

Graphical abstract
Keywords: Derivatives of taxchinin A and brevifoliol, Synthesis, Cytotoxicity, SAR
Abstract
Twenty-one derivatives of taxchinin A (1) and brevifoliol (2) were synthesized and evaluated for cytotoxicity against human non-small lung cancer (A549) cell line. Nine derivatives showed potent activity with IC50 values from 0.48 to 6.22 μM. 5-Oxo-13-TBDMS-taxchinin A (11) and 5-oxo-13,15-epoxy-13-epi-taxchinin A (15) are the most potent derivatives, with IC50 at 0.48 and 0.75 μM, respectively. The structure–activity relationship (SAR) of these compounds established that exocyclic unsaturated ketone at ring C is the key structural element for the activity, while the α,β-unsaturated ketone positioned at ring A has no effect for the activity. The significant cytotoxicity of derivatives 11 and 15 may be due to the conformational change in the taxane rings. The 3D-QSAR study was conducted on this series of compounds, which provided optimal predictive comparative molecular field (CoMFA) model with cross-validated r2 (q2) value of 0.64.
1. Introduction
Paclitaxel, a well-known antitumor drug with novel mechanism of action, has spurred the isolation of more than 400 taxoids from plants of genus Taxus, of which include large number of 11(15 → 1)-abeo-taxoids with 5/7/6 skeleton.1, 2 Most of modification and study of SAR of taxoids were focused on paclitaxel and its analogs with 6/8/6 skeleton and an oxetane ring.3 However, the knowledge base on the modification and biological activities of taxoids with 5/7/6 skeleton still calls for continuing enrichment. Taxchinin A (1), isolated from Taxus chinensis, was the first naturally occurring taxoid to be correctly assigned as the A-nortaxane skeleton,4 although brevifoliol (2) was the first natural taxoid with the A-nortaxane skeleton to be isolated from the leaves of T. brevifolia, it was incorrectly assigned to have a taxa-4(20),11-diene system.5 We have also isolated taxchinin A and brevifoliol from the leaves and stems of T. chinensis and they were found to be the most abundant taxoids in the leaves and stem of this plant (Fig. 1 ).
Figure 1.
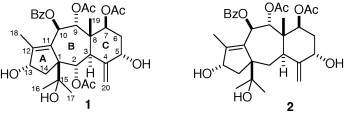
Structure of taxchinin A (1) and brevifoliol (2).
In the previous publication, brevifoliol was reported to show cytotoxicity against CaCo2, MCF-7, KB-403, and COLO-320DM cells.6 And a series of brevifoliol derivatives were prepared, which included the introduction of acetyl, troc, N-benzoyl-(2′R,3′S)-3′-phenyl isoserine, and TES groups at C-5, C-13. However, all of this analogs showed little bioactivity.7, 8 Taxchinin A was also reported to have some cytotoxicity against KB cells,9 but no any modification of taxchinin A could be traced.
Michael reaction acceptors are the functionalities in which the olefins or acetylenes conjugated to electron-withdrawing groups. Michael reaction acceptors are considered as a class of biological activity molecules, directly or indirectly involved in the life processes, are regulators in many signaling pathway in cells, and have played an important role in chemical biology studies. One Michael acceptor CRA-3316 developed to treat Chagas’s disease is in clinical trials.10 Another Michael acceptor AG7088 (Ruprintrivir) has entered phase II clinical studies with human as a potential nasally delivered antirhinoviral agent.11, 12, 13 Moreover, Michael acceptor inhibitors based on the structure of AG7088 drew intense attention in the treatment of SARS (severe acute respiratory syndrome).14 Now Michael acceptors are of great medical interest and have great potential for use as drugs.
Because of the novel skeleton and abundance in the plant, taxchinin A and brevifoliol were then selected as the precursors for the synthesis of taxoids with α,β-unsaturated ketone moiety. And these derivatives, as the Michael reaction acceptors, are proposed to show bioactivity. Therefore the derivatives of taxchinin A and brevifoliol, consisting of the various unsaturated ketones at ring A and ring C, were designed and synthesized. The total 21 synthetic compounds, accompanying the parent compounds 1 and 2, were subjected to a cytotoxic screening of human non-small lung cancer (A549) cell line. The SAR on these compounds was discussed and the CoMFA analysis was performed using the cytotoxic data. The synthetic procedures and the biological assay results are also provided herein.
2. Results and discussion
2.1. Synthesis
Synthesis of derivatives of taxchinin A and brevifoliol (3–23) is shown in Scheme 1, Scheme 2 . In Scheme 1, oxidation of 1 with pyridinium dichromate (PDC) in CH2Cl2 for 1 h selectively produced the 13-oxo-taxchinin A (3), while 5,13-dioxo-taxchinin A (4) could be obtained after 24 h. Compound 2 is very similar to 1 except for the absence of acetoxy group at C-2, however, the oxidation of 2 with PDC in CH2Cl2 is much different from that of 1. Oxidation of 2 with PDC for 1 h gave 13-oxo-brevifoliol (5), whereas 5 accompanying 6 and rearranged product 7 were obtained after 24 h. The rearrangement involving the PDC oxidation of 2 was first reported, and its mechanism was difficult to be explained. All attempts to prepare 5,13-dioxo-brevifoliol were unsuccessful.
Scheme 1.
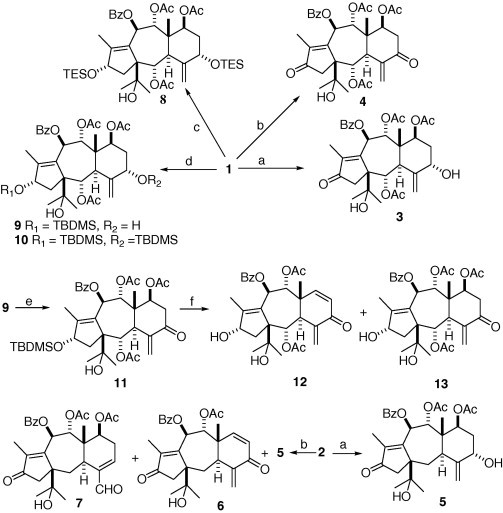
Reagents and conditions: (a) PDC, CH2Cl2, 1 h; (b) PDC, CH2Cl2, 24 h; (c) TESCl, pyridine; (d) TBDMSCl, imidazole, DMF; (e) PDC, CH2Cl2; (f) HF-pyridine, pyridine.
Scheme 2.

Reagents and conditions: (a) TsCl, pyridine; (b) PDC, CH2Cl2; (c) NaH, THF; (d) TESCl, pyridine.
To obtain 5-oxo-taxchinin A (13), strategy of selective protection of OH-13 was employed. Although attempt to prepare 13-TES derivative with chlorotriethylsilane (TESCl) gave only 5,13-bis (TES)-taxchinin A (8), 13-TBDMS-taxchinin A (9) was obtained, along with the byproduct of 5,13-bis (TBDMS)-taxchinin A (10), by treatment with tert-butylchlorodimethylsilane (TBDMSCl) in imidazole and N,N-dimethylformamide (DMF). Further oxidation of 9 with PDC yielded the 5-oxo-13-TBDMS derivative 11 which was treated with the HF-pyridine in pyridine to produce the desired 5-oxo taxchinin A (13) accompanying compound 12 (Scheme 1).
13,15-Epoxy-13-epi-brevifoliol (16) synthesized from 2 was reported,9 which inspired us to prepare 5-oxo-13,15-epoxy-13-epi-taxchinin A (15) and 5-oxo-13,15-epoxy-13-epi-brevifoliol. Treatment of 1 and 2 with tosylchloride (TsCl) yielded 14 and 16, respectively. Further oxidation of 14 with PDC afforded desired product 15, while oxidation of 16 gave the unexpected derivative 17 rather than 5-oxo-13,15-epoxy-13-epi-brevifoliol (Scheme 2).
During our study on the chemistry of 11-(15 → 1)-abeo-taxanes, we found that the 2-hydroxyl-isopropyl group at C-1 of 13-oxo-11-(15 → 1)-abeo-taxanes could be eliminated with sodium hydride (NaH) in THF, which was then employed in our experiment to access more taxoids with unsaturated ketone unit. Reacting 4 with NaH in THF, the elimination of 2-hydroxyl-isopropyl group at C-1 and acetoxy group at C-7 occurred, producing 18 and 19. Treatment of 3 with NaH in THF gave 20 and 21, while treating the 5-TES-13-oxo-taxchinin A with NaH yielded 22. Further oxidation of 20 produced 18 and 23 (Scheme 2).
2.2. Cytotoxicity and structure–activity relationship
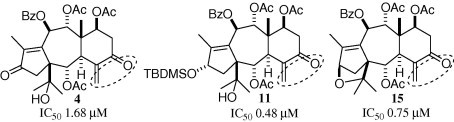
The cytotoxicity of all compounds (1–23) was evaluated against A549 cell line, and the anticancer drug cisplatin (DDP) was used as a reference compound. As shown in Table 1 , nine derivatives exhibited remarkable activity with IC50 values from 0.48 to 6.22 μM. Taxchinin A and its 13-oxo derivative 3 showed no activity, while 5,13-dioxo derivative 4 and 5-oxo derivative 13 exhibited potent activity with IC50 values of 1.68 and 3.16 μM, respectively. Moreover, 5,13-bis (TES) derivative 8, 13-TBDMS derivative 9, and 5,13-bis (TBDMS) derivative 10 showed no activity, but 13-TBDMS-5-oxo derivative 11 exhibited potent activity with IC50 at 0.48 μM. Compound 14, possessing a C-13 (15) tetrahydrofuran ring moiety, exhibited no activity, however, its 5-oxo derivative 15 showed potent activity with IC50 of 0.75 μM.
Table 1.
Cytotoxicity of taxchinin A, brevifoliol and their derivatives against A549 cell linea
| Compound | IC50 (μM) | Compounds | IC50 (μM) |
|---|---|---|---|
| 1 | 60.50 | 13 | 3.16 |
| 2 | NA | 14 | NA |
| 3 | 91.62 | 15 | 0.75 |
| 4 | 1.68 | 16 | 46.47 |
| 5 | NA | 17 | 6.17 |
| 6 | 6.22 | 18 | 2.80 |
| 7 | 31.62 | 19 | 2.48 |
| 8 | 87.33 | 20 | 21.52 |
| 9 | 12.39 | 21 | 80.72 |
| 10 | NA | 22 | 17.37 |
| 11 | 0.48 | 23 | 11.27 |
| 12 | 5.89 | DDP | 2.10 |
NA, not active; IC50 values greater than 100 μM were considered as inactive and represented as NA.
As taxchinin A and its derivative 3, brevifoliol and its 13-oxo derivative 5 were inactive. When the 5-OH of brevifoliol was oxidized to carbonyl group (6), it was found to have activity. Moreover, 13,15-epoxy derivative 16 showed no activity while its oxidized product 17 exhibited marginal activity.
Other derivatives of taxchinin A and brevifoliol, such as 5-oxo-6-ene 12, 5,13-dioxo-1,6-diene 18, and 5,13-dioxo-6-ene 19, showed moderate to good activity, while compounds 7 and 20–23 without the exocyclic unsaturated ketone at ring C showed no activity.
These results indicated that exocyclic unsaturated ketone at ring C is the key structural element for the activity, and all derivatives containing this unit presented potent activity, while the α,β-unsaturated ketone positioned at ring A has no effect for the activity. Moreover, introduction of more double bonds in the molecule gave no remarkable advantage for the activity, and certain derivative possessing the unsaturated aldehyde group at ring C showed weak activity.
In addition, it should be noticed that 5-oxo-13-TBDMS-taxchinin A (11) and 5-oxo-13,15-epoxy-13-epi-taxchinin A (15) are the most potent derivatives among this series of compounds, with IC50 at 0.48 and 0.75 μM, respectively, while 5-oxo-taxchinin A (13) showed only six- and four-fold less active than 11 and 15, with IC50 of 3.16 μM. The significant cytotoxicity of the 5-oxo-13-substituted taxchinin A 11 and 15 may be due to the conformational change in the taxane rings as reflected in certain key J-coupling. The coupling constant between H-2 and H-3 is an important descriptor of the shape of the taxoids, which serves as a reporter of the dihedral angle along the bottom portion of the B ring.15, 16, 17 Among all obtained taxoids (1–23), only compounds 11 and 15 showed similar coupling constant between H-2 and H-3 (Table 2 ). In compound 11 the J 2–3 is 7.6 Hz; in 15 it is 7.1 Hz, which suggested that 11 and 15 might have the similar conformations which are different from those of the other derivatives.
Table 2.
Comparision of the coupling constants between H-2 and H-3 of 11 and 15 in acetone-d6 with those of other obtained taxoids
| Compound | J2–3 (Hz) | Compounds | J2–3 (Hz) |
|---|---|---|---|
| 1 | 9.0 | 13 | 8.2 |
| 2 | br d | 14 | 8.4 |
| 3 | 9.2 | 15 | 7.1 |
| 4 | 8.6 | 16 | br s |
| 5 | br s | 17 | 9.5 |
| 6 | 8.7 | 18 | s |
| 7 | s | 19 | 8.2 |
| 8 | 9.1 | 20 | br s |
| 9 | br s | 21 | s |
| 10 | br s | 22 | s |
| 11 | 7.6 | 23 | s |
| 12 | 8.1 |
2.3. CoMFA analysis
In this study, 18 compounds were employed for the CoMFA analysis. For 3D-QSAR analyses, 13 compounds (unasterisked molecules in Table 3 ) were selected as training set for model construction, and the remaining 5 compounds (asterisked molecules in Table 3) as testing set for model validation.
Table 3.
Predicted activities (PA) from CoMFA models compared with the experimental activities (EA) and the residues (δ)
| Compound | EA | CoMFA |
|
|---|---|---|---|
| PA | δ | ||
| 1 | 4.22 | 4.14 | 0.08 |
| 3 | 4.04 | 4.09 | −0.05 |
| 4 | 5.77 | 5.73 | 0.04 |
| 6a | 5.21 | 4.80 | 0.41 |
| 7 | 4.50 | 4.53 | −0.03 |
| 8 | 4.06 | 4.11 | −0.05 |
| 9 | 4.91 | 4.87 | 0.04 |
| 11 | 6.32 | 6.33 | −0.01 |
| 12 | 5.23 | 5.33 | −0.10 |
| 13a | 5.50 | 5.06 | 0.44 |
| 15 | 6.12 | 6.11 | 0.01 |
| 16a | 4.33 | 4.71 | −0.38 |
| 17 | 5.21 | 5.24 | −0.03 |
| 18 | 5.55 | 5.63 | −0.08 |
| 19 | 5.61 | 5.50 | 0.11 |
| 20a | 4.67 | 4.49 | 0.18 |
| 22 | 4.76 | 4.69 | 0.07 |
| 23a | 4.95 | 4.44 | 0.51 |
Compounds of the testing set.
The inhibition constants were expressed in PIC50 values (PIC50 = −log [IC50]) and correlated with the steric and electrostatic fields (CoMFA) of each compound in training set. The CoMFA results are summarized in Table 4 . The cross-validation with leave-one-out option and the SAMPLS program was carried out to obtain the optimal number of components to be used in the final analysis. After the optimal number of components (four) was determined, a non-cross-validated analysis was performed. The q 2 (cross-validated r 2 of 0.64), SPRESS (cross-validated standard error of prediction of 0.56), r 2 (non-cross-validated r 2 of 0.99), and F values (272.66) were computed according to the definitions in SYBYL. These statistical indexes are reasonably high, indicating that the CoMFA model has a strong predictive ability. The plots of the experimental results versus predicted values of 13 studied compounds are shown in Figure 2 A. The relative contribution to this CoMFA model was 39.5% for the steric field, 60.5% for electrostatic field.
Table 4.
Summary of results from the CoMFA analyses based on the training set and all 18 compounds
| CoMFA |
||
|---|---|---|
| Analyses based on training set | Analyses based on all 18 compounds | |
| PLS statistics | ||
| q2 (CV correlation coefficient) | 0.642 | 0.540 |
| N (number of components) | 4 | 4 |
| S (stand error of prediction) | 0.558 | 0.529 |
| r2 (correlation coefficient) | 0.993 | 0.969 |
| F (F-ratio) | 272.663 | 101.627 |
| Field distribution (%) | ||
| Steric | 39.5 | 42.6 |
| Electrostatic | 60.5 | 57.4 |
Figure 2.
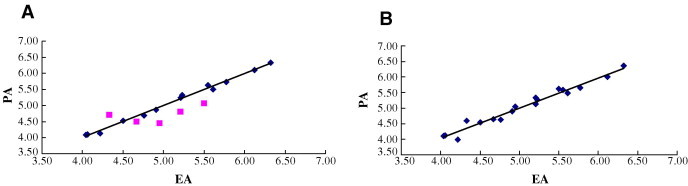
(A) Predicted activities (PA) by CoMFA models versus experimental activities (EA) for the compounds in training set and test set.  , compounds of the training set;
, compounds of the training set;  , compounds of the test set. (B) PA by CoMFA models versus EA. The plot report data obtained via a CoMFA model built using all 18 compounds in Table 3.
, compounds of the test set. (B) PA by CoMFA models versus EA. The plot report data obtained via a CoMFA model built using all 18 compounds in Table 3.
To evaluate the predictive ability of this model, we subsequently calculated the PIC50 values for the five compounds in the test set. As it can be seen in Table 3 and Figure 2A, the calculated values using the constructed CoMFA model are in good agreement with experimental data, and the model exhibits a good predictive ability (r 2 = 0.61).
To evaluate whether the results are biased toward the selected training sets, we have also performed additional CoMFA studies in which all 18 compounds were included in analysis. The resulting cross-validated r 2 is 0.54, with similar relative contributions of the steric, electrostatic fields (Table 4 and Fig. 2B).
The CoMFA result is usually represented as 3D contour maps. It shows regions where variations of steric and electrostatic nature in the structural features of the different molecules contained in the training set lead to increase or decrease in the activity. The CoMFA steric field is presented as contour plots in Figure 3 A. To aid in visualization, compound 11 is displayed in the map as reference structure. A huge sterically favorable region (Fig. 3A, green contour) is located near the atoms C-13, C-14, and C-2, suggesting that bulky groups in this area would increase the cytotoxicity. The yellow region indicates that the bulky substitute near the atoms C-4 and C-5 is not favorable to the activity. CoMFA electrostatic contour map is shown in Figure 3B. To aid in visualization, compound 15 is displayed in the map as reference structure. There is a big blue polyhedron near the atoms C-14, C-1, C-2, indicating that more positive charges in this region should play a favorable role in improving cytotoxicity. There are two small red polyhedra surrounding atom C-5, suggesting that negatively charged substituents in this area lead to an increase of cytotoxicity.
Figure 3.
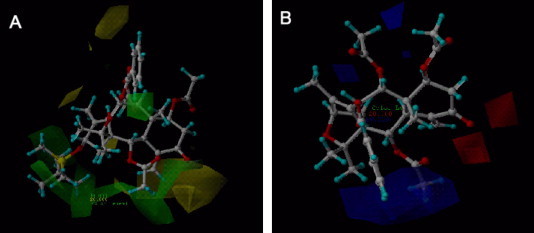
(A) CoMFA steric contour plot; green contours indicate regions where bulky groups increase activity, whereas yellow contours indicate regions where bulky groups decrease activity. (B) CoMFA electrostatic contour plots; blue contours indicate regions where positive charge increases activity, whereas red contours indicate regions where negative charge increases activity.
3. Conclusion
Twenty-one derivatives of taxchinin A and brevifoliol were synthesized and subjected to the screening of A549 tumor cell line. Nine derivatives 4, 6, 11–13, 15, and 17–19 exhibited remarkable activity with IC50 values from 0.48 to 6.22 μM. The observed SAR of these derivatives is very interesting which shows that exocyclic unsaturated ketone at ring C is the key structural element for the activity, while the α,β-unsaturated ketone positioned at ring A has no effect for the activity. Moreover, 5-oxo-13-substituted taxchinin A 11 and 15 are the most potent derivatives among this series of compounds with IC50 of 0.48 and 0.75 μM, respectively, which may be due to the conformational change in the taxane rings, as reflected in the key J-coupling between H-2 and H-3. Further investigation on the SAR utilizing a CoMFA method was carried out. The resulting 3D-QSAR model provides an invaluable tool to design more cytotoxic compounds. With the employment of all of these results, further design, synthesis, and cytotoxic evaluation are ongoing in our laboratory and the results will be reported in due course.
4. Experimental
4.1. General experimental procedures
1H and 13C NMR experiments were performed on a Bruker AM-400 spectrometer at ambient temperature. 2D NMR spectra were recorded on Bruker DRX-500 NMR instrument. IR spectra were recorded on a Bio-Rad FTS-135 spectrometer with KBr pellets. UV spectra were obtained on a UV 2401 PC spectrometer. ESIMS and HRESIMS were taken on a VG Auto Spec-3000 or on a Finnigan MAT 90 instrument. Melting points were determined (uncorrected) on an XRC-1 micro melting point apparatus. Optical rotations were measured with a Horiba SEPA-300 polarimeter. Column chromatography was performed on silica gel (Qingdao Marine Chemical Inc., China).
4.2. In vitro cytotoxicity assay
Exponentially growing human non-small lung cancer cells (A459, obtained from ATCC, 4 × 104/mL) were, respectively, made into the 96-well microplate. Cultures were pre-incubated for 72 h in 37 °C and 5% CO2 incubator. After that, 10 μL controlled or test solution of compounds (100, 10, 1, and 0.1 μM) was put into each well and the plate was incubated for an additional 72 h. At the end of exposure, the cells were fixed by the addition of 50 μL of cold 50% trichloroacetic acid (TCA) at 4 °C for one hour. After washing with tap water, cells of each well were stained with a 0.4% 50 μL SRB (sulforhodamin B) solution in 1% acetic acid for 30 min. Then cultures were rinsed with 1% acetic acid. At last, 10 mM unbuffered Tris solution (150 μL) was added to each well. The OD was then read on a plate reader at a wavelength of 570 nm.
4.3. Synthesis
4.3.1. 13-Oxo-taxchinin A (3)
A solution of taxchinin A 1 (500 mg, 0.81 mmol) in dichloromethane (40 mL) was treated with pyridinium dichromate (PDC, 3.68 g, 9.72 mmol) and stirred at room temperature for 1 h. The reaction mixture was filtered and purified through the column chromatography (petroleum/ethyl acetate, 6.5:3.5) to afford 3 (438 mg, 87.8%) as white amorphous powder: 55.8 (c 0.71, CH3OH); UV (CH3OH) λ max (log ε) 233.8 (4.25) nm; IR (KBr) ν max 3413, 2954, 2925, 1744, 1731, 1696, 1453, 1374, 1240, 1113, 1033, 719 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 6.05 (1H, d, J = 9.2 Hz, H-2β), 2.93 (1H, d, J = 9.2 Hz, H-3α), 4.65 (1H, br s, H-5β), 4.82 (1H, t, J = 9.0 Hz, H-7α), 5.08 (1H, d, J = 3.1 Hz, H-9β), 6.08 (1H, d, J = 3.1 Hz, H-10α), 1.13 (3H, s, H-16), 0.74 (3H, s, H-17), 2.09 (3H, s, H-18), 1.93 (3H, s, H-19), 4.85 (1H, s, H-20a), 5.49 (1H, s, H-20b), 7.97 (2H, d, J = 7.3Hz, Ho-Ph), 7.45 (2H, t, J = 7.6 Hz, Hm-Ph), 7.58 (1H, t, J = 6.9 Hz, Hp-Ph), 2.04 (3H, s, O2CCH3), 1.89 (3H, s, O2CCH3), 1.86 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 65.6 (C, C-1), 69.3 (CH, C-2), 46.1 (CH, C-3), 148.3 (C, C-4), 65.6 (CH, C-5), 36.6 (CH2, C-6), 71.7 (CH, C-7), 44.3 (C, C-8), 75.3 (CH, C-9), 71.4 (CH, C-10), 162.2 (C, C-11), 148.0 (C, C-12), 207.4 (C, C-13), 45.0 (CH2, C-14), 75.9 (C, C-15), 27.1 (CH3,C-16), 28.9 (CH3,C-17), 9.4 (CH3, C-18), 15.2 (CH3, C-19), 114.3 (CH2,C-20), 166.6 (C, O2CC6H5), 131.3 (C, i-O2CC6H5), 130.4 (CH2, o-O2CC6H5), 129.2 (CH2, m-O2CC6H5), 133.7 (CH2, p-O2CC6H5), 170.5 (C, O2CCH3), 170.4 (C, O2CCH3), 170.2 (C, O2CCH3), 21.6 (CH3, O2CCH3), 20.9 (CH3, O2CCH3), 20.7 (CH3, O2CCH3); HRESIMS m/z 635.2466 [M+Na+] (calcd for C33H40O11Na, 635.2468).
4.3.2. 5,13-Dioxotaxchinin A (4)
A solution of taxchinin A 1 (500 mg, 0.81 mmol) in dichloromethane (40 ml) was treated with pyridinium dichromate (PDC, 3.68 g, 9.72 mmol) and stirred at room temperature for 24 h. The reaction mixture was filtered and purified through the column chromatography (petroleum/ethyl acetate, 7.5:2.5) to afford 4 (382 mg, 77.0%) as white amorphous powder: ; UV (CH3OH) λ max (log ε) 233.8 (4.25) nm; IR (KBr) ν max 3523, 2943, 2906, 1745, 1707, 1453, 1372, 1238, 1107, 1049, 1028, 715 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 6.02 (1H, d, J = 8.6 Hz, H-2β), 3.26 (1H, d, J = 8.6 Hz, H-3α), 5.17 (1H, m, H-7α), 5.27 (1H, br s, H-9β), 6.22 (1H, br s, H-10α), 1.21 (3H, s, H-16), 0.82 (3H, s, H-17), 2.08 (3H, s, H-18), 1.90 (3H, s, H-19), 5.30 (1H, s, H-20a), 5.87 (1H, s, H-20b), 8.02 (2H, d, J = 7.6 Hz, Ho-Ph), 7.49 (2H, t, J = 7.5 Hz, Hm-Ph), 7.62 (1H, t, J = 7.4 Hz, Hp-Ph), 2.00 (3H, s, O2CCH3), 1.95 (3H, s, O2CCH3), 1.92 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 62.5 (C, C-1), 69.6 (CH, C-2), 43.4 (CH, C-3), 144.8 (C, C-4), 197.9 (C, C-5), 39.2 (CH2, C-6), 71.5 (CH, C-7), 43.9 (C, C-8), 75.1 (CH, C-9), 70.0 (CH, C-10), 162.0 (C, C-11), 147.9 (C, C-12), 206.5 (C, C-13), 44.8 (CH2, C-14), 75.9 (C, C-15), 27.6 (CH3, C-16), 29.2 (CH3, C-17), 9.5 (CH3, C-18), 13.6 (CH3, C-19), 128.1 (CH2, C-20), 166.7 (C, O2CC6H5), 131.3 (C, i-O2CC6H5), 130.5 (d, o-O2CC6H5), 129.3 (CH, m-O2CC6H5), 133.8 (CH, p-O2CC6H5), 170.3 (C, O2CCH3), 170.2 (C, O2CCH3), 21.9 (CH3, O2CCH3), 20.8 (CH3, O2CCH3), 20.7 (CH3, O2CCH3); HRESIMS m/z 633.2315 [M+Na+] (calcd for C33H38O11Na, 633.2311).
4.3.3. 13-Oxo-brevifoliol (5)
The title compound was synthesized from 2 in 85.1% yield by the same procedure as that for 3: white amorphous powder, (petroleum/ethyl acetate, 7.5:2.5); ; UV (CH3OH) λ max (log ε) 232.2 (4.29) nm; IR (KBr) ν max 3447, 2976, 2937, 1730, 1712, 1630, 1451, 1372, 1260, 1108, 1067, 1028, 748 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 2.86 (1H, br s, H-3α), 4.31 (1H, br s, H-5β), 5.65 (1H, dd,J = 11.4, 5.1 Hz, H-7α), 6.17 (1H, d, J = 3.3 Hz, H-9β), 6.69 (1H, d, J = 3.3 Hz, H-10α), 1.19 (3H, s, H-16), 1.36 (3H, s, H-17), 1.89 (3H, s, H-18), 0.87 (3H, s, H-19), 4.83 (1H, s, H-20a), 5.11 (1H, s, H-20b), 7.98 (2H, br s, Ho-Ph), 7.48 (2H, t, 7.7, Hm-Ph), 7.62 (1H, t, 7.2, Hp-Ph), 2.00 (3H, s, O2CCH3) 1.99 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 59.8 (C, C-1), 28.1 (CH2, C-2), 37.7 (CH, C-3), 151.9 (C, C-4), 72.5 (CH, C-5), 37.2 (CH2, C-6), 70.4 (CH, C-7), 46.0 (C, C-8), 77.9 (CH, C-9), 71.3 (CH, C-10), 166.5 (C, C-11), 146.9 (C, C-12), 209.7 (C, C-13), 50.3 (CH2, C-14), 75.6 (C, C-15), 27.0 (CH3, C-16), 22.9 (CH3, C-17), 9.1 (CH3, C-18), 13.1 (CH3, C-19), 110.4 (CH2, C-20), 165.0 (C, O2CC6H5), 131.3 (C, i-O2CC6H5), 130.4 (CH, o-O2CC6H5), 129.6 (CH, m-O2CC6H5), 134.3 (CH, p-O2CC6H5), 170.3 (C, O2CCH3), 170.0 (C, O2CCH3), 21.3 (CH3, O2CCH3), 20.9 (CH3, O2CCH3); HRESIMS m/z 577.2429 [M+Na+] (calcd for C31H38O9Na, 577.2413).
4.3.4. Oxidation of 2 to compounds 5, 6 and 7
Brevifoliol (2) was treated with pyridinium dichromate (PDC) by the same procedure as that for 4 to produce the mixture which was purified through the column chromatography (petroleum/ethyl acetate, 8:2), affording 5 (8.7%), 6 (18.9%), and 7 (24.4%).
4.3.4.1. Compound 6
White amorphous powder: ; UV (CH3OH) λ max (log ε) 201.0 (4.25) nm; IR (KBr) ν max 3420, 2956, 2938, 1757, 1730, 1719, 1641, 1452, 1372, 1239, 1219, 1110, 1025,738, 709 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 2.97 (1H, d, J = 8.7 Hz, H-3α), 6.04 (1H, d, J = 10.5 Hz, H-6), 7.49 (1H, overlap), 5.97 (1H, d, J = 10.1 Hz, H-9β), 6.31 (1H, d, J = 10.1 Hz, H-10α), 1.12 (3H, s, H-16), 1.29 (3H, s, H-17), 1.72 (3H, s, H-18), 1.50 (3H, s, H-19), 5.46 (1H, s, H-20a), 6.06 (1H, s, H-20b), 8.03 (2H, d, J = 6.7 Hz, Ho-Ph), 7.49 (2H, overlap, Hm-Ph), 7.63 (1H, t, J = 7.4, Hp-Ph), 2.08 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 59.3 (C, C-1), 28.2 (CH2, C-2), 46.5 (CH, C-3), 147.2 (C, C-4), 189.0 (C, C-5), 129.8 (CH, C-6), 155.3 (CH, C-7), 44.9 (C, C-8), 76.1 (CH, C-9), 70.7 (CH, C-10), 165.8 (C, C-11), 146.8 (C, C-12), 206.7 (C, C-13), 50.6 (CH2, C-14), 76.3 (C, C-15), 27.6 (CH3, C-16), 27.1 (CH3, C-17), 8.86 (CH3, C-18), 20.6 (CH3, C-19), 118.4 (CH2, C-20), 164.7 (C, O2CC6H5), 130.7 (C, i-O2CC6H5), 130.6 (CH, o-O2CC6H5), 129.4 (CH, m-O2CC6H5), 134.2 (CH, p-O2CC6H5), 170.0 (C, O2CCH3), 21.6 (CH3, O2CCH3); HRESIMS m/z 515.2036 [M+Na+] (calcd for C29H32O7Na, 515.2045).
4.3.4.2. Compound 7
Yellow amorphous powder: ; UV (CH3OH) λ max (log ε) 231.0 (4.28) nm; IR (KBr) ν max 3446, 2973, 2927, 1744, 1725, 1709, 1636, 1585, 1451, 1371, 1245, 1103, 1027, 712 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 2.65 (1H, s, H-3α), 6.88 (1H, d, J = 5.8 Hz, H-5), 4.91 (1H, dd, J = 10.1, 5.9 Hz, H-7α), 5.26 (1H, d, J = 4.5 Hz, H-9β), 6.17 (1H, d, J = 4.5 Hz, H-10α), 0.82 (3H, s, H-16), 1.14 (3H, s, H-17), 1.82 (3H, s, H-18), 1.62 (3H, s, H-19), 9.43 (1H, s, H-20), 8.01 (2H, d, J = 7.2 Hz, Ho-Ph), 7.47 (2H, t, J = 7.7 Hz, Hm-Ph), 7.60 (1H, t, J = 7.4 Hz, Hp-Ph), 1.94 (3H, s, O2CCH3), 1.93 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 58.9 (C, C-1), 28.5 (CH2, C-2), 35.2 (CH2, C-3), 145.4 (C, C-4), 148.5 (CH, C-5), 27.2 (CH2, C-6), 72.0 (CH, C-7), 44.2 (C, C-8), 74.6 (CH, C-9), 72.1 (CH, C-10), 166.8 (C, C-11), 146.3 (C, C-12), 208.0 (C, C-13), 48.7 (CH2, C-14), 76.0 (C, C-15), 26.3 (CH3, C-16), 27.8 (CH3, C-17), 9.43 (CH3, C-18), 13.6 (CH3, C-19), 194.5 (CH, C-20), 164.8 (C, O2CC6H5), 131.8 (C, i-O2CC6H5), 130.5 (d, o-O2CC6H5), 129.2 (CH, m-O2CC6H5), 133.5 (CH, p-O2CC6H5), 170.7 (C, O2CCH3), 170.4 (C, O2CCH3), 21.0 (CH3, O2CCH3), 20.8 (CH3, O2CCH3); HRESIMS m/z 575.2251 [M+Na+] (calcd for C31H36O9Na, 575.2257).
4.3.5. 5,13-Bis (TES) taxchinin A (8)
Chlorotriethylsilane (22.06 μL, 0.13 mmol) was added to a solution of 1 (40 mg, 0.065 mmol) in dry pyridine (3 ml). The reaction mixture was stirred at room temperature and monitored by TLC until all starting material was consumed. The reaction mixture was then diluted with ethyl acetate and washed with diluted hydrochloric acid and water. The organic layer was dried (Na2SO4) and evaporated. The residue was purified by column chromatography (chloroform/ethyl acetate, 9.5:0.5) to give 8 (40.3 mg, 73.4%) as colorless oil: ; UV (CH3OH) λ max (log ε) 201.8 (4.24); IR (KBr) ν max 3572, 2954, 2924, 1739, 1655, 1456, 1372, 1269, 1248, 1091, 1069, 1029, 744, 712 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 6.03 (1H, d, J = 9.1 Hz, H-2β), 3.58 (1H, d, J = 9.1 Hz, H-3α), 4.53 (1H, br s, H-5β), 5.63 (1H, m, H-7α), 6.12 (1H, d, J = 10.4 Hz, H-9β), 6.57 (1H, d, J = 10.4 Hz, H-10α), 4.63 (1H, m, H-13β), 1.24 (3H, s, H-16), 1.28 (3H, s, H-17), 2.04 (3H, s, H-18), 1.21 (3H, s, H-19), 4.53 (1H, s, H-20a), 5.12 (1H, s, H-20b), 7.90 (2H, d, J = 6.8 Hz, Ho-Ph), 7.48 (2H, t, J = 7.5 Hz, Hm-Ph), 7.61 (1H, t, J = 7.5 Hz, Hp-Ph), 2.04 (3H, s, O2CCH3), 2.03 (3H, s, O2CCH3), 2.01 (3H, s, O2CCH3), 0.68 [12H, J = 7.9 Hz, 2Si(CH2CH3)3], 0.99 [18H, J = 7.9 Hz, 2Si(CH2CH3)3]; 13C NMR (acetone-d 6, 100 MHz) δ 68.1 (C, C-1), 68.7 (CH, C-2), 42.6 (CH, C-3), 146.6 (C, C-4), 75.4 (CH, C-5), 39.7 (CH2, C-6), 69.8 (CH, C-7), 45.8 (C, C-8), 77.1 (CH, C-9), 70.2 (CH, C-10), 133.3 (C, C-11), 153.0 (C, C-12), 77.6 (CH, C-13), 42.1 (CH2, C-14), 76.1 (C, C-15), 26.9 (CH3, C-16), 28.5 (CH3, C-17), 12.3 (CH3, C-18), 14.1 (CH3, C-19), 111.2 (CH2, C-20), 164.9 (C, O2CC6H5), 130.8 (C, i-O2CC6H5), 130.5 (CH, o-O2CC6H5), 129.4 (CH, m-O2CC6H5), 134.0 (CH, p-O2CC6H5), 171.7 (C, O2CCH3), 170.1 (C, O2CCH3), 169.7 (C, O2CCH3), 20.8 (CH3, O2CCH3), 21.7 (CH3, O2CCH3), 21.4 (CH3, O2CCH3), 5.32 (CH2, SiCH2), 5.13 (CH2, SiCH2), 7.24 (CH3, SiCH2CH3), 7.18 (CH3, SiCH2CH3); HRESIMS m/z 865.4500 [M+Na+] (calcd for C49H70O11NaSi2, 865.4354).
4.3.6. 13-TBDMS taxchinin A (9) and 5,13-bis (TBDMS) taxchinin A (10)
Compound 1 (400 mg, 0.65 mmol) was dissolved in N,N-dimethylformamide (3 mL). To this solution was added tert-butylchlorodimethylsilane (589.13 mg, 2.55 mmol) and imidazole (89.6 mg, 1.30 mmol). The reaction mixture was stirred for 5 h at room temperature when all starting material was consumed. The mixture was diluted with ethyl acetate and washed with water and brine. The organic layer was then dried (Na2SO4) and concentrated. The residue was purified by column chromatography (petroleum/ethyl acetate, 9:1) to give 9 (110.9 mg, 23%) and10 (120.0 mg, 22%).
4.3.6.1. Compound 9
White amorphous powder: ; UV (CH3OH) λ max (log ε) 201.6 (4.31) nm; IR (KBr) ν max 3459, 2955, 2930, 1739, 1645, 1370, 1247, 1161, 1104, 1066, 1020, 780, 712 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.98 (1H, br s, H-2β), 3.24 (1H, br s, H-3α), 4.20 (1H, m, H-5β), 5.49 (1H, br s, H-7α), 6.09 (1H, br s, H-9β), 6.56 (1H, br s, H-10α), 4.67 (1H, br s, H-13β), 1.19 (3H, s, H-16), 1.28 (3H, s, H-17), 2.05 (3H, s, H-18), 1.14 (3H, s, H-19), 4.48 (1H, s, H-20a), 5.06 (1H, s, H-20b), 7.90 (2H, d, J = 7.0 Hz, Ho-Ph), 7.49 (2H, t, J = 7.6 Hz, Hm-Ph), 7.61 (1H, t, J = 7.4 Hz, Hp-Ph), 2.08 (3H, s, O2CCH3), 2.04 (3H, s, O2CCH3), 2.03 (3H, s, O2CCH3), 0.16 (6H, s, SiCH3), 0.95 (9H, s, Si(CH3)2C(CH3)3); 13C NMR (acetone-d 6, 100 MHz) δ 69.9 (C, C-1), 65.8 (CH, C-2), 45.5 (CH, C-3), 147.1 (C, C-4), 71.6 (CH, C-5), 36.7 (CH2, C-6), 70.9 (CH, C-7), 44.2 (C, C-8), 75.1 (CH, C-9), 71.0 (CH, C-10), 132.3 (C, C-11), 153.6 (C, C-12), 78.6 (CH, C-13), 42.3 (CH2, C-14), 74.5 (C, C-15), 26.2 (CH3, C-16), 28.2 (CH3, C-17), 13.5 (CH3, C-18), 14.1 (CH3, C-19), 113.1 (CH2, C-20), 166.0 (C, O2CC6H5), 131.3 (C, i-O2CC6H5), 130.4 (CH, o-O2CC6H5), 129.6 (CH, m-O2CC6H5), 133.8 (CH, p-O2CC6H5), 170.6 (C, O2CCH3), 170.4 (C, O2CCH3), 170.0 (C, O2CCH3), 21.7 (CH3, O2CCH3), 20.9 (CH3, O2CCH3), 20.7 (CH3, O2CCH3), −4.6 (CH3, SiCH3), −4.0 [CH3, Si(CH3)2C(CH3)3]; HRESIMS m/z 751.3477 [M+Na+] (calcd for C39H56O11NaSi, 751.3489).
4.3.6.2. Compound 10
White amorphous powder: ; UV (CH3OH) λ max (log ε) 230.0 (4.27) nm; IR (KBr) ν max 3440, 2956, 2931, 1740, 1645, 1370,1246, 1091, 1066, 1030, 777, 711 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.95 (1H, br s H-2β), 3.27 (1H, br s, H-3α), 4.31 (1H, br s, H-5β), 5.49 (1H, br s, H-7α), 6.08 (1H, br s, H-9β), 6.56 (1H, br s, H-10α), 4.68 (1H, t, 6.8, H-13β), 1.19 (3H, s, H-16), 1.28 (3H, s, H-17), 2.08 (3H, s, H-18), 1.14 (3H, s, H-19), 4.57 (1H, s, H-20a), 5.13 (1H, s, H-20b), 7.99 (2H, d, J = 6.9 Hz, Ho-Ph), 7.49 (2H, t, J = 7.6 Hz, Hm-Ph), 7.61 (1H, t, J = 7.4 Hz, Hp-Ph), 1.95 (3H, s, O2CCH3), 1.92 (3H, s, O2CCH3), 1.88 (3H, s, O2CCH3), 0.16 (6H, s, SiCH3), 0.11 (6H, s, SiCH3), 0.95 (9H, s, Si(CH3)2C(CH3)3), 0.92 (9H, s, Si(CH3)2C(CH3)3); 13C NMR (acetone-d 6, 100 MHz) δ 69.9 (C, C-1), 67.1 (CH, C-2), 45.1 (CH, C-3), 147.8 (C, C-4), 71.4 (CH, C-5), 37.9 (CH2, C-6), 70.9 (CH, C-7), 44.1 (C, C-8), 74.9 (CH, C-9), 71.1 (CH, C-10), 132.1 (C, C-11), 152.1 (C, C-12), 78.4 (CH, C-13), 42.3 (CH2, C-14), 75.7 (C, C-15), 26.2 (CH3, C-16), 28.2 (CH3, C-17), 13.5(CH3, C-18), 15.1 (CH3, C-19), 113.8 (CH2, C-20), 166.0 (C, O2CC6H5), 131.3 (C, i-O2CC6H5), 130.4 (CH, o-O2CC6H5), 129.3 (CH, m-O2CC6H5), 133.7 (CH, p-O2CC6H5), 171.7 (C, O2CCH3), 170.4 (C, O2CCH3), 170.0 (C, O2CCH3), 22.0 (CH3, O2CCH3), 20.9 (CH3, O2CCH3), 20.7 (CH3, O2CCH3), −4.6 (CH3, SiCH3), −4.5 (CH3, SiCH3), −4.0 [CH3, Si(CH3)2C(CH3)3], −3.8 [CH3, Si(CH3)2C(CH3)3]; HRESIMS m/z 865.4499 [M+Na+] (calcd for C45H70O11NaSi2, 865.4354).
4.3.7. 5-Oxo-13-TBDMS taxchinin A (11)
The title compound was synthesized from 9 in 88% yield by the same procedure as that for 3: white amorphous powder, (petroleum/ethyl acetate, 9:1); ; UV (MeOH) λ max (log ε) 201.6 (4.31) nm; IR (KBr) ν max 3466, 2955, 2930, 1748, 1602, 1365, 1240, 1088, 1027, 776, 713 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.89 (1H, d, J = 7.6 Hz, H-2β), 3.40 (1H, d, J = 7.6 Hz, H-3α), 5.10 (1H, m, H-7α), 5.19 (1H, br s, H-9β), 6.15 (1H, br s, H-10α), 4.75 (1H, t, J = 6.7 Hz, H-13β), 1.19 (3H, s, H-16), 1.28 (3H, s, H-17), 2.08 (3H, s, H-18), 0.97 (3H, s, H-19), 5.13 (1H, s, H-20a), 5.85 (1H, s, H-20b), 8.01 (2H, d, J = 7.5 Hz, Ho-Ph), 7.49 (2H, t, J = 7.7 Hz, Hm-Ph), 7.61 (1H, t, J = 7.4 Hz, Hp-Ph), 1.99 (3H, s, O2CCH3), 1.94 (3H, s, O2CCH3), 1.90 (3H, s, O2CCH3), 0.17 (6H, s, SiCH3), 0.94 (9H, s, Si(CH3)2C(CH3)3); 13C NMR (acetone-d 6, 100 MHz) δ 66.4 (C, C-1), 70.0 (CH, C-2), 42.8 (CH, C-3), 145.6 (C, C-4), 198.0 (C, C-5), 39.2 (CH2, C-6), 71.2 (CH, C-7), 43.8 (C, C-8), 74.7 (CH, C-9), 70.9 (CH, C-10), 132.4 (C, C-11), 151.8 (C, C-12), 78.9 (CH, C-13), 42.1 (CH2, C-14), 75.5 (C, C-15), 26.2 (CH3, C-16), 28.5 (CH3, C-17), 13.5 (CH3, C-18), 18.5 (CH3, C-19), 126.5 (CH2, C-20), 166.1 (C, O2CC6H5), 131.3 (C, i-O2CC6H5), 130.5 (CH, o-O2CC6H5), 129.3 (CH, m-O2CC6H5), 133.7 (CH, p-O2CC6H5), 170.3 (C, O2CCH3), 170.0 (C, O2CCH3), 170.0 (C, O2CCH3), 22.0 (C, O2CCH3), 20.9 (C, O2CCH3), 20.8 (C, O2CCH3), −4.6 (CH3, SiCH3), −4.0 (CH3, Si(CH3)2C(CH3)3); HRESIMS m/z 749.3351 [M+Na+] (calcd for C39H54O11NaSi, 749.3333).
4.3.8. Conversion of compound 11 to 12 and 13
A solution of 11 (60 mg, 0.083 mmol) in tetrahydrofuran was treated with a 70% solution of hydrogen fluoride in pyridine (0.46 mL) and stirred for 2 h at room temperature. The reaction mixture was diluted with ethyl acetate and the reaction was quenched with aqueous sodium bicarbonate (10 mL). The organic layer was separated, washed with aqueous copper(II) sulfate and brine, dried (Na2SO4), and concentrated. The residue was purified by column chromatography (petroleum/ethyl acetate, 8:2) to give 12 (16.3 mg, 32%) and 13 (30.0 mg, 45.6%).
4.3.8.1. Compound 12
Yellow oil: ; UV (CH3OH) λ max (log ε) 201.2 (4.25) nm; IR (KBr) ν max 3443, 2956, 2925, 1744, 1603, 1464, 1375, 1241, 1068, 1027, 748, 713 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.90 (1H, d, J = 8.1 Hz, H-2β), 3.46 (1H, d, J = 8.1 Hz, H-3α), 5.28 (1H, br s, H-7α), 5.28 (1H, br s, H-9β), 6.17 (1H, br s, H-10α), 4.58 (1H, m, H-13β), 1.17 (3H, s, H-16), 1.28 (3H, s, H-17), 2.08 (3H, s, H-18), 0.93 (3H, s, H-19), 5.17 (1H, s, H-20a), 5.85 (1H, s, H-20b), 8.01 (2H, d, J = 7.8 Hz, Ho-Ph), 7.49 (2H, t, J = 7.7 Hz, Hm-Ph), 7.61 (1H, t, J = 7.4 Hz, Hp-Ph), 1.98 (3H, s, O2CCH3), 1.93 (3H, s, O2CCH3), 1.89 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 65.9 (C, C-1), 70.0 (C, C-2), 42.6 (CH, C-3), 145.4 (C, C-4), 198.1 (C, C-5), 37.7 (CH2, C-6), 71.3 (CH, C-7), 43.7 (C, C-8), 74.9 (CH, C-9), 70.9 (CH, C-10), 134.1 (C, C-11), 153.0 (C, C-12), 77.7 (C, C-13), 42.0 (C, C-14), 75.7 (C, C-15), 27.7 (CH3, C-16), 28.3 (CH3, C-17), 12.0 (CH3, C-18), 14.3 (CH3, C-19), 127.1 (CH2, C-20), 166.0 (C, O2CC6H5), 131.2 (C, i-O2CC6H5), 130.5 (CH, o-O2CC6H5), 129.3 (CH, m-O2CC6H5), 133.8 (CH, p-O2CC6H5), 170.3 (C, O2CCH3), 170.2 (C, O2CCH3), 170.2 (C, O2CCH3), 22.0 (CH3, O2CCH3), 20.8 (CH3, O2CCH3), 20.0 (CH3, O2CCH3); HRESIMS m/z 635.2466 [M+Na+] (calcd for C33H40O11Na, 635.2468).
4.3.8.2. Compound 13
Yellow amorphous powder: ; UV (CH3OH) λ max (log ε) 201.2 (4.18) nm; IR (KBr) ν max 3442, 2976, 2937, 1729, 1712, 1634, 1604, 1451, 1372, 1261, 1108, 1067, 1028, 712 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 6.18 (1H, d, J = 8.2 Hz, H-2β), 4.06 (1H, d, J = 8.2 Hz, H-3α), 6.06 (1H, d, J = 10.1 Hz, H-6), 7.46 (1H, d, J = 10.1 Hz, H-7), 6.14 (1H, d, J = 10.7 Hz, H-9β), 6.38 (1H, d, J = 10.7 Hz, H-10α), 4.47 (1H, m, H-13β), 1.19 (3H, q, H-16), 1.29 (3H, q, H-17), 1.82 (3H, q, H-18), 1.78 (3H, q, H-19), 5.28 (1H, s, H-20a), 5.98 (1H, s, H-20b), 7.98 (2H, d, J = 7.25 Hz, Ho-Ph), 7.49 (2H, t, J = 7.7 Hz, Hm-Ph), 7.63 (1H, t, J = 7.4 Hz, Hp-Ph), 2.04 (3H, s, O2CCH3), 2.03 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 68.1 (C, C-1), 70.4 (CH, C-2), 48.2 (CH, C-3), 144.0 (C, C-4), 189.6 (C, C-5), 129.6 (CH, C-6), 154.9 (CH, C-7), 44.7 (C, C-8), 75.5 (CH, C-9), 70.7 (CH, C-10), 134.7 (C, C-11), 152.9 (C, C-12), 76.8 (CH, C-13), 41.7 (CH2, C-14), 76.2 (C, C-15), 27.2 (CH3, C-16), 28.4 (CH3, C-17), 12.0 (CH3, C-18), 20.5 (CH3, C-19), 120.2 (CH2, C-20), 165.7 (C, O2CC6H5), 130.6 (C, i-O2CC6H5), 130.3 (CH, o-O2CC6H5), 129.4 (CH, m-O2CC6H5), 134.1 (CH, p-O2CC6H5), 171.8 (CH, O2CCH3), 170.0 (C, O2CCH3), 21.8 (CH3, O2CCH3), 21.5 (CH3, O2CCH3). HRESIMS m/z 575.2270 [M+Na+] (calcd for C31H36O9Na, 575.2257).
4.3.9. Conversion of compound 1 to 14
Taxchinin A 1 (100 mg, 0.16 mmol) was dissolved in dry pyridine (4 mL). To this solution was added p-toluenesulfonyl chloride (42.6 mg, 0.372 mmol). The reaction mixture was stirred at room temperature and monitored by TLC until all starting material was consumed. The reaction mixture was diluted with ethyl acetate, and washed with diluted hydrochloric acid, water, and brine. The organic layer was then dried (Na2SO4) and concentrated in vacuo. The residue was purified by column chromatography (petroleum/ethyl acetate, 8:2) to afford 14 (50.38 mg, 51.9%) as yellow oil: ; UV (CH3OH) λ max (log ϵ) 201.2 (3.82) nm; IR (KBr) ν max 3424, 2956, 2853, 1743, 1728, 1463, 1376, 1243, 1090, 1067, 1027, 718 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.68 (1H, d, J = 8.4 Hz, H-2β), 3.25 (1H, d, J = 8.4 Hz, H-3α), 4.63 (1H, br s, H-5β), 4.87 (1H, t, J = 9.1 Hz, H-7α), 4.99 (1H, d, J = 4.3 Hz, H-9β), 6.26 (1H, d, J = 4.3 Hz, H-10α), 4.37 (1H, br s, H-13α), 1.28 (3H, s, H-16), 0.87 (3H, s, H-17), 1.87 (3H, s, H-18), 1.63 (3H, s, H-19), 4.83 (1H, s, H-20a), 5.54 (1H, s, H-20b), 8.05 (2H, d, J = 7.6 Hz, Ho-Ph), 7.56 (2H, t, J = 7.7 Hz, Hm-Ph), 7.67 (1H, t, J = 7.4 Hz, Hp-Ph), 2.05 (3H, s, O2CCH3), 2.03 (3H, s, O2CCH3), 1.95 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 68.4 (C, C-1), 65.9 (CH, C-2), 47.5 (CH, C-3), 148.1 (CH2, C-4), 71.7 (CH, C-5), 36.9 (CH2, C-6), 68.0 (CH, C-7), 43.7 (C, C-8), 74.4 (CH, C-9), 66.8 (CH, C-10), 134.2 (C, C-11), 153.5 (C, C-12), 82.2 (CH, C-13), 48.1 (CH2, C-14), 78.9 (C, C-15), 28.4 (CH3, C-16), 28.0 (CH3, C-17), 14.3 (CH3, C-18), 12.6 (CH3, C-19), 114.0 (CH3, C-20), 165.4 (C, O2CC6H5), 130.2 (C, i-O2CC6H5), 130.4 (CH, o-O2CC6H5), 129.7 (CH, m-O2CC6H5), 134.2 (CH, p-O2CC6H5), 170.7 (C, O2CCH3), 170.4 (C, O2CCH3), 169.9 (C, O2CCH3), 21.5 (CH3, O2CCH3), 20.9 (CH3, O2CCH3), 20.6 (CH3, O2CCH3); HRESIMS m/z 619.2807 [M+Na+] (calcd for C33H40O10Na, 619.2519).
4.3.10. Oxidation of 14 to 15
Compound 15 was synthesized from 14 in 92% yield by the same procedure as that for 3: yellow amorphous powder, (petroleum/ethyl acetate, 7:3); ; UV (CH3OH) λ max (log ϵ) 201.8 (4.19) nm; IR (KBr) ν max 2955, 2925, 1753, 1723, 1678, 1602, 1452, 1374, 1268, 1251, 1094, 1069, 1027, 716 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.64 (1H, d, J = 7.1 Hz, H-2β), 3.52 (1H, d, J = 7.1 Hz, H-3α), 5.19 (1H, m, H-7α), 5.13 (1H, d, J = 4.5 Hz, H-9β), 6.34 (1H, d, J = 4.5 Hz, H-10α), 4.42 (1H, br s, H-13α), 1.28 (3H, s, H-16), 0.77 (3H, s, H-17), 1.72 (3H, s, H-18), 1.19 (3H, s, H-19), 5.22 (1H, s, H-20a), 5.92 (1H, s, H-20b), 8.05 (2H, d, J = 7.4 Hz, Ho-Ph), 7.56 (2H, t, J = 7.6 Hz, Hm-Ph), 7.67 (1H, t, J = 7.3 Hz, Hp-Ph), 2.07 (3H, s, O2CCH3), 2.01 (3H, s, O2CCH3), 1.92 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 67.9 (C, C-1), 66.7 (CH, C-2), 45.3 (CH, C-3), 144.3 (C, C-4), 198.1 (C, C-5), 39.4 (CH2, C-6), 70.1 (CH, C-7), 43.1 (C, C-8), 73.7 (CH, C-9), 67.7 (CH, C-10), 133.8 (C, C-11), 152.9 (C, C-12), 47.7 (CH2, C-14), 79.0 (C, C-15), 29.1 (CH3, C-16), 27.7 (CH3, C-17), 13.5 (CH3, C-18), 12.3 (CH3, C-19), 128.2 (CH2, C-20), 165.5 (C, O2CC6H5), 130.4 (C, i-O2CC6H5), 130.37 (CH, o-O2CC6H5), 129.7 (CH, m-O2CC6H5), 134.4 (CH, p-O2CC6H5), 170.3 (C, O2CCH3), 170.2 (C, O2CCH3), 169.9 (C, O2CCH3), 21.7 (CH3, O2CCH3), 20.8 (CH3, O2CCH3), 20.6 (CH3, O2CCH3); HRESIMS m/z 617.2807 [M+Na+] (calcd for C33H38O10Na, 617.2362).
4.3.11. Conversion of compound 2 to 16
Compound 2 was treated with p-toluenesu- lfonyl chloride by the same procedure as that for 14 to produce crude mixture which was purified by column chromatography (petroleum/ethyl acetate, 8.5:1.5) to afford starting material 2 (52.1%) and compound 16 (41.2%) as colorless oil: ; UV (CH3OH) λ max (log ϵ) 202.0 (4.18) nm; IR (KBr) ν max 3431, 2954, 2922, 1726, 1630, 1463, 1377, 1249, 1111, 1065, 1032, 718 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 3.16 (1H, d, J = 10.9 Hz, H-3α), 4.37 (1H, s, H-5β), 5.64 (1H, dd, J = 12.0, 5.6 Hz, H-7α), 5.13 (1H, d, J = 6.1 Hz, H-9β), 6.28 (1H, d, J = 6.1 Hz, H-10α), 4.17 (1H, br s, H-13α), 1.38 (3H, s, H-16), 0.87 (3H, s, H-17), 1.87 (3H, s, H-18), 1.19 (3H, s, H-19), 4.93 (1H, s, H-20a), 5.12 (1H, s, H-20b), 8.07 (2H, d, J = 7.8 Hz, Ho-Ph), 7.53 (2H, t, J = 7.6 Hz, Hm-Ph), 7.65 (1H, t, J = 7.4 Hz, Hp-Ph), 2.04 (3H, s, O2CCH3), 2.03 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 62.6 (C, C-1), 23.3 (CH2, C-2), 37.2 (CH, C-3), 153.3 (C, C-4), 73.4 (CH, C-5), 32.6 (CH2, C-6), 71.4 (CH, C-7), 47.0 (C, C-8), 74.3 (CH, C-9), 68.0 (CH, C-10), 138.2 (C, C-11), 147.7 (C, C-12), 83.3 (CH, C-13), 52.1 (CH2, C-14), 79.8 (C, C-15), 26.8 (CH3, C-16), 26.0 (CH3, C-17), 12.1 (CH3, C-18), 12.0 (CH3, C-19), 111.4 (CH2, C-20), 165.9 (C, O2CC6H5), 130.8 (C i-O2CC6H5), 130.4 (CH o-O2CC6H5), 129.6 (CH, m-O2CC6H5), 134.1 (CH, p-O2CC6H5), 170.3 (C, O2CCH3), 170.1 (C O2CCH3), 20.9 (CH3, O2CCH3), 20.6 (CH3, O2CCH3); HRESIMS m/z 561.2449 [M+Na+] (calcd for C31H38O8Na, 561.2464).
4.3.12. Oxidation of 16 to 17
Compound 16 was treated with pyridinium dichromate (PDC) by the same procedure as that for 3 to produce crude mixture which was purified by column chromatography (petroleum/ethyl acetate, 8:2) to afford 17 (50.0%) as light yellow foam: ; UV (CH3OH) λ max (log ϵ) 229 (4.37) nm; IR (KBr) ν max 2973, 2926, 1744, 1723, 1677, 1627, 1451,1369, 1268, 1242, 1094, 1068, 1040, 1027, 712 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 3.26 (1H, d, J = 9.5 Hz, H-3α), 6.86 (1H, br s, H-5), 4.95 (1H, dd, J = 10.0, 5.9 Hz, H-7α), 5.17 (1H, d, J = 4.8 Hz, H-9β), 6.25 (1H, d, J = 4.8 Hz, H-10α), 4.31 (1H, br s, H-13α), 1.30 (3H, s, H-16), 0.87 (3H, s, H-17), 1.86 (3H, s, H-18), 1.50 (3H, s, H-19), 9.46 (1H, s, H-20), 8.06 (2H, d, J = 7.5 Hz, Ho-Ph), 7.54 (2H, t, J = 7.3 Hz, Hm-Ph), 7.65 (1H, t, J = 7.1 Hz, Hp-Ph), 2.04 (3H, s, O2CCH3), 2.18 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 64.5 (C, C-1), 22.6 (CH2, C-2), 36.8 (CH, C-3), 144.6 (C, C-4), 146.9 (CH, C-5), 28.0 (CH2, C-6), 71.1 (CH, C-7), 43.7 (C, C-8), 73.0 (CH, C-9), 67.3 (CH, C-10), 136.6 (C, C-11), 149.5 (C, C-12), 82.4 (CH, C-13), 51.6 (CH2, C-14), 78.1 (C, C-15), 25.8 (CH3, C-16), 25.6 (CH3, C-17), 12.8 (CH3, C-18), 12.1 (CH3, C-19), 193.2 (CH, C-20), 165.7 (C, O2CC6H5), 129.9 (C, i-O2CC6H5), 129.5 (CH, o-O2CC6H5), 128.7 (CH, m-O2CC6H5), 133.3 (CH, p-O2CC6H5), 169.7 (C, O2CCH3), 169.2 (C, O2CCH3), 19.7 (CH3, O2CCH3); HRESIMS m/z 559.2299 [M+Na+] (calcd for C31H36O8Na, 559.2307).
4.3.13. Conversion of compound 4 to 18 and 19
Sodium hydride (52.4 mg, 60%, 1.31 mmol) washed with dry ethyl ether firstly was stirred in dry tetrahydrofuran (10 mL). To this mixture was added 4 (200 mg, 0.33 mmol). The reaction mixture was stirred at room temperature and monitored by TLC until all starting material was consumed. The mixture was diluted with ethyl acetate and washed with water and brine. The organic layer was then dried (NaSO4) and concentrated. The residue was purified by column chromatography (chloroform/ethyl acetate, 8:2) to give 18 (44.0 mg, 31%) and 19 (111.6 mg, 62%).
4.3.13.1. Compound 18
Yellow amorphous powder: ; UV (CH3OH) λ max (log ε) 201.2 (4.35) nm; IR (KBr) ν max 2956, 2924, 1750, 1726, 1661, 1628, 1452, 1374, 1262, 1221, 1095, 1069, 1027, 713 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 6.05 (1H, s, H-2β), 4.15 (1H, s, H-3α), 5.90 (1H, d, J = 10.2 Hz, H-6), 6.99 (1H, d, J = 10.2 Hz, H-7), 5.47 (1H, d, J = 4.7 Hz, H-9β), 6.32 (1H, d, J = 4.7 Hz, H-10α), 1.89 (3H, s, H-15), 1.60 (3H, s, H-16), 5.58 (1H, s, H-17a), 6.05 (1H, s, H-17b), 7.93 (2H, d, 7.2, Ho-Ph), 7.44 (2H, t, 7.7, Hm-Ph), 7.57 (1H, t, 7.4, Hp-Ph), 1.96 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 136.9 (C, C-1), 122.4 (CH, C-2), 46.2 (CH, C-3), 146.5 (C, C-4), 188.2 (C, C-5), 128.9 (CH, C-6), 158.8 (CH, C-7), 46.3 (C, C-8), 76.8 (CH, C-9), 70.6 (CH, C-10), 157.9 (C, C-11), 145.2 (C, C-12), 203.3 (C, C-13), 42.3 (CH, C-14), 9.1 (CH3, C-15), 19.3 (CH3, C-16), 120.7 (CH2, C-17), 165.1 (C, O2CC6H5), 129.9 (C, i-O2CC6H5), 130.5 (CH, o-O2CC6H5), 129.7 (CH, m-O2CC6H5), 134.6 (CH, p-O2CC6H5), 169.7 (C, O2CCH3), 20.7 (CH3, O2CCH3); HRESIMS m/z 455.1481 [M+Na+] (calcd for C26H24O6Na, 455.1470).
4.3.13.2. Compound 19
Yellow amorphous powder: ; UV (CH3OH) λ max (log ε) 201.0 (4.42) nm; IR (KBr) ν max 3445, 2925, 1752, 1717, 1679, 1625, 1603, 1451, 1370, 1257, 1281, 1095, 1042, 1026, 753, 712 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 6.38 (1H, d, J = 8.2 Hz, H-2β), 3.45 (1H, d, J = 8.2 Hz, H-3α), 6.05 (1H, d, J = 10.1 Hz, H-6), 7.45 (1H, d, J = 10.1 Hz, H-7), 6.31 (1H, d, J = 10.8 Hz, H-9β), 6.61 (1H, d, J = 10.8 Hz, H-10α), 1.17 (3H, s, H-16), 1.26 (3H, s, H-17), 1.72 (3H, s, H-18), 1.34 (3H, s, H-19), 5.29 (1H, s, H-20a), 5.98 (1H, s, H-20b), 8.02 (2H, d, J = 7.3 Hz, Ho-Ph), 7.49 (2H, t, J = 7.8 Hz, Hm-Ph), 7.65 (1H, t, J = 7.4 Hz, Hp-Ph), 2.15 (3H, s, O2CCH3), 1.79 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 65.0 (C, C-1), 69.6 (CH, C-2), 48.9 (CH, C-3), 143.7 (C, C-4), 189.3 (C, C-5), 130.0 (CH, C-6), 153.8 (CH, C-7), 44.8 (C, C-8), 75.1 (CH, C-9), 69.9 (CH, C-10), 162.2 (C, C-11), 147.1 (C, C-12), 206.8 (C, C-13), 44.4 (CH2, C-14), 76.1 (C, C-15), 28.0 (CH3, C-16), 28.1 (CH3, C-17), 8.9 (CH3 C-18), 20.5 (CH3, C-19), 120.5 (CH2, C-20), 165.7 (C, O2CC6H5), 130.3 (C, i-O2CC6H5), 130.6 (CH, o-O2CC6H5), 129.5 (CH, m-O2CC6H5), 134.4 (CH, p-O2CC6H5), 171.7 (C, O2CCH3), 169.9 (C, O2CCH3), 21.8 (CH3, O2CCH3), 21.2 (CH3, O2CCH3). HRESIMS m/z 455.1481 [M+Na+] (calcd for C26H24O6Na, 455.1470).
4.3.14. Conversion of compound 3 to 20 and 21
Compound 3 was treated with sodium hydride by the same procedure as that for 18 and 19 to produce crude mixture which was purified by column chromatography (chloroform/ethyl acetate, 8:2) to afford 20 (12.0%) and compound 21 (27.5%).
4.3.14.1. Compound 20
Yellow oil: ; UV (CH3OH) λ max (log ε) 201.0 (4.39) nm; IR (KBr) ν max 3437, 2925, 1748, 1726, 1704, 1602, 1452, 1370, 1241, 1094, 1069, 1027, 714 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.85 (1H, br s, H-2), 4.25 (1H, d, br s, H-3α), 4.40 (1H, t, J = 2.6 Hz, H-5β), 5.07 (1H, dd, J = 14.1, 5.1 Hz, H-7α), 5.56 (1H, d, J = 3.7 Hz, H-9β), 6.30 (1H, d, J = 3.7 Hz, H-10α), 1.82 (3H, s, H-15), 1.10 (3H, s, H-16), 4.98 (1H, s, H-17a), 5.21 (1H, s, H-17b), 8.05 (2H, d, J = 7.7 Hz, Ho-Ph), 7.55 (2H, t, J = 7.8 Hz, Hm-Ph), 7.69 (1H, t, J = 7.4 Hz, Hp-Ph), 2.02 (3H, s, O2CCH3), 2.01 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 134.7 (C, C-1), 126.3 (CH, C-2), 42.5 (CH, C-3), 151.0 (C, C-4), 71.8 (CH, C-5), 35.2 (CH2, C-6), 70.4 (CH, C-7), 46.4 (C, C-8), 72.7 (CH, C-9), 71.2 (CH, C-10), 157.9 (C, C-11), 147.5 (C, C-12), 203.5 (C, C-13), 43.1 (CH2, C-14), 8.8 (CH3, C-15), 11.4 (CH3, C-16), 112.6 (CH2, C-17), 165.0 (C, O2CC6H5), 130.1 (C, i-O2CC6H5), 130.5 (CH, o-O2CC6H5), 129.7 (CH, m-O2CC6H5), 134.6 (CH, p-O2CC6H5), 170.0 (C, O2CCH3), 169.6 (C, O2CCH3), 20.9 (CH3, O2CCH3), 20.7 (CH3, O2CCH3); HRESIMS m/z 517.1837 [M+Na+] (calcd for C28H30O8Na, 517.1838).
4.3.14.2. Compound 21
Yellow oil: ; UV (CH3OH) λ max (log ε) 201.0 (4.36) nm; IR (KBr) ν max 3436, 2926, 1736, 1602, 1451, 1247, 1083, 1065, 1026, 712 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.99 (1H, s, H-2), 4.05 (1H, s, H-3α), 4.47 (1H, s, H-5β), 5.39 (1H, dd, J = 11.9, 5.4 Hz, H-7α), 6.08 (1H, s, H-9β), 1.79 (3H, s, H-15), 1.04 (3H, s, H-16), 5.10 (1H, s, H-17a), 5.25 (1H, s, H-17b), 8.11 (2H, d, J = 7.7 Hz, Ho-Ph), 7.57 (2H, t, J = 7.8 Hz, Hm-Ph), 7.70 (1H, t, J = 7.5 Hz, Hp-Ph), 2.04 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 133.1 (C, C-1), 124.5 (CH, C-2), 42.7 (CH, C-3), 150.3 (C, C-4), 73.2 (CH, C-5), 35.9 (CH2, C-6), 74.8 (CH, C-7), 43.6 (C, C-8), 135.8 (CH, C-9), 144.3 (C, C-10), 152.4 (C, C-11), 141.4 (C, C-12), 203.9 (C, C-13), 41.9 (CH2, C-14), 10.3 (CH3, C-15), 13.7 (CH3, C-16), 112.2 (CH2, C-17), 165.3 (C, O2CC6H5), 130.0 (C, i-O2CC6H5), 130.8 (CH, o-O2CC6H5), 129.8 (CH, m-O2CC6H5), 134.7 (CH, p-O2CC6H5), 170.7 (C, O2CCH3), 21.1 (CH3, O2CCH3); HRESIMS m/z 457.1618 [M+Na+] (calcd for C26H26O6Na, 457.1627).
4.3.15. Compound 22
To a solution of compound 3 (100 mg, 0.16 mmol) in dry pyridine (8 mL) was added chlorotriethylsilane (41.51 μL, 0.25 mmol) dropwise. The solution was stirred at room temperature for 12 h when all starting material was consumed. After dilution with ethyl acetate, the solution was washed with diluted hydrochloric acid and water. The organic layer was dried (Na2SO4) and evaporated. The residue was purified by column chromatography (chloroform/ethyl acetate, 19.5:0.5) to give 114 mg of crude product (114 mg). The crude material was dissolved in dry tetrahydrofuran, and treated with sodium hydride by the same procedure as that for 18 and 19 to give compound 22 in 39.8% yield: white amorphous powder, (chloroform/ethyl acetate, 9.5:0.5); ; UV (CH3OH) λ max (log ε) 201.2 (4.55) nm; IR (KBr) ν max 2956, 1753, 1727, 1704, 1638, 1452, 1369, 1234, 1094, 1068, 1027, 1010, 714 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.85 (1H, s, H-2), 4.07 (1H, s, H-3α), 4.49 (1H, br s, H-5β), 5.07 (1H, dd, J = 11.5, 4.8 Hz, H-7α), 5.55 (1H, J = 3.6 Hz, H-9β), 6.30 (1H, J = 3.6 Hz, H-10α), 1.82 (3H, s, H-15), 1.11 (3H, s, H-16), 5.00 (1H, s, H-17a), 5.26 (1H, s, H-17b), 8.05 (2H, d, J = 7.8 Hz, Ho-Ph), 7.55 (2H, t, J = 7.9 Hz, Hm-Ph), 7.69 (1H, t, J = 7.4 Hz, Hp-Ph), 2.04 (2H, s, O2CCH3), 2.03 (3H, s, O2CCH3), 0.62 (6H, q, 7.9, SiCH2), 0.97 (9H, t, 7.9, SiCH2CH3); 13C NMR (acetone-d 6, 100 MHz) δ 134.9 (C, C-1), 126.0 (CH, C-2), 42.4 (CH, C-3), 150.8 (C, C-4), 71.7 (CH, C-5), 36.0 (CH2, C-6), 68.9 (CH, C-7), 46.3 (C, C-8), 73.8 (CH, C-9), 70.2 (CH, C-10), 157.8 (C, C-11), 147.6 (C, C-12), 203.5 (C, C-13), 43.0 (CH2, C-14), 8.7 (CH3, C-15), 11.2 (CH3, C-16), 112.3 (CH2, C-17), 164.9 (C, O2CC6H5), 129.9 (C, i-O2CC6H5), 130.4 (CH, o-O2CC6H5), 129.7 (CH, m-O2CC6H5), 134.6 (CH, p-O2CC6H5), 169.9 (C, O2CCH3), 169.5 (C, O2CCH3), 20.9 (CH3, O2CCH3), 20.7 (CH3, O2CCH3), 5.3 (CH2, SiCH2), 7.1 (CH3, SiCH2CH3); HRESIMS m/z 631.2695 [M+Na+] (calcd for C34H44O8NaSi, 631.2703).
4.3.16. Compound 23
To a solution of pyridinium chlorochromate (130.90 mg, 0.61 mmol) in dichloromethane (5 mL) was added 20 (50 mg, 0.10 mmol) in dichloromethane (3 mL). The mixture was stirred at room temperature for 6 h. The reaction mixture was filtered and purified through the column chromatography (petroleum ether/ethyl acetate, 8.5:1.5) and concentrated to afford 18 (17.49 mg, 17.5%) and 23 (22.51 mg, 45.2%).
4.3.16.1. Compound 23
Yellow oil: ; UV (CH3OH) λ max (log ε) 201.0 (4.19) nm; IR (KBr) ν max 2956, 2925, 1750, 1728, 1707, 1641, 1550, 1453, 1375, 1261, 1100, 1070, 1042, 1025, 714 cm−1; 1H NMR (acetone-d 6, 400 MHz) δ 5.50 (1H, s, H-2), 4.06 (1H, s, H-3α), 7.02 (1H, d, J = 5.7 Hz, H-5), 5.05 (1H, dd, J = 10.6, 5.2 Hz, H-7α), 5.51 (1H, d, J = 4.2 Hz, H-9β), 6.27 (1H, d, J = 4.2 Hz, H-10α), 1.81 (3H, s, H-15), 1.74 (3H, s, H-16), 9.56 (1H, s, H-17), 8.05 (2H, d, J = 8.1 Hz, Ho-Ph), 7.56 (2H, t, J = 7.7 Hz, Hm-Ph), 7.70 (1H, t, J = 7.5 Hz, Hp-Ph), 2.03 (3H, s O2CCH3), 1.96 (3H, s, O2CCH3); 13C NMR (acetone-d 6, 100 MHz) δ 137.3 (C, C-1), 126.2 (CH, C-2), 39.1 (CH, C-3), 141.2 (C, C-4), 174.6 (CH, C-5), 28.0 (CH2, C-6), 76.8 (CH, C-7), 43.7 (C, C-8), 76.9 (CH, C-9), 71.5 (CH, C-10), 157.5 (C, C-11), 143.0 (C, C-12), 203.5 (C, C-13), 40.7 (CH2, C-14), 9.2 (CH3, C-15), 16.2 (CH3, C-16), 193.5 (CH, C-17), 165.7 (C, O2CC6H5), 129.9 (C, i-O2CC6H5), 130.7 (CH, o-O2CC6H5), 129.7 (CH, m-O2CC6H5), 134.7 (CH, p-O2CC6H5), 170.7 (C, O2CCH3), 170.0 (C, O2CCH3), 21.2 (CH3, O2CCH3), 20.9 (CH3, O2CCH3); HRESIMS m/z 515.1692 [M+Na+] (calcd for C28H28O8Na, 515.1681).
4.4. CoMFA
Structures of 18 compounds were built using SYBYL 7.0 molecular modeling software. The structural energy minimization was performed using standard TRIPOS force field and Gasteiger–Hückel charge with an energy gradient convergence criterion of 0.001 kcal/mol and a distance-dependent dielectric constant. Systematic conformational searches were carried out to find the lowest energy structures.
Steric and electrostatic interactions were calculated using an sp3 carbon atom as steric probe and a +1 charge as electrostatic probe with Tripos force field. The CoMFA grid spacing was 2.0 Å in the x, y, and z directions. The default value of 30 kcal/mol was set as the maximum steric and electrostatic energy cutoff. Minimum-sigma (column filtering) was set to be 2.0 kcal/mol. The regression analysis was carried out using the full cross-validated partial least-squares (PLS) method (leave-one-out) with CoMFA standard options for scaling of variables. The final model (non-cross-validated conventional analysis) was developed with the optimum number of components equal to that yielding the highest q 2.
References and notes
- 1.Baloglu E., Kingston D.G.I. J. Nat. Prod. 1999;62:1448. doi: 10.1021/np990176i. [DOI] [PubMed] [Google Scholar]
- 2.Shi Q.W., Zhao Y.M., Si X.-T., Li Z.P., Yamada T., Kiyota H. J. Nat. Prod. 2006;69:280. doi: 10.1021/np0503451. [DOI] [PubMed] [Google Scholar]
- 3.Kingston D.G.I. Chem. Commun. 2001:867. [Google Scholar]
- 4.Fuji K., Tanaka K., Li B., Shingu T., Taga T. Tetrohedron Lett. 1992;33:7915. [Google Scholar]
- 5.Chen R., Kingston D.G.I. J. Nat. Prod. 1994;57:1017. doi: 10.1021/np50109a025. [DOI] [PubMed] [Google Scholar]
- 6.Khanuja, S. P. S.; Santha, T. R. A.; Kumar, Garg A.; Misra, R. K.; Chattopadhyay, S. K.; Srivastva, S.; Negi, A. S. U.S. Patent 20040127561 A1, 2002.
- 7.Georg G.I., Cheruvallath Z.S., Vander V.D., Ye Q.M., Mitscher L.A., Himes R.H. Bioorg. Med. Chem. Lett. 1993;3:1349. [Google Scholar]
- 8.Tremblay S., Soucy C., Towers N., Gunning P.J., Breau L. J. Nat. Prod. 2004;67:838. doi: 10.1021/np0304565. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi J., Hosoyama H., Wang X.X., Shigemori H., Koiso Y., Iwasaki S., Sasaki T., Naito M., Tsuruo T. Bioorg. Med. Chem. Lett. 1997;7:393. [Google Scholar]
- 10.Palmer, J. T.; Rasnick, D.; Klaus, J. L.; U.S. Patent 6287840, 2001.
- 11.Matthews D.A., Dragovich P.S., Webber S.E., Fuhrman S.A., Patick A.K., Zalman L.S., Hendrickson T.F., Love R.A., Prins T.J., Marakovits J.T., Zhou R., Tikhe J., Ford C.E., Meador J.W., Ferre R.A., Brown E.L., Binford S.L., Brothers M.A., DeLisle D.M., Worland S.T. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11000. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang K.E., Hee B., Lee C.A., Liang B., Potts B.C. Drug Metab. Dispos. 2001;29:729. [PubMed] [Google Scholar]
- 13.Dragovich P.S., Prins T.J., Zhou R., Brown E.L., Maldonado F.C., Fuhrman S.A., Zalman L.S., Tuntland T., Lee C.A., Patick A.K., Matthews D.A., Hendrickson T.F., Kosa M.B., Liu B., Batugo M.R., Gleeson J.P., Sakata S.K., Chen L., Guzman M.C., Meador J.W., 3rd, Ferre R.A., Worland S.T. J. Med. Chem. 2002;45:1607. doi: 10.1021/jm010469k. [DOI] [PubMed] [Google Scholar]
- 14.Sirois S., Wei D.Q., Du Q., Chou K.C. J. Chem. Inf. Comput. Sci. 2004;44:1111. doi: 10.1021/ci034270n. [DOI] [PubMed] [Google Scholar]
- 15.Barboni L., Datta A., Dutta D., Georg G.I., Vander Velde D.G., Himes R.H., Wang M., Snyder J.P. J. Org. Chem. 2001;66:3321. doi: 10.1021/jo0015467. [DOI] [PubMed] [Google Scholar]
- 16.Deka V., Dubois J., Thoret S., Guéritte F., Guénard D. Org. Lett. 2003;5:5031. doi: 10.1021/ol036043c. [DOI] [PubMed] [Google Scholar]
- 17.Barboni L., Giarlo G., Ricciutelli M., Ballini R., Georg G., Vander Velde D.G., Himes R.H., Wang M., Lakdawala A., Synder J.P. Org. Lett. 2004;6:461. doi: 10.1021/ol036204c. [DOI] [PubMed] [Google Scholar]