains AD-169 and Davis in HEL cell cultures, VSV, Coxsackie B4 and respiratory syncytial virus (RSV) in HeLa cell cultures, parainfluenza-3, reovirus-1, Sindbis virus, Coxsackie B4 and Punta Toro virus in Vero cell cultures, influenza A (H1N1, H3N2) influenza B virus in MDCK cell cultures and feline coronavirus (FIPV) and feline herpes virus in CrFK cell cultures and their activities were compared with those of ganciclovir and cidofovir. Unfortunately, none of the compounds tested were endowed with appreciable inhibitory activity at subtoxic concentrations except for compound 1b that showed the highest antiviral activity against HCMV (EC50: 2.9–15.3 μM). By comparison of the ganciclovir potency against HCMV we can conclude that 1b was 3-fold less active against HCMV AD-169 strain and almost equally potent against Davis strain as ganciclovir (EC50: 5.0 μM for AD-169 strain, EC50: 3.4 μM for Davis strain). However, it should be mentioned that this compound was also cytostatic at CC50 value of 13.3 μM in HEL cell cultures. Therefore, the observed anti-HCMV activity may well be due to an indirect cytostatic activity of the compounds rather than a specific antiviral activity. No specific antiviral effect was noted for any compounds evaluated against other viruses (data not given).
4. Conclusions
We have prepared a series of 5-substitutedthiophene-2-carboxaldeyhde thiosemicarbazones and their platinum(II) and palladium(II) complexes. In conclusion, none of the new compounds showed antiviral potency at subtoxic concentrations except for 1b that showed slight inhibitory activity against HCMV at 2.9–15.3 μM. Several compounds in particular 4a were endowed with significant cytostatic potency (IC50: 1.7 μM for HeLa cell cultures) and can be viewed as new lead compounds for further modifications. The other ligands and their platinum complexes were showed only moderate cytostatic activity in the lower micromolar range.
5. Experimental
5.1. Chemicals and instruments
All reactions were monitored by thin layer chromatography (TLC) using Merck silica gel 60 F254 plates. Chemicals and solvents purchased from Aldrich, Fluka and Riedel de Haen. Melting points were determined by EZ-Melt melting point apparatus and were uncorrected. Electronic spectra were recorded in DMF on a PG Instruments T80+ UV–visible Spectrophometer. IR spectra on KBr disks were recorded on a Perkin Elmer 1620 model FT-IR spectrophotometer. Elemental analyses (C,H,N,S) were performed on a VarioMICRO elemental analyzer. 1H and 13C NMR spectra were obtained at room temperature with a Brucker AVANC-DPX-400 MHz NMR spectrometer in DMSO-d 6 using TMS as an internal standard. The chemical shift values are given in ppm and following abbreviations were used: s = singlet, d = doublet, dd = double doublet, t = triplet, m = multiplet. The coupling constants (J) are given in Hertz. The mass spectra of all compounds were recorded on an Agilent 1100 MSD spectrometer in the electro spray mode. Thermogravimetric analysis of the complexes were performed by a Shimadzu TGA 50 thermogravimetric analyzer under nitrogen atmosphere with a heating rate of 10 °C/min.
5.1.1. Synthesis of thiosemicarbazones (1–5): a general method
To a hot solution of 4-substituted- N-phenylthiosemicarbazide (5.98 mmol) in methanol (30 mL) was added dropwise a solution of the corresponding 5-substituted-2-thiophene carboxaldehydes (5.98 mmol) in methanol (5 mL) during 15 min. The reaction mixture was refluxed and the progress of the reaction was followed by TLC (about 10 h) and cooled. After cooling the product was filtered and the filtrate was concentrated under reduced pressure. The crude product was recrystallized from methanol.
5.1.2. Thiophene-2-carboxaldehyde-N(4)phenyl thiosemicarbazone (1)
Yellow solid (methanol). Yield: 85%. m.p.: 188–190 °C. Anal. calc. (C12H11N3S2): C, 55.14; H, 4.24; N, 16.08; S, 24.54; found: C, 55.29; H, 4.28; N, 16.16; S, 24.32%. ES-MS (m/z) 261 [M+], 262 [M + H]+. UV/Vis λ max(nm): 260, 344. IR v max(cm−1): 3295, 3132 (N–H); 1588 (C N); 1170, (C–N); 1042 (N–N); 783 (C S). 1H NMR (DMSO-d 6, δ ppm): 11.83 (s, 1H, CSNHN); 9.81 (s, 1H, PhNH); 8.36 (s, 1H, HC N); 7.70 (d, 1H, J = 4.99 Hz, C2 proton of thiophene ring); 7.58 (d, 2H, J = 7.82 Hz, ArH ortho position); 7.54 (d, 1H, J = 3.29 Hz C4 proton of thiophene ring); 7.37 (t, 2H, ArH meta position); 7.20 (t, 1H, ArH para position); 7.15 (dd, 1H, J 1 = 3.73 Hz, J 2 = 4.85 Hz, C3 proton of thiophene ring). 13C NMR (DMSO-d 6, δ ppm): 174.22 (C S), 144.68 (ArC), 140.65 (C N), 138.52, 130.05, 129.13, 128.44, 127.91, 127.16, 126.55 (ArC).
5.1.3. 5-Chlorothiophene-2-carboxaldehyde-N(4)phenyl thiosemicarbazone (2)
Yellow solid (methanol). Yield: 67%. m.p.: 130–132 °C. Anal. calc. (C12H10ClN3S2): C, 47.86; H, 3.21; N, 12.69; S, 22.62; found: C, 47.72; H, 3.41; N, 12.21; S, 22.58%. ES-MS (m/z) 296 [M + H+]. UV/Vis λ max(nm): 258, 351. IR ν max(cm−1): 3316, 3132 (N-H), 1591 (C N), 1198 (C–N), 1064 (N–N), 787 (C S). 1H NMR (DMSO-d 6, δ ppm): 11.89 (s, 1H, CSNHN); 9.86 (s,1H, PhNH); 8.24 (s,1H, HC N); 7.54 (t, 2H, J = 8.42 Hz, ArH meta position) 7.40 (d,1H, J = 4.05 Hz, C3 proton of thiophene ring); 7.36 (d, 2H, J = 8.23 Hz, ArH ortho position); 7.20 (t, 1H, ArH para position); 7.16 (d, 1H, J = 3.94 Hz, C4 proton of thiophene ring). 13C NMR (DMSO-d 6, δ ppm): 176.14 (C S), 156.22 (ArC), 139.42 (C N), 138.30, 137.90, 137.65, 134.37, 133.78, 131.59, 131.20, 128.55, 126.08 (ArC).
5.1.4. 5-Phenylthiophene-2-carboxaldeyde-N(4)phenyl thiosemicarbazone (3)
Yellow solid (methanol). Yield: 83%. m.p.: 182–184 °C. Anal. calc. (C18H15N3S2): C, 64.06; H, 4.48; N, 12.45; S, 19.00; found: C, 64.72; H, 4.41; N, 12.21; S, 19.28%. ES-MS (m/z) 337 [M+]. UV/Vis λ max(nm): 266, 378. IR ν max(cm−1): 3304, 3115 (N–H); 1592 (C N); 1160 (C–N); 1028 (N–N); 804 (C S). 1H NMR (DMSO-d 6, δ ppm): 11.90 (s, 1H, CSNHN); 9.84 (s, 1H, PhNH); 8.33 (s,1H, HC N); 7.73 (d, 2H, J = 7.29 Hz, ArH, ortho position of phenyl bearing to thiophene ring); 7.60 (d, 2H, J = 7.45 Hz, ArH, ortho position); 7.58 (d, 1H, J = 3.87 Hz, C3 proton of thiophene ring); 7.55 (d,1H, J = 3.90 Hz, C4 proton of thiophene ring); 7.34–7.48 (m, 5H, ArH, meta position and para and meta position phenyl bearing to thiophene ring); 7.21 ( t, 1H, ArH, para position). 13C NMR (DMSO-d 6, δ ppm): 175.42 (C S), 147.56 (ArC), 137.28 (C N), 136.91, 133.46, 132.54, 131.17, 129.02, 128.75, 128.43, 126.12, 125.89, 124.38, 123.60 (ArC).
5.1.5. 5-Nitrothiophene-2-carboxaldeyde-N(4)phenyl thiosemicarbazone (4)
Yellow solid (methanol). Yield: 82%. m.p.: 190–192 °C. Anal. calc. (C12H10N4O2S2): C, 47.04; H, 3.29; N, 18.29; S, 20.93; found: C, 47.22; H, 3.34; N, 18.28; S, 20.79%. ES-MS (m/z) 306 [M+]. UV/Vis λ max(nm): 281, 433. IR ν max(cm−1): 3342, 3140 (N–H); 1595 (C N); 1348 (–NO2); 1162 (C–N); 1110 (N–N); 825 (C S). 1H NMR (DMSO-d 6, δ ppm): 12.19 (s, 1H, CSNHN); 10.19 (s, 1H, PhNH); 8.31 (s,1H, HC N); 8.11 (d, 1H, J = 4.36 Hz, C3 proton of thiophene ring); 7.62 (d, 1H, J = 4.40 Hz, C4 proton of thiophene ring); 7.51 (d, 2H, J = 7.42 Hz, ArH ortho position); 7.38 (t, 2H, J 1 = 7.55 Hz ve J 2 = 8.17 Hz, ArH meta position); 7.24 (t, 1H, J = 8.46 Hz, ArH para position). 13C NMR (DMSO-d 6, δ ppm): 175.84 (C S), 144.22 (ArC), 138.25 (C N), 137.29, 130.48, 129.36, 128.34, 127.98, 127.02, 126.13 (ArC).
5.1.6. Thiophene-2-carboxaldehyde-N(4)nitrophenyl thiosemicarbazone (5)
Yellow solid (methanol). Yield: 83%; m.p.: 188–190 °C. Anal. calc. (C12H10N4O2S2): C, 47.04; H, 3.29; N, 18.29; S, 20.93; found: C, 47.16; H, 3.25; N, 18.32; S, 20.91%. ES-MS (m/z) 306 [M+]. UV/Vis λ max(nm): 260, 353. IR: ν max(cm−1): 3360, 3142 (N–H); 1582 (C N); 1342 (–NO2); 1171 (C–N); 1108 (N–N); 800 (C S). 1H NMR (DMSO-d 6, δ ppm): 11.91 (s, 1H, CSNHN); 10.23 (s,1H, PhNH); 8.40 (s,1H, HC N); 8.25 (d, 2H, J = 7.15 Hz, ArH ortho position of nitro group); 8.04 (d, 2H, J = 7.11 Hz, ArH, meta position of nitro group); 7.74 (d, 1H, J = 4.99 Hz, C2 proton of thiophene ring); 7.58 (d, 1H, J = 3.60 Hz, C4 proton of thiophene ring); 7.17 (dd, 1H, J 1 = 3.64 Hz, J 2 = 4.93 Hz, C3 proton of thiophene ring). 13C NMR (DMSO-d 6, δ ppm): 176.21 (C S), 145.18 (ArC), 139.42 (C N), 138.76, 131.17, 130.49, 129.01, 128.00, 127.51, 126.88, 126.25 (ArC).
5.1.7. Preparation of Pt(II) and Pd(II) complexes (1a–5a): a general method
A solution of K2PtCl4 or K2PdCl4 (0.38 mmol) in 10 mL distilled water was added to appropriate thiosemicarbazone derivative (0.38 mmol) in 10 mL ethanol. The reaction mixture was stirred for 24 h at room temperature. The solid precipitated was filtered and washed with EtOH and diethyl ether and dried in vacou over silica gel.
5.1.8. Pt(C12H10N3S2)Cl2 (1a)
Orange solid (ethanol). Yield: 69%. m.p.: 212 °C (with decomposition). Anal. calc. (C12H10N3S2Cl2Pt): C, 27.38; H, 1.91; N, 7.98; S, 12.18; found: C, 27.43; H, 1.98; N, 7.82; S, 12.67%. ES-MS (m/z) 526 [M+]. UV/Vis λ max(nm): 265, 371. IR ν max(KBr, cm−1): 3272 (N-H); 1510 (C N); 1095 (N-N); 748 (C S); 521 (Pt–N); 400 (Pt–S). 1H NMR (DMSO-d 6, δ ppm): 9.94 (s, 1H, PhNH); 8.48 (s, 1H, HC N); 8.00–6.92 (m, 8H, ArH).
5.1.9. Pt(C12H9ClN3S2)Cl2 (2a)
Dark yellow solid (ethanol). Yield: 53%. m.p.: 243 °C (with decomposition). Anal. calc. (C12H9N3S2Cl3Pt): C, 25.65; H, 1.79; N, 7.48; S, 11.41; found: C, 25.43; H, 1.58; N, 7.82; S, 11.67%. ES-MS (m/z) 561 [M+]. UV/Vis λ max(nm): 264, 386. IR ν max(KBr, cm−1): 3298 (N-H); 1524 (C N); 1088 (N–N); 747 (C–S); 513 (Pt–N); 402 (Pt–S). 1H NMR (DMSO-d 6, δ ppm): 9.85 (s, 1H, PhNH); 8.62 (s, 1H, HC N); 7.92–6.93 (m, 7H, ArH).
5.1.10. Pt(C18H14N3S2)Cl2 (3a)
Orange solid (ethanol). Yield: 56%. m.p.: 256 °C (with decomposition). Anal. calc. (C18H14N3S2Cl2Pt): C, 35.89; H, 2.34; N, 6.97; S, 10.64; found: C, 35.62; H, 2.02; N, 6.73; S, 10.48%. ES-MS (m/z) 602 [M+]. UV/Vis λ max(nm): 272, 403. IR ν max(KBr, cm−1): 3288 (N–H); 1528 (C N); 1049 (N–N); 791 (C–S); 507 (Pt–N); 418 (Pt–S). 1H NMR (DMSO-d 6, δ ppm): 9.81 (s, 1H, PhNH); 8.62 (s, 1H, HC N); 7.78–6.98 (m, 12H, ArH).
5.1.11. Pt(C12H9N4O2S2)Cl2 (4a)
Dark brown solid (ethanol). Yield: 52%. m.p.: 220 °C (with decomposition). Anal. calc. (C12H9N4O2S2Cl2Pt): C, 25.23; H, 1.59; N, 9.81; S, 11.22; found: C, 25.43; H, 1.19; N, 9.66; S, 11.83%. ES-MS (m/z) 571 [M+]. UV/Vis λ max(nm): 295, 467. IR ν max(KBr, cm−1): 3320 (N-H); 1565 (C N); 1046 (N–N); 807 (C–S); 518 (Pt–N); 411 (Pt-S). 1H NMR (DMSO-d 6, δ ppm): 10.12 (s, 1H, PhNH); 8.63 (s, 1H, HC N); 8.18–7.02 (m, 7H, ArH).
5.1.12. Pt(C12H9N4O2S2)Cl2 (5a)
Orange solid (ethanol). Yield 62%. m.p.: 225 °C (with decomposition). Anal. calc. (C12H9N4O2S2Cl2Pt): C, 25.23; H, 1.59; N, 9.81; S, 11.22; found: C, 25.61; H, 1.65; N, 9.96; S, 11.43%. ES-MS (m/z) 571 [M+]. UV/Vis λ max(nm): 344, 419. IR ν max(KBr, cm−1): 3289 (N–H); 1533 (C N); 1172 (N–N); 776 (C–S); 516 (Pt–N); 409 (Pt–S). 1H NMR (DMSO-d 6, δ ppm): 10.69 (s, 1H, PhNH); 9.09 (s, 1H, HC N); 8.12–7.64 (m, 7H, ArH).
5.1.13. Pd(C12H10N3S2)Cl2 (1b)
Brown solid (ethanol). Yield: 58%. m.p.: 272 °C (with decomposition). Anal. calc. (C12H10N3S2Cl2Pd): C, 32.93; H, 2.30; N, 9.60; S, 14.65; found: C, 32.27; H, 2.35; N, 9.53; S, 14.45%. ES-MS (m/z) 438 [M+]. UV/Vis λ max(nm): 262, 368. IR ν max(KBr, cm−1): 3242 (N-H); 1527 (C N); 1073 (N–N); 717 (C–S); 518 (Pd–N); 403 (Pd–S). 1H NMR (DMSO-d 6, δ ppm): 9.83 (s, 1H, PhNH); 8.66 (s, 1H, HC N); 8.02–7.09 (m, 8H, ArH).
5.1.14. Pd(C12H9ClN3S2)Cl2 (2b)
Brown solid (ethanol). Yield: 62%. m.p.: 250 °C (with decomposition). Anal. calc. (C12H9N3S2Cl3Pd): C, 30.53; H, 1.92; N, 8.90; S, 13.58; found: C, 30.28; H, 1.81; N, 8.62; S, 13.85%. ES-MS (m/z) 438 [M–Cl]+. UV/Vis λ max(nm): 261, 372. IR ν max(KBr, cm−1): 3205 (N–H); 1533 (C N); 1105 (N–N); 748 (C–S); 511 (Pd–N); 405 (Pd–S). 1H NMR (DMSO-d 6, δ ppm): 9.80 (s, 1H, PhNH); 8.55 (s, 1H, HC N); 7.68–7.18 (m, 7H, ArH).
5.1.15. Pd(C18H14N3S2)Cl2 (3b)
Brown solid (ethanol). Yield: 59%. m.p.: 307 °C (with decomposition). Anal. calc. (C18H14N3S2Cl2Pd): C, 42.08; H, 2.75; N, 8.18; S, 12.48; found: C, 42.19; H, 2.82; N, 8.33; S, 12.82%. ES-MS (m/z) 514 [M+]. UV/Vis λ max(nm): 270, 389. IR ν max(KBr, cm−1): 3241 (N-H); 1527 (C N); 1072 (N–N); 749 (C–S), 502 (Pd–N), 412 (Pd–S). 1H NMR (DMSO-d 6, δ ppm): 9.79 (s, 1H, PhNH); 8.64 (s, 1H, HC N); 8.00–7.33 (m, 12H, ArH).
5.1.16. Pd(C12H9N4O2S2)Cl2 (4b)
Dark brown solid (ethanol). Yield: 54%. m.p.: 302 °C (with decomposition). Anal. calc. (C12H9N4O2S2Cl2Pd): C, 29.86; H, 1.88; N, 11.61; S, 13.29; found: C, 29.43; H, 1.89; N, 11.66; S, 13.72%. ES-MS (m/z) 483 [M+]. UV/Vis λ max(nm): 294, 452. IR ν max(KBr, cm−1): 3255 (N-H); 1538 (C N); 1320 (–NO2); 1099 (N–N); 778 (C–S), 510 (Pd–N), 408 (Pd–S). 1H NMR (DMSO-d 6, δ ppm): 10.27 (s, 1H, PhNH); 8.36 (s, 1H, HC N); 8.31–7.20 (m, 7H, ArH).
5.1.17. Pd(C12H9N4O2S2)Cl2 (5b)
Brown solid (ethanol). Yield 61%. m.p.: 296 °C (with decomposition). Anal. calc. (C12H9N4O2S2Cl2Pd): C, 29.86; H, 1.88; N, 11.61; S, 13.29; found: C, 29.67; H, 1.75; N, 11.84; S, 13.34%. ES-MS (m/z) 483 [M+]. UV/Vis λ max(nm): 335, 412. IR ν max(KBr, cm−1): 3262 (N–H); 1547 (C N); 1329 (–NO2); 1181 (N–N); 769 (C–S), 509 (Pd–N), 410 (Pd–S). 1H NMR (DMSO-d 6, δ ppm): 10.26 (s, 1H, PhNH); 8.91 (s, 1H, HC N); 8.38–7.22 (m, 7H, ArH).
5.2. Biological assays
The antiviral assays were based on the inhibition of virus-induced cytopathicity in confluent cell cultures, and the cytostatic assays on inhibition of tumor cell proliferation in exponentially growing tumor cell cultures according to previously described methodology [55], [56], [57].
5.2.1. Antiviral activity assays
The antiviral assays, other than the anti-HIV assays, were based on inhibition of virus-induced cytopathicity in HEL [herpes simplex virus type 1 (HSV-1) (KOS), HSV-2 (G), vaccinia virus, vesicular stomatitis virus, cytomegalovirus (HCMV) and varicella-zoster virus (VZV)], Vero (parainfluenza-3, reovirus-1, Sindbis virus and Coxsackie B4), HeLa (vesicular stomatitis virus, Coxsackie virus B4, and respiratory syncytial virus), MDCK [influenza A (H1N1; H3N2) and influenza B] or CrFK (feline coronavirus (FIPV) and feline herpes virus) cell cultures. Confluent cell cultures (or nearly confluent for MDCK cells) in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) in the presence of varying concentrations (100, 20, 4, … μM) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. The minimal cytotoxic concentration (MCC) of the compounds was defined as the compound concentration that caused a microscopically visible alteration of cell morphology. The inhibitory concentration-50 (IC50) was defined as the compound concentration required to inhibit cell proliferation by 50%. The methodology of the anti-HIV assays was as follows: human CEM (∼3 × 105 cells/cm3) cells were infected with 100 CCID50 of HIV(IIIB) or HIV-2(ROD)/ml and seeded in 200 μL wells of a microtiter plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37 °C, HIV-induced CEM giant cell formation was examined microscopically. Antiviral activity was expressed as the concentration required to reduce virus-induced cytopathogenicity by 50% (EC50).
5.2.2. Cytostatic/toxic activity assays
Cytostatic activity against CEM (human T-lymphocytes) and HeLa (human cervix carcinoma) cell lines were measured essentially as originally described for the mouse leukemia cell lines [8]. Human lymphocyte CEM and human cervix carcinoma HeLa cells were seeded in 96-well microtiter plates at 75,000 (CEM) or 20,000 (HeLa) cells per 200 μL-well in the presence of different concentrations of the test compounds. After 3 (CEM) or 4 (HeLa) days, the viable cell number was counted using a Coulter counter apparatus. The 50% cytostatic concentration (CC50) was defined as the compound concentration required to inhibit tumor cell proliferation by 50%.
Cytotoxicity measurements were based on the inhibition of MDCK, HEL, CRFK, Vero and HeLa cell growth. Cells were seeded at 5 × 103 cells/well into 96-well microtiter plates. Then, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37 °C, the alteration of morphology of the cell cultures was recorded microscopically. Cytotoxicity was expressed as minimum cytotoxic concentration (MCC) or the compound concentration that causes a microscopically detectable alteration of normal cell morphology.
Acknowledgments
This work was supported by Scientific Research Projects Governing Unit Council of Scientific Research Projects (Grant no. FEF. 09.09), Gaziantep, Turkey and the Concerted Action of the K.U. Leuven (GOA no. 10/014). We thank Mrs. Leentje Persoons, Mrs. Frieda De Meyer, Mrs. Anita Camps, Mrs. Lies Van den Heurck, Mr. Steven Carmans, Mrs. Leen Ingels, Mrs. Kristien Erven and Mr. Kris Uyttersprot for excellent technical asistance.
References
- 1.Rosenberg B., Van Camp L., Trosko J.E., Mansour V.H. Nature. 1969;222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 2.Orvig C., Abrams M.J. Chem. Rev. 1999;99:2201–2203. doi: 10.1021/cr980419w. [DOI] [PubMed] [Google Scholar]
- 3.Kovala-Demertzi D., Boccarelli A., Demertzis M.A., Coluccia M. Chemotherapy. 2007;53:148–152. doi: 10.1159/000099986. [DOI] [PubMed] [Google Scholar]
- 4.Sakurai H., Kojima Y., Yoshikawa Y., Kawabe K., Yasui H. Coord. Chem. Rev. 2002;226:187–198. [Google Scholar]
- 5.Louie A.Y., Meade T.J. Chem. Rev. 1999;99:2711–2734. doi: 10.1021/cr9804285. [DOI] [PubMed] [Google Scholar]
- 6.Rosu T., Pahontu E., Pasculescu S., Georgescu R., Stanica N., Curaj A., Popescu A., Leabu M. Eur. J. Med. Chem. 2010;45:1627–1634. doi: 10.1016/j.ejmech.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Kovala-Demertzi D., Papageorgiou A., Papathanasis L., Alexandratos A., Dalezis P., Miller J.R., Demertzis M.A. Eur. J. Med. Chem. 2009;44:1296–1302. doi: 10.1016/j.ejmech.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Rebolledo A.P., Vieites M., Gambino D., Piro O.E., Castellano E.E., Zani C.L., Souza-Fagundes E.M., Teixeira L.R., Batista A.A., Beraldo H. J. Inorg. Biochem. 2005;99:698–706. doi: 10.1016/j.jinorgbio.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 9.Kostova I. Recent Pat. Anticancer Drug Discov. 2006;1:1–22. doi: 10.2174/157489206775246458. [DOI] [PubMed] [Google Scholar]
- 10.Iakovidou Z., Papageorgiou A., Demertzis M.A., Mioglou E., Mourelatos D., Kotsis A., Nath Yadav P., Kovala-Demertzi D. Anti-Cancer Drugs. 2001;12:65–70. doi: 10.1097/00001813-200101000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Huang R., Wallqvist A., Covell D.G. Biochem. Pharmacol. 2005;69:1009–1039. doi: 10.1016/j.bcp.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Galanski M., Jakupec M.A., Keppler B.K. Curr. Med. Chem. 2005;12:2075–2094. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 13.Gielen M., Tiekink E.R.T. Metallotherapeutic drugs and metal-based diagnostic agents: the use of metals in medicine. Wiley; 2005. [Google Scholar]
- 14.Karki S.S., Thota S., Darj S.Y., Balzarini J., De Clercq E. Bioorg. Med. Chem. 2007;15:6632–6641. doi: 10.1016/j.bmc.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 15.Deegan C., Coyle B., McCann M., Devereux M., Egan D. Chem. Biol. Interact. 2006;164:115–125. doi: 10.1016/j.cbi.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 16.Afrasiabi Z., Sinn E., Chen J., Ma Y., Rheingold A.L., Zakharov L.N., Rath N., Padhye S. Inorg. Chim. Acta. 2004;357:271. [Google Scholar]
- 17.Alderden R.A., Hall M.D., Hambley T.W. J. Chem. Ed. 2006;83:728–734. [Google Scholar]
- 18.Gomez A., Quiroga C., Navarro Ranninger Coord. Chem. Rev. 2004;248:119. [Google Scholar]
- 19.Nath Yadav P., Demertzis M.A., Kovala-Demertzi D., Skoulika S., West D.X. Inorg. Chim. Acta. 2003;349:30. [Google Scholar]
- 20.Wong E., Giandomenico C.M. Chem. Rev. 1999;99:2451. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 21.Hambley T.W. Coord. Chem. Rev. 1997;166:181–223. [Google Scholar]
- 22.Stordal B., Pavlakis N., Davey R. Cancer Treat. Rev. 2007;33:688–703. doi: 10.1016/j.ctrv.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Gojo I., Tidwell M.L., Greer J., Takebe N., Seiter K., Pochron M.F., Johnson B., Sznol M., Karp J.E. Leuk. Res. 2007;31:1165–1173. doi: 10.1016/j.leukres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Quiroga A.G., Perez J.M., Lopez-Solera I., Montero E.I., Masaguer J.R., Alonso C., Navarro-Ranninger C. J. Inorg. Biochem. 1998;69:275–281. doi: 10.1016/s0162-0134(98)00003-8. [DOI] [PubMed] [Google Scholar]
- 25.Kovala-Demertzi D., Demertzis M.A., Filiou E., Pantazaki A.A., Yadav P.N., Miller J.R., Zheng Y., Kyriakidis D.A. Biometals. 2003;16:411–418. doi: 10.1023/a:1022543718598. [DOI] [PubMed] [Google Scholar]
- 26.Dilovic I., Rubbic M., Vrdoljak V., Pavelic S.K., Kralj M., Piantanida I., Cindric M. Bioorg. Med. Chem. 2008;16:5189–5198. doi: 10.1016/j.bmc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Finch R.A., Liu M.C., Gril S.P., Rose W.C., Loomis R., Vasquez K.M., Cheng Y.C., Sartorelli A.C. Biochem. Pharmacol. 2000;59:983–991. doi: 10.1016/s0006-2952(99)00419-0. [DOI] [PubMed] [Google Scholar]
- 28.Domagk G., Behnisch R., Mietzsch F., Schmidt H. Naturwissenschaften. 1946;56:315. [Google Scholar]
- 29.Kasuga N.C., Sekino K., Ishikawa M., Honda A., Yokoyama M., Nakano S., Shimada N., Koumo C., Nomiya K. J. Inorg. Biochem. 2003;96:298–310. doi: 10.1016/s0162-0134(03)00156-9. [DOI] [PubMed] [Google Scholar]
- 30.Feun L., Modiano M., Lee K., Mao J., Marini A., Savaraji N., Plezia P., Almassian B., Colacino E., Fischer J., MacDonal S. Cancer Chemother. Pharmacol. 2002;50:223–229. doi: 10.1007/s00280-002-0480-0. [DOI] [PubMed] [Google Scholar]
- 31.Bharti N., Husain K., Gonzalez Garza M.T., Cruz-Vega D.E., Castro-Garza J., Benito D., Cardenas M., Naqvi F., .Azam A. Bioorg. Med. Chem. Lett. 2002;12:3475–3478. doi: 10.1016/s0960-894x(02)00703-5. [DOI] [PubMed] [Google Scholar]
- 32.Oliveira R., Souza-Fagundes E.M., Soares R.P., Andrade A.A., Krettli A.U., Zani C.L. Eur. J. Med. Chem. 2008;43:1983–1988. doi: 10.1016/j.ejmech.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Abid M., Agarwal S.M., Azam A. Eur. J. Med. Chem. 2008;43:2035–2039. doi: 10.1016/j.ejmech.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 34.Quenelle D.C., Keith K.A., Kern E.R. Antivir. Res. 2006;71:24–30. doi: 10.1016/j.antiviral.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Vieites M., Otero L., Santos D., Toloza J., Figueroa R., Normbuena E., Olea-Azar C., Aguirre G., Cerecetto H., Gonzalez M., Morello A., Maya J.D., Garat B., .Gambino D. J. Inorg. Biochem. 2008;102:1033–1043. doi: 10.1016/j.jinorgbio.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 36.Yogeeswari P., Sriram D., Jit L.R.J.S., Kumar S.S., Stables J.P. Eur. J. Med. Chem. 2002;37:231–236. doi: 10.1016/s0223-5234(02)01338-7. [DOI] [PubMed] [Google Scholar]
- 37.Vieites M., Otero L., Santos D., Olea-Azar C., Norambuena E., Aguirre G., Cerecetto H., Gonzalez M., Kemmerling U., Morello A., Maya J.D., Gambino D. J. Inorg. Biochem. 2009;103:411–418. doi: 10.1016/j.jinorgbio.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Vermeulen M., Zwanenburg B., Chittenden G., Verhagen H. Eur. J. Med. Chem. 2003;38:729–737. doi: 10.1016/s0223-5234(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 39.Rineh A., Mahmood N., Abdollahi M., Foroumad A., Sorkhi M., Sharfiee A. Archiv der Pharmazie Chem. Life Sci. 2007;340:409–415. doi: 10.1002/ardp.200700045. [DOI] [PubMed] [Google Scholar]
- 40.Krishnan K., Prathiba K., Jayaprakash V., Basu A., Mishra N., Zhou B., Hu S., Yen Y. Bioorg. Med. Chem. Lett. 2008;18:6248–6250. doi: 10.1016/j.bmcl.2008.09.097. [DOI] [PubMed] [Google Scholar]
- 41.Wiles M., Suprunchuk T. J. Med. Chem. 1971;14(3):252–254. doi: 10.1021/jm00285a023. [DOI] [PubMed] [Google Scholar]
- 42.Raposo M.M.M., Garcia-Acosta B., Abalos T., Calero P., Martinez-Manez R., Ros-Lis J.V., Soto J. J. Org. Chem. 2010;75:2922–2933. doi: 10.1021/jo100082k. [DOI] [PubMed] [Google Scholar]
- 43.Husain K., Abid M., Azam A. Eur. J. Med. Chem. 2007;42:1300–1308. doi: 10.1016/j.ejmech.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 44.Papathanasis L., Demertzis M.A., Yadav P.N., Kovala-Demertzi D., Prentjas C., Castineiras A., Skoulika S., West D.X. Inorg. Chim. Acta. 2004;357:4113–4120. [Google Scholar]
- 45.Li Y., Yang Z.-Y., Wu J.-C. Eur. J. Med. Chem. 2010;45:5692–5701. doi: 10.1016/j.ejmech.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 46.Vazquez-Garcia D., Fernandez A., Fernandez J.J., Lopez-Torres M., Suarez A., Ortigueira J.M., Vila J.M., Adams H. J. Organomet. Chem. 2000;595:199–207. [Google Scholar]
- 47.Gambino D., Otero L., Vieites M., Boiani M., Gonzalez M., Baran E.J., Cerecetto H. Spectrochim. Acta A. 2007;68:341–348. doi: 10.1016/j.saa.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 48.Husain K., Bhat A.R., Azam A. Eur. J. Med. Chem. 2008;43:2016–2028. doi: 10.1016/j.ejmech.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 49.Hernandez W., Paz J., Vaisberg A., Spodine E., Richter R., Beyer Bioinorg L. Chem. Appl. 2008;6:1–9. doi: 10.1155/2008/690952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suni V., Kurup M.R.P., Nethaji M. Spectrochim. Acta Part A. 2006;63:174–181. doi: 10.1016/j.saa.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Singh S., Bharti N., Naqvi F., Azam A. Eur. J. Med. Chem. 2004;39:459–465. doi: 10.1016/j.ejmech.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Finkielsztein L.M., Castro E.F., Fabian L.E., Moltrasio G.Y., Campos R.H., Cavallaro L.V., Moglioni A.G. Eur. J. Med. Chem. 2008;43:1767–1773. doi: 10.1016/j.ejmech.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 53.Matesanz A.I., Souza P. J. Inor. Biochem. 2007;101:1354–1361. doi: 10.1016/j.jinorgbio.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 54.Bharti N., Shailendra, Sharma S., Naqvi F., Azam A. Bioorg. Med. Chem. 2003;11:2923–2929. doi: 10.1016/s0968-0896(03)00213-x. [DOI] [PubMed] [Google Scholar]
- 55.Manta S., Tzioumaki N., Tsoukala E., Panagiotopoulou A., Pelecanou M., Balzarini J., Komiotis D. Eur. J. Med. Chem. 2009;44:4764–4771. doi: 10.1016/j.ejmech.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 56.Tsoukala E., Tzioumaki N., Manta S., Riga A., Balzarini J., Komiotis D. Bioorg. Chem. 2010;38:285–293. doi: 10.1016/j.bioorg.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dzimbeg G., Zorc B., Kralj M., Ester K., Pavelic K., Andrei G., Snoeck R., Balzarini J., De Clercq E., Mintas M. Eur. J. Med. Chem. 2008;43:1180–1187. doi: 10.1016/j.ejmech.2007.09.001. [DOI] [PubMed] [Google Scholar]