

Abstract
Equine arteritis virus (EAV) is the causative agent of equine viral arteritis (EVA), a respiratory and reproductive disease of equids. There has been significant recent progress in understanding the molecular biology of EAV and the pathogenesis of its infection in horses. In particular, the use of contemporary genomic techniques, along with the development and reverse genetic manipulation of infectious cDNA clones of several strains of EAV, has generated significant novel information regarding the basic molecular biology of the virus. Therefore, the objective of this review is to summarize current understanding of EAV virion architecture, replication, evolution, molecular epidemiology and genetic variation, pathogenesis including the influence of host genetics on disease susceptibility, host immune response, and potential vaccination and treatment strategies.
Keywords: Equine arteritis virus, EAV, EVA
1. Introduction
Equine arteritis virus (EAV) was first isolated from the lung of an aborted fetus following an extensive outbreak of respiratory disease and abortion on a Standardbred breeding farm near Bucyrus, Ohio, USA, in 1953 (Doll et al., 1957a, Doll et al., 1957b). After isolation of the causative virus and description of characteristic vascular lesions, equine viral arteritis (EVA) was identified as an etiologically distinct disease of the horse (Doll et al., 1957a). EAV is a small enveloped, positive-sense, single-stranded RNA virus that is the prototype virus in the family Arteriviridae (genus: Arterivirus), order Nidovirales, a taxonomic grouping that includes porcine reproductive and respiratory syndrome virus (PRRSV), simian hemorrhagic fever virus (SHFV), and lactate dehydrogenase-elevating virus (LDV) of mice (Cavanagh, 1997). Pioneering work on the distinctive replication strategy utilized by EAV originally led to the taxonomic designation of the Order Nidovirales (Cavanagh, 1997), a grouping of morphologically distinct viruses included in the families Arterviridae, Coronaviridae, and Roniviridae that all utilize a similar replication strategy that involves the generation of a nested set of subgenomic RNAs (de Vries et al., 1997, den Boon et al., 1991, Gorbalenya et al., 2006, Snijder and Spaan, 2006). The order Nidovirales has been expanded recently to include several newly identified plus-stranded RNA viruses including wobbly possum disease virus (WPDV), a close relative to the other members of the family Arteriviridae and the cause of neurologic disease among free-ranging Australian brushtail possums (Trichosurus vulpecula) in New Zealand (Dunowska et al., 2012). Similarly, two new genetically divergent SHFV variants (SHFV-krc1 and SHFV-krc2) were recently identified in a single male colobus monkey (Procolobus rufomitratus tephrosceles; Lauck et al., 2011). Both SHFV-krc1 and SHFV-krc2 are highly divergent from the prototypic LVR 42-0/6941 strain of SHFV (52.0% and 58.1% nucleotide diversity, respectively) and, interestingly, the two variants are also significantly different from one another and share only 51.9% nucleotide sequence identity. Subsequently, two additional highly divergent variants of SHFV (SHFV-krtg-1a/b and SHFV-krtg-2a/b [79.4% nucleotide identity]) were isolated from African red-tailed (guenon) monkeys (Cercopithecus ascanius) from Kibale National Park, Uganda (Lauck et al., 2013). These two variants were also genetically distinct from the prototypic LVR 42-0/6941 strain of SHFV (54.1%) and the SHFV-krc1 and SHFV-krc2 (50.1%) variants. Additional novel nidoviruses have also been isolated recently from mosquitoes, including Cavally virus (CAVV) and Nam Dinah virus (NDiV). These newly identified arthropod-borne nidoviruses are provisionally placed in a new family Mesoniviridae, which is an intermediate between the families Arteriviridae and Coronaviridae and more closely related to the family Roniviridae (Lauber et al., 2012). The recent recognition of these related but distinct viruses that share similar replication strategies indicates an increasing need for reclassification of the order Nidovirales.
Like the other arteriviruses, EAV infection is highly species-specific and exclusively limited to members of the family Equidae, which includes horses, donkeys, mules, and zebras (Stadejek et al., 2006, Timoney and McCollum, 1993). The EAV associated disease, EVA, is a respiratory and reproductive disease of horses that occurs worldwide (Bell et al., 2006, Glaser et al., 1996, Timoney and McCollum, 1993). Although there is only one known EAV serotype, field strains of the virus differ in their virulence and neutralization phenotype (Balasuriya et al., 1999b, Balasuriya et al., 2002a, Balasuriya et al., 2007, Balasuriya and Maclachlan, 2004, Go et al., 2012, MacLachlan et al., 1996, McCollum et al., 1998, Patton et al., 1999, Pronost et al., 2010, Vairo et al., 2012, Zhang et al., 2010b, Zhang et al., 2012). The clinical signs exhibited by individual EAV-infected horses depend on a variety of factors including the age and physical condition of the animal, challenge dose and route of infection, strain of virus, and environmental factors. With the sole and notable exception of the experimentally derived and highly horse-adapted, virulent Bucyrus strain, other strains and field isolates of EAV very rarely cause fatal infection in adult horses (McCollum and Timoney, 1998, Pronost et al., 2010). The vast majority of EAV infections are subclinical, but acutely infected animals may develop a wide range of clinical signs including pyrexia, depression, anorexia, dependent edema (scrotum, ventral trunk, and limbs), stiffness of gait, conjunctivitis, lacrimation and swelling around the eyes (periorbital and supraorbital edema), respiratory distress, urticaria, and leukopenia (Timoney and McCollum, 1993). The incubation period of 3–14 days (typically 6–8 days following venereal exposure) is followed by pyrexia of up to 41 °C (105.8 °F) that may persist for 2–9 days. The virus can cause abortion of pregnant mares, with abortion rates during field outbreaks varying from approximately 10% to 70%, depending on the virus strain (Timoney and McCollum, 1993). EAV-induced abortions can occur at any time between 3 and 10 months of gestation. Infection of neonatal foals can cause a severe fulminating interstitial pneumonia, and in 1–3 months old foals a progressive “pneumo-enteric” syndrome (Vaala et al., 1992). A variable proportion of acutely infected stallions (10–70%) become persistently infected and shed the virus exclusively in their semen (Timoney and McCollum, 1993 and references therein). There is no evidence of EAV causing persistent infection in mares, geldings, or foals. The virus persists mainly in the ampulla of the stallion's reproductive tract, and the establishment and maintenance of the carrier state in the stallion is testosterone-dependent.
Serologic surveys have shown that EAV infection has occurred among horses in North and South America, Europe, Australia, Africa, and Asia (Echeverria et al., 2003, Eichhorn et al., 1995, Szeredi et al., 2005, Timoney and McCollum, 1993). Other countries such as Iceland and Japan are apparently free of the virus. Recent studies have shown that New Zealand is also free of active EAV infection (McFadden et al., 2013). However, the seroprevalence of EAV infection of horses varies between countries and among horse breeds within a country. For example, the seroprevalence of EAV infection varies among horses of different breeds and ages in the United States (US), with marked disparity between the prevalence of infection of Standardbred and Thoroughbred horses (Timoney and McCollum, 1993). EAV infection is considered endemic in Standardbred but not Thoroughbred horses in the US, with 77.5% to 84.3% of all Standardbreds but only up to 5.4% of Thoroughbreds being seropositive to the virus (Hullinger et al., 2001, McCollum and Bryans, 1973, McCue et al., 1991, McKenzie, 1996, Moraillon and Moraillon, 1978, Timoney and McCollum, 1988, Timoney and McCollum, 1993). Similarly, the seroprevalence of EAV infection of Standardbred horses in California was 68.5% in 1991, versus less than 2% in all other breeds tested (McCue et al., 1991). The 1998 National Animal Health Monitoring System (NAHMS) equine survey showed that only 0.6% of the US American Quarter Horse (AQH) population was seropositive to EAV (Anonymous, 2000). However, the extensive US outbreak of EVA in 2006–2007 mainly involved AQHs and this very likely significantly increased the seroprevalence of EAV within this breed. The seroprevalence of EAV infection of Warmblood stallions is also very high in a number of European countries, with some 55 to 93% of Austrian Warmblood stallions being seropositive to EAV (Burki et al., 1992). Similarly, there is high seroprevalence among mares and stallions of Hucul horses in Poland, 53.2% and 68.2%, respectively (Rola et al., 2011).
Transmission of EAV between horses occurs via either the respiratory or venereal route (Cole et al., 1986, Doll et al., 1957b, McCollum et al., 1971, Timoney and McCollum, 1993, Timoney et al., 1986, Timoney et al., 1987). Horizontal respiratory transmission occurs after aerosolization of infected respiratory tract secretions from acutely infected horses; high titers of EAV are present in respiratory secretions for some 7–14 days during acute infection (McCollum et al., 1971). However, direct and close contact is necessary for aerosol transmission of EAV between horses (Collins et al., 1987, Timoney and McCollum, 1988). EAV can also be transmitted by aerosol from urine and other body secretions of acutely infected horses, aborted fetuses and their membranes, and the masturbates of acutely or chronically infected stallions (Burki et al., 1992, Glaser et al., 1996, Glaser et al., 1997, Guthrie et al., 2003, McCollum, 1981, McCollum et al., 1971, McCollum et al., 1995). Venereal transmission of EAV contained in the semen of stallions that are either acutely or chronically infected with the virus is an especially important route of natural transmission of the virus (Timoney et al., 1986, Timoney et al., 1987). Recently, it has been demonstrated that EAV can be experimental transmitted to naïve recipient mares via embryo transfer from a donor mare inseminated with EAV-infective semen to a naïve recipient mare (Broaddus et al., 2011a).
2. Virion structure
The virion of EAV is spherical with a diameter of 40–60 nm and consists of a genome-containing nucleocapsid core (25–35 nm in diameter) composed of nucleocapsid (N) protein that is surrounded by a relatively smooth lipid envelope containing several membrane proteins (Deshpande et al., 2007, Horzinek et al., 1971, Magnusson et al., 1970). The envelope lacks large surface projections (Fig. 1 ). The EAV N protein appears to be organized as dimers of dimers (i.e. tetramers), which may reflect the arrangement of the protein in the viral nucleocapsid. The dimeric N-protein structure is similar to the previously determined structure of the N protein of PRRSV. Recent cryo-electron tomographic studies suggest that the core of the PRRSV virion is composed of a double-layered chain of N proteins bundled into a hollow ball with an asymmetric, linear arrangement, rather than the isometric core that was previously described (Spilman et al., 2009, Dokland, 2010). The structural proteins of the EAV virion include seven envelope proteins (E, GP2, GP3, GP4, ORF5a protein, GP5, and M) and the N protein, which respectively are encoded by ORFs 2a, 2b, 3–4, 5a, 5b, and 6–7 that are located at the 3′ proximal quarter of the genome (Fig. 2 ; de Vries et al., 1992, Firth et al., 2011, Snijder and Spaan, 2007, Snijder et al., 1999, Wieringa et al., 2004). These structural proteins are expressed from six subgenomic viral messenger RNAs (sg mRNAs) that form a 3′-co-terminal nested set and contain a common leader sequence encoded by the 5′-end of the genome (Fig. 2 and Table 1 ; den Boon et al., 1996).
Fig. 1.
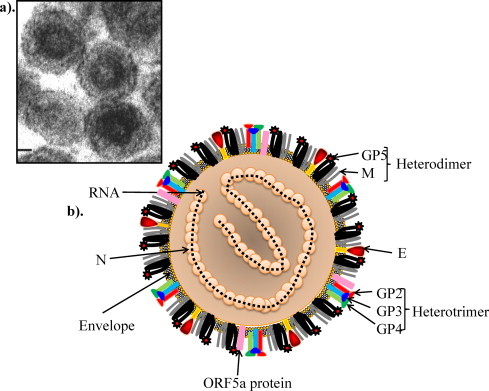
Equine arteritis virus particle: (a) electron micrograph of EAV (adapted from Snijder et al., Chapter 20, Topley & Wilson's Microbiology and Microbial Infections. London, UK with permission), (b) schematic presentation of EAV particle.
Fig. 2.
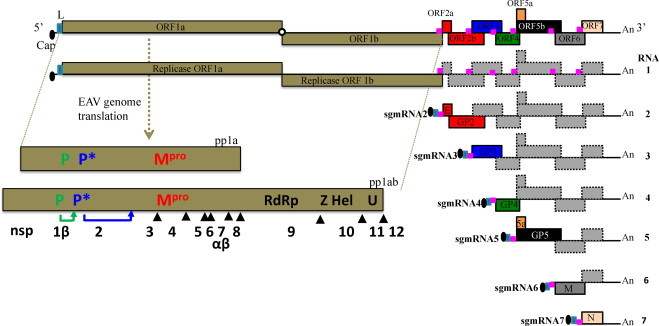
The genome organization and polycistronic nature of the EAV genome. The genomic open reading frames (ORFs) are indicated and the names of the corresponding proteins are depicted. The pink boxes represent the body transcription regulatory sequences (TRSs). The nested set of mRNAs that is found in infected cells is depicted below the genome, with RNA1 being identical to the viral genome and sg mRNAs 2–7 being used to express the structural protein genes located in the 3′-proximal quarter of the genome. The light blue box at the 5′ end of each sg mRNA represents the common leader sequence, which is derived from the 5′ end of the genome. With the exception of the bicistronic sg mRNAs 2 and 5, the sg mRNAs are functionally monocistronic. Translation of proteins from sg mRNAs 2 (E and GP2 proteins) and 5 (ORF5a protein and GP5) occurs by leaky scanning of the 5′-proximal end of these sg mRNAs (Firth et al., 2011, Snijder et al., 1999). The ORFs 1a and 1b located at the 5′ end of the genome and are translated into two polyproteins (pp1a and pp1ab) that are further processed into 12–13 nonstructural proteins by three viral proteases (nsps 1, 2, and 4).
Table 1.
Nonstructural and structural proteins of EAV.a
| ORF | Protein | Nucleotide position (bp) | Protein length (aa) | Predicted M.W. (kDa) |
|---|---|---|---|---|
| ORF1a | 1a polyprotein | 225–5399 | 1725 | 190 |
| ORF1b | – | 5405–9751 | – | – |
| ORF1ab | 1ab polyprotein | 225–9751 | 3176 | 349.3 |
| nsp1 | 255–1004 | 260 | 28.6 | |
| nsp2 | 1005–2717 | 571 | 61.4 | |
| nsp3 | 2718–3416 | 233 | 25 | |
| nsp4 | 3417–4028 | 204 | 21 | |
| nsp5 | 4029–4514 | 162 | 18.1 | |
| nsp6 | 4515–4580 | 22 | 2.3 | |
| nsp7 | 4581–5255 | 225 | 25.2 | |
| nsp8 | 5256–5405 | 50 | 5.5 | |
| nsp9 | 5256–7333 | 693 | 76.8 | |
| nsp10 | 7334–8734 | 467 | 50.5 | |
| nsp11 | 8735–9391 | 219 | 24.2 | |
| nsp12 | 9392–9748 | 119 | 12.5 | |
| ORF2a | E | 9751–9954 | 67 | 7.4 |
| ORF2b | GP2 | 9824–10,507 | 227 | 25.0 |
| ORF3 | GP3 | 10,306–10,797 | 163 | 17.9 |
| ORF4 | GP4 | 10,700–11,158 | 142 | 15.6 |
| ORF5a | ORF5a | 11,112–11,291 | 59 | 6.5 |
| ORF5b | GP5 | 11,146–11,913 | 255 | 28.0 |
| ORF6 | M | 11,901–12,389 | 162 | 17.8 |
| ORF7 | N | 12,313–12,645 | 110 | 12.1 |
Based on EAV VBS (ATCC VR-796) published sequence (GenBank accession number DQ846750).
The non-glycosylated membrane protein M and glycosylated GP5 protein are the most abundant envelope proteins, and these major envelope proteins are presented as disulfide-linked heterodimers in virus particles. The envelope (E) protein is the third most abundant protein in the viral membrane (Snijder et al., 1999). The GP2, GP3, and GP4 proteins are minor envelope glycoproteins that exist in equimolar amounts in virus particles (de Vries et al., 1992, Wieringa et al., 2002). The minor envelope glycoproteins are covalently associated and form a heterotrimeric complex on the surface of the virion (Wieringa et al., 2002, Wieringa et al., 2003a, Wieringa et al., 2003b, Wieringa et al., 2004). The major envelope proteins (GP5 and M) and the N protein are essential for EAV particle formation, whereas neither E protein nor the minor envelope proteins are required for production of viral particles since absence of any of these proteins does not inhibit incorporation of viral genomic RNA or change buoyant density or microscopic appearance of the virus (Molenkamp et al., 2000). In contrast, with the exception of the ORF5a protein, all major (N, GP5, and M) and minor (E, GP2, GP3, and GP4) structural proteins are required for production of infectious progeny virus, as shown by individually knocking out the expression of each structural protein (Wieringa et al., 2004). Furthermore, it has been shown by reverse genetic studies that elimination of ORF5a protein expression cripples EAV, leading to production of progeny virus with a small plaque phenotype and significantly reduced virus titer (Firth et al., 2011).
3. Overview of the EAV life cycle
EAV replicates primarily in equine macrophages and endothelial cells lining small blood vessels, but also in selected epithelia, mesothelium, and smooth muscle cells of the tunica media of smaller arteries, venules, and the myometrium (Balasuriya and Snijder, 2008, Duan et al., 1997, Kim et al., 1993, Lawson et al., 1997, MacLachlan et al., 1996, Del Piero, 2000b, Plagemann and Moennig, 1992, Rossow, 1998). Unlike other arteriviruses, EAV replicates in vitro in a variety of primary cell cultures, including equine pulmonary artery endothelial, horse kidney, rabbit kidney, and hamster kidney cells, and in a number of continuous cell lines such as baby hamster kidney (BHK-21), rabbit kidney-13 (RK-13), African green monkey kidney (VERO), rhesus monkey kidney (LLC-MK2), MARC-145, hamster lung (HmLu), SV-40 transformed equine ovary and canine hepatitis virus-transformed hamster tumor cells (HS and HT-7; Hyllseth, 1969, Konishi et al., 1975, Lu et al., 2012, Maess et al., 1970, Radwan and Burger, 1973). Enveloped viruses typically have surface molecules that bind to cell surface receptors that mediate the process of cell attachment and membrane fusion with the host cell membrane. The viral envelope protein(s) involved in virus attachment and entry of EAV has not been fully characterized. Dobbe et al. (2001) demonstrated that EAV expressing the ectodomain of GP5 of PRRSV IAF-Klop strain (Pirzadeh et al., 1998) did not change the cellular tropism of the virus. In a recent study, Lu et al. (2012) unequivocally demonstrated that the ectodomains of GP5 and M of PRRSV are not the major determinants of cellular tropism, further supporting the conclusion that the minor envelope proteins are the critical proteins in mediating cellular tropism in arteriviruses (Tian et al., 2012).
While receptors for EAV have not yet been definitively identified, it is proposed that EAV is taken up via clathrin-dependent endocytosis and then delivered to acidic endosomal compartments (Nitschke et al., 2008). Following entry and release into the cytosol, the plus-stranded RNA genome is uncoated and translated into two replicase polyproteins (pp1a and pp1ab) from which at least 13 non-structural proteins (nsps) are released by autoproteolytic processing mediated by the viral proteases (Fig. 3, Fig. 4, Fig. 5 ; Fang and Snijder, 2010, Ziebuhr et al., 2000). The nsps assemble into a membrane-bound replication/transcription complex (RTC) (Knoops et al., 2012, Monastyrska et al., 2013). Genome replication and sg mRNA transcription occur in the RTC and sg mRNAs are then translated into the viral structural proteins. The newly synthesized genome is encapsidated into the N protein to form the nucleocapsid, which becomes enveloped by budding through the endoplasmic reticulum–Golgi intermediate compartment (ERGIC) that contains membranes that include the viral envelope proteins. Newly formed virions mature in the Golgi complex during their movement through the exocytic pathway and are ultimately released from infected cells.
Fig. 3.
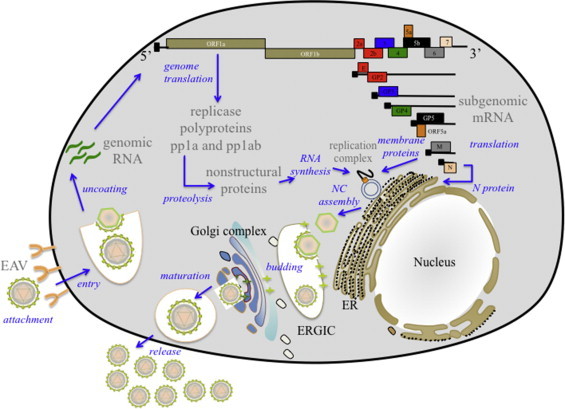
Schematic overview of EAV life cycle. ER: endoplasmic reticulum; ERGIC: ER–Golgi intermediate compartment; NC: nucleocapsid.
Fig. 4.
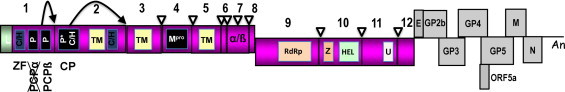
Schematic representation of the processing of the EAV replicase polyproteins (pp1a and pp1ab) and generation of individual nsps. The papain-like cysteine protease (PCPβ) and cysteine protease (CP) cleavage sites are indicated by black arrows. The Mpro cleavage sites are indicated by open arrowheads. The genes encoding structural proteins are depicted in gray. P and P* are, PCP and CP, respectively; Mpro, 3C-like main protease; Z, zinc-binding domain; C/H, cysteine/histidine-rich clusters; TM, predicted transmembrane domains; U, nidoviral uridylate-specific endoribonuclease (NendoU). Figure modified from Nidoviruses book chapter on the arterivirus replicase (Van Hemert and Snijder, 2008) with permission from ASM press.
Fig. 5.
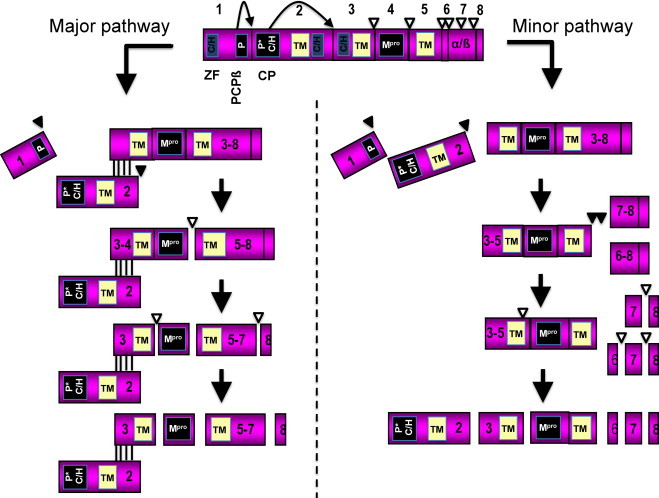
Proteolytic processing of EAV replicase polyprotein pp1a through the major and minor pathways. The papain-like cysteine protease (PCPβ) and cysteine protease (CP) cleavage sites are indicated by black arrows. The Mpro cleavage sites are indicated by open arrowheads. P and P* are, PCP and CP, respectively; Mpro, 3C-like main protease; C/H, cysteine/histidine-rich clusters; TM, predicted transmembrane domains.
Figure modified from Nidoviruses book chapter (Van Hemert and Snijder, 2008) with permission from the ASM Press.
4. Genome organization, replication, and transcription
4.1. Genome organization
The EAV genome is a linear positive-sense single-stranded RNA molecule with a 5′-methylated nucleotide cap and a 3′-untranslated region followed by a poly (A) tail (Snijder and Spaan, 2007, Snijder and Meulenberg, 1998). There are some 35 EAV full-length genome sequences available currently in GenBank (Table 2 ). The genome of individual virus strains varies between 12,704 and 12,731 bp, and includes a 5′ leader sequence (224 nucleotides [nts]) and at least ten open reading frames (ORFs, Fig. 2; Balasuriya et al., 2004b, Balasuriya et al., 2007, den Boon et al., 1991, Miszczak et al., 2012, van Dinten et al., 1997, Zhang et al., 2010b). The two most 5′-proximal ORFs (1a and 1b) occupy approximately three fourths of the genome (Fig. 2). The replicase ORFs 1a and 1b are translated to produce polyproteins pp1a (1725 amino acids) and pp1ab (3176 amino acids; based on the published sequence of the EAV VBS strain [ATCC VR-796], GenBank accession number DQ846750), with the latter being a C-terminally extended version of the former (den Boon et al., 1991). ORF 1b translation depends on a −1 ribosomal frameshift (estimated efficiency 15–20%) just before termination of ORF 1a translation (den Boon et al., 1991). The ORF 1a/1b overlap region contains signals that promote frameshifting: a so-called “slippery” sequence, the actual ribosomal frameshift site, and a downstream RNA pseudoknot structure. These two precursor proteins are extensively processed after translation into at least 13 nsps (nsp1–12, including nsp7 α/β; Fig. 4, Fig. 5, Table 1) by three viral proteases (nsp1, nsp2, and nsp4) (Barrette-Ng et al., 2002, den Boon et al., 1995, Snijder et al., 1995a, Snijder and Meulenberg, 1998, Snijder et al., 1994, Snijder et al., 1995b, Snijder et al., 2001, Tijms et al., 2001, van Aken et al., 2006a, van Aken et al., 2006b, van Aken et al., 2006c, van den Born et al., 2007, van Dinten et al., 1996, van Dinten et al., 1999, Wassenaar et al., 1997, Ziebuhr et al., 2000). The greatest variation in the EAV replicase gene occurs in the portion of ORF1a encoding the nsp2 protein, with considerable variation at amino acids 388–488 (Balasuriya et al., 2004b, Miszczak et al., 2012, Zhang et al., 2008a, Zhang et al., 2008b, Zhang et al., 2008c). The remaining eight ORFs (2a, 2b, 3, 4, 5a, 5b, 6 and 7) are located in the 3′ quarter of the genome and encode eight structural proteins (seven envelope proteins and the nucleocapsid protein) of the virus (see below). Secondary structure analysis by chemical and enzymatic probing of 3′-end of the genome has identified two RNA domains that are essential for virus replication and most likely play a key role in viral RNA synthesis (Beerens and Snijder, 2006). The first domain, located directly upstream of the 39 untranslated region (UTR; nt 12,610–12,654 of the genome), is mainly single-stranded but contains one small stem–loop structure. The second domain is located within the 3′ UTR (nt 12,661–12,690) and folds into a prominent stem–loop structure with a large loop region. This second stem–loop structure may act as a recognition signal during the initiation of minus-strand RNA synthesis (see below).
Table 2.
Full-length genome sequences of EAV strains available in the GenBank.
| Strain (size base pairs) | Source | GenBank accession number |
|---|---|---|
| EAV Utr (12,704)f | Laboratory strain | NC_002532 |
| EAV030 (12,704)c | EAV infectious cDNA clone | Y07862 |
| EAV Bucyrus (12,704)d | EAV virulent Bucyrus strain (VBS) horse passage 15 plural fluid (EAV ATCC VR-796) | DQ846750 |
| EAVrVBS (12,704)d | First EAV VBS infectious cDNA clone | DQ846751 |
| EAV HK25 (12,704)c | EAV VBS passage 25 in horse kidney | EU586273 |
| EAV HK116 (12,704)c | EAV VBS passage 116 in horse kidney | EU586274 |
| EAV ARVAC (12,704)c | MLV Vaccine | EU586275 |
| pEAVrMLV (12,704)g | EAV MLV vaccine strain infectious cDNA clone | FJ798195 |
| Hela-EAVP35 (12,704)c | EAV VBS passage 35 in HeLa cells (laboratory strain) | EU252113 |
| Hela-EAVP80 (12,704)c | EAV VBS passage 80 in HeLa cells (laboratory strain) | EU252114 |
| EAV CW96 (12,708)e | Semen (EU-1) | AY349167 |
| EAV CW01 (12,708)e | Semen (EU-1) | AY349168 |
| EAV F63 (12,722)a | Semen (EU-2) | JN211320 |
| EAV F61 (12,722)a | Semen (EU-2) | JN211318 |
| EAV F27 (12,710)a | Semen (EU-2) | JN211316 |
| EAV F62 (12,722)a | Semen (EU-2) | JN211319 |
| EAV F60 (12,710)a | Semen (EU-2) | JN211317 |
| EAV S4216 (12,731)b | Semen (EU-1) | GQ903811 |
| EAV S3583 (12,731)b | Semen (EU-1) | GQ903809 |
| EAV S3943 (12,731)b | Semen (EU-1) | GQ903807 |
| EAV S3886 (12,731)b | Semen (EU-1) | GQ903805 |
| EAV S3854 (12,731)b | Semen (EU-1) | GQ903803 |
| EAV S3711 (12,731)b | Semen (EU-1) | GQ903801 |
| EAV S3712 (12,731)b | Semen (EU-1) | GQ903799 |
| EAV S3961 (12,731)b | Semen (EU-1) | GQ903797 |
| EAV S3861 (12,731)b | Semen (EU-1) | GQ903795 |
| EAV S3817 (12,731)b | Semen (EU-1) | GQ903810 |
| EAV S4445 (12,731)b | Semen (EU-1) | GQ903808 |
| EAV S4421 (12,731)b | Semen (EU-1) | GQ903806 |
| EAV S4333 (12,731)b | Semen (EU-1) | GQ903804 |
| EAV S4227 (12,731)b | Semen (EU-1) | GQ903802 |
| EAV S4007 (12,731)b | Semen (EU-1) | GQ903800 |
| EAV S4417 (12,731)b | Semen (EU-1) | GQ903798 |
| EAV S3699 (12,731)b | Semen (EU-1) | GQ903796 |
| EAV S3685 (12,731)b | Semen (EU-1) | GQ903794 |
4.2. Genome replication and transcription
The 5′ untranslated region (UTR) of the EAV genome, comprising approximately 225 nts functions as a protective element against degradation by exonucleases (Gallie, 1998). According to the nidovirus transcription model proposed by Sawicki and Sawicki (1995), which is the most widely accepted model, sg mRNAs are produced by a discontinuous transcription mechanism controlled by the transcription regulatory sequence (TRS; “discontinuous extension of minus-strand RNA model” [Fig. 6 ]). The TRS are short and conserved sequence elements (5′-UCAACU-3′ in EAV) that determine a base-pairing interaction between positive and nascent minus-strand RNA and are essential for leader-to-body joining (Pasternak et al., 2001, Pasternak et al., 2003, van Marle et al., 1999a). Similar to other nidoviruses, replication and transcription are processed through different minus-strand intermediates: a full-length minus-strand template is used for replication, while subgenome-sized minus strands produced during a process of discontinuous RNA synthesis are used to synthesize sg mRNAs (Fig. 6; Pasternak et al., 2006, Sawicki and Sawicki, 1995, Sawicki et al., 2007, van Marle et al., 1999a). The initiation of full-length minus-strand RNA (or anti-genome) synthesis, which is also used as template for new genome RNA replication, occurs after recognition of RNA signals near the 3′ end of the viral genome by the RNA-dependent RNA polymerase (RdRp) complex (Snijder and Spaan, 2006). Recognition signals located close to the 3′ end of the anti-genome are apparently used for production of new genomic RNA (Beerens et al., 2007, Beerens and Snijder, 2006, Siddell et al., 2005).
Fig. 6.
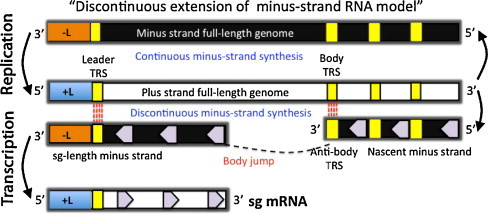
Illustration of the transcription model in nidoviruses. The “discontinuous extension of minus-strand RNA model” proposes that an sg-length negative strand is produced that functions as a template for generation of sg mRNA (Sawicki and Sawicki, 1995). The anti-body TRS in minus-strand may serve as a “jump” signal of the nascent minus strand to leader TRS located at the 5′-end of the plus-strand full-length genome. After the anti-leader (-L) is added to the nascent minus strand, the sg-length minus strand functions as template for positive strand sg mRNA synthesis.
Figure modified from Pasternak et al. (2006) with permission from the Journal of General Virology.
The EAV sg mRNAs contain a common 5′ sequence of 211 nt called “leader”, which is identical to the 5′ end of the viral genomic RNA (Snijder and Meulenberg, 1998). The leader TRS is located in a hairpin loop structure at the 3′ end of the leader (van den Born et al., 2005a) and the body TRSs are present upstream of every structural protein gene except ORFs 2b and 5a (de Vries et al., 1990, den Boon et al., 1996, Firth et al., 2011, Snijder et al., 1999). The EAV genome contains multiple UCAACU sequences that could be transcriptionally silent. Therefore, only a single TRS has been identified as an active site for each of the six sg mRNAs. Moreover, a number of 5′-UCAAC-3′ sequences were also identified in the replicase gene (den Boon et al., 1996). However, multiple active TRSs per sg mRNA have been described and discontinuous RNA synthesis depends not only on base pairing between sense leader TRS and antisense body TRS, but also on the primary sequence of the body TRS (Pasternak et al., 2000, Pasternak et al., 2001, Pasternak et al., 2003, Pasternak et al., 2004, Pasternak et al., 2006). Introduction of site-specific mutations into the mRNA leader-body junctions of TRS strongly suggested that the discontinuous step occurs during minus-strand synthesis. While the leader TRS merely plays a targeting role for strand transfer, the body TRS fulfills multiple functions. Minus-strand RNA synthesis, initiated at the 3′-end of the viral genome, is attenuated at one of the body TRS regions (Pasternak et al., 2006, Sawicki et al., 2007). Subsequently, the nascent minus-strand carrying the body TRS complement at its 3′ end is translocated to the 5′ end of the genomic template. In EAV, the genomic leader TRS serves as a base-pairing target for the 3′ end of the nascent minus-strand that is facilitated by its presence in the loop of an RNA hairpin. When minus-strand synthesis resumes, nascent strands are extended with the complement of the genomic leader sequence, yielding a nested set of subgenome-length minus-strand templates that are used for the subsequent synthesis of the various sg mRNAs. If attenuation does not occur, minus-strand RNA synthesis proceeds to yield a full-length complement of the genome, the intermediate being required for its replication. During the discontinuous transcription mechanism, the TRS at the 3′ end of the leader transcript base pairs with the complement of the body TRS in the negative-stranded template allowing extension of the complement of the genomic leader sequence in the nascent negative strand (Sawicki and Sawicki, 1995). The resulting sg negative-stranded RNA serves as template for the synthesis of the corresponding sg mRNA. With the exception of the last one, each sg mRNA, is structurally polycistronic, although they are functionally monocistronic and only the 5′-proximal ORF is translated from each sg mRNA. However, there are two exceptions, where sg mRNAs 2 and sg mRNA 5 are functionally bicistronic and each encode two structural proteins per sg mRNA (Firth et al., 2011, Snijder et al., 1999).
Formation of paired membranes and double-membrane vesicles (DMVs) at 3–6 h post-infection is a characteristic feature of arterivirus replication (Breese and McCollum, 1970, Pol and Wagenaar, 1992, Pol et al., 1997, Stueckemann et al., 1982, Wood et al., 1970). The transmembrane nsps are incorporated into cellular organelles, particularly the ER, where early viral RNA synthesis occurs and results in increased expression of replicase proteins. Interaction of accumulated nsps with membranes results in membrane pairing and vesiculation (Pedersen et al., 1999). It is assumed that formation of DMVs that are derived from the endoplasmic reticulum (ER) is based on interactions between luminal domains of transmembrane proteins of either virus and/or cellular origin to bring opposite membranes together. The outer membranes of EAV-induced DMVs are interconnected with each other and with the ER, thus forming a reticulovesicular network (RVN) resembling that previously described in severe acute respiratory syndrome (SARS) coronavirus infected cells (Knoops et al., 2012). Electron spectroscopic imaging of DMVs has revealed the presence of phosphorus in amounts equaling on average a few dozen copies of the EAV RNA genome. Furthermore, electron tomography has identified a network of nucleocapsid protein-containing protein tubules that appear to be intertwined with the RVN, which indicates there is a connection between EAV induced RVN and nucleocapsid assembly. Recently, it was also shown that autophagy marker protein microtubule-associated protein 1 light chain 3 (LC3) and ER degradation-enhancing alpha-mannosidase-like1 (EDEM1) are associated with DMV in EAV infected cells (Monastyrska et al., 2013).
In nidovirus-infected cells, newly synthesized viral RNA and many replicase subunits are present in the perinuclear region together with a large number of DMVs (Pedersen et al., 1999, Snijder et al., 2006, van der Meer et al., 1999). Immunoelectron microscopy revealed that viral nsps that are part of the replication/transcription complexes (RTC) and nascent viral RNA are associated with DMVs (Gosert et al., 2002, Knoops et al., 2008, Knoops et al., 2012, Pedersen et al., 1999, Stertz et al., 2007, van der Meer et al., 1999). Therefore, DMVs are presumed to carry the enzyme complex responsible for virus replication and sg mRNA synthesis. Expression of EAV nsp2–3 is critical in inducing formation of DMVs (Pedersen et al., 1999, Snijder et al., 2001). Among the EAV replicase subunits, the nsp2, nsp3, and nsp5 encode a large number of hydrophobic regions that are presumed to have a structural function by inducing DMV formation (Snijder et al., 2001) and anchoring the RTC to the membrane (Snijder et al., 1994, van der Meer et al., 1998, Wassenaar et al., 1997). More recently, isolated RTCs were shown to contain replicase subunits involved in DMV formation (nsp2 and nsp3) or viral replication (nsp9 and nsp10), and were co-sedimented with newly synthesized viral RNA in a heavy membrane fraction (van Hemert et al., 2008), which confirmed their association with DMVs and their essential role in RTC function.
4.3. Nonstructural proteins of EAV
The post-translational processing of the EAV replicase pp1a and pp1ab has been studied extensively (Siddell et al., 2005, Ziebuhr et al., 2000 and references therein). In particular, pp1a and pp1ab undergo proteolytic processing at 11 cleavage sites by three virus-encoded proteinases (nsp1, nsp2, and nsp4) resulting in 13 nsps (1–12, including nsp7α and 7β; Fig. 2, Fig. 4, Table 1; van Aken et al., 2006c, Ziebuhr et al., 2000). Of these, the nsp9 to nsp12 subunits are encoded by ORF1b, although nsp9, the viral RNA-dependent RNA polymerase (RdRp), includes a small N-terminal ORF1a-encoded domain, which is identical to nsp8 and is encoded just upstream of the ribosomal frameshift site. The ORF1a encodes two papain-like cysteine protease domains (“accessory proteases” located in nsp1 and nsp2) and a chymotrypsin-like serine protease (“main protease”) in nsp4. The ORF1a protein is cleaved at seven sites resulting in eight cleaved end products, nsp1 to nsp8, plus a number of processing intermediates (Snijder et al., 1994, Snijder et al., 1996, Wassenaar et al., 1997). The ORF1b is processed into four major end products by nsp4 protease, including the RNA-dependent RNA polymerase (RdRp) and NTPase/RNA helicase domains essential for viral RNA replication and mRNA transcription (den Boon et al., 1991, Gorbalenya et al., 1989b). Immunofluorescence studies have shown that the ORF1b-encoded replicase subunits are localized to the perinuclear region of EAV-infected cells suggesting that they may be associated with intracellular membranous compartments (van Dinten et al., 1996), most likely the ER. These observations were further confirmed by in-depth ultrastructural analysis of EAV-infected cells using electron tomography (see above; Knoops et al., 2012). The mature nsps are involved in the formation of a membrane-anchored RTC that directs viral genome replication and synthesis of a nested set of sg mRNAs (Pasternak et al., 2006, Sawicki et al., 2007). These three virus-encoded proteinases and their corresponding cleavage sites are well conserved in other arteriviruses.
Nsp1 – The 29-kDa nsp1 is the first viral protein expressed and is the most N-terminal replicase cleavage product produced. It contains an “accessory protease” that is not involved directly in proteolytic processing of the RdRp and helicase. Three domains have been identified in the nsp1 region: (1) A predicted zinc finger (ZF) domain; (2) A papain-like cysteine protease (PCP) α; and (3) PCPβ. The predicted ZF domain present near the N terminus of nsp1 contains four Cys and His residues (Van Hemert and Snijder, 2008). ZFs in many cellular transcription factors are involved in protein–nucleic acid and protein–protein interactions (Laity et al., 2001). The nsp1 is released from the replicase polyprotein by the autocatalytic activity of PCPβ, which is responsible for cleavage of nsp1/2, located in its C-terminal domain (Snijder et al., 1992, Snijder et al., 1994). The cleavage and release of nsp1 from the replicase polyprotein is essential for viral RNA synthesis (Tijms et al., 2007). In EAV, the PCPα is functionally inactive due to its loss of an active Cys site during virus evolution (den Boon et al., 1995). However, PCPα may possess RNA-binding and/or protein-binding activities (along with PCPβ), serving as a platform for the ZF during transcription. Therefore, the structural integrity of nsp1 is essential since all three domains are required for trans-activation of sg RNA synthesis (Tijms et al., 2001). It has been reported that nsp1 is essential for sg RNA transcription but dispensable for genome replication (Tijms et al., 2007). The nsp1 not only plays a crucial role in processing of the replicase polyprotein and production of sg mRNA, but it is also important for virion biogenesis (Tijms et al., 2007). Nsp1 is the only EAV replicase subunit that localizes both in the nucleus and cytoplasm where it associates with modified cytoplasmic membranes of the replication complex (Pedersen et al., 1999, Tijms et al., 2002). During the early stages of infection, nsp1 is primarily located in the nucleus while its cytoplasmic localization, especially in the perinuclear region, becomes evident later in infection (Tijms et al., 2002). Nsp1 protein is also implicated in controlling the balance between genome replication and sg mRNA synthesis (Nedialkova et al., 2010). Reverse and forward genetics studies have shown that nsp1 tightly controls the relative abundance of viral mRNA species in infected cells. In summary, nsp1 plays a major role in fine-tuning virus replication, sg mRNA synthesis, and virus production, and in coordinating the replicative cycle of arteriviruses.
Nsp2 – The 61-kDa nsp2 serves as a cofactor in the processing of the downstream part of the polyprotein directed by the nsp4 protease (Snijder et al., 2001). The nsp2 contains two hydrophobic regions around aa 450 and 490 and a main hydrophobic region between aa 520 and 640, which are sufficiently large to span the lipid bilayer several times. However, the exact topology of this protein awaits further characterization (Snijder et al., 2001). The N-terminal domain of nsp2 contains cysteine protease activity responsible for cleavage of the nsp2/3 site (Snijder et al., 1995b). Therefore, nsp2 is released from the polyprotein by internal cysteine autoprotease activity (Snijder et al., 1994). In the major processing pathway, the mature nsp2 acts as cofactor for the processing of the nsp4/5 site by the nsp4 serine protease (SP; Fig. 5). The nsp2 interacts strongly with nsp3 or nsp3-containing precursors (e.g. nsp3–8), a property that may be required for processing the nsp4/5 site by the nsp4 SP. When there is no interaction between nsp2 and nsp3–8, its internal site nsp4/5 cannot be cleaved and further processing of the replicase subunits occurs via the minor processing pathway (Fig. 5).
Recently it has been shown that the nsp2 of arteriviruses has deubiquitinating enzyme (DUB) activity (van Kasteren et al., 2012). The DUB enzymes remove ubiquitin from innate immunity signaling factors and provide a potential opportunity for viruses to evade host innate defense system. The DUBs of arteriviruses resemble the ovarian tumor domain-containing (OTU) family of DUBs. The arterivirus DUB domain resides in the N-terminal region of nsp2 and was previously shown to direct the proteolytic processing of the nsp2/3 site. In addition to its resemblance to OTU DUBs, the nsp2 protease belongs to the papain-like protease (PLP) family and therefore, it has been referred to as PLP2-DUB. Using both in vitro and cell-based assays, it has been shown that PLP2-DUB activity is conserved in all members of the arterivirus family and that DUB can inhibit RIG-I-mediated innate immune signaling when overexpressed in mouse embryonic fibroblast cells (van Kasteren et al., 2013). These studies have also shown that the production of beta interferon (IFN-β) depends on the recognition of arterivirus RNA by the pattern-recognition receptor MDA5, and EAV PLP2-mutant viruses that lack DUB activity exhibit strikingly enhanced innate immune signaling in infected cells. Compared with wild type virus infection, IFN-β mRNA levels in equine cells infected with PLP2 mutants were increased by nearly a log 10 order of magnitude. The crystal structure of EAV PLP2 in complex with ubiquitin has been resolved and it has been shown that PLP2 binds ubiquitin using a zinc finger that is uniquely integrated into an exceptionally compact OTU-domain fold that represents a new subclass of zinc dependent OTU DUBs (van Kasteren et al., 2013). The ubiquitin-binding surface is distant from the catalytic site, which explains how mutation(s) on this surface can reduce DUB activity without affecting polyprotein cleavage. These new findings not only established PLP2 DUB activity as a critical factor in innate immune evasion of EAV (and other arteriviruses), but the selective inactivation of DUB activity also offers an innovative strategy for developing improved MLV vaccines against arteriviruses encoding similar dual-specificity proteases.
Nsp3 – The 22-kDa nsp3 is highly hydrophobic and is presumed to be a tetra-spanning transmembrane molecule (Posthuma et al., 2008). It contains four conserved cysteine residues in the predicted first luminal domain (aa 853–902) that are likely to be involved in membrane pairing and DMV formation (Posthuma et al., 2008). Due to the multiple spanning nature of the protein, nsp3 is likely to play a key role in the interaction between the RTC and host cell membranes. Site-directed mutagenesis studies have shown that mutation of nsp3 impairs the interaction of the protein with the ER, disrupts the formation of DMVs, affects replicase polyprotein processing, and inhibits viral RNA synthesis (Posthuma et al., 2008, Snijder et al., 2001). It is possible that the N-terminal domain (aa 833–993) of nsp3 could function as a signal sequence for membrane insertion of nsp2/3 polyprotein before its cleavage by nsp2 (Posthuma et al., 2008). However, the mechanism of interaction between nsp3 and the membranes or with other replicase subunits involved in DMV formation remains poorly understood.
Nsp4 – The 21-kDa nsp4 has chymotrypsin-like SP activity at the N-terminal domain (Barrette-Ng et al., 2002). The nsp4 includes a unique C-terminal domain (CTD) connected to the catalytic two-barrel structure by aa residues 155 and 156 that are poorly conserved among arteriviruses (van Aken et al., 2006a, van Aken et al., 2006b). The three-dimensional structure of EAV nsp4 has been determined to 2.0 Å resolution, showing that nsp4 adopts the smallest known chymotrypsin-like fold with a canonical catalytic triad of Ser-120, His-39, and Asp-65, and a novel alpha/beta C-terminal extension domain that may play a role in mediating protein–protein interactions (Barrette-Ng et al., 2002). As the protease name indicates, the main catalytic residue of this C-like protease in arteriviruses is Ser. Nsp4 is the “main protease” that controls the production of the viral RNA-dependent RNA polymerase and RNA helicase (Gorbalenya et al., 1991, Ziebuhr et al., 2000). It is responsible for processing the polypeptides (nsp3–8 and nsp3–12) that remain after nsp1 and nsp2 have been autocatalytically released from pp1a and pp1ab (Barrette-Ng et al., 2002, Snijder et al., 1992, Snijder et al., 1994, Snijder et al., 1995b, Snijder et al., 1996, van Dinten et al., 1999, Wassenaar et al., 1997). The nsp4 SP cleaves at five sites in the ORF1a protein (Snijder et al., 1996) and three sites in the ORF1b protein (Wassenaar et al., 1997). The nsp4 SP processes the C-terminal half of the ORF1a (nsp3–8) protein via two alternative pathways (Fig. 5). In the major processing pathway, nsp2 associates with nsp3–8 (96 kDa) as a cofactor that triggers nsp4 SP to cleave the majority of nsp3–8 precursors at the nsp4/5 site resulting in nsp3–4 (50 kDa) and nsp5–8 (44 kDa) intermediates. Subsequently, the nsp5–8 is cleaved at the nsp7/8 site while the nsp5/6 and nsp6/7 are inaccessible to the protease (Wassenaar et al., 1997). In the minor processing pathway, the nsp4/5 site remains intact while the nsp5/6 and nsp6/7 sites are cleaved (Fig. 5; Wassenaar et al., 1997). Furthermore, it was also shown using reverse genetic analysis that all nsp4-mediated cleavages in EAV pp1a are critical for viral RNA synthesis (Barrette-Ng et al., 2002). In addition, these studies identified a novel cleavage site for the nsp4 protease, which is located within the nsp7 subunit and appears to be conserved among arteriviruses (van Aken et al., 2006c).
Nsp5, nsp6, nsp7α/β, and nsp8 – The processing intermediate nsp5/6/7/8 is a result of cleavage of the nsp4/5 junction upon association of nsp2 as a cofactor with nsp3–8. In the major pathway, nsp3/4 and nsp7/8 junctions are cleaved, but not the nsp5/6 and nsp6/7 (Fig. 5); therefore, cleaved nsp6 and nsp7 are not generated. In the minor pathway, nsp4/5 is not cleaved; instead, nsp4 cleaves the nsp3/4, nsp5/6, nsp6/7, and nsp7/8 junctions of the nsp3–8 intermediate producing cleaved nsp6 and nsp7 (Wassenaar et al., 1997). Fully processed nsp5, nsp6, nsp7, and nsp8 are approximately 41, 3, 25, and 5.5 kDa, respectively (Snijder et al., 1994).
The nsp5 contains a significant portion of the highly hydrophobic domain suggesting a role in the membrane association of the EAV replication complex (Snijder et al., 1994, van der Meer et al., 1998). Moreover, the presence of hydrophobic nsp5 in the N-terminus of processing intermediates, like nsp5–8, nsp5–7 and even larger intermediates such as nsp5–12, supports its probable association with the intracellular membrane (van der Meer et al., 1998). There is little published information regarding the nsp6 and nsp8 proteins (Snijder et al., 1994). An internal cleavage site (nsp7α/β) is present in mature nsp7 that is cleaved by nsp4 protease to produce nsp7α (123 aa; ∼13.5 kDa) and nsp7β fragments, respectively, corresponding to the N- and C-terminal regions of nsp7 (van Aken et al., 2006c). Most recently, the structure of nsp7α, the most conserved region of nsp7, has been determined by nuclear magnetic resonance (NMR) spectroscopy (Manolaridis et al., 2011). The nsp7α contains four hydrophobic regions needed for correct folding of the proteins and has a unique conformation consisting of three α-helices packed against a mixed β-sheet. Although this study did not provide a definite conclusion regarding the function of nsp7, it confirmed that nsp7α is critical for arterivirus RNA synthesis.
Nsp9 – The 80-kDa EAV nsp9 (aa 1678–2370 of pp1ab) contains the RdRp domain that functions as the catalytic subunit of the viral RTC and directs viral RNA synthesis in conjunction with other viral proteins and host cellular proteins. The EAV nsp9/RdRp initiates RNA synthesis by a de novo mechanism involving homopolymeric templates in a template-specific manner. Increased intracellular concentration of Zn2+ can block the initial step of viral RNA replication carried out by nsp9/RdRp (te Velthuis et al., 2010).
Nsp10 – The 51-kDa EAV nsp10 contains a predicted zinc-binding domain (ZBD) in its N-terminus and the nucleoside triphosphate-binding/helicase (Hel) motif in its C-terminal domain (den Boon et al., 1991, Gorbalenya et al., 1989b, van Dinten et al., 2000). The nsp10 is the most conserved replicase subunit of the viral RNA synthesis machinery (Gorbalenya et al., 1988, Gorbalenya et al., 1989a, Gorbalenya and Koonin, 1989). The nsp10 has ATPase and nucleic acid duplex-unwinding activities that require the presence of 5′ single-stranded regions indicating 5′–3′ polarity of the reaction (Seybert et al., 2000). The ZBD region, especially the 13 conserved Cys and His residues that are most likely to be associated with zinc binding, is critical for ATPase and duplex-unwinding activities of the C-proximal helicase domain (den Boon et al., 1991, Gorbalenya et al., 1989b, Seybert et al., 2005, van Dinten et al., 2000). Reverse genetic studies have shown that mutation of conserved Cys/His residues in ZBD disrupted interactions between the ZF and enzymatic domains, thus completely blocking viral RNA synthesis and virion biogenesis (Seybert et al., 2005, van Dinten et al., 2000). Taken together, the nsp10-associated ZF domain has multiple functions specifically involved in viral RNA replication, sg mRNA transcription and virion biogenesis (van Dinten et al., 2000).
Nsp11 – The 26-kDa nsp11 is a multifunctional protein with a crucial role in viral RNA synthesis. The nsp11 contains a highly conserved endoribonuclease (NendoU; nidoviral-endonuclease specific for U) domain that is not present in other RNA viruses and is therefore considered a unique genetic marker of the order Nidovirales (Ivanov et al., 2004, Posthuma et al., 2006). The NendoU possesses pyrimidine-specific (uridylates in particular) endoribonuclease activity independent of divalent cations (e.g. Mn2+) in vitro (Nedialkova et al., 2009). Studies using reverse genetics have shown the importance of NendoU activity for viral RNA synthesis (Nedialkova et al., 2009, Posthuma et al., 2006). However, identification of substrate and molecular characterization of NendoU remain unclear.
Nsp12 – There is no published information regarding either the structure or role of the 12-kDa nsp12 of arteriviruses.
4.4. Structural proteins of EAV
4.4.1. Major envelope proteins
GP5 – The 30- to 42-kDa GP5 protein is an abundant virion protein encoded by ORF5. GP5 typically contains either one or two potential N-glycosylation sites (Asn-56 and Asn-81), and the majority of laboratory adapted EAV strains contain only the N-linked sugar moiety at position Asn-56 (Fig. 7 ; Balasuriya et al., 1997, Glaser et al., 1995, Stadejek et al., 1999). A third potential N-glycosylation site at aa 73 (Asn-73) was identified in five EAV isolates from clinical specimens obtained during the extensive 2006–2007 outbreak of EVA in the US (Zhang et al., 2010b). The GP5 protein is a type IV integral membrane protein with triple-membrane spanning domains from aa 116 to 181 (Fig. 7) (de Vries et al., 1992). The ectodomain of GP5 includes 95 aa and contains an N-terminal signal sequence (aa 1–18) that is cleaved (Snijder et al., 2003). The smear appearance of the GP5 protein after analysis by denaturing SDS-PAGE is due to the addition of poly-N-acetyllactosamine during its transport through the secretory pathway (de Vries et al., 1992, de Vries et al., 1995a). However, the function of poly-N-acetyllactosamine modification of the GP5 protein is unclear.
Fig. 7.
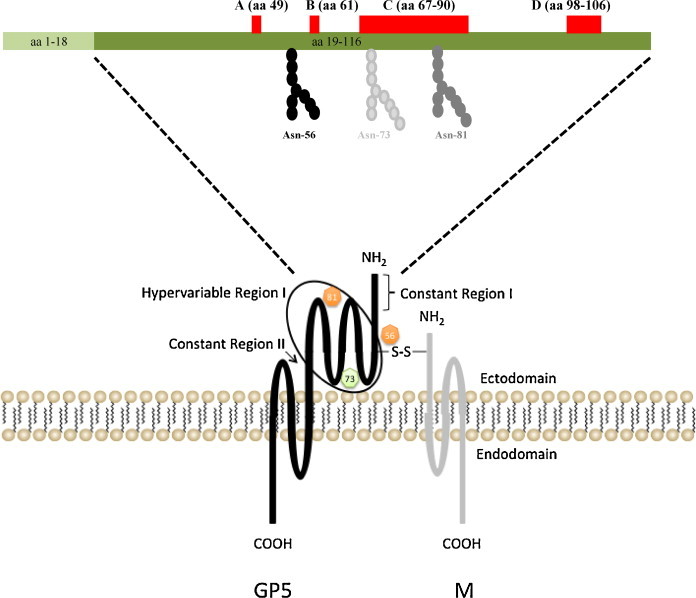
Predicted structure of GP5 and M heterodimer. The predicted N-glycosylation sites (Asn-56 and Asn-81) are depicted with an orange circle. The green circle represents the N-glycosylation site (Asn-73) found in isolates of an extensive recent EVA outbreak in North America. The GP5 and M proteins are covalently linked by a disulfide bond (S—S) formed between Cys-34 in the GP5 protein and Cys-8 in the M protein. The major neutralization determinants of EAV that are located in the N-terminal ectodomain of the GP5 major envelope glycoprotein of EAV are depicted. The signal sequence (aa 1–18), four major neutralization sites (A, B, C and D) and putative glycosylation sites (aa positions 56, 73 and 81) are identified.
Equine EAV-specific polyclonal antisera and EAV-neutralizing monoclonal antibodies bind to the N-terminal hydrophilic ectodomain (aa 19–115) of GP5 (Balasuriya et al., 1997, Chirnside et al., 1995a, Deregt et al., 1994, Glaser et al., 1995, Nugent et al., 2000). Four distinct neutralization determinants of the virus have been identified in GP5: aa 49 (site A), 61 (site B), 67–90 (site C), and 98–106 (site D; Fig. 7; Balasuriya et al., 1993, Balasuriya et al., 1995a, Balasuriya et al., 1997, Chirnside et al., 1995a, Deregt et al., 1994, Glaser et al., 1995, Zhang et al., 2008b). Except for site A located in the C1 region, these sites are located in the V1 variable region (aa 61–121) within the second half of the N-terminal ectodomain. Site D expresses several overlapping linear epitopes in the protein that may possibly interact with the three other sites to form conformational epitopes (Balasuriya et al., 1995a, Balasuriya et al., 1997, Balasuriya et al., 2004a, Glaser et al., 1995).
Membrane (M) protein – The 16-kDa M protein is encoded by ORF6. The M protein is the most highly conserved envelope protein of arteriviruses (de Vries et al., 1992). The M protein has three internal membrane-spanning segments and a short amino terminus exposed at the virion surface (aa 10–18), and its carboxy-terminus (approximately aa 72) is buried within the virus’ interior (Fig. 7; de Vries et al., 1992, Snijder and Meulenberg, 1998). It lacks N-glycosylation sites and contains five methionine residues and one cysteine. The M protein forms a disulfide-linked heterodimer with the GP5 protein in the virus particle (de Vries et al., 1995a). The M protein also forms covalently linked homodimers, but only the GP5/M heterodimers are incorporated into virus particles. It is possibly involved in virus budding.
4.4.2. GP5/M heterodimeric complex
The GP5 and M proteins form a disulfide-linked heterodimer in the virus particle and occur in equimolar amounts in the virion (Fig. 7; de Vries et al., 1995a). These proteins associate with each other with different kinetics and efficiencies in infected cells. Newly synthesized GP5 proteins rapidly interact with M protein, while newly produced M proteins are incorporated slowly into the heterodimer. Presence of a high concentration of M protein in the ER is a prerequisite for efficient recruitment of GP5 protein into the GP5/M heterodimeric complex (de Vries et al., 1995a). The interaction between the two major envelope proteins is determined by the formation of the disulfide bridge between the cysteine residue at position 8 (Cys-8) of M and Cys-34 of GP5 since abolition of either cysteine residue completely inhibited heterodimerization of the two proteins (Snijder et al., 2003). Heterodimerization of the two major envelope proteins is essential for EAV infectivity. Studies using chimeric viruses have shown that replacement of the EAV GP5 ectodomain with that of different arterivirus species did not alter the cell tropism of EAV, indicating that GP5 is not involved in receptor binding (Dobbe et al., 2001).
The GP5/M heterodimeric complex is essential for EAV assembly where the interaction between nucleocapsid and viral envelope is most likely mediated by the disulfide-bonded GP5/M heterodimers since the minor envelope proteins are not required for generation of virus particles. Covalent association of the GP5 and M proteins is indispensable for the production of infectious arteriviruses (Snijder et al., 2003, Verheije et al., 2002). It has been clearly demonstrated that heterodimerization of the GP5 and M proteins is critical for the authentic post-translational modification (glycosylation) and conformational maturation of the neutralization determinants in GP5 (Balasuriya et al., 2000, Balasuriya et al., 2004a). The M protein may act as an essential scaffold on which the GP5 protein folds to form the epitopes that induce neutralizing antibodies in virus-infected animals. Nonetheless, peptides derived from portions of the GP5 ectodomain can induce EAV-neutralizing antibodies in horses (Castillo-Olivares et al., 2001, Chirnside et al., 1995a).
4.4.3. Minor envelope proteins
E protein – The EAV E protein is a small 8 kDa protein encoded by ORF2a (Snijder et al., 1999). ORF2a and ORF2b are both expressed from a bicistronic mRNA2. The E protein lacks N-linked oligosaccharide side chains and its single cysteine residue does not form intermolecular disulfide bonds. E protein is stable, highly hydrophobic, and predicted to be an integral membrane protein with an uncleaved signal-anchor sequence in the central part of the molecule (Fig. 8 ). E protein could be either a type III integral membrane protein or it may span the lipid bilayer twice with both termini located in the cytoplasmic face of the membrane (Snijder et al., 1999). It contains a conserved site for myristoylation in the N-terminal domain followed by a phosphorylation site for casein kinase II (Thaa et al., 2009). The E protein occurs in intermediate amounts in infected cells, as compared to the major structural proteins (Snijder et al., 1999). It has been suggested that the E protein is noncovalently associated with the GP2, GP3 and GP4 trimeric complex. In the absence of the minor envelope protein complex, the amount of E protein in viral particles is diminished by 60–80%. Similarly, the absence of E protein completely blocks the incorporation of the minor envelope proteins (GP2, GP3, and GP4) into viral particles (Wieringa et al., 2004). These findings suggest that the E protein along with GP2 is essential for the production of infectious progeny virus but dispensable for virus assembly and release (Snijder et al., 1999). The E protein may have an intermediate receptor binding and/or virus entry function with the GP2/GP3/GP4 complex (Das et al., 2011). It has been also shown that the porcine arterivirus (PRRSV) E protein possesses ion channel protein-like properties and is essential for virus infection but dispensable for virion assembly (Lee and Yoo, 2006, Wu et al., 2005).
Fig. 8.
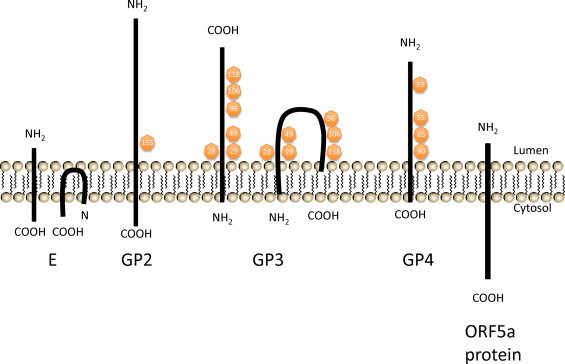
Predicted membrane topology of the minor envelope proteins: E, GP2, GP3, GP4, and ORF5a protein. Two alternative predictions of E protein and GP3 membrane topology are shown. Putative N-glycosylation sites are indicated in yellow circles with corresponding amino acid positions.
GP2 – The GP2 protein is a 25 kDa minor envelope protein encoded by ORF2b (Fig. 8). GP2 is a type I membrane glycoprotein with a single N-glycosylation site at position 155 (Asn-155) (de Vries et al., 1995b). It has three amino-terminal (extracellular) cysteine residues likely to be important for dimerization and protein function, and one in its transmembrane anchor domain (de Vries et al., 1995b). The Cys-102 is responsible for the cystine bridge with the GP4 protein while Cys-48 and Cys-137 form an intramolecular disulfide bond (Wieringa et al., 2003a). The GP2 protein occurs in several distinct conformations, including as four monomers as a result of different disulfide-bonded structures and as a disulfide-linked dimer (de Vries et al., 1995b). However, only the dimeric form of GP2, which is further associated with GP3 and GP4 proteins as a heterotrimeric complex, is assembled into viral particles whereas the GP2 monomers are retained in the endoplasmic reticulum of infected cells (de Vries et al., 1995b). GP2 is abundant in EAV-infected cells, but is expressed in low amounts in viral particles. The GP2 protein is not essential for viral RNA replication and transcription (Molenkamp et al., 2000, Snijder et al., 1999). Sequence comparison analysis indicates that the GP2 protein is highly conserved between EAV isolates (Hedges et al., 1996).
GP3 – The GP3 is encoded by ORF3 and is a membrane-associated protein of 36–42 kDa with six potential N-glycosylation sites, Asn-28, Asn-29, Asn-49, Asn-96, Asn-106, and Asn-118 (Hedges et al., 1999a, Wieringa et al., 2002). The membrane topology of GP3 proteins has still not been clearly demonstrated. The protein could be either a class II protein, anchored into the lipid bilayer by its uncleaved amino-terminal signal sequence, or a class IV protein, possibly embedded by both sides of the hydrophobic terminal domains (Fig. 8; Hedges et al., 1999a, Wieringa et al., 2002). The GP3 protein has two putative N-myristoylation sites and three casein kinase II phosphorylation motifs but it is unclear whether they are active sites. Little is known about the function of this protein; however, inactivation of ORF3 expression inhibited the production of infectious virus particles indicating that GP3 protein has an essential role in the viral replication cycle.
GP4 – The GP4 protein, encoded by ORF4, is a typical class I integral membrane glycoprotein with amino-terminal signal sequence and its predicted carboxy-terminal hydrophobic membrane anchor (Fig. 8; Wieringa et al., 2002). Three of the four predicted N-glycosylation sites (Asn-33, Asn-55, Asn-65, and Asn-90) are functional and produce a fully glycosylated protein with a molecular mass of 28 kDa (Wieringa et al., 2002). However, Wieringa et al. (2002) reported that N-glycosylation of the GP4 protein is an inefficient process so that only some of the protein molecules acquire N-linked glycans in EAV-infected cells. Moreover, only the fully glycosylated form of GP4 is present in virus particles, which indicates that only a small fraction of the ORF4 products produced in EAV-infected cells are incorporated into virus particles (Wieringa et al., 2002). The function of GP4, like that of the GP3 protein, is not well characterized, but both are essential in the viral replication cycle.
ORF5a protein – A detailed computational analysis revealed an additional ORF (ORF5a) that overlaps the 5′ end of ORF5, and is conserved in all arterivirus species (Firth et al., 2011, Johnson et al., 2011). The ORF5a protein is 59 aa and is likely to be expressed from the same subgenomic mRNA (sg mRNA5) as the GP5 protein, possibly involving leaky ribosomal scanning. The ORF5a protein is predicted to be a type III membrane protein with a short (5–12 aa) amino terminal domain into the lumen, a central signal-transmembrane sequence and a carboxy-terminal domain into the cytosol (Fig. 8; Johnson et al., 2011). The function of this protein is yet to be characterized, but reverse genetic studies suggest that this novel protein may be the eighth structural protein of arteriviruses and important for arterivirus infection.
4.4.4. GP2/GP3/GP4 heterotrimeric complex
The GP2, GP3, and GP4 proteins are incorporated into virions as a covalently associated heterotrimeric complex and lack of any one of the components blocks the successful incorporation of the other minor envelope proteins (Wieringa et al., 2004). After the release of virus particles from infected cells, GP3 becomes disulfide-linked to the GP2/GP4 heterodimers which results in a 66-kDa complex consisting of covalently bound GP2, GP3, and GP4 (Wieringa et al., 2003b). Due to this post-assembly modification, both GP2/GP4 heterodimers and GP2/GP3/GP4 heterotrimers are detected in EAV particles (Wieringa et al., 2003b). Cystine bridges play essential roles in the formation and stabilization of the trimeric complex. The cysteine residues at positions 48 and 137 (Cys-48 and Cys-137, respectively) of GP2 are linked by an intra-chain disulfide bond while the cysteine residue at position 102 (Cys-102) of GP2 forms an intermolecular cystine bridge with one of the cysteines of the GP4 protein (Wieringa et al., 2003a). The covalent association of GP3 with the GP2/GP4 heterodimer likely occurs through the GP4 protein; however, the precise cysteine residue forming the intermolecular disulfide bond between GP4 and GP3 has not yet been determined (Fig. 9 ). Similarly, the cysteine residue of GP4 that interacts with Cys-102 of GP2 has not been identified. It has been shown that folding and oligomerization of GP2, GP3, and GP4 expressed in E. coli are independent processes (Kabatek, 2013). However, it remains to be shown whether this is true in virus-infected cells. The GP2/GP3/GP4/E heterotrimeric complex is most likely involved in the virus entry process into the cell based on the observation that it is not required for EAV virus particle formation, but is essential for production of infectious progeny virus.
Fig. 9.
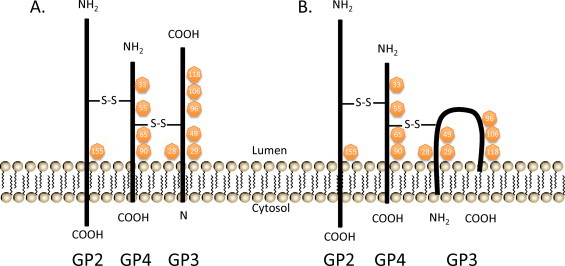
Two predicted models for the disulfide-bonded structure of the covalently linked GP2—GP3—GP4 heterotrimer. These alternative models differ in membrane topology of GP3, which is currently uncertain. The GP3 protein may be either a class II membrane protein (A) or a class IV membrane protein (B). The intermolecular cystine bridges are depicted arbitrarily with disulfide bonds (S—S) since the Cys positions have not yet been determined. N-glycosylation sites are indicated in orange circles with corresponding amino acid positions.
Modified from Wieringa et al. (2003b) with permission.
4.4.5. Nucleocapsid (N) protein
The 14-kDa N protein, encoded by ORF7, is the most abundant viral protein in EAV-infected cells. The N protein is synthesized in the cytoplasm of EAV-infected cells and possibly binds to envelope protein domains exposed to the cytoplasm during the budding process. Most of the N protein localizes in the cytoplasm of infected cells, whereas a small amount resides in the nucleus where it apparently accumulates in nucleoli. This suggests a possible post-translational modification of some molecules of N protein, such as phosphorylation. The N-terminal domain (aa 1–47) of the N protein, the least conserved region, is thought to interact with the viral genomic RNA during nucleocapsid assembly (Tijms et al., 2002). The C-terminal (aa 49–110) of the N protein forms a dimer consisting of a β-sheet that is capped and flanked by α-helices. In the crystal structure, the N protein forms a dimer–dimer (i.e. a tetramer), organized via a quasi-twofold axis (Deshpande et al., 2007). The N protein comprises more than 30% of the protein molecules in EAV particles.
5. Host–virus interactions
5.1. Pathogenesis
Although it is abundantly clear that many of the clinical manifestations of EVA result from vascular injury, the pathogenesis of EVA has not yet been comprehensively defined. Studies with the highly virulent, horse-adapted Bucyrus strain of EAV confirm that death in horses inoculated with this virus is a consequence of severe vascular damage potentially leading to disseminated intravascular coagulation (Del Piero, 2000a, Del Piero, 2000b, Lopez et al., 1996, MacLachlan et al., 1996). Similarly, endothelial infection is characteristic of EVA in horses naturally infected with the virus (Del Piero, 2000a, Del Piero, 2000b). The characteristic vascular lesions of EVA have been compared to those of Aleutian disease of mink and other immune-mediated vascular diseases (Bishop, 1989, Henson and Crawford, 1974), however, the lesions of EVA do not appear to be the result of immune-mediated injury because they develop at only 4–5 days after experimental inoculation, which is not consistent with a typical immune-mediated process. Furthermore, arteries larger than 1 mm may be affected, and neither immunoglobulin G (IgG) nor complement (C3) are present in the lesions, as would be expected if immune complexes were responsible. Therefore, vascular injury in EVA likely results from direct virus-mediated injury to the lining (endothelium) of affected vessels perhaps exacerbated by the activity of virus-induced, host-derived procoagulant and proinflammatory mediators. In muscular arteries, EAV infects and replicates in endothelial cells (ECs) and causes extensive damage to the endothelium and the subjacent internal elastic lamina, then gains access to the media of the wall of affected vessels. Increased vascular permeability and leukocyte infiltration resulting from generation of chemotactic factors lead to hemorrhage and edema around these vessels (Coignoul and Cheville, 1984, Estes and Cheville, 1970).
EAV infects alveolar macrophages (AMΦ) after respiratory infection of horses, but recent in vitro studies using cultured equine AMΦ confirmed that although these cells were susceptible to infection, they were refractory to productive replication of EAV (Moore et al., 2003). In contrast, productive replication occurred following EAV infection of cultured blood-derived equine macrophages (BMΦ; Moore et al., 2003). EAV infection of both equine AMΦ and BMΦ resulted in increased transcription of genes encoding proinflammatory mediators, including IL-1β, IL-6, IL-8, and TNF-α, with the release of substantial quantities of TNF-α into the culture medium (Moore et al., 2003). Furthermore, virulent and avirulent strains of EAV induced different quantities of TNF-α and other proinflammatory cytokines (IL-1b, IL-6, IL-8), and the magnitude of the cytokine response of equine AMΦ and BMΦ to EAV infection reflected the virulence of the infecting virus strain (Moore et al., 2003). In addition, expression of leukocyte adhesion molecules is upregulated following EAV infection. Not only may these factors be important in inciting inflammation to limit initial virus replication, they may also contribute to the vascular and tissue injury that characterizes EVA. In summary, these in vitro studies clearly showed that cytokine mediators are produced by EAV infected equine cells and, presumably, these mediators can play an important role in determining the nature and severity of the outcome of infection. However, full characterization of the innate and adaptive immune responses of horses to EAV infection have not yet been done and, in particular, the likely central role of dendritic cell infection should be addressed. Similarly, although EAV causes lytic infection in susceptible cells, the mechanism(s) of cell death remains poorly characterized. EAV infection induces apoptosis by activation of caspase 8 and caspase 9 after infection of Vero cells (Archambault and St-Laurent, 2000, St-Louis and Archambault, 2007), and apoptosis in EAV infected BHK-21 cells is mediated through the intrinsic signaling pathway (Cholleti et al., 2013). Taken together, these data provide evidence that EAV is able to induce apoptotic cell death in vitro. However, in-depth mechanism(s) of apoptosis or the significance of apoptosis in pathogenesis of EVA during natural infection has not been investigated.
Abortion after EAV infection of pregnant mares likely is the result of a lethal fetal infection, rather than myometritis or placental damage that impairs progesterone synthesis, leading to fetal expulsion (MacLachlan et al., 2000). The tissues of aborted fetuses contain higher titers of virus than those of the dams from which they abort, indicating that substantial virus replication can occur in the fetus itself. The stress that results from fetal infection would be expected to activate the fetal hypothalamic-pituitary axis, thus inducing abortion.
5.2. Use of infectious cDNA clones of EAV and reverse genetics
The naked genome of a positive-strand RNA virus is infectious: it serves as an mRNA for translation of viral replicase, thus, infection can be initiated by a simple transfection of the genome into the host cell. Construction of full-length cDNA clones of plus-stranded RNA viruses have facilitated studies in which site-specific nucleotide changes (site-directed mutagenesis) are introduced into the viral genome to study the mechanisms of virus replication and gene expression at the molecular level. Similarly, entire portions of the genome of different virus strains, or related viruses, can be exchanged to produce recombinant chimeric viruses. Infectious RNA transcripts are produced in vitro from linearized full-length infectious cDNA clones transfected into mammalian cells. Such infectious cDNA clones of RNA viruses are used not only as basic research tools, but also to develop rational vaccines against viral pathogens.
The original infectious cDNA clones of EAV were derived from a highly cell culture adapted strain of the virus (Table 2; de Vries et al., 2000, Glaser et al., 1998, van Dinten et al., 1997) and the recombinant progeny virus derived from the first EAV infectious cDNA clone (GenBank accession number: Y07862; van Dinten et al., 1997), pEAV030 is attenuated in horses (Balasuriya et al., 1999b). Snijder and colleagues did pioneering work to characterize the molecular biology of EAV and other nidoviruses using the pEAV030 infectious cDNA clone (Siddell et al., 2005, Snijder et al., 1995a, Snijder et al., 2005, Snijder et al., 2006, Snijder, 1998). Specifically, they used this infectious cDNA clone to characterize the mechanism of EAV replication along with processing of viral non-structural proteins (den Boon et al., 1995, Snijder, 1998, Snijder, 2001, van Aken et al., 2006a, van Aken et al., 2006b, van Dinten et al., 1999), transcription and subgenomic mRNA synthesis (Tijms et al., 2001, Tijms et al., 2007, Tijms and Snijder, 2003, van den Born et al., 2005a, van Dinten et al., 2000, van Marle et al., 1999a, van Marle et al., 1999b), and the viral replication complex (Snijder et al., 2001, Snijder et al., 2006). This clone also apparently contributed to the publications of other groups (de Vries et al., 2000, Glaser et al., 1998). Balasuriya et al., 2004a, Balasuriya et al., 2004b employed site-directed mutagenesis of this same infectious cDNA clone to generate a panel of chimeric and mutant viruses to characterize the neutralization determinants of EAV (Balasuriya et al., 2004a). The third infectious cDNA clone of EAV was derived from the horse-adapted, experimentally derived, highly virulent Bucyrus strain (VBS) of the virus (Balasuriya et al., 2007). The recombinant virus generated from this infectious cDNA clone (prVBS) causes severe disease and high lethality in experimentally infected horses (Balasuriya et al., 2007). Recently, a full-length cDNA clone of the MLV vaccine strain (prMLV) that was originally derived by extended cell culture passage of the Bucyrus strain of EAV (VBS) has also been constructed (Zhang et al., 2012). These two related infectious cDNA clones have been used to generate a panel of chimeric viruses by swapping genes encoding minor and major envelope viral proteins from virulent and avirulent viruses to characterize the tropism of EAV to equine peripheral blood mononuclear cells (Go et al., 2010).
Reverse genetic manipulation of infectious cDNA clones of EAV has greatly increased the understanding of the replication of the virus and its molecular biology and pathogenesis, however, manipulation of the genome of arteriviruses is complicated due to the compact and overlapping gene arrangement. de Vries et al. (2000) investigated the importance of the overlapping gene organization in the virus’ life-cycle by constructing a series of mutant full-length cDNA clones in which EAV ORFs 4/5 or ORFs 5/6 or ORFs 4/5/6 were separated (de Vries et al., 2000). RNA transcribed from each of these plasmids was infectious, indicating that the overlapping gene arrangement is not essential for viability of the virus. The fact that small changes in the spacing between ORFs 4/5 and ORFs 5/6 in the mutant viruses had only a limited effect on virus yields allowed the generation of recombinant chimeric viruses between virulent and attenuated strains of EAV. Thus, genes encoding structural and nonstructural proteins can readily be interchanged between strains of EAV of differing virulence to horses, and these chimeric viruses have then been used to study virulence determinants, mechanisms of pathogenesis and persistent infection (Go et al., 2010, Zhang et al., 2008a, Zhang et al., 2008c). A similar approach has been used to construct a panel of EAV/PRRSV chimeric viruses by swapping GP5 and M proteins (ectodomains and full-length) of PRRSV into pVBS cDNA clone backbone (Lu et al., 2012).
EAV cDNA clones have also been used as a vector for heterologous gene expression (de Vries et al., 2000), such as enhanced green fluorescence protein (eGFP; de Vries et al., 2000, van den Born et al., 2007). The infectious recombinant EAV expressing GFP (EAV-GFP2) from its replicase had similar growth characteristics to those of the wild-type virus. The recombinant virus EAV-GFP2 is a convenient tool for basic and applied research studies. The same approach, namely insertion of a foreign gene in the arterivirus replicase gene for intracellular expression, could also be used for vaccine development.
5.3. Viral factors
Using the reverse genetics approach, the virulence determinants of EAV have been mapped to genes encoding both nonstructural (nsp1, nsp2, nsp7, and nsp10) and structural proteins (GP2, GP4, GP5, and M; Zhang et al., 2008a). However, it appears that the most important virulence determinants of EAV are located in the structural protein genes of the virus. The interaction among the GP2, GP3, GP4, GP5, and M envelope proteins plays a major role in determining the tropism of EAV for CD14+ monocytes, whereas tropism for CD3+ T lymphocytes is determined by the GP2, GP4, GP5, and M envelope proteins but not the GP3 protein (Go et al., 2010). Using an in vitro cell culture model of persistent EAV infection, it has been shown that combined amino acid substitutions in E, GP2, GP3, and GP4 proteins or a single amino acid substitution in the GP5 protein could facilitate persistent EAV infection in HeLa cells (Zhang et al., 2008c). However, the viral protein(s) involved in establishment of persistent infection in the stallion's reproductive tract are yet to be definitively identified. In summary, the virulence determinants of EAV appear to be complex and to involve multiple genes encoding both envelope and nonstructural proteins (multigenic).
5.4. Application of equine genomics
Viruses constantly adapt to and modulate the host environment to their advantage during replication and propagation. Therefore, virus interaction with host target cells is complex and it is possible that almost every step of the virus infection cycle may rely on the recruitment and co-opting of cellular proteins and basic machineries by viral proteins. Moreover, as a virus that induces persistent infection in some stallions as a virus-reservoir mechanism, EAV has likely developed strategies to avoid host antiviral factors by using its own viral proteins to modulate the cellular environment. With the advent of genomic tools, it has become possible to better study virus-host interactions at the molecular level. In particular, a variety of new “genomic and proteomic” methodologies have been developed to identify and characterize host molecules that closely interact with viruses during their life cycles. Thus, the “genomics revolution” has provided high-throughput tools with which to study complex virus-host interactions and, further, to provide information that will facilitate vaccine development and the design of efficacious therapeutic drugs.
It is abundantly clear that host genetic polymorphisms can affect microbial pathogenesis in animals. However, most host-pathogen interactions are complex and affected by several host genetic determinants. Until recently, emphasis was placed exclusively on identifying the viral factors affecting EVA pathogenesis, with no attention to the potential effect of host genetics on disease outcome. A high-quality draft of the genome of the horse (Equus caballus) was published recently (Wade et al., 2009), and the 55K equine single nucleotide polymorphisms (SNPs) chip was made available soon thereafter (Illumina Equine SNP50 chip, Illumina Inc, San Diego, CA). SNPs are variations of a single base-pair in the host's DNA; they are now used as genetic markers to identify chromosome regions associated with hereditary aspects of susceptibility to human or animal diseases (Chowdhary et al., 2008, Davila and Hibberd, 2009, Hill, 2006, Swinburne, 2009). The availability of the equine SNP chip provides a means of investigating genetic aspects of common horse diseases that was not previously possible.
5.5. Host factors
Recent studies have demonstrated that the clinical outcome of EAV infection is determined by host genetic factors. Specifically, based on the in vitro susceptibility of CD3+ T lymphocytes to EAV infection, horses can be segregated into susceptible and resistant phenotypic groups (Fig. 10 ; Go et al., 2010, Go et al., 2011a). Genome-wide association studies of these horses (GWAS) identified a common, genetically dominant haplotype associated with the in vitro susceptible phenotype in the region of ECA11 (49,572,804–49,643,932; Go et al., 2011a). Biological pathways analysis identified a variety of cellular genes within this region of ECA11 that encode proteins previously associated with virus attachment and entry, cytoskeletal organization, and NF-κB pathways. These cellular proteins are likely to be important in the pathogenesis of EVA infection in horses, so they clearly warrant further investigation to determine their precise roles (Go et al., 2011a). Furthermore, host cellular factors that are involved in formation of the replication complex and other membrane structures during virus replication, as well as cellular proteins involved in immune modulation during acute and persistent infection, should also be investigated using contemporary molecular biology techniques (e.g. siRNA, transcriptome analysis, etc.).
Fig. 10.
Dual-color immunofluorescence flow cytometry analysis of T cells from carrier and seropositive non-carrier stallions following in vitro infection with EAV. (A) Carrier status was confirmed by repeatedly isolating EAV from semen over an extended period of time. (B) Non-carrier stallions were uninfected but seropositive to EAV following natural infection, not vaccination. Lymphocytes from each stallion that were labeled with both anti-CD3 and anti-nsp1 (12A4) MAbs are shown in the upper right quadrant of each dot plot. Note significant double labeling of lymphocytes from the carrier stallion but not the non-carrier stallion for both CD3 and EAV after in vitro infection.
Modified from Go et al. (2012) with permission from the Journal of Virology.
Experimental EAV infection of horses with either the in vitro CD3+ T cell susceptibility or resistance phenotype showed a significant difference between the two groups in terms of proinflammatory and immunomodulatory cytokine mRNA expression. Of considerable importance, clinical signs of disease were enhanced in horses possessing the in vitro CD3+ T cell resistant phenotype (Go et al., 2011b). These studies provide direct evidence for a correlation in individual horses between their genotype and the extent of viral replication, occurrence and severity of clinical signs, and cytokine gene expression after experimental EAV infection. Furthermore, CD3+ T lymphocytes from EAV persistently infected (carrier) stallions are susceptible to in vitro EAV infection (Go et al., 2012), whereas stallions that did not become long-term carriers after infection with EAV did not possess the CD3+ T lymphocyte-susceptible phenotype (Fig. 10). Taken together, these data suggested that stallions with EAV susceptible CD3+ T lymphocytes are at a higher risk of becoming persistently infected as compared to stallions that lack this phenotype and GWAS analysis confirmed that five of seven proven carrier stallions had the dominant ECA11 haplotype previously associated with the CD3+ T lymphocyte susceptible phenotype. In summary, these data support the hypothesis that the long-term EAV carrier status in stallions is likely predisposed by specific host genetic factors.
6. Equine immune response to EAV
6.1. Innate immune response
Natural EAV infection of horses occurs via either the respiratory tract following exposure to infective secretions/excretions, or the reproductive tract following venereal exposure (Balasuriya et al., 2001, Timoney and McCollum, 1993). Thus, it is the innate immune response of the mucosal lining of the respiratory and genital tracts that constitute the first line of defense that EAV encounters following natural exposure to the virus. There are few detailed studies that describe the innate immune response of horses to EAV, thus information pertaining to innate antiviral immunity is largely derived from the study of PRRSV and other viruses of the order Nidovirales (Beura et al., 2010, Beura et al., 2012, Fang et al., 2012, Jung et al., 2009, Subramaniam et al., 2010, Sun et al., 2012). However, preliminary studies have shown that EAV inhibits type I IFN production in infected cells and recent studies confirm that nsps 1, 2, and 11 are capable of inhibiting type I IFN activity. Of these three nsps, nsp1 has the strongest inhibitory effect on IFN synthesis. Clearly, failure to induce type I IFN in EAV infected cells may allow the virus to subvert the equine innate immune response. More recent work has further shown that the deubiquitinase function of the papain-like protease 2 suppresses the innate immune response in arterivirus-infected cells (reviewed under the section on nsp2; van Kasteren et al., 2012, van Kasteren et al., 2013).
6.2. Antibody (humoral) immune response
EAV infection in horses induces long-lasting immunity that likely protects against reinfection with most if not all strains of the virus (Balasuriya and Maclachlan, 2004, Bryans et al., 1957, Doll et al., 1968, McCollum, 1970, McCollum, 1986). The humoral immune response to EAV is characterized by the development of both complement-fixing and virus-specific neutralizing (VN) antibodies (Fukunaga and McCollum, 1977, McCollum, 1969). Complement-fixing antibodies develop 1–2 weeks after infection, peak after 2–3 weeks, and steadily decline to disappear by 8 months, whereas VN antibodies are detected within 1–2 weeks after exposure, peak at 2–4 months, and persist 3 years or more (Balasuriya et al., 1999b, Balasuriya et al., 2002a, Balasuriya et al., 2005, Fukunaga and McCollum, 1977, Timoney and McCollum, 1993). Foals born to immune mares are protected against clinical EVA by passive transfer of VN antibodies in the colostrum. VN antibodies appear a few hours after colostrum feeding, peak at 1 week of age, and gradually decline to extinction between 2 and 6, and rarely 7, months of age. The mean biological half-life of maternal antibodies in serum from foals is 32 days (Hullinger et al., 1998). Furthermore, passively acquired antibodies protected young foals that received colostrum from EAV seropositive mares against subsequent EAV infection, confirming the importance of antibodies in protection against the disease (Hullinger et al., 1998, McCollum, 1976). However, VN antibodies are not detected in foal sera prior to nursing, confirming that the passive immunity does not occur via the placenta in utero (McCollum, 1976). Foals that received colostrum should be vaccinated after maternal antibodies disappear since these passive antibodies can neutralize the vaccine virus (McCollum, 1976).
Appearance of neutralizing antibodies coincides with the disappearance of virus from the circulation of infected horses (Fukunaga et al., 1981, McCollum, 1969). However, virus persists in the reproductive tract of the carrier stallion for a variable period despite the presence of high titers of VN antibodies in serum (Timoney and McCollum, 1993). Vaccination of horses with either modified-live or inactivated virus vaccines also can induce a robust VN antibody response (Fukunaga et al., 1990, Fukunaga et al., 1991, McCollum, 1986).
GP5 expresses the major neutralization determinants of EAV and although considerable variation exists in the sequence of the GP5 protein of field strains of the virus, there is only one serotype of EAV and all strains evaluated thus far are neutralized by polyclonal antiserum raised against the prototype virulent Bucyrus strain (Balasuriya et al., 1993, Balasuriya et al., 1995b, Balasuriya et al., 1997, Balasuriya et al., 2004a, Balasuriya and Maclachlan, 2004, Chirnside et al., 1995a, Deregt et al., 1994, Glaser et al., 1995, Zhang et al., 2010a, Zhang et al., 2010b). However, field strains of EAV are frequently distinguished on the basis of differences in their neutralization phenotype with polyclonal antisera and monoclonal antibodies and, similarly, geographically and temporally distinct strains of EAV can differ in the severity of the clinical disease they induce and in their abortigenic potential (Balasuriya et al., 1998, Balasuriya et al., 1999a, McCollum and Timoney, 1998, McCollum et al., 1998, Murphy et al., 1992, Patton et al., 1999, Timoney and McCollum, 1993). Most of the neutralization epitopes of EAV (with the exception of aa 49) are located in the variable region V1 (aa 61–121) of the GP5 protein (Balasuriya et al., 1997). Studies using MAbs and reverse genetics have shown that aa changes at aa 61 (site B) and 67–90 (site C) of the GP5 protein influence the expression of conformational neutralizing epitopes and similar changes are likely to be responsible for antigenic variation among EAV field strains (Balasuriya et al., 1997, Balasuriya et al., 2004a). Therefore, it is likely that aa changes in the GP5 N-terminal ectodomain, especially if they occur during persistent infection in carrier stallions, contribute to the emergence of EAV strains with different neutralization phenotypes (Balasuriya et al., 2004a). Horses also produce non-neutralizing anti-GP5 antibodies in response to EAV infection or immunization with inactivated whole-virus vaccine (Chirnside et al., 1995b, Weiland et al., 2000).
The humoral immune response to structural EAV proteins in naturally or experimentally infected horses varies widely with the infecting EAV strain, the interval after infection, and the individual horse (Hedges et al., 1998, MacLachlan et al., 1998, Nugent et al., 2000). The variation in the antibody response to EAV structural proteins was also observed in horses immunized with the modified live EAV vaccine (Hedges et al., 1998, MacLachlan et al., 1998). Non-neutralizing antibodies that recognize the N and M proteins are detected as early as 14 days post-infection (dpi) and can last at least 145 days in sequential sera from horses experimentally infected with EAV (Chirnside et al., 1988, Chirnside et al., 1995a, Chirnside et al., 1995b, Chirnside et al., 1995c, Chirnside et al., 1995a, Hedges et al., 1998, Kheyar et al., 1997, MacLachlan et al., 1998, Weiland et al., 2000). Of the EAV structural proteins, the M protein is the most consistently recognized by immunoblotting or ELISA analysis of convalescent sera from EAV-infected horses (Hedges et al., 1998, MacLachlan et al., 1998), whereas responses to the N and GP5 proteins are more variable. The C-terminal domain of the M protein (aa 88–162) contains linear epitopes and is recognized by all EAV-specific horse antisera (Jeronimo and Archambault, 2002). The GP2 protein was poorly recognized by equine antisera (MacLachlan et al., 1998). The ORF7 encoding the N protein is highly conserved among strains (Chirnside et al., 1994). The portion between aa 1 and 69 of the N protein has been identified as the most immunoreactive region. Sera from persistently infected stallions consistently recognized GP5, N, and M proteins. In contrast, most convalescent sera from mares, geldings, and stallions that did not become carriers after exposure to EAV recognized the carboxy-terminal region of the M protein (MacLachlan et al., 1998). Sera from horses experimentally or persistently infected with EAV strongly reacted with nsp2, nsp4, nsp5 and nsp12, whereas horses vaccinated with the MLV vaccine did not react with nsp5 and reacted only weakly with nsp4 (Go et al., 2011c).
6.3. Cell-mediated immune response
The cell-mediated immune response (CMI) to EAV is poorly characterized. Castillo-Olivares et al. (2003) described EAV-specific cytotoxic T lymphocyte (CTL) responses using peripheral blood mononuclear cells (PBMCs) from convalescent EAV-infected (experimental) ponies. The data showed that cytotoxicity induced by EAV-stimulated PBMCs was virus-specific, genetically restricted, and mediated by CD8+ T cells, and that EAV-specific CTL precursors persist for at least 1 year after infection. There are no other comprehensive studies to date that describe CMI response to the MLV vaccine or to any other strains of EAV in horses, nor any published studies that definitively identify the specific viral protein(s) targeted by the CTL response of EAV-infected horses.
7. Genetic diversity, molecular epidemiology and evolution of EAV
Original phylogenetic analyses based on ORF5 clustered field strains of EAV into two distinct clades, reflecting their origins in either North American (NA) or Europe (EU) (Balasuriya et al., 1995b, Hornyak et al., 2005, Stadejek et al., 1999). The EAV isolates in the European group could be further divided into two subgroups, European subgroup-1 (EU-1) and European subgroup-2 (EU-2). Comparison of sequence data from individual EU and NA isolates showed movement of viruses between the two continents from movement/transport of carrier stallions and/or their semen (Balasuriya et al., 1995b, Hornyak et al., 2005, Miszczak et al., 2012, Surma-Kurusiewicz et al., 2012, Zhang et al., 2010b). However, EAV field isolates from North America and Europe are closely related and differ only by 15% at the nucleotide level. The major nucleotide and amino acid differences between NA and EU EAV strains occur in the nsp2-encoding portion of ORF 1a, as well as in ORFs 3 and 5 that encode the GP3 and GP5 envelope proteins respectively (Balasuriya and Maclachlan, 2004, Hedges et al., 2001, Miszczak et al., 2012, Rola et al., 2011, Zhang et al., 2010b). Comparative nucleotide and amino acid full-length sequence analysis of EAV strains (Table 1) clearly demonstrated that there is considerable variation in nsp2, with aa substitutions being concentrated in the central segment (aa 388–496) of the protein. The biological significance of this extensive variation of nsp2 among field strains of EAV remains uncertain.
During persistent infection of carrier stallions, EAV infection is restricted to the reproductive tract and is localized principally in the ampulla of the vas deferens (McCollum et al., 1994, Timoney and McCollum, 1993, Timoney et al., 1986). Notwithstanding high neutralizing antibody titers in their serum, carrier stallions continuously shed infectious virus in their semen. Carrier stallions are the essential virus reservoir that maintains and perpetuates EAV in the equine population between breeding seasons. Most importantly, the carrier stallion clearly is responsible for generating the genetic heterogeneity that distinguishes individual field strains of EAV (Hedges et al., 1999a, Hedges et al., 1999b). Sequence analyses of the variable ORF5 gene of strains of EAV sequentially present in the semen of carrier stallions showed that EAV behaves as a quasispecies (population of genetically related viral variants) during persistent infection, leading to both genetic and phenotypic divergence of the virus (Balasuriya et al., 1999a, Balasuriya et al., 2004b, Hedges et al., 1999b, Miszczak et al., 2012, Zhang et al., 2008b, Zhang et al., 2010b). Outbreaks of EVA result from the emergence and spread of specific variants of EAV that are present in the quasispecies virus population in the semen of individual carrier stallions; however, the mechanisms involved in selection and emergence of virulent viral variants remain unclear. It is also clear that novel variants with distinct neutralization phenotypes arise during persistent infection of carrier stallions, and that the altered neutralization phenotype of these variants correlates with amino acid changes in specific regions of the GP5 envelope glycoprotein (Balasuriya et al., 1997, Balasuriya et al., 1998, Balasuriya et al., 1999a, Balasuriya et al., 2004a, Balasuriya and Maclachlan, 2004, Hedges et al., 1999b). However, despite this diversity all of the variants that arise in the course of persistent infection of carrier stallions are neutralized by high-titer polyclonal equine sera, which suggests that immune evasion is not responsible for the establishment of persistent EAV infection of carrier stallions. There also is no evidence to date that positive selective pressures, which would be expected if there was immune-selection, are responsible for establishment of persistent EAV infection of stallions (Balasuriya et al., 2004b, Hedges et al., 1999b).
The recent advent of molecular techniques has helped greatly to increase understanding of the epidemiology of EAV infection and occurrence of outbreaks of EVA. For example, investigation of an extensive outbreak of EVA on a breeding farm showed that a single virus variant present in the semen of a carrier stallion was selected and then efficiently transmitted by aerosol among other horses on the farm (Balasuriya et al., 1999a). Thus, it appears that the considerable genetic heterogeneity afforded by the viral quasispecies likely facilitates persistence of EAV in the reproductive tract of carrier stallions. However, only some members (variants) within the quasispecies appear to be capable of efficient aerosol transmission to other horses, perhaps because of an enhanced ability to replicate within the respiratory tract. The individual strains of EAV that circulated during this restricted EVA outbreak were genetically stable during repeated horizontal and vertical passage in horses, unlike the diverse, quasispecies virus population contained in the semen of the carrier stallions on the farm. In contrast, the 2006–2007 multistate EVA outbreak in the US spanned a period of more than 10 months of active circulation of EAV in horses of several breeds on multiple farms in different states. Characterization of viruses isolated during this extensive and protracted outbreak showed that genomic variability increased with extensive horizontal and vertical virus transmission over an extended time period (Zhang et al., 2010a, Zhang et al., 2010b). Moreover, one of the isolates from an aborted fetus had a very distinct neutralization phenotype and was not neutralized by monoclonal antibodies that neutralized the other EAV isolates from the outbreak. Similarly this virus was neutralized to a significantly lower titer by polyclonal antisera as compared to other isolates. In summary, genetic divergence of EAV occurs in the course of persistent infection of the reproductive tract of carrier stallions, leading to the emergence of novel strains of EAV, and likely compensating for the minimal virus diversity that is generated during small/restricted outbreaks of EVA when the virus can be transmitted by respiratory and/or venereal routes. Therefore, the persistently infected carrier stallion serves as the natural reservoir that harbors EAV between breeding seasons and also provides the environment in which genetic diversification of the virus occurs.
8. Diagnosis of EAV infection
Confirmation of a diagnosis of EVA currently is based on virus detection by either virus isolation (VI) or polymerase chain reaction (PCR), and/or serological demonstration of rising neutralizing antibody titers (4-fold or greater) in paired serum samples taken at a 21- to 28-day interval using the virus neutralization test (VNT). The VNT is the principal serological assay used to detect evidence of EAV infection by most laboratories around the world, and it continues to be the current World Organization for Animal Health (OIE) prescribed standard test for EVA (Anonymous, 2012a). Several laboratories have developed and evaluated enzyme-linked immunosorbent assays (ELISAs) to detect antibodies to EAV using whole virus, synthetic peptides, or recombinant viral proteins (e.g. GP5, M, and N) as antigens (Castillo-Olivares et al., 2003, Cook et al., 1989, Duthie et al., 2008, Hedges et al., 1998, Kondo et al., 1998, Nugent et al., 2000, Starik et al., 2001, Wagner et al., 2003). Various studies have shown that the source of antigen as well as the sera evaluated can markedly influence the results obtained with EAV protein specific ELISAs and competitive ELISA. Recently, a competitive blocking ELISA using a GP5-specific monoclonal antibody has been described and this assay reportedly has very high specificity and sensitivity (Anonymous, 2012b). However, none of these ELISAs or recently described microsphere immunoassay (Luminex) has yet been shown to be of equivalent sensitivity and specificity to the VNT (Duthie et al., 2008, Go et al., 2007). Thus, VNT assay remains the “gold standard” for detection of serum antibodies to EAV.
EAV can be isolated in cell cultures, typically RK-13 cells (Anonymous, 2012a), from nasal swabs or anticoagulated blood collected from adult horses with signs of EVA, or the tissues of aborted equine fetuses (lung, spleen, lymph nodes, and placenta). Carrier stallions are first identified by serology as they are always seropositive, and persistent infection is confirmed by VI from semen in cell culture or by test-breeding seronegative mares which are monitored for seroconversion to EAV after breeding.
Several reverse transcriptase-polymerase chain reaction (RT-PCR) assays (e.g. standard RT-PCR, RT-nested PCR [RT-nPCR] and real-time RT-PCR [rRT-PCR]) for detection of EAV nucleic acids in cell culture supernatants and clinical specimens have been developed (Balasuriya et al., 2002b, Chirnside and Spaan, 1990, Gilbert et al., 1997, Ramina et al., 1999, Sekiguchi et al., 1995, St-Laurent et al., 1994, Westcott et al., 2003). These assays target different genes (ORFs 1b, 3, 4, 5, 6, and 7), and their sensitivity and specificity vary considerably. The sensitivity of RT-PCR-based assays is significantly increased by using either RT-nPCR that incorporates two primer pairs specific for ORF 1b or real-time TaqMan RT-PCR that uses primers and a probe specific for a highly conserved region of ORF7 (Balasuriya et al., 2002b, Lu et al., 2008, Miszczak et al., 2011). Although VI is currently the OIE-approved gold standard for the detection of EAV in semen and is the prescribed test for international trade, it has been demonstrated that at least one published rRT-PCR assay has equal or even higher sensitivity than VI for the detection of EAV nucleic acid in semen samples from carrier stallions (Miszczak et al., 2011). Very clearly, rRT-PCR has significant advantages over VI in terms of reproducibility between laboratories, ease and speed of completion, and cost, thus it is logical that the most accurate rRT-PCR assay would replace VI as the prescribed test for international trade. Histopathologic examination coupled with immunohistochemical staining is also useful for diagnosis of abortion in particular (Del Piero, 2000a, Del Piero, 2000b, Hornyak et al., 2004, Lopez et al., 1996, Szeredi et al., 2003), as are standard RT-PCR or rRT-PCR assays.
9. Vaccines
The modified live virus (MLV) vaccine, ARVAC® (Fort Dodge Animal Health, Fort Dodge, IA [now Zoetis Animal Health, Kalamazoo, MI]) is currently used widely in North America, whereas an adjuvanted killed virus vaccine, ARTERVAC®, is approved for use in several European countries. While the current MLV vaccine against EVA is safe and believed to be efficacious, the vaccine is not recommended by the manufacturer for use in pregnant mares, especially during the last 2 months of gestation, or in foals less than 6 weeks of age unless they are at high risk of natural exposure (Balasuriya and Maclachlan, 2004, Broaddus et al., 2011b, Timoney and McCollum, 1993). Furthermore, horses that are vaccinated with the MLV vaccine cannot be serologically distinguished from naturally infected animals. Vaccination studies using the MLV showed that a single dose of vaccine stimulates only low, transient levels of VN antibodies; however, additional doses boost antibody levels with titers maintained for at least 9–12 months (Fukunaga et al., 1982, McKinnon et al., 1986, Timoney and McCollum, 1988). Similarly, a formalin-inactivated vaccine without adjuvant was shown to induce high titers of VN antibodies after repeated doses (Fukunaga et al., 1990, Fukunaga et al., 1991, Fukunaga et al., 1994, Fukunaga et al., 1996).
A stable full-length cDNA clone of the current MLV vaccine strain of EAV has been recently developed (Zhang et al., 2012). RNA transcripts generated from this plasmid (pEAVrMLV) were infectious upon transfection into mammalian cells and the resultant recombinant virus (rMLV) had 100% nucleotide identity to the parental MLV vaccine strain of EAV. A single silent nucleotide substitution was introduced into the nucleocapsid gene (pEAVrMLVB), enabling the cloned vaccine virus (rMLVB) to be distinguished from the parental MLV vaccine as well as other field and laboratory strains of EAV using an allelic discrimination rRT-PCR assay. In vivo studies confirm that the cloned vaccine virus is safe and induces high titers of neutralizing antibodies against EAV in experimentally immunized horses. However, when challenged with the heterologous KY84 strain of EAV, the rMLVB vaccine virus protected immunized horses in terms of reducing the magnitude and duration of viremia and virus shedding, but did not totally prevent clinical signs of EVA, although these were reduced in severity. While it is believed there is only one known serotype of EAV, field strains clearly differ in their neutralization phenotype (Balasuriya et al., 1995a, Balasuriya et al., 1997, Balasuriya et al., 2004a, MacLachlan and Balasuriya, 2006, Miszczak et al., 2012, Zhang et al., 2008b, Zhang et al., 2010b). It has been shown previously that the serum from horses vaccinated with the MLV vaccine strain neutralizes some EAV field strains such as the KY84 strain to a lower titer (≤1:8–1:64) (Balasuriya et al., 1997, Balasuriya et al., 2004a, Go, 2011, Zhang et al., 2010b). This likely explains why rMLVB vaccinated horses were not fully protected against EVA following challenge with the heterologous KY84 strain, and also further confirms the importance of high titer neutralizing antibodies (≥1:64) in protecting against the clinical signs of EAV infection (Fukunaga and McCollum, 1977, Timoney, 1988, Timoney et al., 1988). These recent data also emphasize the need for additional in-depth cross-neutralization studies using more recent EAV isolates representing all three phylogenetic clades of EAV (North American and two European [EU-1 and EU-2]).
The infectious cDNA clone of the MLV vaccine strain of EAV could potentially be used to design and to develop more broadly protective recombinant MLV vaccines by systematically incorporating key neutralization epitopes from various EAV isolates of significantly distinct neutralization phenotypes. Furthermore, the vaccine clone pEAVrMLVB could be further manipulated to improve the vaccine efficacy as well as to develop a marker vaccine for serological differentiation of EAV naturally infected from vaccinated animals.
Other experimental EAV vaccine constructs using recombinant DNA technology have been described, including a subunit vaccine, a Venezuelan equine encephalitis [VEE] replicon particle (VRP)-based vaccine, an infectious clone-based live-marker vaccine, and a DNA vaccine (reviewed in Balasuriya and Maclachlan, 2004). The N-terminal ectodomain (aa 18–122) of GP5 expressed in bacterial cells induced higher neutralizing antibody titers compared to other GP5 polypeptides in ponies (Castillo-Olivares et al., 2001). Balasuriya et al. (2002a) demonstrated that alphavirus VRP co-expressing GP5/M from the VBS strain of EAV can induce complete protective immunity in horses to challenge with the heterologous KY84 strain of EAV. DNA vaccination using genes encoding the GP5 and N proteins of EAV has been tested in mice (Tobiasch et al., 2001). The highest VN antibodies were induced in animals vaccinated with plasmid DNA encoding ORF5 (Tobiasch et al., 2001). Later, a DNA vaccine expressing ORFs 2, 5, and 7 in combination with equine interleukin-2 was evaluated in horses (Giese et al., 2002). After the initial immunization, three boosters were administered at 2-week intervals. All vaccinated horses developed high VN antibody titers regardless of age and VN antibodies were still detectable over a year later (Giese et al., 2002). Castillo-Olivares et al. (2003) described creation of a candidate live marker vaccine for EAV by deletion of the major neutralization domain (aa 66–112) in the GP5 protein in an infectious cDNA clone. Similarly, the EAV030 cDNA clone has been used to develop disabled infectious single-cycle (DISC) mutants using complementing cell lines expressing minor structural proteins (GP2 [formally GS], GP3, and GP4) (Zevenhoven-Dobbe et al., 2004). However, vaccines based on recombinant DNA technology have not yet been adopted for commercial use.
10. Antivirals and other therapeutics
The antiviral potential of phosphorodiamidate morpholino oligomers (PMOs) directed against EAV has been evaluated previously (van den Born et al., 2005b, Zhang et al., 2010a). PMOs are single-stranded DNA analogs (Summerton and Weller, 1997), usually 20–25 bases in length, water-soluble, and nuclease-resistant (Summerton, 1999). PMOs can base pair with a complementary RNA target sequence and form a steric blockade to interfere with gene expression, particularly with translation of the genome (Stein et al., 1997, Summerton, 1999). Peptide-conjugation to the PMO (P-PMO) greatly improves delivery of PMO into cells (Abes et al., 2006). The 5′ UTR of the EAV genome contains the most sensitive targets for inhibition with P-PMOs (van den Born et al., 2005b). Treatment with an antisense P-PMO targeting the EAV 5′-terminus at a concentration of 5–10 μM eliminated the virus from persistently infected HeLa cells (Zhang et al., 2010a). However, the efficacy of P-PMO against EAV infection in vivo remains to be evaluated. Furthermore, horse cells transfected with small interfering RNA (siRNAs) targeting the ORF1ab prior to infection with EAV markedly reduced subsequent virus titers in the cell cultures (Heinrich et al., 2009). In other studies, it was reported that horse cells treated with 100 U/ml recombinant equine IFN-γ were protected against infection with 100 TCID50 of infectious EAV (Sentsui et al., 2010). Several carbohydrate-binding agents, such as plant and non-plant lectins, have also been shown to have antiviral activities against nidoviruses (van der Meer et al., 2007). Although the antiviral effects shown in vitro are promising, in vivo application of antiviral agents and therapy may be limited due to the large mass of horses and the inherent costs involved.
The EAV carrier state is clearly testosterone-dependent, and transient suppression of testosterone production in carrier stallions may offer therapeutic promise in the elimination of EAV infection (Glaser et al., 1996, Holyoak et al., 1994, Little et al., 1991, Timoney and McCollum, 1993). There are preliminary data to support the strategy that GnRH (Gonadotropin-releasing hormone) vaccines or antagonists can temporarily limit the shedding of the virus in the semen of carrier stallions.
11. Summary and perspectives
There has been significant recent progress in understanding the molecular biology of EAV and the pathogenesis of its infection in horses. The application of cutting edge molecular biology techniques along with development of reverse genetic systems to manipulate the viral genome has facilitated these advancements. EAV has been used as a model system with which to study the replication and transcription of nidoviruses, in part because of its small genome size and similar genome organization and replication strategy to coronaviruses and toroviruses. However, the exact site and location of EAV RNA synthesis and RNA-synthesizing enzyme complexes in infected cells remains to be identified. Furthermore, the subcellular localization of the individual EAV nsps and the various membrane structures they appear to induce warrants further investigation.
Although, the key viral determinants of virulence and persistence have been identified using laboratory strains of EAV, this data should be carefully extrapolated to better identify how virulent virus strains emerge during persistent infection in stallions. Similarly, most field strains of EAV are naturally attenuated and the amino acid changes involved in attenuation of these viruses may not be the same as those in vaccine viruses. Furthermore, the amino acid substitutions that are associated with the establishment of persistent infection in HeLa cells may differ from those involved in the establishment of persistent infection in the stallion. Thus, further whole genome sequence analysis of the quasispecies populations of field strains of EAV from disease outbreaks and from the semen of persistently infected stallions is critical to further refine the viral determinants of virulence and persistence.
Future development of efficacious vaccines that will engender broad heterotypic immunity to all strains of EAV will require ongoing comparative sequence analysis of ORF5 of field isolates of the virus from around the world. Recent studies indicate that the existing vaccines may not induce complete protection against all field strains of EAV. Logically, future vaccines will be developed utilizing recombinant DNA technology, such as the use of infectious clones as a stable genetic background to develop marker (DIVA) vaccines that distinguish naturally infected horses from vaccinated ones.
The equine innate and cellular immune response to EAV infection has also not yet been fully investigated. For example, the viral proteins that express CTL epitopes are yet to be identified and characterized. Similarly, aspects of the pathogenesis of EAV infection of horses require further characterization. Notably, the EAV cellular receptor is yet to be identified, and mechanisms of virus attachment and entry have yet to be elucidated. The recent availability of contemporary genomic techniques will also facilitate the identification of the cellular factors and pathways associated with assembly of EAV particles in infected cells. Of particular importance is the fact that neither the location of EAV persistence in the male reproductive tract nor the mechanisms of persistence have been examined in depth using contemporary molecular biology techniques. Such studies are prerequisite to the logical design and development of antiviral drugs to clear virus from the reproductive tract of carrier stallions. It has been shown that establishment of the carrier state may correlate with susceptibility of CD3+ T cell populations to infection with EAV, which is associated with a genetically dominant haplotype on ECA11. Characterization of the genetic basis of the EAV carrier state in stallions could lead to development of genetic tests capable of identifying stallions that are at most risk of becoming long-term carriers following EAV exposure and so offer important new insights into effective treatment options.
Current OIE (World Organization for Animal Health) approved diagnostic assays are largely based on outdated “classical virology” techniques that should quickly be replaced with more appropriate contemporary assays. Specifically, it has been shown that at least one rRT-PCR assay has equal or higher sensitivity than VI in RK-13 cells for the detection of EAV nucleic acid in semen samples. The rRT-PCR has enormous advantage in terms of assay standardization between laboratories as well as in terms of operational time and cost and should be considered equivalent or an alternative to VI for EAV detection in samples of raw semen. Similarly, the VN assay remains the OIE prescribed regulatory test for serologic detection of EAV infection of the horse, despite the fact that this assay is expensive, labor-intensive, and time-consuming to perform. Furthermore, results of the VN assay can vary markedly among laboratories when insufficient attention is paid to standardization of both test reagents and procedures, thus there is an urgent need to develop highly specific and sensitive ELISA or microsphere immunoassay (Luminex) for the detection of EAV antibodies in serum samples. Incorporation of the immunogenic nsps in such an assay is likely to improve both its sensitivity and specificity.
Acknowledgements
The authors gratefully acknowledge the intellectual and creative input of Drs. Peter Timoney, Eric J. Snijder, Ernest Bailey, Jianqiang Zhang and Zhengchun Lu to the studies included in this review article. This work was partially supported by Agriculture and Food Research Initiative competitive grant no. 2013-68004-20360 from the USDA National Institute of Food and Agriculture. Yun Young Go was supported by a Geoffrey C. Hughes Foundation graduate fellowship from the Maxwell H. Gluck Equine Research Center.
References
- Abes S., Moulton H.M., Clair P., Prevot P., Youngblood D.S., Wu R.P., Iversen P.L., Lebleu B. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control. Release. 2006;116:304–313. doi: 10.1016/j.jconrel.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Anonymous . Equine Viral Arteritis (EVA) and the U.S. Horse Industry. USDA-APHIS, VS, CEAH, National Animal Health Monitoring System; Fort Collins, CO: 2000. NAHMS. [Google Scholar]
- Anonymous . OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals (Mammals, Birds and Bees) 2012. 2.5.10. Equine viral arteritis. [Google Scholar]
- Anonymous . Press Release, Inc.; 2012. Equine Arteritis Virus cELISA Correlates Well with SN. [Google Scholar]
- Archambault D., St-Laurent G. Induction of apoptosis by equine arteritis virus infection. Virus Genes. 2000;20:143–147. doi: 10.1023/a:1008122715387. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Dobbe J.C., Heidner H.W., Smalley V.L., Navarrette A., Snijder E.J., MacLachlan N.J. Characterization of the neutralization determinants of equine arteritis virus using recombinant chimeric viruses and site-specific mutagenesis of an infectious cDNA clone. Virology. 2004;321:235–246. doi: 10.1016/j.virol.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Evermann J.F., Hedges J.F., McKeirnan A.J., Mitten J.Q., Beyer J.C., McCollum W.H., Timoney P.J., MacLachlan N.J. Serologic and molecular characterization of an abortigenic strain of equine arteritis virus isolated from infective frozen semen and an aborted equine fetus. J. Am. Vet. Med. Assoc. 1998;213:1586–1589. [PubMed] [Google Scholar]
- Balasuriya U.B., Hedges J.F., MacLachlan N.J. Molecular epidemiology and evolution of equine arteritis virus. Adv. Exp. Med. Biol. 2001;494:19–24. doi: 10.1007/978-1-4615-1325-4_2. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Hedges J.F., Nadler S.A., McCollum W.H., Timoney P.J., MacLachlan N.J. Genetic stability of equine arteritis virus during horizontal and vertical transmission in an outbreak of equine viral arteritis. J. Gen. Virol. 1999;80(Pt 8):1949–1958. doi: 10.1099/0022-1317-80-8-1949. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Hedges J.F., Smalley V.L., Navarrette A., McCollum W.H., Timoney P.J., Snijder E.J., MacLachlan N.J. Genetic characterization of equine arteritis virus during persistent infection of stallions. J. Gen. Virol. 2004;85:379–390. doi: 10.1099/vir.0.19545-0. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Heidner H.W., Davis N.L., Wagner H.M., Hullinger P.J., Hedges J.F., Williams J.C., Johnston R.E., David Wilson W., Liu I.K., James MacLachlan N. Alphavirus replicon particles expressing the two major envelope proteins of equine arteritis virus induce high level protection against challenge with virulent virus in vaccinated horses. Vaccine. 2002;20:1609–1617. doi: 10.1016/s0264-410x(01)00485-6. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Heidner H.W., Hedges J.F., Williams J.C., Davis N.L., Johnston R.E., MacLachlan N.J. Expression of the two major envelope proteins of equine arteritis virus as a heterodimer is necessary for induction of neutralizing antibodies in mice immunized with recombinant Venezuelan equine encephalitis virus replicon particles. J. Virol. 2000;74:10623–10630. doi: 10.1128/jvi.74.22.10623-10630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasuriya U.B., Leutenegger C.M., Topol J.B., McCollum W.H., Timoney P.J., MacLachlan N.J. Detection of equine arteritis virus by real-time TaqMan reverse transcription-PCR assay. J. Virol. Methods. 2002;101:21–28. doi: 10.1016/s0166-0934(01)00416-5. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Maclachlan N.J. The immune response to equine arteritis virus: potential lessons for other arteriviruses. Vet. Immunol. Immunopathol. 2004;102:107–129. doi: 10.1016/j.vetimm.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Maclachlan N.J., De Vries A.A., Rossitto P.V., Rottier P.J. Identification of a neutralization site in the major envelope glycoprotein (GL) of equine arteritis virus. Virology. 1995;207:518–527. doi: 10.1006/viro.1995.1112. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Patton J.F., Rossitto P.V., Timoney P.J., McCollum W.H., MacLachlan N.J. Neutralization determinants of laboratory strains and field isolates of equine arteritis virus: identification of four neutralization sites in the amino-terminal ectodomain of the G(L) envelope glycoprotein. Virology. 1997;232:114–128. doi: 10.1006/viro.1997.8551. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Rossitto P.V., DeMaula C.D., MacLachlan N.J. A 29K envelope glycoprotein of equine arteritis virus expresses neutralization determinants recognized by murine monoclonal antibodies. J. Gen. Virol. 1993;74(Pt 11):2525–2529. doi: 10.1099/0022-1317-74-11-2525. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Snijder E.J. Arterivirus. In: Mettenleiter T.C., Sobrino F., editors. Animal Viruses: Molecular Biology. Caister Academic Press; Norwich, United Kingdom: 2008. pp. 97–148. [Google Scholar]
- Balasuriya U.B., Snijder E.J., Heidner H.W., Zhang J., Zevenhoven-Dobbe J.C., Boone J.D., McCollum W.H., Timoney P.J., MacLachlan N.J. Development and characterization of an infectious cDNA clone of the virulent Bucyrus strain of Equine arteritis virus. J. Gen. Virol. 2007;88:918–924. doi: 10.1099/vir.0.82415-0. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Snijder E.J., van Dinten L.C., Heidner H.W., Wilson W.D., Hedges J.F., Hullinger P.J., MacLachlan N.J. Equine arteritis virus derived from an infectious cDNA clone is attenuated and genetically stable in infected stallions. Virology. 1999;260:201–208. doi: 10.1006/viro.1999.9817. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B., Timoney P.J., McCollum W.H., MacLachlan N.J. Phylogenetic analysis of open reading frame 5 of field isolates of equine arteritis virus and identification of conserved and nonconserved regions in the GL envelope glycoprotein. Virology. 1995;214:690–697. doi: 10.1006/viro.1995.0087. [DOI] [PubMed] [Google Scholar]
- Balasuriya U.B.R., Dobbeb J.C., Heidner H.W., Snijder E.J., MacLachlan N.J. Characterization of the virulence determinants of equine arteritis virus using an infectious cDNA clone derived from the virulent Bucyrus strain. Xth International Nidovirus Symposium; Cheyenne Mountain Resort, Colorado Springs, CO, June 25–30; 2005. [Google Scholar]
- Barrette-Ng I.H., Ng K.K., Mark B.L., Van Aken D., Cherney M.M., Garen C., Kolodenko Y., Gorbalenya A.E., Snijder E.J., James M.N. Structure of arterivirus nsp4. The smallest chymotrypsin-like proteinase with an alpha/beta C-terminal extension and alternate conformations of the oxyanion hole. J. Biol. Chem. 2002;277:39960–39966. doi: 10.1074/jbc.M206978200. [DOI] [PubMed] [Google Scholar]
- Beerens N., Selisko B., Ricagno S., Imbert I., van der Zanden L., Snijder E.J., Canard B. De novo initiation of RNA synthesis by the arterivirus RNA-dependent RNA polymerase. J. Virol. 2007;81:8384–8395. doi: 10.1128/JVI.00564-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N., Snijder E.J. RNA signals in the 3′ terminus of the genome of equine arteritis virus are required for viral RNA synthesis. J. Gen. Virol. 2006;87:1977–1983. doi: 10.1099/vir.0.81750-0. [DOI] [PubMed] [Google Scholar]
- Bell S.A., Balasuriya U.B.R., MacLachlan N.J. Equine viral arteritis. Clin. Tech. Equine Pract. 2006;5:233–238. [Google Scholar]
- Beura L.K., Sarkar S.N., Kwon B., Subramaniam S., Jones C., Pattnaik A.K., Osorio F.A. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol. 2010;84:1574–1584. doi: 10.1128/JVI.01326-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura L.K., Subramaniam S., Vu H.L., Kwon B., Pattnaik A.K., Osorio F.A. Identification of amino acid residues important for anti-IFN activity of porcine reproductive and respiratory syndrome virus non-structural protein 1. Virology. 2012;433:431–439. doi: 10.1016/j.virol.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop S.P. Animal models of vasculitis. Toxicol. Pathol. 1989;17:109–117. doi: 10.1177/019262338901700106. [DOI] [PubMed] [Google Scholar]
- Breese S.S., Jr., McCollum W.H. Electron microscopic characterization of equine arteritis virus. 2nd International Conference on Equine Infectious Diseases; Basel, Switzerland; 1970. [Google Scholar]
- Broaddus C.C., Balasuriya U.B., Timoney P.J., White J.L., Makloski C., Torrisi K., Payton M., Holyoak G.R. Infection of embryos following insemination of donor mares with equine arteritis virus infective semen. Theriogenology. 2011;76:47–60. doi: 10.1016/j.theriogenology.2011.01.017. [DOI] [PubMed] [Google Scholar]
- Broaddus C.C., Balasuriya U.B., White J.L., Timoney P.J., Funk R.A., Holyoak G.R. Evaluation of the safety of vaccinating mares against equine viral arteritis during mid or late gestation or during the immediate postpartum period. J. Am. Med. Assoc. 2011;38(6):741–750. doi: 10.2460/javma.238.6.741. [DOI] [PubMed] [Google Scholar]
- Bryans J.T., Crowe M.E., Doll E.R., McCollum W.H. Isolation of a filterable agent causing arteritis of horses and abortion by mares; its differentiation from the equine abortion (influenza) virus. Cornell Vet. 1957;47:3–41. [PubMed] [Google Scholar]
- Burki F., Hofer A., Nowotny N. Objective data plead to suspend import-bans for seroreactors against equine arteritis virus except for breeder stallions. J. Appl. Anim. Res. 1992;1:31–42. [Google Scholar]
- Castillo-Olivares J., de Vries A.A., Raamsman M.J., Rottier P.J., Lakhani K., Westcott D., Tearle J.P., Wood J.L., Mumford J.A., Hannant D., Davis-Poynter N.J. Evaluation of a prototype sub-unit vaccine against equine arteritis virus comprising the entire ectodomain of the virus large envelope glycoprotein GL: induction of virus-neutralizing antibody and assessment of protection in ponies. J. Gen. Virol. 2001;82:2425–2435. doi: 10.1099/0022-1317-82-10-2425. [DOI] [PubMed] [Google Scholar]
- Castillo-Olivares J., Wieringa R., Bakonyi T., de Vries A.A., Davis-Poynter N.J., Rottier P.J. Generation of a candidate live marker vaccine for equine arteritis virus by deletion of the major virus neutralization domain. J. Virol. 2003;77:8470–8480. doi: 10.1128/JVI.77.15.8470-8480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch. Virol. 1997;142:629–633. [PubMed] [Google Scholar]
- Chirnside E.D., Cook R.F., Lock M.W., Mumford J.A. Monoclonal antibodies to equine arteritis virus. The Fifth International Conference of Equine Infectious Diseases; Lexington, KY; 1988. pp. 262–267. [Google Scholar]
- Chirnside E.D., de Vries A.A., Mumford J.A., Rottier P.J. Equine arteritis virus-neutralizing antibody in the horse is induced by a determinant on the large envelope glycoprotein GL. J. Gen. Virol. 1995;76(Pt 8):1989–1998. doi: 10.1099/0022-1317-76-8-1989. [DOI] [PubMed] [Google Scholar]
- Chirnside E.D., Francis P.M., de Vries A.A., Sinclair R., Mumford J.A. Development and evaluation of an ELISA using recombinant fusion protein to detect the presence of host antibody to equine arteritis virus. J. Virol. Methods. 1995;54:1–13. doi: 10.1016/0166-0934(95)00020-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirnside E.D., Francis P.M., Mumford J.A. Expression cloning and antigenic analysis of the nucleocapsid protein of equine arteritis virus. Virus Res. 1995;39:277–288. doi: 10.1016/0168-1702(95)00098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirnside E.D., Spaan W.J. Reverse transcription and cDNA amplification by the polymerase chain reaction of equine arteritis virus (EAV) J. Virol. Methods. 1990;30:133–140. doi: 10.1016/0166-0934(90)90014-7. [DOI] [PubMed] [Google Scholar]
- Chirnside E.D., Wearing C.M., Binns M.M., Mumford J.A. Comparison of M and N gene sequences distinguishes variation amongst equine arteritis virus isolates. J. Gen. Virol. 1994;75(Pt 6):1491–1497. doi: 10.1099/0022-1317-75-6-1491. [DOI] [PubMed] [Google Scholar]
- Cholleti H., Paidikondala M., Munir M., Hakhverdyan M., Baule C. Equine arteritis virus induced cell death is associated with activation of the intrinsic apoptotic signalling pathway. Virus Res. 2013;171:222–226. doi: 10.1016/j.virusres.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Chowdhary B.P., Paria N., Raudsepp T. Potential applications of equine genomics in dissecting diseases and fertility. Anim. Reprod. Sci. 2008;107:208–218. doi: 10.1016/j.anireprosci.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Coignoul F.L., Cheville N.F. Pathology of maternal genital tract, placenta, and fetus in equine viral arteritis. Vet. Pathol. 1984;21:333–340. doi: 10.1177/030098588402100311. [DOI] [PubMed] [Google Scholar]
- Cole J.R., Hall R.F., Gosser H.S., Hendricks J.B., Pursell A.R., Senne D.A., Pearson J.E., Gipson C.A. Transmissibility and abortogenic effect of equine viral arteritis in mares. J. Am. Vet. Med. Assoc. 1986;189:769–771. [PubMed] [Google Scholar]
- Collins J.K., Kari S., Ralston S.L., Bennet D.G., Traub-Dargatz J.L., McKinnon A.O. Equine viral arteritis in a veterianry teaching hospital. Prev. Vet. Med. 1987;4:389–397. [Google Scholar]
- Cook R.F., Gann S.J., Mumford J.A. The effects of vaccination with tissue culture-derived viral vaccines on detection of antibodies to equine arteritis virus by enzyme-linked immunosorbent assay (ELISA) Vet. Microbiol. 1989;20:181–189. doi: 10.1016/0378-1135(89)90041-2. [DOI] [PubMed] [Google Scholar]
- Das P.B., Vu H.L., Dinh P.X., Cooney J.L., Kwon B., Osorio F.A., Pattnaik A.K. Glycosylation of minor envelope glycoproteins of porcine reproductive and respiratory syndrome virus in infectious virus recovery, receptor interaction, and immune response. Virology. 2011;410:385–394. doi: 10.1016/j.virol.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Davila S., Hibberd M.L. Genome-wide association studies are coming for human infectious diseases. Genome Med. 2009;1:19. doi: 10.1186/gm19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries A.A., Chirnside E.D., Bredenbeek P.J., Gravestein L.A., Horzinek M.C., Spaan W.J. All subgenomic mRNAs of equine arteritis virus contain a common leader sequence. Nucleic Acids Res. 1990;18:3241–3247. doi: 10.1093/nar/18.11.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries A.A., Chirnside E.D., Horzinek M.C., Rottier P.J. Structural proteins of equine arteritis virus. J. Virol. 1992;66:6294–6303. doi: 10.1128/jvi.66.11.6294-6303.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries A.A., Glaser A.L., Raamsman M.J., de Haan C.A., Sarnataro S., Godeke G.J., Rottier P.J. Genetic manipulation of equine arteritis virus using full-length cDNA clones: separation of overlapping genes and expression of a foreign epitope. Virology. 2000;270:84–97. doi: 10.1006/viro.2000.0245. [DOI] [PubMed] [Google Scholar]
- de Vries A.A., Post S.M., Raamsman M.J., Horzinek M.C., Rottier P.J. The two major envelope proteins of equine arteritis virus associate into disulfide-linked heterodimers. J. Virol. 1995;69:4668–4674. doi: 10.1128/jvi.69.8.4668-4674.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries A.A., Raamsman M.J., van Dijk H.A., Horzinek M.C., Rottier P.J. The small envelope glycoprotein (GS) of equine arteritis virus folds into three distinct monomers and a disulfide-linked dimer. J. Virol. 1995;69:3441–3448. doi: 10.1128/jvi.69.6.3441-3448.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries A.A.F., Horzinek M.C., Rottier P.J.M., de Groot R.J. The genome organization of the Nidovirales: similarities and differences between arteri-, toro-, and coronaviruses. Semin. Virol. 1997;8:33–47. doi: 10.1006/smvy.1997.0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Piero F. Diagnosis of equine arteritis virus infection in two horses by using monoclonal antibody immunoperoxidase histochemistry on skin biopsies. Vet. Pathol. 2000;37:486–487. doi: 10.1354/vp.37-5-486. [DOI] [PubMed] [Google Scholar]
- Del Piero F. Equine viral arteritis. Vet. Pathol. 2000;37:287–296. doi: 10.1354/vp.37-4-287. [DOI] [PubMed] [Google Scholar]
- den Boon J.A., Faaberg K.S., Meulenberg J.J., Wassenaar A.L., Plagemann P.G., Gorbalenya A.E., Snijder E.J. Processing and evolution of the N-terminal region of the arterivirus replicase ORF1a protein: identification of two papainlike cysteine proteases. J. Virol. 1995;69:4500–4505. doi: 10.1128/jvi.69.7.4500-4505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon J.A., Kleijnen M.F., Spaan W.J., Snijder E.J. Equine arteritis virus subgenomic mRNA synthesis: analysis of leader-body junctions and replicative-form RNAs. J. Virol. 1996;70:4291–4298. doi: 10.1128/jvi.70.7.4291-4298.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon J.A., Snijder E.J., Chirnside E.D., de Vries A.A., Horzinek M.C., Spaan W.J. Equine arteritis virus is not a togavirus but belongs to the coronavirus-like superfamily. J. Virol. 1991;65:2910–2920. doi: 10.1128/jvi.65.6.2910-2920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deregt D., de Vries A.A., Raamsman M.J., Elmgren L.D., Rottier P.J. Monoclonal antibodies to equine arteritis virus proteins identify the GL protein as a target for virus neutralization. J. Gen. Virol. 1994;75(Pt 9):2439–2444. doi: 10.1099/0022-1317-75-9-2439. [DOI] [PubMed] [Google Scholar]
- Deshpande A., Wang S., Walsh M.A., Dokland T. Structure of the equine arteritis virus nucleocapsid protein reveals a dimer–dimer arrangement. Acta Crystallogr. D: Biol. Crystallogr. 2007;63:581–586. doi: 10.1107/S0907444907008372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbe J.C., van der Meer Y., Spaan W.J., Snijder E.J. Construction of chimeric arteriviruses reveals that the ectodomain of the major glycoprotein is not the main determinant of equine arteritis virus tropism in cell culture. Virology. 2001;288:283–294. doi: 10.1006/viro.2001.1074. [DOI] [PubMed] [Google Scholar]
- Dokland T. The structural biology of PRRSV. Virus Res. 2010;154:86–97. doi: 10.1016/j.virusres.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll E.R., Bryans J.T., McCollum W.H., Crowe M.E. Isolation of a filterable agent causing arteritis of horses and abortion by mares; its differentiation from the equine abortion (influenza) virus. Cornell Vet. 1957;47:3–41. [PubMed] [Google Scholar]
- Doll E.R., Bryans J.T., Wilson J.C., McCollum W.H. Immunization against equine viral arteritis using modified live virus propagated in cell cultures of rabbit kidney. Cornell Vet. 1968;48:497–524. [PubMed] [Google Scholar]
- Doll E.R., Knappenberger R.E., Bryans J.T. An outbreak of abortion caused by the equine arteritis virus. Cornell Vet. 1957;47:69–75. [PubMed] [Google Scholar]
- Duan X., Nauwynck H.J., Pensaert M.B. Effects of origin and state of differentiation and activation of monocytes/macrophages on their susceptibility to porcine reproductive and respiratory syndrome virus (PRRSV) Arch. Virol. 1997;142:2483–2497. doi: 10.1007/s007050050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunowska M., Biggs P.J., Zheng T., Perrott M.R. Identification of a novel nidovirus associated with a neurological disease of the Australian brushtail possum (Trichosurus vulpecula) Vet. Microbiol. 2012;156:418–424. doi: 10.1016/j.vetmic.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie S., Mills H., Burr P. The efficacy of a commercial ELISA as an alternative to virus neutralisation test for the detection of antibodies to EAV. Equine Vet. J. 2008;40:182–183. doi: 10.2746/042516408X276951. [DOI] [PubMed] [Google Scholar]
- Echeverria M.G., Pecoraro M.R., Galosi C.M., Etcheverrigaray M.E., Nosetto E.O. The first isolation of equine arteritis virus in Argentina. Rev. Sci. Tech. 2003;22:1029–1033. doi: 10.20506/rst.22.3.1458. [DOI] [PubMed] [Google Scholar]
- Eichhorn W., Heilmann M., Kaaden O.R. Equine viral arteritis with abortions: serological and virological evidence in Germany. Z. Veterinarmed. B. 1995;42:573–576. doi: 10.1111/j.1439-0450.1995.tb00750.x. [DOI] [PubMed] [Google Scholar]
- Estes P.C., Cheville N.F. The ultrastructure of vascular lesions in equine viral arteritis. Am. J. Pathol. 1970;58:235–253. [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Fang L., Wang Y., Lei Y., Luo R., Wang D., Chen H., Xiao S. Porcine reproductive and respiratory syndrome virus nonstructural protein 2 contributes to NF-kappaB activation. Virol. J. 2012;9:83. doi: 10.1186/1743-422X-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Snijder E.J. The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res. 2010;154:61–76. doi: 10.1016/j.virusres.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth A.E., Zevenhoven-Dobbe J.C., Wills N.M., Go Y.Y., Balasuriya U.B., Atkins J.F., Snijder E.J., Posthuma C.C. Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production. J. Gen. Virol. 2011;92:1097–1106. doi: 10.1099/vir.0.029264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga Y., Imagawa H., Tabuchi E., Akiyama Y. Clinical and virological findings on experimental equine viral arteritis in horses. Bull. Equine Res. Inst. 1981;18:110–118. [Google Scholar]
- Fukunaga Y., McCollum W.H. Complement-fixation reactions in equine viral arteritis. Am. J. Vet. Res. 1977;38:2043–2046. [PubMed] [Google Scholar]
- Fukunaga Y., Wada R., Hirasawa K., Kamada M., Kumanomido T., Akiyama Y. Effect of the modified Bucyrus strain of equine arteritis virus experimentally inoculated into horses. Bull. Equine Res. Inst. 1982;19:97–101. [Google Scholar]
- Fukunaga Y., Wada R., Kanemaru T., Imagawa H., Kamada M., Samejima T. Immune potency of lyophilized, killed vaccine for equine viral arteritis and its protection against abortion in pregnant mares. J. Equine Vet. Sci. 1996;16:217–221. [Google Scholar]
- Fukunaga Y., Wada R., Kanemaru T., Imagawa H., Kamada M., Samejima T., Nakajima H., Plowright W. Protection against abortion in pregnant mares vaccinated with killed vaccine after exposure to equine arteritis virus. 7th International Conference of Equine Infectious Diseases; Newmarket; 1994. p. 340. [Google Scholar]
- Fukunaga Y., Wada R., Matsumura T., Sugiura T., Imagawa H. Induction of immune response and protection from equine viral arteritis (EVA) by formalin inactivated-virus vaccine for EVA in horses. Z. Veterinarmed. B. 1990;37:135–141. doi: 10.1111/j.1439-0450.1990.tb01036.x. [DOI] [PubMed] [Google Scholar]
- Fukunaga Y., Wada R., Matsumura T., Sugiura T., Imagawa H. An attempt to protect against persistent infection of equine viral arteritis in the reproductive tract of stallions using formalin inactivated-virus vaccine. Sixth International Conference on Equine Infectious Diseases; Cambridge; 1991. pp. 239–244. [Google Scholar]
- Gallie D.R. A tale of two termini: a functional interaction between the termini of an mRNA is a prerequisite for efficient translation initiation. Gene. 1998;216:1–11. doi: 10.1016/s0378-1119(98)00318-7. [DOI] [PubMed] [Google Scholar]
- Giese M., Bahr U., Jakob N.J., Kehm R., Handermann M., Muller H., Vahlenkamp T.H., Spiess C., Schneider T.H., Schusse G., Darai G. Stable and long-lasting immune response in horses after DNA vaccination against equine arteritis virus. Virus Genes. 2002;25:159–167. doi: 10.1023/a:1020109801925. [DOI] [PubMed] [Google Scholar]
- Gilbert S.A., Timoney P.J., McCollum W.H., Deregt D. Detection of equine arteritis virus in the semen of carrier stallions by using a sensitive nested PCR assay. J. Clin. Microbiol. 1997;35:2181–2183. doi: 10.1128/jcm.35.8.2181-2183.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser A.L., de Vries A.A., Dubovi E.J. Comparison of equine arteritis virus isolates using neutralizing monoclonal antibodies and identification of sequence changes in GL associated with neutralization resistance. J. Gen. Virol. 1995;76(Pt 9):2223–2233. doi: 10.1099/0022-1317-76-9-2223. [DOI] [PubMed] [Google Scholar]
- Glaser A.L., de Vries A.A., Raamsman M.J., Horzinek M.C., Rottier P.J.M. An infectious cDNA clone of equine arteritis virus: a tool for future fundamental studies and vaccine development. In: Wernery U., Wade J.F., Mumford J.A., Kaaden O.R., editors. The Eight International Conference on Equine Infectious Diseases. R&W Publications; Newmarket, UK: 1998. pp. 166–176. [Google Scholar]
- Glaser A.L., de Vries A.A., Rottier P.J., Horzinek M.C., Colenbrander B. Equine arteritis virus: a review of clinical features and management aspects. Vet. Q. 1996;18:95–99. doi: 10.1080/01652176.1996.9694625. [DOI] [PubMed] [Google Scholar]
- Glaser A.L., de Vries A.A., Rottier P.J., Horzinek M.C., Colenbrander B. Equine arteritis virus: clinical symptoms and prevention. Tijdschr. Diergeneeskd. 1997;122:2–7. [PubMed] [Google Scholar]
- Go Y.Y. University of Kentucky; Lexington: 2011. Molecular and Genomic Approaches to Understanding Host–Virus Interactions in Shaping the Outcome of Equine Arteritis Virus Infection. [Google Scholar]
- Go Y.Y., Bailey E., Cook F.R., Coleman S.J., Macleod J.N., Chen K.C., Timoney P.J., Balasuriya U.B. Genome-wide association study among four horse breeds identifies a common haplotype associated with in vitro CD3+ T cell susceptibility/resistance to equine arteritis virus infection. J. Virol. 2011;85:13174–13184. doi: 10.1128/JVI.06068-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y.Y., Bailey E., Timoney P.J., Shuck K.M., Balasuriya U.B. Evidence that in vitro susceptibility of CD3+ T lymphocytes to equine arteritis virus infection reflects genetic predisposition of naturally infected stallions to become carriers of the virus. J. Virol. 2012 doi: 10.1128/JVI.01698-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y.Y., Cook R.F., Fulgencio J.Q., Campos J.R., Henney P., Timoney P.J., Horohov D.W., Balasuriya U.B. Assessment of correlation between in vitro CD3(+) T cell susceptibility to EAV infection and clinical outcome following experimental infection. Vet. Microbiol. 2011 doi: 10.1016/j.vetmic.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Go Y.Y., Snijder E.J., Timoney P.J., Balasuriya U.B. Characterization of equine humoral antibody response to the nonstructural proteins of equine arteritis virus. Clin. Vaccine Immunol. 2011;18:268–279. doi: 10.1128/CVI.00444-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y.Y., Wong S.J., Branscum A., Demarset V.L., Shuck K.M., Vickers M.L., Zhang J., McCollum W.H., Timoney P.J., Balasuriya U.B.R. Development of a Fluorescent Microsphere Immunoassay for Detection of Antibodies Specific to Equine Arteritis Virus and Comparison with the Virus Neutralization Test. Clin. Vaccine Immunol. 2007;15:12. doi: 10.1128/CVI.00388-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y.Y., Zhang J., Timoney P.J., Cook R.F., Horohov D.W., Balasuriya U.B. Complex interactions between the major and minor envelope proteins of equine arteritis virus determine its tropism for equine CD3+ T lymphocytes and CD14+ monocytes. J. Virol. 2010;84:4898–4911. doi: 10.1128/JVI.02743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Blinov V.M., Donchenko A.P., Koonin E.V. An NTP-binding motif is the most conserved sequence in a highly diverged monophyletic group of proteins involved in positive strand RNA viral replication. J. Mol. Evol. 1989;28:256–268. doi: 10.1007/BF02102483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V. Viral proteins containing the purine NTP-binding sequence pattern. Nucleic Acids Res. 1989;17:8413–8440. doi: 10.1093/nar/17.21.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. A novel superfamily of nucleoside triphosphate-binding motif containing proteins which are probably involved in duplex unwinding in DNA and RNA replication and recombination. FEBS Lett. 1988;235:16–24. doi: 10.1016/0014-5793(88)81226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Lai M.M. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. 1991;288:201–205. doi: 10.1016/0014-5793(91)81034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie A.J., Howell P.G., Hedges J.F., Bosman A.M., Balasuriya U.B., McCollum W.H., Timoney P.J., MacLachlan N.J. Lateral transmission of equine arteritis virus among Lipizzaner stallions in South Africa. Equine Vet. J. 2003;35:596–600. doi: 10.2746/042516403775467162. [DOI] [PubMed] [Google Scholar]
- Hedges J.F., Balasuriya U.B., Ahmad S., Timoney P.J., McCollum W.H., Yilma T., MacLachlan N.J. Detection of antibodies to equine arteritis virus by enzyme linked immunosorbant assays utilizing GL M and N proteins expressed from recombinant baculoviruses. J. Virol. Methods. 1998;76:127–137. doi: 10.1016/s0166-0934(98)00131-1. [DOI] [PubMed] [Google Scholar]
- Hedges J.F., Balasuriya U.B., MacLachlan N.J. The open reading frame 3 of equine arteritis virus encodes an immunogenic glycosylated, integral membrane protein. Virology. 1999;264:92–98. doi: 10.1006/viro.1999.9982. [DOI] [PubMed] [Google Scholar]
- Hedges J.F., Balasuriya U.B., Timoney P.J., McCollum W.H., MacLachlan N.J. Genetic variation in open reading frame 2 of field isolates and laboratory strains of equine arteritis virus. Virus Res. 1996;42:41–52. doi: 10.1016/0168-1702(96)01294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges J.F., Balasuriya U.B., Timoney P.J., McCollum W.H., MacLachlan N.J. Genetic divergence with emergence of novel phenotypic variants of equine arteritis virus during persistent infection of stallions. J. Virol. 1999;73:3672–3681. doi: 10.1128/jvi.73.5.3672-3681.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges J.F., Balasuriya U.B., Topol J.B., Lee D.W., MacLachlan N.J. Genetic variation of ORFs 3 and 4 of equine arteritis virus. Adv. Exp. Med. Biol. 2001;494:69–72. doi: 10.1007/978-1-4615-1325-4_10. [DOI] [PubMed] [Google Scholar]
- Heinrich A., Riethmuller D., Gloger M., Schusser G.F., Giese M., Ulbert S. RNA interference protects horse cells in vitro from infection with Equine Arteritis Virus. Antiviral Res. 2009;81:209–216. doi: 10.1016/j.antiviral.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Henson J.B., Crawford T.B. The pathogenesis of virus-induced arterial disease – Aleutian disease and equine viral arteritis. Adv. Cardiol. 1974;13:183–191. doi: 10.1159/000395537. [DOI] [PubMed] [Google Scholar]
- Hill A.V. Aspects of genetic susceptibility to human infectious diseases. Annu. Rev. Genet. 2006;40:469–486. doi: 10.1146/annurev.genet.40.110405.090546. [DOI] [PubMed] [Google Scholar]
- Holyoak G.R., Little T.V., Vernon M., McCollum W.H., Timoney P.J. Correlation between ultrasonographic findings and serum testosterone concentration in prepubertal and peripubertal colts. Am. J. Vet. Res. 1994;55:450–457. [PubMed] [Google Scholar]
- Hornyak A., Denes B., Szeredi L., Dencso L., Rusvai M. Diagnostic application of immunoperoxidase monolayer assay using monoclonal antibodies produced against equine arteritis virus 14-kDa nucleocapsid protein. Hybrid. Hybridomics. 2004;23:368–372. doi: 10.1089/hyb.2004.23.368. [DOI] [PubMed] [Google Scholar]
- Hornyak A., Bakonyi T., Tekes G., Szeredi L., Rusvai M. A novel subgroup among genotypes of equine arteritis virus: genetic comparison of 40 strains. J. Vet. Med. B Infect. Dis. Vet. Public Health. 2005;52:112–118. doi: 10.1111/j.1439-0450.2005.00833.x. [DOI] [PubMed] [Google Scholar]
- Horzinek M., Maess J., Laufs R. Studies on the substructure of togaviruses. II. Analysis of equine arteritis, rubella, bovine viral diarrhea, and hog cholera viruses. Arch. Gesamte Virusforsch. 1971;33:306–318. [PubMed] [Google Scholar]
- Hullinger P.J., Gardner I.A., Hietala S.K., Ferraro G.L., MacLachlan N.J. Seroprevalence of antibodies against equine arteritis virus in horses residing in the United States and imported horses. J. Am. Vet. Med. Assoc. 2001;219:946–949. doi: 10.2460/javma.2001.219.946. [DOI] [PubMed] [Google Scholar]
- Hullinger P.J., Wilson W.D., Rossitto P.V., Patton J.F., Thurmond M.C., MacLachlan N.J. Passive transfer, rate of decay, and protein specificity of antibodies against equine arteritis virus in horses from a Standardbred herd with high seroprevalence. J. Am. Vet. Med. Assoc. 1998;213:839–842. [PubMed] [Google Scholar]
- Hyllseth B. A plaque assay of equine arteritis virus in BHK-21 cells. Arch. Gesamte Virusforsch. 1969;28:26–33. doi: 10.1007/BF01250842. [DOI] [PubMed] [Google Scholar]
- Ivanov K.A., Hertzig T., Rozanov M., Bayer S., Thiel V., Gorbalenya A.E., Ziebuhr J. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12694–12699. doi: 10.1073/pnas.0403127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C., Archambault D. Importance of M-protein C terminus as substrate antigen for serodetection of equine arteritis virus infection. Clin. Diagn. Lab. Immunol. 2002;9:698–703. doi: 10.1128/CDLI.9.3.698-703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C.R., Griggs T.F., Gnanandarajah J., Murtaugh M.P. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J. Gen. Virol. 2011;92:1107–1116. doi: 10.1099/vir.0.030213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K., Renukaradhya G.J., Alekseev K.P., Fang Y., Tang Y., Saif L.J. Porcine reproductive and respiratory syndrome virus modifies innate immunity and alters disease outcome in pigs subsequently infected with porcine respiratory coronavirus: implications for respiratory viral co-infections. J. Gen. Virol. 2009;90:2713–2723. doi: 10.1099/vir.0.014001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabatek A.a.V.M. Folding and oligomerization of the gp2b/gp3/gp4 spike proteins of equine arteritis virus in vitro. Viruses. 2013;4:10. doi: 10.3390/v4030414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheyar A., Martin S., St-Laurent G., Timoney P.J., McCollum W.H., Archambault D. Expression cloning and humoral immune response to the nucleocapsid and membrane proteins of equine arteritis virus. Clin. Diagn. Lab. Immunol. 1997;4:648–652. doi: 10.1128/cdli.4.6.648-652.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.S., Kwang J., Yoon I.J., Joo H.S., Frey M.L. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch. Virol. 1993;133:477–483. doi: 10.1007/BF01313785. [DOI] [PubMed] [Google Scholar]
- Knoops K., Barcena M., Limpens R.W., Koster A.J., Mommaas A.M., Snijder E.J. Ultrastructural characterization of arterivirus replication structures: reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 2012;86:2474–2487. doi: 10.1128/JVI.06677-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., Kikkert M., Worm S.H., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T., Fukunaga Y., Sekiguchi K., Sugiura T., Imagawa H. Enzyme-linked immunosorbent assay for serological survey of equine arteritis virus in racehorses. J. Vet. Med. Sci. 1998;60:1043–1045. doi: 10.1292/jvms.60.1043. [DOI] [PubMed] [Google Scholar]
- Konishi S., Akashi H., Sentsui H., Ogata M. Studies on equine viral arteritis. I. Characterization of the virus and trial survey on antibody with Vero cell cultures. Nihon Juigaku Zasshi. 1975;37:259–267. doi: 10.1292/jvms1939.37.5_259. [DOI] [PubMed] [Google Scholar]
- Laity J.H., Lee B.M., Wright P.E. Zinc finger proteins: new insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001;11:39–46. doi: 10.1016/s0959-440x(00)00167-6. [DOI] [PubMed] [Google Scholar]
- Lauber C., Ziebuhr J., Junglen S., Drosten C., Zirkel F., Nga P.T., Morita K., Snijder E.J., Gorbalenya A.E. Mesoniviridae: a proposed new family in the order Nidovirales formed by a single species of mosquito-borne viruses. Arch. Virol. 2012;157:1623–1628. doi: 10.1007/s00705-012-1295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauck M., Hyeroba D., Tumukunde A., Weny G., Lank S.M., Chapman C.A., O’Connor D.H., Friedrich T.C., Goldberg T.L. Novel, divergent simian hemorrhagic fever viruses in a wild Ugandan red colobus monkey discovered using direct pyrosequencing. PLoS ONE. 2011;6:e19056. doi: 10.1371/journal.pone.0019056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauck M., Sibley S.D., Hyeroba D., Tumukunde A., Weny G., Chapman C.A., Ting N., Switzer W.M., Kuhn J.H., Friedrich T.C., O’Connor D.H., Goldberg T.L. Exceptional simian hemorrhagic fever virus diversity in a wild African primate community. J. Virol. 2013;87:688–691. doi: 10.1128/JVI.02433-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson S.R., Rossow K.D., Collins J.E., Benfield D.A., Rowland R.R. Porcine reproductive and respiratory syndrome virus infection of gnotobiotic pigs: sites of virus replication and co-localization with MAC-387 staining at 21 days post-infection. Virus Res. 1997;51:105–113. doi: 10.1016/s0168-1702(97)00086-5. [DOI] [PubMed] [Google Scholar]
- Lee C., Yoo D. The small envelope protein of porcine reproductive and respiratory syndrome virus possesses ion channel protein-like properties. Virology. 2006;355:30–43. doi: 10.1016/j.virol.2006.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little T.V., Holyoak G.R., McCollum W.H., Timoney P.J. Output of equine arteritis virus from persistently infected stallions is testosterone dependent. Proceedings of the 6th international conference on equine infectious diseases; Cambridge, 1992; 1991. pp. 225–229. [Google Scholar]
- Lopez J.W., del Piero F., Glaser A., Finazzi M. Immunoperoxidase histochemistry as a diagnostic tool for detection of equine arteritis virus antigen in formalin fixed tissues. Equine Vet. J. 1996;28:77–79. doi: 10.1111/j.2042-3306.1996.tb01593.x. [DOI] [PubMed] [Google Scholar]
- Lu Z., Branscum A.J., Shuck K.M., Zhang J., Dubovi E.J., Timoney P.J., Balasuriya U.B. Comparison of two real-time reverse transcription polymerase chain reaction assays for the detection of Equine arteritis virus nucleic acid in equine semen and tissue culture fluid. J. Vet. Diagn. Invest. 2008;20:147–155. doi: 10.1177/104063870802000202. [DOI] [PubMed] [Google Scholar]
- Lu Z., Zhang J., Huang C.M., Go Y.Y., Faaberg K.S., Rowland R.R., Timoney P.J., Balasuriya U.B. Chimeric viruses containing the N-terminal ectodomains of GP5 and M proteins of porcine reproductive and respiratory syndrome virus do not change the cellular tropism of equine arteritis virus. Virology. 2012;432:99–109. doi: 10.1016/j.virol.2012.05.022. [DOI] [PubMed] [Google Scholar]
- MacLachlan N.J., Balasuriya U.B. Equine viral arteritis. Adv. Exp. Med. Biol. 2006;581:429–433. doi: 10.1007/978-0-387-33012-9_77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLachlan N.J., Balasuriya U.B., Hedges J.F., Schweidler T.M., McCollum W.H., Timoney P.J., Hullinger P.J., Patton J.F. Serologic response of horses to the structural proteins of equine arteritis virus. J. Vet. Diagn. Invest. 1998;10:229–236. doi: 10.1177/104063879801000302. [DOI] [PubMed] [Google Scholar]
- MacLachlan N.J., Balasuriya U.B., Rossitto P.V., Hullinger P.A., Patton J.F., Wilson W.D. Fatal experimental equine arteritis virus infection of a pregnant mare: immunohistochemical staining of viral antigens. J. Vet. Diagn. Invest. 1996;8:367–374. doi: 10.1177/104063879600800316. [DOI] [PubMed] [Google Scholar]
- MacLachlan N.J., Conley A.J., Kennedy P.C. Bluetongue and equine viral arteritis viruses as models of virus-induced fetal injury and abortion. Anim. Reprod. Sci. 2000;60–61:643–651. doi: 10.1016/s0378-4320(00)00105-6. [DOI] [PubMed] [Google Scholar]
- Maess J., Reczko E., Bohm H.O. Equine arteritis virus: multiplication in BHK 21-cells buoyant density and electron microscopical demonstration. Arch. Gesamte Virusforsch. 1970;30:47–58. [PubMed] [Google Scholar]
- Magnusson P., Hyllseth B., Marusyk H. Morphological studies on equine arteritis virus. Arch. Gesamte Virusforsch. 1970;30:105–112. doi: 10.1007/BF01250177. [DOI] [PubMed] [Google Scholar]
- Manolaridis I., Gaudin C., Posthuma C.C., Zevenhoven-Dobbe J.C., Imbert I., Canard B., Kelly G., Tucker P.A., Conte M.R., Snijder E.J. Structure and genetic analysis of the arterivirus nonstructural protein 7alpha. J. Virol. 2011;85:7449–7453. doi: 10.1128/JVI.00255-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollum W.H. Development of a modified virus strain and vaccine for equine viral arteritis. J. Am. Vet. Med. Assoc. 1969;155:318–322. [PubMed] [Google Scholar]
- McCollum W.H. Vaccination for equine viral arteritis. Proceedings of the Second International Conference on Equine Infectious Diseases; Paris; 1970. pp. 143–151. [Google Scholar]
- McCollum W.H. Studies of passive immunity in foals to equine viral arteritis. Vet. Microbiol. 1976;1:45–54. [Google Scholar]
- McCollum W.H. Pathologic features of horses given avirulent equine arteritis virus intramuscularly. Am. J. Vet. Res. 1981;42:1218–1220. [PubMed] [Google Scholar]
- McCollum W.H. Responses of horses vaccinated with avirulent modified-live equine arteritis virus propagated in the E. Derm (NBL-6) cell line to nasal inoculation with virulent virus. Am. J. Vet. Res. 1986;47:1931–1934. [PubMed] [Google Scholar]
- McCollum W.H., Bryans J.T. Serological identification of infection by equine arteritis virus in horses of several countries. Proceedings of the 3rd International Conference of Equine Infectious Diseases; Paris, 1972; 1973. pp. 256–263. [Google Scholar]
- McCollum W.H., Little T.V., Timoney P.J., Swerczek T.W. Resistance of castrated male horses to attempted establishment of the carrier state with equine arteritis virus. J. Comp. Pathol. 1994;111:383–388. doi: 10.1016/s0021-9975(05)80096-9. [DOI] [PubMed] [Google Scholar]
- McCollum W.H., Prickett M.E., Bryans J.T. Temporal distribution of equine arteritis virus in respiratory mucosa, tissues and body fluids of horses infected by inhalation. Res. Vet. Sci. 1971;12:459–464. [PubMed] [Google Scholar]
- McCollum W.H., Timoney P.J., Tengelsen L.A. Clinical, virological and serological responses of donkeys to intranasal inoculation with the KY-84 strain of equine arteritis virus. J. Comp. Pathol. 1995;112:207–211. doi: 10.1016/s0021-9975(05)80062-3. [DOI] [PubMed] [Google Scholar]
- McCollum W.H., Timoney P.J. Experimental observations on the virulence of isolates of equine arteritis virus. Proceeding of the 8th International Conference on Equine Infectious Diseases; Dubai; 1998. pp. 558–559. [Google Scholar]
- McCollum W.H., Timoney P.J., Lee J.W., Jr., Habacker P.L., Balasuriya U.B.R., MacLachlan N.J. Features of an outbreak of equine viral arteritis on a breeding farm associated with abortion and fatal interstitial pneumonia in neonatal foals. Proceeding of the 8th International Conference on Equine Infectious Diseases; Dubai; 1998. pp. 559–560. [Google Scholar]
- McCue P.M., Hietala S.K., Spensely M.S., Mihalyi J., Hughes J.P. Prevalence of equine viral arteritis in California horses. Calif. Vet. 1991;45:24–26. [Google Scholar]
- McFadden A.M., Pearce P.V., Orr D., Nicoll K., Rawdon T.G., Pharo H., Stone M. Evidence for absence of equine arteritis virus in the horse population of New Zealand. N. Z. Vet. J. 2013 doi: 10.1080/00480169.2012.755664. [DOI] [PubMed] [Google Scholar]
- McKenzie J. Survey of stallions for equine arteritis virus. Surveillance. 1996:17–18. [Google Scholar]
- McKinnon A.O., Colbern G.T., Collins J.K., Bowen R.A., Voss J.L., Umphenour N.W. Vaccination of stallions with modified live equine viral arteritis virus. J. Equine Vet. Sci. 1986;6:66–69. [Google Scholar]
- Miszczak F., Legrand L., Balasuriya U.B., Ferry-Abitbol B., Zhang J., Hans A., Fortier G., Pronost S., Vabret A. Emergence of novel equine arteritis virus (EAV) variants during persistent infection in the stallion: origin of the 2007 French EAV outbreak was linked to an EAV strain present in the semen of a persistently infected carrier stallion. Virology. 2012;423:165–174. doi: 10.1016/j.virol.2011.11.028. [DOI] [PubMed] [Google Scholar]
- Miszczak F., Shuck K.M., Lu Z., Go Y.Y., Zhang J., Sells S., Vabret A., Pronost S., Fortier G., Timoney P.J., Balasuriya U.B. Evaluation of two magnetic-bead-based viral nucleic acid purification kits and three real-time reverse transcription-PCR reagent systems in two TaqMan assays for equine arteritis virus detection in semen. J. Clin. Microbiol. 2011;49:3694–3696. doi: 10.1128/JCM.01187-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenkamp R., van Tol H., Rozier B.C., van der Meer Y., Spaan W.J., Snijder E.J. The arterivirus replicase is the only viral protein required for genome replication and subgenomic mRNA transcription. J. Gen. Virol. 2000;81:2491–2496. doi: 10.1099/0022-1317-81-10-2491. [DOI] [PubMed] [Google Scholar]
- Monastyrska I., Ulasli M., Rottier P.J., Guan J.L., Reggiori F., de Haan C.A. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy. 2013;9:164–174. doi: 10.4161/auto.22743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore B.D., Balasuriya U.B., Watson J.L., Bosio C.M., MacKay R.J., MacLachlan N.J. Virulent and avirulent strains of equine arteritis virus induce different quantities of TNF-alpha and other proinflammatory cytokines in alveolar and blood-derived equine macrophages. Virology. 2003;314:662–670. doi: 10.1016/s0042-6822(03)00506-3. [DOI] [PubMed] [Google Scholar]
- Moraillon A., Moraillon R. Results of an epidemiological investigation on viral arteritis in France and some other European and African countries. Ann. Rech. Vet. 1978;9:43–54. [PubMed] [Google Scholar]
- Murphy T.W., McCollum W.H., Timoney P.J., Klingeborn B.W., Hyllseth B., Golnik W., Erasmus B. Genomic variability among globally distributed isolates of equine arteritis virus. Vet. Microbiol. 1992;32:101–115. doi: 10.1016/0378-1135(92)90099-f. [DOI] [PubMed] [Google Scholar]
- Nedialkova D.D., Gorbalenya A.E., Snijder E.J. Arterivirus Nsp1 modulates the accumulation of minus-strand templates to control the relative abundance of viral mRNAs. PLoS Pathogens. 2010;6:e1000772. doi: 10.1371/journal.ppat.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedialkova D.D., Ulferts R., van den Born E., Lauber C., Gorbalenya A.E., Ziebuhr J., Snijder E.J. Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. J. Virol. 2009;83:5671–5682. doi: 10.1128/JVI.00261-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke M., Korte T., Tielesch C., Ter-Avetisyan G., Tunnemann G., Cardoso M.C., Veit M., Herrmann A. Equine arteritis virus is delivered to an acidic compartment of host cells via clathrin-dependent endocytosis. Virology. 2008;377:248–254. doi: 10.1016/j.virol.2008.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent J., Sinclair R., deVries A.A., Eberhardt R.Y., Castillo-Olivares J., Davis Poynter N., Rottier P.J., Mumford J.A. Development and evaluation of ELISA procedures to detect antibodies against the major envelope protein (G(L)) of equine arteritis virus. J. Virol. Methods. 2000;90:167–183. doi: 10.1016/s0166-0934(00)00231-7. [DOI] [PubMed] [Google Scholar]
- Pasternak A.O., Gultyaev A.P., Spaan W.J., Snijder E.J. Genetic manipulation of arterivirus alternative mRNA leader-body junction sites reveals tight regulation of structural protein expression. J. Virol. 2000;74:11642–11653. doi: 10.1128/jvi.74.24.11642-11653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., Spaan W.J., Snijder E.J. Regulation of relative abundance of arterivirus subgenomic mRNAs. J. Virol. 2004;78:8102–8113. doi: 10.1128/JVI.78.15.8102-8113.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., Spaan W.J., Snijder E.J. Nidovirus transcription: how to make sense…? J. Gen. Virol. 2006;87:1403–1421. doi: 10.1099/vir.0.81611-0. [DOI] [PubMed] [Google Scholar]
- Pasternak A.O., van den Born E., Spaan W.J., Snijder E.J. Sequence requirements for RNA strand transfer during nidovirus discontinuous subgenomic RNA synthesis. EMBO J. 2001;20:7220–7228. doi: 10.1093/emboj/20.24.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., van den Born E., Spaan W.J., Snijder E.J. The stability of the duplex between sense and antisense transcription-regulating sequences is a crucial factor in arterivirus subgenomic mRNA synthesis. J. Virol. 2003;77:1175–1183. doi: 10.1128/JVI.77.2.1175-1183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J.F., Balasuriya U.B., Hedges J.F., Schweidler T.M., Hullinger P.J., MacLachlan N.J. Phylogenetic characterization of a highly attenuated strain of equine arteritis virus from the semen of a persistently infected standardbred stallion. Arch. Virol. 1999;144:817–827. doi: 10.1007/s007050050547. [DOI] [PubMed] [Google Scholar]
- Pedersen K.W., van der Meer Y., Roos N., Snijder E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999;73:2016–2026. doi: 10.1128/jvi.73.3.2016-2026.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirzadeh B., Gagnon C.A., Dea S. Genomic and antigenic variations of porcine reproductive and respiratory syndrome virus major envelope GP5 glycoprotein. Can. J. Vet. Res. 1998;62:170–177. [PMC free article] [PubMed] [Google Scholar]
- Plagemann P.G., Moennig V. Lactate dehydrogenase-elevating virus, equine arteritis virus, and simian hemorrhagic fever virus: a new group of positive-strand RNA viruses. Adv. Virus Res. 1992;41:99–192. doi: 10.1016/S0065-3527(08)60036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol J.M., Wagenaar F. American Association of Swine Practitioners Newsletter. 1992. Morphogenesis of Lelystad virus in porcine lung alveolar macrophages; p. 29. [Google Scholar]
- Pol J.M., Wagenaar F., Reus J.E.G. Comparative morphogenesis of three PRRS virus strains. Vet. Microbiol. 1997;55:203–208. doi: 10.1016/s0378-1135(96)01329-6. [DOI] [PubMed] [Google Scholar]
- Posthuma C.C., Nedialkova D.D., Zevenhoven-Dobbe J.C., Blokhuis J.H., Gorbalenya A.E., Snijder E.J. Site-directed mutagenesis of the Nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J. Virol. 2006;80:1653–1661. doi: 10.1128/JVI.80.4.1653-1661.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posthuma C.C., Pedersen K.W., Lu Z., Joosten R.G., Roos N., Zevenhoven-Dobbe J.C., Snijder E.J. Formation of the arterivirus replication/transcription complex: a key role for nonstructural protein 3 in the remodeling of intracellular membranes. J. Virol. 2008;82:4480–4491. doi: 10.1128/JVI.02756-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronost S., Pitel P.H., Miszczak F., Legrand L., Marcillaud-Pitel C., Hamon M., Tapprest J., Balasuriya U.B., Freymuth F., Fortier G. Description of the first recorded major occurrence of equine viral arteritis in France. Equine Vet. J. 2010;42:713–720. doi: 10.1111/j.2042-3306.2010.00109.x. [DOI] [PubMed] [Google Scholar]
- Radwan A.I., Burger D. The complement-requiring neutralization of equine arteritis virus by late antisera. Virology. 1973;51:71–77. doi: 10.1016/0042-6822(73)90366-8. [DOI] [PubMed] [Google Scholar]
- Ramina A., Dalla Valle L., De Mas S., Tisato E., Zuin A., Renier M., Cuteri V., Valente C., Cancellotti F.M. Detection of equine arteritis virus in semen by reverse transcriptase polymerase chain reaction-ELISA. Comp. Immunol. Microbiol. Infect. Dis. 1999;22:187–197. doi: 10.1016/s0147-9571(98)00136-2. [DOI] [PubMed] [Google Scholar]
- Rola J., Larska M., Rola J.G., Belak S., Autorino G.L. Epizotiology and phylogeny of equine arteritis virus in hucul horses. Vet. Microbiol. 2011;148:402–407. doi: 10.1016/j.vetmic.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Rossow K.D. Porcine reproductive and respiratory syndrome. Vet. Pathol. 1998;35:1–20. doi: 10.1177/030098589803500101. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol. 1995;380:499–506. doi: 10.1007/978-1-4615-1899-0_79. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L., Siddell S.G. A contemporary view of coronavirus transcription. J. Virol. 2007;81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi K., Sugita S., Fukunaga Y., Kondo T., Wada R., Kamada M., Yamaguchi S. Detection of equine arteritis virus (EAV) by polymerase chain reaction (PCR) and differentiation of EAV strains by restriction enzyme analysis of PCR products. Arch. Virol. 1995;140:1483–1491. doi: 10.1007/BF01322675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentsui H., Wu D., Murakami K., Kondo T., Matsumura T. Antiviral effect of recombinant equine interferon-gamma on several equine viruses. Vet. Immunol. Immunopathol. 2010;135:93–99. doi: 10.1016/j.vetimm.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Seybert A., Posthuma C.C., van Dinten L.C., Snijder E.J., Gorbalenya A.E., Ziebuhr J. A complex zinc finger controls the enzymatic activities of nidovirus helicases. J. Virol. 2005;79:696–704. doi: 10.1128/JVI.79.2.696-704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybert A., van Dinten L.C., Snijder E.J., Ziebuhr J. Biochemical characterization of the equine arteritis virus helicase suggests a close functional relationship between arterivirus and coronavirus helicases. J. Virol. 2000;74:9586–9593. doi: 10.1128/jvi.74.20.9586-9593.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddell S.G., Ziebuhr J., Snijder E.J. Coronaviruses, toroviruses and arteriviruses. In: Mahy B.W.J., ter Meulen V., editors. Topley & Wilson's Microbiology and Microbial Infections, Virology. Hodder Arnold; 2005. pp. 823–856. [Google Scholar]
- Snijder E., Wassenaar A.L., Den Boon J.A., Spaan W.J. Proteolytic processing of the arterivirus replicase. Adv. Exp. Med. Biol. 1995;380:443–451. doi: 10.1007/978-1-4615-1899-0_71. [DOI] [PubMed] [Google Scholar]
- Snijder E.J. The arterivirus replicase. The road from RNA to protein(s), and back again. Adv. Exp. Med. Biol. 1998;440:97–108. [PubMed] [Google Scholar]
- Snijder E.J. Arterivirus RNA synthesis dissected. Nucleotides, membranes, amino acids, and a bit of zinc. Adv. Exp. Med. Biol. 2001;494:241–253. [PubMed] [Google Scholar]
- Snijder E.J., Spaan W.J.M. Arteriviruses. In: Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E., editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia, PA: 2007. pp. 1337–1355. [Google Scholar]
- Snijder E.J., Dobbe J.C., Spaan W.J. Heterodimerization of the two major envelope proteins is essential for arterivirus infectivity. J. Virol. 2003;77:97–104. doi: 10.1128/JVI.77.1.97-104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Meulenberg J.J. The molecular biology of arteriviruses. J. Gen. Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Siddell S.G., Gorbalenya A.E. The order Nidovirales. In: Mahy W., ter Meulen V., editors. Topley and Wilson's Microbiology and Microbial Infections; Virology Volume. Hodder Arnold; London: 2005. pp. 390–404. [Google Scholar]
- Snijder E.J., Spaan W.J. Arteriviruses. In: Knipe D.M., Howley P.M., editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2006. pp. 1337–1355. [Google Scholar]
- Snijder E.J., van der Meer Y., Zevenhoven-Dobbe J., Onderwater J.J., van der Meulen J., Koerten H.K., Mommaas A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006;80:5927–5940. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., van Tol H., Pedersen K.W., Raamsman M.J., de Vries A.A. Identification of a novel structural protein of arteriviruses. J. Virol. 1999;73:6335–6345. doi: 10.1128/jvi.73.8.6335-6345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., van Tol H., Roos N., Pedersen K.W. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J. Gen. Virol. 2001;82:985–994. doi: 10.1099/0022-1317-82-5-985. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Wassenaar A.L., Spaan W.J. The 5′ end of the equine arteritis virus replicase gene encodes a papainlike cysteine protease. J. Virol. 1992;66:7040–7048. doi: 10.1128/jvi.66.12.7040-7048.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Wassenaar A.L., Spaan W.J. Proteolytic processing of the replicase ORF1a protein of equine arteritis virus. J. Virol. 1994;68:5755–5764. doi: 10.1128/jvi.68.9.5755-5764.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Wassenaar A.L., Spaan W.J., Gorbalenya A.E. The arterivirus Nsp2 protease. An unusual cysteine protease with primary structure similarities to both papain-like and chymotrypsin-like proteases. J. Biol. Chem. 1995;270:16671–16676. doi: 10.1074/jbc.270.28.16671. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Wassenaar A.L., van Dinten L.C., Spaan W.J., Gorbalenya A.E. The arterivirus nsp4 protease is the prototype of a novel group of chymotrypsin-like enzymes, the 3C-like serine proteases. J. Biol. Chem. 1996;271:4864–4871. doi: 10.1074/jbc.271.9.4864. [DOI] [PubMed] [Google Scholar]
- Spilman M.S., Welbon C., Nelson E., Dokland T. Cryo-electron tomography of porcine reproductive and respiratory syndrome virus: organization of the nucleocapsid. J. Gen. Virol. 2009;90:527–535. doi: 10.1099/vir.0.007674-0. [DOI] [PubMed] [Google Scholar]
- St-Laurent G., Morin G., Archambault D. Detection of equine arteritis virus following amplification of structural and nonstructural viral genes by reverse transcription-PCR. J. Clin. Microbiol. 1994;32:658–665. doi: 10.1128/jcm.32.3.658-665.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Louis M.C., Archambault D. The equine arteritis virus induces apoptosis via caspase-8 and mitochondria-dependent caspase-9 activation. Virology. 2007;367:147–155. doi: 10.1016/j.virol.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Stadejek T., Bjorklund H., Bascunana C.R., Ciabatti I.M., Scicluna M.T., Amaddeo D., McCollum W.H., Autorino G.L., Timoney P.J., Paton D.J., Klingeborn B., Belak S. Genetic diversity of equine arteritis virus. J. Gen. Virol. 1999;80(Pt 3):691–699. doi: 10.1099/0022-1317-80-3-691. [DOI] [PubMed] [Google Scholar]
- Stadejek T., Mittelholzer C., Oleksiewicz M.B., Paweska J., Belak S. Highly diverse type of equine arteritis virus (EAV) from the semen of a South African donkey: short communication. Acta Vet. Hung. 2006;54:263–270. doi: 10.1556/AVet.54.2006.2.12. [DOI] [PubMed] [Google Scholar]
- Starik E., Ginter A., Coppe P. ELISA and direct immunofluorescence test to detect equine arteritis virus (EAV) using a monoclonal antibody directed to the EAV-N protein. J. Vet. Med. B Infect. Dis. Vet. Public Health. 2001;48:1–9. doi: 10.1046/j.1439-0450.2001.00420.x. [DOI] [PubMed] [Google Scholar]
- Stein D., Foster E., Huang S.B., Weller D., Summerton J. A specificity comparison of four antisense types: morpholino, 2′-O-methyl RNA, DNA, and phosphorothioate DNA. Antisense Nucleic Acid Drug Dev. 1997;7:151–157. doi: 10.1089/oli.1.1997.7.151. [DOI] [PubMed] [Google Scholar]
- Stertz S., Reichelt M., Spiegel M., Kuri T., Martinez-Sobrido L., Garcia-Sastre A., Weber F., Kochs G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology. 2007;361:304–315. doi: 10.1016/j.virol.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stueckemann J.A., Holth M., Swart W.J., Kowalchyk K., Smith M.S., Wolstenholme A.J., Cafruny W.A., Plagemann P.G. Replication of lactate dehydrogenase-elevating virus in macrophages. 2. Mechanism of persistent infection in mice and cell culture. J. Gen. Virol. 1982;59:263–272. doi: 10.1099/0022-1317-59-2-263. [DOI] [PubMed] [Google Scholar]
- Subramaniam S., Kwon B., Beura L.K., Kuszynski C.A., Pattnaik A.K., Osorio F.A. Porcine reproductive and respiratory syndrome virus non-structural protein 1 suppresses tumor necrosis factor-alpha promoter activation by inhibiting NF-kappaB and Sp1. Virology. 2010;406:270–279. doi: 10.1016/j.virol.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim. Biophys. Acta. 1999;1489:141–158. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- Summerton J., Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- Sun Z., Li Y., Ransburgh R., Snijder E.J., Fang Y. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J. Virol. 2012;86:3839–3850. doi: 10.1128/JVI.06466-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surma-Kurusiewicz K., Winiarczyk S., Adaszek L. Comparative analysis of ORF5 nucleotide sequences and amino acid sequences of the GP5 protein of equine arteritis virus (EAV) detected in the semen of stallions from Eastern Poland. Res. Vet. Sci. 2012 doi: 10.1016/j.rvsc.2012.09.017. [DOI] [PubMed] [Google Scholar]
- Swinburne J. Inherited disease in the horse: mapping complex disease variants is on the horizon. Vet. J. 2009;179:317–318. doi: 10.1016/j.tvjl.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Szeredi L., Hornyak A., Denes B., Rusvai M. Equine viral arteritis in a newborn foal: parallel detection of the virus by immunohistochemistry, polymerase chain reaction and virus isolation. J. Vet. Med. B Infect. Dis. Vet. Public Health. 2003;50:270–274. doi: 10.1046/j.1439-0450.2003.00684.x. [DOI] [PubMed] [Google Scholar]
- Szeredi L., Hornyak A., Palfi V., Molnar T., Glavits R., Denes B. Study on the epidemiology of equine arteritis virus infection with different diagnostic techniques by investigating 96 cases of equine abortion in Hungary. Vet. Microbiol. 2005;108:235–242. doi: 10.1016/j.vetmic.2005.04.013. [DOI] [PubMed] [Google Scholar]
- te Velthuis A.J., van den Worm S.H., Sims A.C., Baric R.S., Snijder E.J., van Hemert M.J. Zn(2+) inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathogens. 2010;6:e1001176. doi: 10.1371/journal.ppat.1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaa B., Kabatek A., Zevenhoven-Dobbe J.C., Snijder E.J., Herrmann A., Veit M. Myristoylation of the arterivirus E protein: the fatty acid modification is not essential for membrane association but contributes significantly to virus infectivity. J. Gen. Virol. 2009;90:2704–2712. doi: 10.1099/vir.0.011957-0. [DOI] [PubMed] [Google Scholar]
- Tian D., Wei Z., Zevenhoven-Dobbe J.C., Liu R., Tong G., Snijder E.J., Yuan S. Arterivirus minor envelope proteins are a major determinant of viral tropism in cell culture. J. Virol. 2012;86:3701–3712. doi: 10.1128/JVI.06836-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijms M.A., Nedialkova D.D., Zevenhoven-Dobbe J.C., Gorbalenya A.E., Snijder E.J. Arterivirus subgenomic mRNA synthesis and virion biogenesis depend on the multifunctional nsp1 autoprotease. J. Virol. 2007;81:10496–10505. doi: 10.1128/JVI.00683-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijms M.A., Snijder E.J. Equine arteritis virus non-structural protein 1, an essential factor for viral subgenomic mRNA synthesis, interacts with the cellular transcription co-factor p100. J. Gen. Virol. 2003;84:2317–2322. doi: 10.1099/vir.0.19297-0. [DOI] [PubMed] [Google Scholar]
- Tijms M.A., van der Meer Y., Snijder E.J. Nuclear localization of non-structural protein 1 and nucleocapsid protein of equine arteritis virus. J. Gen. Virol. 2002;83:795–800. doi: 10.1099/0022-1317-83-4-795. [DOI] [PubMed] [Google Scholar]
- Tijms M.A., van Dinten L.C., Gorbalenya A.E., Snijder E.J. A zinc finger-containing papain-like protease couples subgenomic mRNA synthesis to genome translation in a positive-stranded RNA virus. Proc. Natl. Acad. Sci. U. S. A. 2001;98:1889–1894. doi: 10.1073/pnas.041390398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timoney P.J. Equine viral arteritis: epidemiology and control. J. Equine Vet. Sci. 1988;8:54–59. [Google Scholar]
- Timoney P.J., McCollum W.H. Equine viral arteritis: epidemiology and control. J. Equine Vet. Sci. 1988;8:54–59. [Google Scholar]
- Timoney P.J., McCollum W.H. Equine viral arteritis. Vet. Clin. North Am. Equine Pract. 1993;9:295–309. doi: 10.1016/S0749-0739(17)30397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timoney P.J., McCollum W.H., Murphy T.W., Roberts A.W., Willard J.G., Carswell G.D. The carrier state in equine arteritis virus infection in the stallion with specific emphasis on the venereal mode of virus transmission. J. Reprod. Fertil. Suppl. 1987;35:95–102. [PubMed] [Google Scholar]
- Timoney P.J., McCollum W.H., Roberts A.W., Murphy T.W. Demonstration of the carrier state in naturally acquired equine arteritis virus infection in the stallion. Res. Vet. Sci. 1986;41:279–280. [PubMed] [Google Scholar]
- Timoney P.J., Umphenour N.W., McCollum W.H. Safety evaluation of a commercial modified live equine arteritis virus vaccine for use in stallions. In: Powell D.G., editor. Proceedings of the fifth International Conference on Equine Infectious Diseases; Lexington, KY, 1987; 1988. pp. 19–27. [Google Scholar]
- Tobiasch E., Kehm R., Bahr U., Tidona C.A., Jakob N.J., Handermann M., Darai G., Giese M. Large envelope glycoprotein and nucleocapsid protein of equine arteritis virus (EAV) induce an immune response in Balb/c mice by DNA vaccination; strategy for developing a DNA-vaccine against EAV-infection. Virus Genes. 2001;22:187–199. doi: 10.1023/a:1008175525254. [DOI] [PubMed] [Google Scholar]
- Vaala W.E., Hamir A.N., Dubovi E.J., Timoney P.J., Ruiz B. Fatal, congenitally acquired infection with equine arteritis virus in a neonatal thoroughbred. Equine Vet. J. 1992;24:155–158. doi: 10.1111/j.2042-3306.1992.tb02803.x. [DOI] [PubMed] [Google Scholar]
- Vairo S., Vandekerckhove A., Steukers L., Glorieux S., Van den Broeck W., Nauwynck H. Clinical and virological outcome of an infection with the Belgian equine arteritis virus strain 08P178. Vet. Microbiol. 2012;157:12. doi: 10.1016/j.vetmic.2012.01.014. [DOI] [PubMed] [Google Scholar]
- van Aken D., Benckhuijsen W.E., Drijfhout J.W., Wassenaar A.L., Gorbalenya A.E., Snijder E.J. Expression, purification, and in vitro activity of an arterivirus main proteinase. Virus Res. 2006;120:97–106. doi: 10.1016/j.virusres.2006.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Aken D., Snijder E.J., Gorbalenya A.E. Mutagenesis analysis of the nsp4 main proteinase reveals determinants of arterivirus replicase polyprotein autoprocessing. J. Virol. 2006;80:3428–3437. doi: 10.1128/JVI.80.7.3428-3437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Aken D., Zevenhoven-Dobbe J., Gorbalenya A.E., Snijder E.J. Proteolytic maturation of replicase polyprotein pp1a by the nsp4 main proteinase is essential for equine arteritis virus replication and includes internal cleavage of nsp7. J. Gen. Virol. 2006;87:3473–3482. doi: 10.1099/vir.0.82269-0. [DOI] [PubMed] [Google Scholar]
- van den Born E., Posthuma C.C., Gultyaev A.P., Snijder E.J. Discontinuous subgenomic RNA synthesis in arteriviruses is guided by an RNA hairpin structure located in the genomic leader region. J. Virol. 2005;79:6312–6324. doi: 10.1128/JVI.79.10.6312-6324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Born E., Posthuma C.C., Knoops K., Snijder E.J. An infectious recombinant equine arteritis virus expressing green fluorescent protein from its replicase gene. J. Gen. Virol. 2007;88:1196–1205. doi: 10.1099/vir.0.82590-0. [DOI] [PubMed] [Google Scholar]
- van den Born E., Stein D.A., Iversen P.L., Snijder E.J. Antiviral activity of morpholino oligomers designed to block various aspects of Equine arteritis virus amplification in cell culture. J. Gen. Virol. 2005;86:3081–3090. doi: 10.1099/vir.0.81158-0. [DOI] [PubMed] [Google Scholar]
- van der Meer F.J., de Haan C.A., Schuurman N.M., Haijema B.J., Peumans W.J., Van Damme E.J., Delputte P.L., Balzarini J., Egberink H.F. Antiviral activity of carbohydrate-binding agents against Nidovirales in cell culture. Antiviral Res. 2007;76:21–29. doi: 10.1016/j.antiviral.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer Y., Snijder E.J., Dobbe J.C., Schleich S., Denison M.R., Spaan W.J., Locker J.K. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 1999;73:7641–7657. doi: 10.1128/jvi.73.9.7641-7657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer Y., van Tol H., Locker J.K., Snijder E.J. ORF1a-encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. J. Virol. 1998;72:6689–6698. doi: 10.1128/jvi.72.8.6689-6698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinten L.C., den Boon J.A., Wassenaar A.L., Spaan W.J., Snijder E.J. An infectious arterivirus cDNA clone: identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc. Natl. Acad. Sci. U. S. A. 1997;94:991–996. doi: 10.1073/pnas.94.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinten L.C., Rensen S., Gorbalenya A.E., Snijder E.J. Proteolytic processing of the open reading frame 1b-encoded part of arterivirus replicase is mediated by nsp4 serine protease and is essential for virus replication. J. Virol. 1999;73:2027–2037. doi: 10.1128/jvi.73.3.2027-2037.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinten L.C., van Tol H., Gorbalenya A.E., Snijder E.J. The predicted metal-binding region of the arterivirus helicase protein is involved in subgenomic mRNA synthesis, genome replication, and virion biogenesis. J. Virol. 2000;74:5213–5223. doi: 10.1128/jvi.74.11.5213-5223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinten L.C., Wassenaar A.L., Gorbalenya A.E., Spaan W.J., Snijder E.J. Processing of the equine arteritis virus replicase ORF1b protein: identification of cleavage products containing the putative viral polymerase and helicase domains. J. Virol. 1996;70:6625–6633. doi: 10.1128/jvi.70.10.6625-6633.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hemert M.J., de Wilde A.H., Gorbalenya A.E., Snijder E.J. The in vitro RNA synthesizing activity of the isolated arterivirus replication/transcription complex is dependent on a host factor. J. Biol. Chem. 2008;283:16525–16536. doi: 10.1074/jbc.M708136200. [DOI] [PubMed] [Google Scholar]
- Van Hemert M.J., Snijder E.J. The arterivirus replicase. In: Perlman S., Gallagher T., Snijder E.J., editors. Nidoviruses. American Society for Microbiology; Washington, DC: 2008. pp. 83–101. [Google Scholar]
- van Kasteren P.B., Bailey-Elkin B.A., James T.W., Ninaber D.K., Beugeling C., Khajehpour M., Snijder E.J., Mark B.L., Kikkert M. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E838–E847. doi: 10.1073/pnas.1218464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kasteren P.B., Beugeling C., Ninaber D.K., Frias-Staheli N., van Boheemen S., Garcia-Sastre A., Snijder E.J., Kikkert M. Arterivirus and nairovirus ovarian tumor domain-containing deubiquitinases target activated RIG-I to control innate immune signaling. J. Virol. 2012;86:773–785. doi: 10.1128/JVI.06277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G., Dobbe J.C., Gultyaev A.P., Luytjes W., Spaan W.J., Snijder E.J. Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription-regulating sequences. Proc. Natl. Acad. Sci. U. S. A. 1999;96:12056–12061. doi: 10.1073/pnas.96.21.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G., van Dinten L.C., Spaan W.J., Luytjes W., Snijder E.J. Characterization of an equine arteritis virus replicase mutant defective in subgenomic mRNA synthesis. J. Virol. 1999;73:5274–5281. doi: 10.1128/jvi.73.7.5274-5281.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheije M.H., Welting T.J., Jansen H.T., Rottier P.J., Meulenberg J.J. Chimeric arteriviruses generated by swapping of the M protein ectodomain rule out a role of this domain in viral targeting. Virology. 2002;303:364–373. doi: 10.1006/viro.2002.1711. [DOI] [PubMed] [Google Scholar]
- Wade C.M., Giulotto E., Sigurdsson S., Zoli M., Gnerre S., Imsland F., Lear T.L., Adelson D.L., Bailey E., Bellone R.R., Blocker H., Distl O., Edgar R.C., Garber M., Leeb T., Mauceli E., MacLeod J.N., Penedo M.C., Raison J.M., Sharpe T., Vogel J., Andersson L., Antczak D.F., Biagi T., Binns M.M., Chowdhary B.P., Coleman S.J., Della Valle G., Fryc S., Guerin G., Hasegawa T., Hill E.W., Jurka J., Kiialainen A., Lindgren G., Liu J., Magnani E., Mickelson J.R., Murray J., Nergadze S.G., Onofrio R., Pedroni S., Piras M.F., Raudsepp T., Rocchi M., Roed K.H., Ryder O.A., Searle S., Skow L., Swinburne J.E., Syvanen A.C., Tozaki T., Valberg S.J., Vaudin M., White J.R., Zody M.C., Lander E.S., Lindblad-Toh K. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science (New York, NY) 2009;326:865–867. doi: 10.1126/science.1178158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner H.M., Balasuriya U.B., James MacLachlan N. The serologic response of horses to equine arteritis virus as determined by competitive enzyme-linked immunosorbent assays (c-ELISAs) to structural and non-structural viral proteins. Comp. Immunol. Microbiol. Infect. Dis. 2003;26:251–260. doi: 10.1016/S0147-9571(02)00054-1. [DOI] [PubMed] [Google Scholar]
- Wassenaar A.L., Spaan W.J., Gorbalenya A.E., Snijder E.J. Alternative proteolytic processing of the arterivirus replicase ORF1a polyprotein: evidence that NSP2 acts as a cofactor for the NSP4 serine protease. J. Virol. 1997;71:9313–9322. doi: 10.1128/jvi.71.12.9313-9322.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland E., Bolz S., Weiland F., Herbst W., Raamsman M.J., Rottier P.J., De Vries A.A. Monoclonal antibodies directed against conserved epitopes on the nucleocapsid protein and the major envelope glycoprotein of equine arteritis virus. J. Clin. Microbiol. 2000;38:2065–2075. doi: 10.1128/jcm.38.6.2065-2075.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott D.G., King D.P., Drew T.W., Nowotny N., Kindermann J., Hannant D., Belak S., Paton D.J. Use of an internal standard in a closed one-tube RT-PCR for the detection of equine arteritis virus RNA with fluorescent probes. Vet. Res. 2003;34:165–176. doi: 10.1051/vetres:2002063. [DOI] [PubMed] [Google Scholar]
- Wieringa R., De Vries A.A., Post S.M., Rottier P.J. Intra- and intermolecular disulfide bonds of the GP2b glycoprotein of equine arteritis virus: relevance for virus assembly and infectivity. J. Virol. 2003;77:12996–13004. doi: 10.1128/JVI.77.24.12996-13004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieringa R., de Vries A.A., Raamsman M.J., Rottier P.J. Characterization of two new structural glycoproteins, GP(3) and GP(4), of equine arteritis virus. J. Virol. 2002;76:10829–10840. doi: 10.1128/JVI.76.21.10829-10840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieringa R., de Vries A.A., Rottier P.J. Formation of disulfide-linked complexes between the three minor envelope glycoproteins (GP2b, GP3, and GP4) of equine arteritis virus. J. Virol. 2003;77:6216–6226. doi: 10.1128/JVI.77.11.6216-6226.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieringa R., de Vries A.A., van der Meulen J., Godeke G.J., Onderwater J.J., van Tol H., Koerten H.K., Mommaas A.M., Snijder E.J., Rottier P.J. Structural protein requirements in equine arteritis virus assembly. J. Virol. 2004;78:13019–13027. doi: 10.1128/JVI.78.23.13019-13027.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood O., Tauraso N., Liebhaber H. Electron microscopic study of tissue cultures infected with simian haemorrhagic fever virus. J. Gen. Virol. 1970;7:129–136. doi: 10.1099/0022-1317-7-2-129. [DOI] [PubMed] [Google Scholar]
- Wu W.H., Fang Y., Rowland R.R., Lawson S.R., Christopher-Hennings J., Yoon K.J., Nelson E.A. The 2b protein as a minor structural component of PRRSV. Virus Res. 2005;114:177–181. doi: 10.1016/j.virusres.2005.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zevenhoven-Dobbe J.C., Greve S., van Tol H., Spaan W.J., Snijder E.J. Rescue of disabled infectious single-cycle (DISC) equine arteritis virus by using complementing cell lines that express minor structural glycoproteins. J. Gen. Virol. 2004;85:3709–3714. doi: 10.1099/vir.0.80443-0. [DOI] [PubMed] [Google Scholar]
- Zhang J., Go Y.Y., Huang C.M., Meade B.J., Lu Z., Snijder E.J., Timoney P.J., Balasuriya U.B. Development and characterization of an infectious cDNA clone of the modified live virus vaccine strain of equine arteritis virus. Clin. Vaccine Immunol. 2012;19:1312–1321. doi: 10.1128/CVI.00302-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Go Y.Y., MacLachlan N.J., Meade B.J., Timoney P.J., Balasuriya U.B. Amino acid substitutions in the structural or nonstructural proteins of a vaccine strain of equine arteritis virus are associated with its attenuation. Virology. 2008;378:355–362. doi: 10.1016/j.virol.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Zhang J., Stein D.A., Timoney P.J., Balasuriya U.B. Curing of HeLa cells persistently infected with equine arteritis virus by a peptide-conjugated morpholino oligomer. Virus Res. 2010;150:138–142. doi: 10.1016/j.virusres.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Timoney P.J., MacLachlan N.J., McCollum W.H., Balasuriya U.B. Persistent equine arteritis virus infection in HeLa cells. J. Virol. 2008;82:8456–8464. doi: 10.1128/JVI.01249-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Timoney P.J., Shuck K.M., Seoul G., Go Y.Y., Lu Z., Powell D.G., Meade B.J., Balasuriya U.B. Molecular epidemiology and genetic characterization of equine arteritis virus isolates associated with the 2006–2007 multi-state disease occurrence in the USA. J. Gen. Virol. 2010;91:2286–2301. doi: 10.1099/vir.0.019737-0. [DOI] [PubMed] [Google Scholar]
- Zhang J., Timoney P.J., MacLachlan N.J., Balasuriya U.B.R. Identification of an additional neutralization determinant of equine arteritis virus. Virus Res. 2008;138(1–2):4. doi: 10.1016/j.virusres.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]