

Abstract
The crystal structure of a conserved domain of nonstructural protein 3 (nsP3) from severe acute respiratory syndrome coronavirus (SARS-CoV) has been solved by single-wavelength anomalous dispersion to 1.4 Å resolution. The structure of this “X” domain, seen in many single-stranded RNA viruses, reveals a three-layered α/β/α core with a macro-H2A-like fold. The putative active site is a solvent-exposed cleft that is conserved in its three structural homologs, yeast Ymx7, Archeoglobus fulgidus AF1521, and Er58 from E. coli. Its sequence is similar to yeast YBR022W (also known as Poa1P), a known phosphatase that acts on ADP-ribose-1″-phosphate (Appr-1″-p). The SARS nsP3 domain readily removes the 1″ phosphate group from Appr-1″-p in in vitro assays, confirming its phosphatase activity. Sequence and structure comparison of all known macro-H2A domains combined with available functional data suggests that proteins of this superfamily form an emerging group of nucleotide phosphatases that dephosphorylate Appr-1″-p.
Introduction
Severe acute respiratory syndrome (SARS) emerged as the first severe and readily transmissible new disease of the 21st century and caused 8000 infections and more than 800 deaths in 2003 (Groneberg et al., 2003). The causative organism is a new coronavirus (SARS-CoV) that is distantly related to group II coronaviruses. The virus has a single-stranded RNA genome of ∼29.7 kb that encodes at least 14 putative open reading frames (ORFs) (Peiris et al., 2003, Drosten et al., 2003) ( Figure 1A). Two-thirds of the viral genome at the 5′ end is organized as a single highly conserved ORF, known as ORF-1a/1ab, that is translated into two large polyproteins, pp1a and pp1ab (Ksiazek et al., 2003). Translation of pp1ab involves ribosomal frameshifting, a feature also seen in many other coronaviruses (Baranov et al., 2005, Snijder et al., 2003). Termed as the “replicase polyproteins,” pp1a and pp1ab are subsequently posttranslationally cleaved by two virus-encoded proteases, the 3C-like protease (the main protease or 3CL-Pro) and the papain-like cysteine protease (PLP), into 16 mature protein products (Snijder et al., 2003) (Figure 1A). These “nonstructural proteins” or nsPs (nsP1–nsP16) form a giant replicase complex that participates in numerous functions during viral infection, such as replication of the RNA genome, processing of subgenomic RNA, and packaging of newly budding virions (Ziebuhr et al., 2001).
Figure 1.

Genomic Location of the X Domain of SARS nsP3
(A) Schematic of the SARS genome and proteome showing the location of SARS nsp3 and its putative functional domains. The predicted functions of different nsPs of ORF1a/ORF1ab are highlighted along with the structural and accessory genes. Abbreviations used are: P65, P65 protein homolog of murine hepatitis virus; 3CL-Pro, SARS main protease; RdRp, RNA-dependent RNA polymerase; Hel, Zn2+-dependent helicase; ExoN, homolog of exonuclease; NendoU, uridylate-specific endonuclease; 2′-O-MT, methyltransferase; E, small envelope glycoprotein; M, matrix; N, nucleocapsid; ADRP, ADP-ribose-1″-phosphate phosphatase; SUD, SARS-specific unique domain; PL2-Pro, papain-like protease; TM, transmembrane domain. (Figure modified from Snijder et al., 2003).
(B) Sequence alignment of known macrodomains. Group I: ADRP domain found in nsPs of different coronaviruses: Sars_nsP3, SARS coronavirus-Tor2; R1AB_CVBM, bovine coronavirus; AAR01012, human coronavirus OC43; Q66WN5, murine hepatitis virus; Q6Q1S3, human coronavirus NL63; R1AB_PEDV7, porcine epidemic diarrhea virus; R1AB_CVPPU, transmissible gastroenteritis virus; R1AB_IBVBC, avian infectious bronchitis virus. Group II: ADRP homologs from other related viruses: Q6X2U4, rubella virus; O90370, Igbo Ora virus; Q8JUX6, Chikungunya virus; O10380, Semliki forest virus; Q8QHM4, Mayaro virus; Q9JGK9 Ross River virus; P87515, Barmah forest virus; Q87644, Sindbis virus; Q86924, Aura virus; Q88791, Western equine encephalomyelitis virus; Q66580, Eastern equine encephalitis virus; O90163, Venezuelan equine encephalitis virus; Q8QL53, sleeping disease virus; O90368, O'nyong-nyong virus; Q8JJX1, salmon pancreatic disease virus; and Group III: macrodomain hypothetical proteins of the A1pp superfamily: gi|20178242, E. coli ymdB; gi|20178260, Deinococcus radiodurans; gi|20178146, Ralstonia solanacearum; gi|20178157, Mesorhizobium loti; gi|20178167, Pseudomonas aeruginosa; gi|20090472, Methanosarcina acetivorans; gi|20178156, Thermoplasma volcanium; gi|16082127, Thermoplasma acidophilum; gi|19705253, Fusobacterium nucleatum; gi|20178176, Pyrococcus abyssi; gi|20178181, Pyrococcus horikoshii; gi|11499116, Archaeoglobus fulgidus (AF1521); gi|20178224, Pyrobaculum aerophilum; gi|20178255, Sulfolobus solfataricus; gi|20178177, Thermotoga maritima; gi|20094386, Methanopyrus kandleri; gi|20178182, Aquifex aeolicus; gi|20178236, Aeropyrum pernix; gi|20178237, Mycobacterium tuberculosis. The two structurally characterized members of group III are highlighted in red.
The third of these nonstructural proteins, nsP3, is a large multidomain protein of 1922 amino acids that spans residues 819–2740 of pp1a (NP_828862; gi:34555776) (Thiel et al., 2003). Mature nsP3 results due to proteolytic cleavage of pp1a at two sites (818GA819 and 2740GK2741) by the papain-like proteinase (Thiel et al., 2003). nsP3 has conserved sequence motifs of six independent domains: (1) an N-terminal glu-rich acidic domain; (2) an X domain with predicted Appr-1″-p processing activity; (3) a SUD domain (SARS-specific unique domain); (4) a peptidase C-16 domain that contains the papain-like protease; (5) a transmembrane domain; and (6) the Y domain (Figure 1A).
The conserved X domain of nsP3 has been predicted to house a putative adenosine diphosphate ribose 1″ phosphatase (ADRP) function and is annotated in domain classification databases such as SMART (Letunic et al., 2004) and Interpro (Mulder et al., 2003) as a member of the A1pp superfamily that includes more than 300 proteins from archaea, bacteria, eukaryotes, and single-stranded, positive-sense RNA viruses. Structures of three homologs from this superfamily, yeast Ymx7, E. coli Er58, and a conserved C-terminal domain of nonhistone macro-H2A from Archeoglobus fulgidus (AF1521), show that they all adopt a generic macro-H2A-like fold with minor variations. While the function of some members of this superfamily, like the human poly-(ADP-ribose) polymerase, has been experimentally characterized (D'Amours et al., 1999), that of many others is yet to be determined.
As a part of an integrated program to study emerging infectious diseases, we have undertaken the structural and functional characterization of all of the major protein products in SARS-CoV by using a multipronged approach. This included a detailed bioinformatics analysis of the SARS-Tor2 genome by using sensitive profile-based methods like PSI-BLAST (Altschul et al., 1997), FFAS (Rychlewski et al., 2000), and HMMER (Eddy, 1996) for detection of remote homologs, identification of domain boundaries in multidomain proteins, and functional annotation. Based on this analysis, 173 constructs that cover the entire proteome were designed and cloned into vectors for overexpression in E. coli and baculovirus systems (http://sars.scripps.edu).
In this study, we present the first of the crystal structures from this effort, that of the highly conserved putative phosphatase domain of nsP3. To our knowledge, this is the first crystal structure of this domain from positive-sense, single-stranded RNA viruses. It reveals a close structural relationship with prototypical macro-H2A-like fold proteins. One of its sequence homologs, Poa1p (YBR022) from Saccharomyces cerevisiae, was recently functionally characterized as a highly specific phosphatase that removes the 1″ phosphate group of ADP-ribose-1″-phosphate (Appr-1″-p) in the latter half of the tRNA splicing pathway in yeast (Shull et al., 2005), hinting at a similar substrate specificity for SARS ADRP. Using an in vitro assay, we experimentally validate that this ADRP domain of SARS nsP3 is indeed a phosphatase that removes the terminal 1″ phosphate from Appr-1″-p. To our knowledge, these results, combined with recently elucidated structures of two hypothetical proteins, suggest that a majority of macro-H2A fold members form a new family of nucleotide phosphatases.
Results and Discussion
Description of the ADRP Domain of SARS nsP3
The cloned insert contains 182 residues from nsP3 of SARS-CoV-Tor2 and has a monomer molecular weight of 19,523 Da and a pI of 6.9. The final structural model refined against crystallographic data to 1.4 Å resolution has four subunits in the asymmetric unit in very similar conformations (rmsds < 0.4 Å for 166 Cα atoms). We do not observe electron density for a few residues at the C terminus of each of the four monomers. These include 5 residues in chain D, 15 residues in the chain A, and 9 residues each in the B and C monomers. The final refinement statistics and stereochemical parameters of the structure are listed in Table 1. Overall, each subunit consists of eight β strands and six α helices ( Figure 2A). Strands 2–8 form a central seven-stranded β sheet that has a strand order of 2387465. The outermost strands on either side are antiparallel to the rest. The six helices straddle the central β sheet to form a three-layered α/β/α topology. Two of the subunits in the asymmetric unit form a loosely packed head-to-head dimer (Figure 2B). A short loop connecting strand 6 and helix H4 is involved in weak interfacial contacts with the conserved Gly-rich segment of the other monomer. The interface is fairly small at ∼870 Å2 (435 Å2 per monomer) and predominantly nonpolar (60%). Residues that form the putative active site lie close to the dimer interface. The enzyme elutes as a homodimer in gel-filtration studies (data not shown), indicating that the physiologically relevant form of this protein may be dimeric.
Table 1.
Data Collection and Refinement Statistics
| SeMet (Peak λ) | Native | |
|---|---|---|
| Data Collection | ||
| Space group | P212121 | P212121 |
| Unit cell parameters | a = 76.920 Å, b = 81.224 Å, c = 125.695 Å | a = 76.495 Å, b = 81.585 Å, c = 125.465 Å |
| Wavelength (Å) | 0.97941 | 0.9794 |
| Resolution range (Å) | 50.0–2.2 | 50.0–1.40 |
| Number of observations | 1,909,845 | 2,836,583 |
| Number of unique reflections | 41,023 | 145,609 |
| Completeness (%) | 100.0 (99.8) | 99.20 (98.9) |
| Redundancy | 7.6 | 3.7 |
| Mean I/σ (I) | 30.13 (6.04) | 32.93 (2.82) |
| Rsyma on I | 0.091 (0.507) | 0.043 (0.590) |
| Highest resolution shell (Å) | 2.24–2.20 | 1.42–1.40 |
| Figure of merit after RESOLVE | 0.54 | |
| Refinement | ||
| Rworkb | 16.4 (22.2) | |
| Rfreec | 19.0 (25.0) | |
| Protein atoms (average B factor) | 5,485 (18.1) | |
| Solvent atoms (average B factor) | 950 (36.91) | |
| Hetero atoms (average B factor) | 72 (35.28) | |
| Rmsd bond length (Å) | 0.018 | |
| Rmsd bond angle (°) | 1.69 | |
| Ramachandran statistics | ||
| Most favored (%) | 90.8 | |
| Additionally allowed (%) | 8.4 | |
| Generously allowed (%) | 0.8 | |
Values in parentheses are for data corresponding to the outermost shell.
Rsym = Σhkl[(Σj|Ij − <I>|)/Σj|Ij|].
Rwork = Σhkl |Fo − Fc|/Σhkl|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
5% of the reflections (7,683 reflections) was used in the calculation of Rfree.
Figure 2.

Structure of SARS ADRP
(A) Ribbon representation of the SARS nsP3 ADRP monomer. The two glycine-rich loops are shown in yellow. Secondary structures are colored from blue (N) to red (C terminus). Helices are numbered H1–H6, and β strands are numbered from 1 to 8.
(B) The SARS ADRP dimer observed between the B and D subunits in the asymmetric unit. The four conserved segments are colored red in each subunit; the conserved histidines and asparagines at the active site are shown as ball-and-sticks.
SARS ADRP Belongs to the Macro-H2A Fold
Comparison of one of the chains of SARS ADRP against all known structures in the PDB by using DALI (Holm and Sander, 1993) revealed the presence of two structural homologs: a hypothetical protein from Archeoglobus fulgidus, AF1521 (PDB code: 1HJZ; z score of 18.1; rmsd of 2.4 Å for 152 superimposed Cα atoms; pairs with a z score > 3.0 are considered structurally similar), and the N-terminal domain of bovine lens leucine aminopeptidase (PDB code: 1LAM; z score of 8.0; rmsd of 2.6 Å for 119 Cα atoms; Sträter and Lipscomb, 1995). Both structures are members of the “macrodomain-like” fold as defined in the SCOP database (Murzin et al., 1995). This fold includes two other structural homologs from E. coli, aminopeptidase A (PepA) and a hypothetical protein ymbD (Northeast Structural Genomics Consortium target Er58; PDB code: 1SPV). The topological connectivity of the secondary structural elements of these four proteins along with the ADRP domain of SARS nsP3 is shown in a similar orientation in Figure 3 (A, B, D, E, and F). All of them share the same three-layered α/β/α core, with minor variations. They have a mixed β sheet of six strands with strand order 165243. The first strand and the first helix are absent in bovine lens leucine aminopeptidase (BlLAP, Figure 3A). AF1521 has two insertions to this core, a β strand inserted at the N terminus and an α helix between strands 3 and 4 (Figure 3E). The SARS ADRP domain has two β strands inserted at the N terminus, one of which forms part of the central β sheet (Figure 3F). The sixth protein structure in Figure 3, Ymx7 from yeast (PDB code: 1TXZ), is a member of this fold and has a circular permutation (Figure 3C). The first strand and the first helix of this protein occupy structural positions that correspond to the last β strand and the C-terminal helix (H6) of a canonical macro-like fold.
Figure 3.

Fold Classification of the SARS ADRP Domain
(A–F) (A) Bovine Lens Leu-aminopeptidase (1LAM). (B) E. coli PepA (1GYT). (C) yeast Appr phosphatase homolog (1TXZ). (D) E. coli hypothetical protein Er58 (1SPV). (E) Archeoglobus fuldiges AF1521. (F) ADRP domain of SARS nsp3. The helices are colored cyan, and the strands are colored yellow in the core macrodomain. The inserted secondary structural elements that are not part of the main core are highlighted in red. The circular permutation seen in yeast Ymx7 (1TXZ) is marked in green, and the C-terminal helical domain is shown in white.
Function of Macro-like Fold Proteins
BlLAP is an exopeptidase that cleaves amino acids from the N terminus of polypeptides (Burley et al., 1990). E. coli PepA is a DNA binding protein that is involved in Xer site-specific recombination and transcriptional control of the carAB operon (Sträter et al., 1999). Although both share significant similarities in sequence (31% identity) and structure (both are homohexameric with a dinuclear Zn2+ in their active site), they have widely different functions. The peptidase activity is not needed by PepA to function during Xer-specific recombination (McCulloch et al., 1994) or during repression of carAB transcription (Charlier et al., 1995). On the other hand, blLAP does not have any demonstrated DNA binding function.
AF1521 is a stand-alone macrodomain from Archeoglobus fulgidus and is a close homolog of the C-terminal nonhistone domain of the largest variant of histone H2A (Pehrson and Fried, 1992, Pehrson and Fuji, 1998). It is evolutionarily related to P loop-containing nucleotide triphosphate hydrolases. The structure has been solved in its apo form (Allen et al., 2003) and in complex with two ligands, Mg2+-ADP (PDB code: 2BFR, unpublished) and ADP-ribose (PDB code: 2BFQ; unpublished). Yeast Ymx7 is a conserved hypothetical protein from the ADH3-RCA1 intergenic region. Although its function has not been experimentally demonstrated, it has been annotated as an ADP-ribose-1″-monophosphatase (ADRP) based on its sequence similarity to known ADRPs (Kumaran et al., 2005). Finally, the structure of a conserved hypothetical protein, Er58, from E. coli that was solved by the Northeast Structural Genomics Consortium (PDB code: 1SPV, unpublished) reveals a canonical macro-like fold. Its function remains unknown.
It would thus appear that the five known members of this fold fall into two broad functional groups, one containing BlLAP and E. coli PepA and the second containing the other three hypothetical proteins. All members of the second group not only share a similar global architecture, but also share conserved active site features. Although all of these proteins can be picked up by PSI-BLAST by using SARS ADRP as a query template, it is clear that the SARS domain is closer to phosphatases of the second group.
The Putative Active Site
The ADRP domain of SARS nsP3 has a deep solvent-exposed cleft on the protein surface that is very similar to that seen in AF1521, yeast Ymx7, and E. coli Er58. Surface representations showing the distribution of electrostatic potential on SARS ADRP and on the structures of ligand bound forms of AF1521 and yeast Ymx7 (shown in Figure 4A) clearly indicate that the putative active site cleft is similar in the three structures. Repeated soaking and cocrystallization attempts failed to yield cocrystals of SARS ADRP with ADP-ribose, perhaps because the active site is occluded by the dimer interface. However, the availability of the product (ADP-ribose) bound forms of AF1521 and yeast Ymx7 facilitates a detailed structure comparison of these two homologs with SARS ADRP. Many of the residues that interact with the ligand are conserved in the three structures. A view of the proposed active site of SARS ADRP along with the superimposed structures of AF1521 and yeast Ymx7 are shown in Figure 4B, highlighting the interactions that are likely between residues of the protein with the ligand. A structure-based sequence alignment of SARS ADRP with four of its structural homologs is shown in Figure 4C. The BlLAP sequence was omitted in this alignment.
Figure 4.

Structure Comparison of SARS ADRP with YMX7 and AF1521
(A) Surface of SARS ADRP showing the distribution of electrostatic potential. The ADP-ribose bound complexes of AF1521 (PDB code: 2BFQ; 1.5 Å) and Yeast Ymx7 (PDB code: 1TXZ; 2.0 Å) are shown for comparison. The bound ligands in the two structures are shown as ball-and-sticks.
(B) Superposition of the three structures (SARS ADRP is in green, AF1521 is in cyan, and Ymx7 is in purple). The bound ADP and ADP-ribose are shown as ball-and-sticks. The residues from SARS ADRP that are proposed to interact with the ligand are shown in ball-and-sticks, and the putative interactions are highlighted as dotted lines.
(C) Structure-based sequence alignment of SARS ADRP with its four structure homologs: AF1521 ADP-ribose complex (2BFQ); E. coli hypothetical protein Er58 (1SPV); E. coli PepA (1GYT); and yeast Ymx7 ADP-ribose complex (1TXZ). Helical regions are in cyan, and β strands are in yellow. Regions that can be confidently aligned are in capital letters, and those that align poorly or do not align at all are in small letters. The four conserved segments are highlighted in rectangular blocks. The circular permutation of yeast Ymx7 is marked in red.
Most macro-like fold proteins, including the ADRP domain from RNA viruses, show the presence of four conserved stretches of residues (Figure 1B). The first motif “XXNAAN,” where XX are any two hydrophobic amino acids, is highly conserved across the superfamily. This is immediately followed by a Gly-rich region (GGGVAG) that is reminiscent of the Walker A motif seen in many P loop nucleotide hydrolases (Walker et al., 1982). A notable feature is that the invariant lysine of the Walker A motif is an arginine in some coronaviruses and is absent in others (Figure 1B). The third stretch, “XVVGP,” where X is often a conserved histidine, is in the middle of the polypeptide. Finally, a stretch of 4 residues mainly consisting of small hydrophilic amino acids and a glycine is present near the C terminus of the polypeptide chain (Figure 1B). Residues from the third motif occupy structurally similar positions to the Walker B motif in classical P loop hydrolases. These four regions line the putative active site of the ADRP domain of the SARS nsP3 structure. The first motif forms part of the fourth β strand (Figure 4C), while the Gly-rich segment is part of a loop that connects strand 4 with the second helix. The third motif connects strand 6 to helix H4.
Description of the Putative Active Site of SARS ADRP
The active site can be broadly divided into the adenine binding cleft, the first ribose, and the bisphosphate binding site, followed by the terminal ribose-phosphate binding pocket that is the center of catalysis. As anticipated, the adenine binding pocket consists of largely hydrophobic residues. It is less conserved in the three structures than the other two pockets. In SARS ADRP, residues Ile23, Ala52, Pro125, and Ala154 form the walls of the putative adenine binding cleft. In the AF1521-ADP-ribose complex structure, the adenine ring is stabilized by two hydrogen bonds. One of the side chain carbonyl oxygens of Asp20 is within hydrogen bonding distance to the N1 and N6 of the adenine rings. In SARS ADRP, the equivalent residue is Asp22 and is likely to play a similar role. The other hydrogen bond is between the N7 and the backbone carbonyl group of Gly42. The binding site of the first ribose ring is a highly hydrated solvent-exposed cleft in which multiple water-mediated interactions are seen between the ribose and residues Asp177 and Ser180 in AF1521. In SARS ADRP, residues Asn156 and Asp157 that lie in a loop between strand 6 and helix H6 are likely to stabilize the ribose by forming similar polar interactions.
The α and β phosphates of the ADP moiety are mainly stabilized by backbone hydrogen bonds with the two Gly-rich motifs in a manner similar to that observed in P loop hydrolases. While the α phosphate is stabilized by hydrogen bonds with the backbone of the three consecutive glycines of motif II, the β phosphate interacts with the amides of Gly130 and Ile131. This loop also helps to stabilize the β phosphate, as the Walker B motif does in P loop hydrolases.
The terminal ribose moiety of the ADP-ribose-1″-phosphate lies on a cleft that is approximately perpendicular to the adenine binding pocket. This is the putative site of catalysis. The side chain amide nitrogen of the conserved asparagine (N80) forms hydrogen bonds with the O3 and O4 of the ribose in the yeast Ymx7-ADP-ribose complex (Figure 4D). This residue from the first motif superimposes almost perfectly with Asn40 in SARS ADRP and Asn34 in AF1521 and is invariant among all macro-like fold members (Figure 1B). Asp90 and His145, two residues that have been implicated in catalysis in yeast Ymx7, lie embedded underneath the loop that connects strand 4 and helix H2 (Figure 4D).
Enzymatic Activity of SARS ADRP
Given the close similarity between the three structures (SARS ADRP, AF1521, and Ymx7) and the similarity at the sequence level between SARS ADRP and yeast Poa1p (an enzyme with demonstrated ADRP activity), it was apparent that their function was likely to be similar as well. We therefore tested the ability of SARS ADRP to dephosphorylate Appr-1″-p in vitro. We employed a generic assay that monitors the liberation of inorganic phosphate in solution during catalysis (Webb, 1992). The results are shown in Figure 5. We observed a sustained release of phosphate after the addition of increasing amounts of the substrate (Appr-1″-p) to the assay containing fixed amounts of the enzyme (Figure 5A). Upon overnight incubation, the amount of phosphate released was proportional to the amount of the substrate added, suggesting that SARS ADRP indeed had the ability to dephosphorylate Appr-1″-p into ADP-ribose and inorganic phosphate (Figure 5B). Further kinetic characterization of the enzyme shows that the dephosphorylation is relatively low, with a KM of 52.7 ± 8.2 μM and a kcat of 5.19 min−1. While the observed catalytic efficiency of this enzyme is not very high, it is comparable to the values reported for Poa1p. In a TLC-based assay with radiolabeled substrates, both Poa1p and Hal2p, a known 3′ phosphatase of 5′,3′-pAp, showed similar low catalytic yields (KM = 2.8 μM; kcat = 1.7 min−1 for Poa1p), but both enzymes were highly specific for the Appr-1″-p substrate (Shull et al., 2005). A few well-known phosphatases whose activity has been monitored by the same assay (Wang et al., 1995) also show similar levels of activity.
Figure 5.
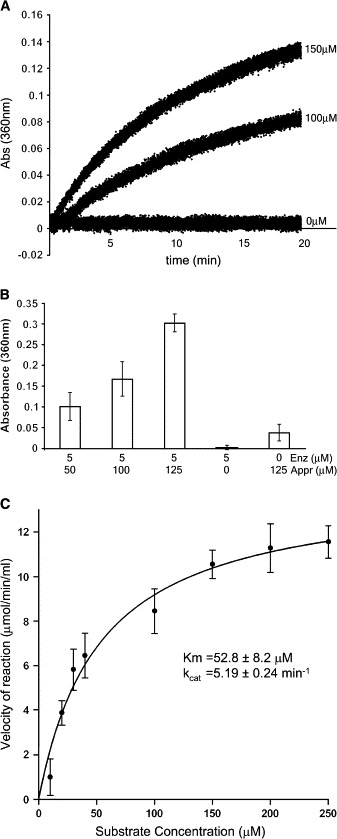
Enzymatic Activity of the SARS ADRP Domain
(A) Continuous release of inorganic phosphate monitored by an increase in absorbance for the initial 20 min of the reaction in two substrate concentrations.
(B) Amount of phosphate released at three different substrate concentrations after overnight incubation with 5 μM enzyme when incubated with three different concentrations of the substrate. The error bars correspond to the standard deviations of three independent measurements at each concentration.
(C) Michaelis-Menton kinetics (rate plot) showing the activity of the enzyme at different concentrations of the substrate (Appr-1″-p) and the obtained values of KM and kcat.
There might be multiple reasons for the low activity levels detected in these assays. It might be intrinsic for this class of enzymes, as seen in the case of yeast Poa1p. Moreover, the released product, ADP-ribose, is a competitive inhibitor of this reaction (Shull et al., 2005). The proposed active site is occluded at the dimer interface in the crystal structure (Figure 2B) and might be hindering access to the substrate in our in vitro assay. The in vivo scenario might be different, where enzyme activity might be regulated by other components of the replicase complex.
Catalytic Mechanism
Yeast Ymx7, one of the structural homologs of SARS ADRP, has been proposed to perform the same reaction. It is a remote homolog of the macrodomain superfamily, albeit with a circular permutation (Figure 3C). It also has a different set of catalytic residues at the active site when compared to classical macrodomains. Based on the structure of the ADP-ribose bound Ymx7, Kumaran et al. (2005) have speculated on a catalytic mechanism that involves three residues: Asp90, His145, and Asn80. While the histidine and asparagine residues are conserved in all three of the structures, Ymx7, AF1521, and SARS nsP3, the equivalent position of Asp90 of Ymx7 is an alanine in the other two (Ala50 in ADRP and Ala44 in AF1521-ADP-ribose complex; Figure 4D). This would imply that while the proposed mechanism might be correct in the case of yeast Ymx7, it cannot be the mode of dephosphorylation for either ADRP or AF1521. Instead, these two enzymes have a histidine (His45 in ADRP and His39 in AF1521) residue that might be in close proximity to the terminal 1″ phosphate of the substrate and might therefore be involved in catalysis. Alternately, it might be speculated that the role of the predominant nucleophile might be played by one of the aspartates or glutamates from the loop 101NAGEDIQ107 in SARS and other coronaviral ADRPs. This loop shows large conformational changes in the apo and ligand bound forms of AF1521 and Ymx7 and is rich in acidic residues (Figure 1B). Further mechanistic studies, cocrystallization experiments, and mutagenesis of these residues that are implicated here are necessary to elucidate the catalytic mechanism of this enzyme. Despite repeated attempts at soaking and cocrystallization, we have not been able to observe density of the bound substrate. A possible reason might be the limited accessibility of the active site, as it is buried in the dimer interface during crystal packing (Figure 2B).
Discussion
The demonstrated function of SARS ADRP as an Appr-1″-p phosphatase has important functional implications in the SARS life cycle. While the manner in which the virus infects the human host is fairly well characterized, many of the postinfection events that occur in the intracellular milieu of the host remain poorly understood. The infection process begins when the spike glycoprotein present on the viral coat recognizes one of two receptors present on the human cell surface: angiotensin-converting enzyme-2 (ACE-2) (Li et al., 2003, Kuhn et al., 2004) or a C-type lectin known as L-SIGN or CD209L (Jeffers et al., 2004). In arteri- and coronaviruses, an early postinfection event is the transcription of a nested set of subgenomic RNA (Lai and Holmes, 2001, Thiel et al., 2003). The resulting mRNAs contain a short 5′-terminal “leader” sequence derived from the 5′ end of the genome. The fusion of the two noncontiguous RNA segments is achieved by a discontinuous step in the synthesis of the minus strand and involves transcription regulatory sequences or TRSs (Thiel et al., 2003, Pasternak et al., 2001). Very few experimental details exist on the processing, maturation, and subsequent roles of these important molecules in the viral life cycle.
This process has parallels in the eukaryotic tRNA splicing pathway that has been well studied in yeast and plants (Culver et al., 1994, Phizicky and Greer, 1993, Peebles et al., 1983). In these organisms, pre-tRNA splicing is initiated by cleavage at the splice site by an endonuclease. The resulting tRNA halves are then ligated to yield mature tRNA that retains the 2′ phosphomonoester group at the splice site (Phizicky and Greer, 1993, McCraith and Phizicky, 1990). Using NAD as an acceptor, a phosphotransferase removes the 2′ phosphate to yield ADP-ribose-1″-2″ cyclic phosphate or Appr>p (Culver et al., 1993). A cyclic phosphodiesterase hydrolyzes Appr>p to yield Appr-1″-p (Culver et al., 1994, Martzen et al., 1999). The terminal step in this pathway is a phosphatase-catalyzed conversion of Appr-1″-p into ADP-ribose and inorganic phosphate, which are channeled through various cellular metabolic pathways.
There is increasing evidence that the NendoU (nsP15) in SARS functions in a manner analogous to the endonuclease of the tRNA splicing pathway. It is a Mn2+-dependent enzyme that also releases products with 2″-3″ cyclic phosphorylated ends (Ivanov et al., 2004, Bhardwaj et al., 2004). While work on this enzyme was in progress, the eukaryotic homolog of NendoU, the XendoU from Xenopus laevis, was functionally characterized, highlighting the existence of an orthologous pathway in higher eukaryotes (Gioia et al., 2005). Details of this process are only beginning to emerge. It is noteworthy that, although orthologs of cyclic phosphodiesterase (CPD), the enzyme that catalyzes the previous step in the tRNA splicing pathway, has been found in group II coronaviruses along with toro- and rotaviruses, it is absent in the SARS virus (Thiel et al., 2003).
SARS NendoU specifically recognizes uridylate bases at GUU sites of RNA (Ivanov et al., 2004). The virus protects its own RNA by methylating its 5′ termini CAP by using an Ado-Met-dependent RNA methyltransferase (von Grotthuss et al., 2003), a process that is imperative during coronaviral replication and an active area of therapeutic intervention (Bach et al., 1995, Vlot et al., 2002). The possibility that SARS ADRP, NendoU, and the methyltransferase might be acting in concert and might therefore be functionally linked has been the subject of previous speculation (Thiel et al., 2003). The precise role of these three enzymes along with 3′-5′ exonuclease and RNA polymerase and their possible interaction with each other as integral components of the replicase complex remain poorly understood. It is becoming increasingly clear that coronaviruses not only differ from other related viruses in having a bigger genome size, but they also have an uncanny similarity with DNA-based life forms in their ability to maintain, synthesize, and regulate the proteomic and genomic components of their life cycle in hitherto unforeseen ways. The work presented here further reinforces this view and hints at the possibility of a tRNA splicing pathway-like process by which the generation of subgenomic RNA and its subsequent translation to yield mature viral proteins is regulated.
Orthologs of SARS ADRP are found embedded in nonstructural proteins of many related ssRNA viruses, especially in alphaviruses of togaviridae (group II of Figure 1B). These include, among others, nsP2 of Sindbis virus (Strauss et al., 1984), nsP3 of O'nyong-nyong virus (Lanciotti et al., 1998), nsP3 of Ross River virus (Shirako and Yamaguchi, 2000), P150 of the lone nsP in Rubella virus (Zheng et al., 2003), nsP3 of Mayaro virus (Anderson et al., 1954), and nsP3 of Semliki Forest virus (Tuittila et al., 2000). Many of these viruses have a greatly reduced genome size (∼10 kb), with only about 4–5 ORFs. On the other hand, the five known human coronaviruses, HCoV-OC43, HCoV-229E, HCoV-NL63, HCoV-HKU1, and SARS-CoV, have genome sizes of 27–32 kb. The occurrence of this phosphatase as part of their replicative machinery underscores the importance of this enzyme in their life cycle and hints at a similar mechanism by which their genomic and subgenomic RNA could be processed inside their respective host cells. However, given the greatly reduced proteome size and reliance of some togaviridae members on host enzymes to meet their replication needs, this process may be somewhat different from that seen in SARS and the other human coronaviruses.
Conclusions
To our knowledge, this study provides the first structural characterization of a highly specific phosphatase from an RNA virus. The experimental demonstration of phosphatase activity on Appr-1″-p, combined with its structural relationship with other known macro-fold members, strongly hints at the possibility that many “hypothetical” proteins of this superfamily might in fact be phosphatases that act on similar substrates. The unique differences between the active site of SARS ADRP and yeast Ymx7, both of which dephosphorylate the same substrate, imply that, while being structurally and functionally homologous, they probably employ different catalytic mechanisms. Further studies are needed to fully explore the functional significance of this enzyme in the larger context of the membrane bound replicase complex and its regulation of translation and replication of viral RNA. If true, the functional link between SARS ADRP and other nsPs highlighted here could provide new avenues for investigation of the replication process of the virus in infected cells, with the hope of developing therapeutic agents aimed at inhibiting viral replication.
Experimental Procedures
Cloning, Expression, and Purification
The sequence corresponding to residues 184–365 (182 aa) of SARS nonstructural protein nsP3 (gi:34555776; NP_828862) of poly-protein pp1a was amplified by polymerase chain reaction (PCR) from genomic cDNA of SARS-Tor2 strain with Taq polymerase and primer pairs encoding the predicted 5′ and 3′ ends (forward: 5′-CCAGTTAATCAGTTTACTGGTTATTTAAAACTTACTGAC-3′; reverse: 5′-CTCCTCTTGTTTAGGTGCTTCC-3′). The PCR product was cloned into plasmid pMH1f, which encodes an expression and purification tag (MGSDKIHHHHHH) at the amino terminus. Protein expression was performed on a sequence-verified clone in native 2xYT or selenomethionine (SeMET)-containing media by using the E. coli methionine auxotrophic strain DL41. Bacteria were lysed by sonication in lysis buffer (50 mM KPO4 [pH 7.8], 300 mM NaCl, 10% glycerol, 5 mM imidazole, two Roche EDTA-free protease inhibitor tablets) with 0.5 mg/ml lysozyme. Cell debris was clarified by ultracentrifugation at 45,000 rpm for 20 min (4°C), and the soluble fraction was applied onto a metal chelate column (Talon resin charged with cobalt; Clontech). The column was washed in 20 mM Tris (pH 7.8), 300 mM NaCl, 10% glycerol, 5 mM imidazole and was eluted with 25 mM Tris (pH 7.8), 300 mM NaCl, 150 mM imidazole. The resultant protein was further purified by using anion exchange chromatography on a Poros HQ column with elution buffer containing 25 mM Tris (pH 8.0) and 1 M NaCl. The pure fractions of the protein were pooled, and the buffer was exchanged into crystallization buffer (10 mM Tris [pH 7.8], 150 mM NaCl) and concentrated by centrifugal ultrafiltration. The final concentration of native and SeMET protein was 1.0 mM and 1.4 mM, respectively. The protein was either frozen in liquid nitrogen for later use or used immediately for crystallization trials.
Crystallization and Data Collection
The protein was crystallized with the nanodroplet vapor diffusion method (Santarsiero et al., 2002) by using standard JCSG crystallization protocols (Lesley et al., 2002). Thick, rectangular, rod-like crystals (∼200 μm × ∼100 μm × ∼75 μm) appeared after 10 days in 0.4 μl drops containing 0.2 μl each of protein and crystallization well solution containing 1.5 M sodium malonate (pH 7.0). A higher concentration (1.8 M) of sodium malonate with 25% glycerol was used as cryoprotectant. A native 1.4 Å dataset (at a wavelength of 0.9794 Å) was collected at beamline 11.1 of the Stanford Synchrotron Radiation Laboratory by using Blu-ICE (McPhillips et al., 2002). Anomalous diffraction data were collected at the Advanced light Source (ALS, Berkeley, CA) on beamline 8.2.1 at a wavelength of 0.97941 Å, corresponding to the peak wavelength of a selenium SAD experiment. Reflections were indexed in a primitive orthorhombic lattice (Space group P212121), integrated, and scaled by using the HKL2000 suite (Otwinowski and Minor, 1997).
Structure Determination and Refinement
The initial phases were obtained by the single wavelength anomalous dispersion (SAD) phasing method with data collected to 2.2 Å at the selenium peak wavelength by using the program SOLVE (Terwilliger and Berendzen, 1999). All 12 selenium sites were located, and the resulting phases had a figure of merit of 0.54 after density modification procedures by using RESOLVE (Terwilliger, 2003). The resultant phases from SAD were merged, improved, and extended for a native data set to 1.4 Å by using the programs CAD and DM as implemented in the CCP4 package (CCP4, 1994) assuming four monomers in the ASU with Matthews coefficient 2.6 and a solvent content of 51% (Cowtan, 1994). Automated model building with Arp/wARP (ver 6.0; Lamzin and Wilson, 1997) traced ∼80% of the backbone and docked 65% of the sequence, including the side chains. The rest of the sequence was manually built into the density with O (Jones et al., 1991) and was refined against the high-resolution native data to 1.4 Å with iterative rounds of model building and refinement by using Refmac5 (Murshudov et al., 1997) of CCP4. Although RESOLVE did identify the presence of NCS among the monomers, it was not used at any stage of refinement. A summary of data collection and refinement statistics is shown in Table 1. The stereochemical quality of the final refined model was checked with Procheck (Laskowski et al., 1993), and the dimer interface was calculated by using the protein-protein interaction server. The ribbon diagrams were made with Pymol (DeLano, 2002).
Enzyme Assays
The substrate Appr-1″-p was a kind gift from Prof. Phyzicky (Rochester Univ, USA) and was enzymatically prepared by reacting the precursor Appr>p with cyclic phosphodiesterase (CPD) by using procedures described in Shull et al. (2005). Phosphate release was monitored by the Enzchek assay (Molecular Probes Inc, Eugene OR, USA) by following the manufacturer's instructions. The assay uses the method of Webb (1992), which monitors the release of inorganic phosphate by coupling the phosphatase reaction with the enzymatic conversion of 2-amino-6-mercapto-7-methyl-purine riboside (MESG) to 2 amino-6-mercapto-7-methyl purine and ribose-1-phosphate by purine nucleoside phosphorylase. The substrate MESG has an absorbance maximum of 330 nm, whereas that of the product is 360 nm. Each 1 ml reaction mixture contains 50 mM Tris (pH 7.5), 1 mM MgCl2, 0.1 mM sodium azide, 200 μM MESG, 1 U purine nucleoside phosphorylase, and 2.7 μM enzyme. Increasing amounts of the substrate were added to the reaction mixture, and the ADRP reaction was monitored by changes in absorbance at 360 nm in a UV spectrophotometer. To check for phosphate contamination, appropriate control reactions were performed in the presence of enzyme, but with no substrate and vice versa. No measurable phosphate contamination was detected either from the enzyme preparation, substrate degradation, or from the buffers. The assay components were checked with known amounts of phosphate standard supplied by the manufacturer. A molar extinction coefficient of 11,000 M−1 cm−1 of the product of the PNP reaction at 360 nm was used to quantitate the amount of released inorganic phosphate (Etzkorn et al., 1994).
Acknowledgments
The authors gratefully acknowledge Alexei Brooun and Amy Houle for helping with cloning SARS targets. Bioinformatics support for this project was provided by Enrique Abola, Anand Kolatkar, and Sophie Coon of The Scripps Research Institute and Weizhong Li and Adam Godzik of the Burnham Institute. The authors thank Neil Shull and Prof. Eric M. Phizicky from Rochester University for providing the substrate Appr-1″-p. We also acknowledge the helpful support of the beamline staff at the Advanced Photon Source (GM/CA-CAT), Advanced Light Source (BL-8.2.1), and Stanford Synchrotron Radiation Laboratory (SSRL) (BL-11.1) synchrotron facilities for help in data collection. SSRL BL11-1 is supported by the National Institutes of Health (NIH) National Center for Research Resources, NIH National Institutes of General Medical Sciences, Department of Energy, Office of Biological and Environmental Research, Stanford University, and The Scripps Research Institute (TSRI). The General Medicine and Cancer Institutes Collaborative Access Team is supported by the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). This study was supported by National Institutes of Allergy and Infectious Disease/NIH Contract # HHSN 266200400058C “Functional and Structural Proteomics of the SARS-CoV” to P. K. TSRI manuscript 17502-CB.
Published: November 8, 2005
Accession Numbers
The final refined coordinates and the structure factors have been deposited in the Protein Data Bank under ID code 2ACF.
References
- Allen M.D., Buckle A.M., Cordell S.C., Lowe J., Bycroft M. The crystal structure of AF1521, a protein from Archaeoglobus fulgidus with homology to the non-histone domain of macroH2A. J. Mol. Biol. 2003;330:503–511. doi: 10.1016/s0022-2836(03)00473-x. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson C.R., Downs W.G., Wattley G.H., Ahin N.W., Reese A.A. Mayaro virus: a new human disease agent. II. Isolation from blood of patients in Trinidad. Am. J. Trop. Med. Hyg. 1954;6:1012–1016. doi: 10.4269/ajtmh.1957.6.1012. [DOI] [PubMed] [Google Scholar]
- Bach C., Cramer A., Scholtissek C. Effect of methyltransferase inhibitors on the regulation of baculovirus protein synthesis. J. Gen, Virol. 1995;76:1025–1032. doi: 10.1099/0022-1317-76-4-1025. [DOI] [PubMed] [Google Scholar]
- Baranov P.V., Henderson C.M., Anderson C.B., Gesteland R.F., Atkins J.F., Howard M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology. 2005;332:498–510. doi: 10.1016/j.virol.2004.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj K., Guarino L., Kao C.C. The severe acute respiratory syndrome coronavirus Nsp15 protein is an endoribonuclease that prefers manganese as a cofactor. J. Virol. 2004;78:12218–12224. doi: 10.1128/JVI.78.22.12218-12224.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley S.K., David P.R., Taylor A., Lipscomb W.N. Molecular structure of leucine aminopeptidase at 2.7-Å resolution. Proc. Natl. Acad. Sci. USA. 1990;87:6878–6882. doi: 10.1073/pnas.87.17.6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project, Number 4) The CCP4 Suite: programs for Protein Crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Charlier D., Hassanzadeh G., Kholti A., Gigot D., Pierard A., Glansdorff N. CarP, involved in pyrimidine regulation of the Escherichia coli carbamoyl-phosphate synthetase operon encodes a sequence-specific DNA-binding protein identical to XerB and PepA, also required for resolution of ColEI multimers. J. Mol. Biol. 1995;250:392–406. doi: 10.1006/jmbi.1995.0385. [DOI] [PubMed] [Google Scholar]
- Cowtan, K. (1994). DM: an automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography 31, 34–38.
- Culver G.M., McCraith S.M., Zillmann M., Kierzek R., Michaud N., LaReau R.D., Turner D.H., Phizicky E.M. An NAD derivative produced during transfer RNA splicing: ADP-ribose 1″-2″ cyclic phosphate. Science. 1993;261:206–208. doi: 10.1126/science.8392224. [DOI] [PubMed] [Google Scholar]
- Culver G.M., Consaul S.A., Tycowski K.T., Filipowicz W., Phizicky E.M. tRNA splicing in yeast and wheat germ. A cyclic phosphodiesterase implicated in the metabolism of ADP-ribose 1″-2″ cyclic phosphate. J. Biol. Chem. 1994;269:24928–24934. [PubMed] [Google Scholar]
- D'Amours D., Desnoyers S., D'Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- DeLano, W.L. (2002). The PyMOL Molecular Graphics System (http://pymol.sourceforge.net).
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Eddy S.R. Hidden Markov models. Curr. Opin. Struct. Biol. 1996;6:361–365. doi: 10.1016/s0959-440x(96)80056-x. [DOI] [PubMed] [Google Scholar]
- Etzkorn F.A., Chang Z.Y., Stoltz L.A., Walsh C.T. Cyclophilin residues that affect the noncompetitive inhibition of the protein serine phosphatase activity of Calcineurin by the cyclophilin-cyclophorin A complex. Biochemistry. 1994;33:2380–2388. doi: 10.1021/bi00175a005. [DOI] [PubMed] [Google Scholar]
- Gioia U., Laneve P., Dlakic M., Arceci M., Bozzoni I., Caffarelli E. Functional characterisation of XendoU, the endoribonuclease involved in small nucleolar RNA biosynthesis. J. Biol. Chem. 2005;280:18996–19002. doi: 10.1074/jbc.M501160200. [DOI] [PubMed] [Google Scholar]
- Groneberg D.A., Zhang L., Welte T., Zabel P., Chung K.F. Severe acute respiratory syndrome: global initiatives for disease diagnosis. QJM. 2003;96:845–852. doi: 10.1093/qjmed/hcg146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Sander C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Ivanov K.A., Hertzig T., Rozanov M., Bayer S., Thiel V., Gorbalenya A.E., Ziebuhr J. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl. Acad. Sci. USA. 2004;101:12694–12699. doi: 10.1073/pnas.0403127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers S.A., Tusell S.M., Gillim-Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Jr., Thackray L.B., Young M.D., Mason R.J., et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou J.-Y., Cowan S.W., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kuhn J.H., Li W., Choe H., Farzan M. Angiotensin-converting enzyme 2: a functional receptor for SARS coronavirus. Cell. Mol. Life Sci. 2004;61:2738–2743. doi: 10.1007/s00018-004-4242-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran D., Eswaramoorthy S., Studier F.W., Swaminathan S. Structure and mechanism of ADP-ribose-1″-monophosphatase (Appr-1″-pase), a ubiquitous cellular processing enzyme. Protein Sci. 2005;14:719–726. doi: 10.1110/ps.041132005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C., Holmes K.V. In: Fields Virology. Knipe D.M., Howley P.M., editors. Lippincott; Philadelphia, PA: 2001. Coronaviruses; pp. 1163–1185. [Google Scholar]
- Lamzin V.S., Wilson K.S. In: Methods in Enzymology. Carter C., Sweet B., editors. Academic Press; Orlando, FL: 1997. Automated refinement for protein crystallography; pp. 269–305. [DOI] [PubMed] [Google Scholar]
- Lanciotti R.S., Ludwig M.L., Rwaguma E.B., Lutwama J.J., Kram T.M., Karabatsos N., Cropp B.C., Miller B.R. Emergence of epidemic O'nyong-nyong fever in Uganda after a 35-year absence: genetic characterization of the virus. Virology. 1998;252:258–268. doi: 10.1006/viro.1998.9437. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- Lesley S.A., Kuhn P., Godzik A., Deacon A.M., Mathews I., Kreusch A., Spraggon G., Klock H.E., McMullan D., Shin T., et al. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc. Natl. Acad. Sci. USA. 2002;99:11664–11669. doi: 10.1073/pnas.142413399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Copley R.R., Schmidt S., Ciccarelli F.D., Doerks T., Schultz J., Ponting C.P., Bork P. SMART 4.0: towards genomic data integration. Nucleic Acids Res. 2004;32:D142–D144. doi: 10.1093/nar/gkh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martzen M.R., McCraith S.M., Spinelli S.L., Torres F.M., Fields S., Grayhack E.J., Phizicky E.M. A biochemical genomics approach for identifying genes by the activity of their products. Science. 1999;286:1153–1155. doi: 10.1126/science.286.5442.1153. [DOI] [PubMed] [Google Scholar]
- McCraith S.M., Phizicky E.M. A highly specific phosphatase from Saccharomyces cerevisiae implicated in tRNA splicing. Mol. Cell. Biol. 1990;10:1049–1055. doi: 10.1128/mcb.10.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch R., Burke M.E., Sherratt D.J. Peptidase activity of Escherichia coli aminopeptidase A is not required for its role in Xer site-specific recombination. Mol. Microbiol. 1994;12:241–251. doi: 10.1111/j.1365-2958.1994.tb01013.x. [DOI] [PubMed] [Google Scholar]
- McPhillips T.M., McPhillips S.E., Chiu H.J., Cohen A.E., Deacon A.M., Ellis P.J., Garman E., Gonzalez A., Sauter N.K., Phizackerley R.P., et al. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at Macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Barrell D., Bateman A., Binns D., Biswas M., Bradley P., Bork P., Bucher P., Copley R.R., Courcelle E., Das U., et al. The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res. 2003;31:315–318. doi: 10.1093/nar/gkg046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Murzin A.G., Brenner S.E., Hubbard T., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pasternak A.O., van den Born E., Spaan W.J., Snijder E.J. Sequence requirements for RNA strand transfer during nidovirus discontinuous subgenomic RNA synthesis. EMBO J. 2001;20:7220–7228. doi: 10.1093/emboj/20.24.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peebles C.L., Gegenheimer P., Abelson J. Precise excision of intervening sequences from precursor tRNAs by a membrane-associated yeast endonuclease. Cell. 1983;32:525–536. doi: 10.1016/0092-8674(83)90472-5. [DOI] [PubMed] [Google Scholar]
- Pehrson J.R., Fried V.A. MacroH2A, a core histone containing a large nonhistone region. Science. 1992;257:1398–1400. doi: 10.1126/science.1529340. [DOI] [PubMed] [Google Scholar]
- Pehrson J.R., Fuji R.N. Evolutionary conservation of histone macroH2A subtypes and domains. Nucleic Acids Res. 1998;26:2837–2842. doi: 10.1093/nar/26.12.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky E.M., Greer C.L. Pre-tRNA splicing: variation on a theme or exception to the rule? Trends Biochem. Sci. 1993;18:31–34. doi: 10.1016/0968-0004(93)90085-2. [DOI] [PubMed] [Google Scholar]
- Rychlewski L., Jaroszewski L., Li W., Godzik A. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 2000;9:232–241. doi: 10.1110/ps.9.2.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarsiero B.D., Yegian D.T., Lee C.C., Spraggon G., Gu J., Scheibe D., Uber D.C., Cornell E.W., Nordmeyer R.A., Kolbe W.F., et al. An approach to rapid protein crystallization using nanodroplets. J. Appl. Crystallogr. 2002;35:278–281. [Google Scholar]
- Shirako Y., Yamaguchi Y. Genome structure of Sagiyama virus and its relatedness to other alphaviruses. J. Gen. Virol. 2000;81:1353–1360. doi: 10.1099/0022-1317-81-5-1353. [DOI] [PubMed] [Google Scholar]
- Shull N.P., Spinelli S.L., Phizicky E.M. A highly specific phosphatase that acts on ADP-ribose 1″-phosphate, a metabolite of tRNA splicing in Saccharomyces cerevisiae. Nucleic Acids Res. 2005;33:650–660. doi: 10.1093/nar/gki211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L.M., Guan Y., Rozanov M., Spaan W.J.M., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sträter N., Lipscomb W.N. Two-metal ion mechanism of bovine lens leucine aminopeptidase: active site solvent structure and binding mode of L-leucinal, a gem-diolate transition state analogue, by X-ray crystallography. Biochemistry. 1995;34:14792–14800. doi: 10.1021/bi00045a021. [DOI] [PubMed] [Google Scholar]
- Sträter N., Sherratt D.J., Colloms S.D. X-ray structure of aminopeptidase A from Escherichia coli and a model for the nucleoprotein complex in Xer site-specific recombination. EMBO J. 1999;18:4513–4522. doi: 10.1093/emboj/18.16.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss E.G., Rice C.M., Strauss J.H. Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology. 1984;133:92–110. doi: 10.1016/0042-6822(84)90428-8. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. Statistical density modification using local pattern matching. Acta Crystallogr. 2003;D59:1688–1701. doi: 10.1107/S0907444903015142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C., Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weißbrich B., Snijder E.J., Rabenau H., Doerr H.W., et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2313. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Tuittila M.T., Santagati M.G., Roytta M., Maeaettae J.A., Hinkkanen A.E. Replicase complex genes of Semliki Forest virus confer lethal neurovirulence. J. Virol. 2000;74:4579–4589. doi: 10.1128/jvi.74.10.4579-4589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlot A.C., Menard A., Bol J.F. Role of the alfalfa mosaic virus methyltransferase-like domain in negative-strand RNA synthesis. J. Virol. 2002;76:11321–11328. doi: 10.1128/JVI.76.22.11321-11328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Grotthuss M., Wyrwicz L.S., Rychlewski L. mRNA cap-1 methyltransferase in the SARS genome. Cell. 2003;113:701–702. doi: 10.1016/S0092-8674(03)00424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.E., Saraste M., Runswick M.J., Gay N.J. Distantly related sequences in the α-subunits and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.-X., Cheng Q., Killilea S.D. A continuous spectrophotometric assay for phosphorylase kinase. Anal. Biochem. 1995;230:55–61. doi: 10.1006/abio.1995.1437. [DOI] [PubMed] [Google Scholar]
- Webb M.R. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc. Natl. Acad. Sci. USA. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D.-P., Zhou Y.M., Zhao K., Han Y.-R., Frey T.K. Characterization of genotype II Rubella virus strains. Arch. Virol. 2003;148:1835–1850. doi: 10.1007/s00705-003-0132-7. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J., Thiel V., Gorbalenya A.E. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J. Biol. Chem. 2001;276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]