

Abstract
Affinity technologies have been applied at several stages of the drug discovery process, ranging from target identification and purification to the identification of preclinical candidates. The detection of ligand–macromolecule interactions in lead discovery is the best studied and most powerful of these techniques. Although affinity methods have been in widespread use for about a decade, only recently have many reports emerged on their utility. Primary affinity screens of large libraries of small molecules or fragments have begun to produce results for challenging targets. Furthermore, in secondary assays affinity methods are opening new avenues to tackle important medicinal chemistry tasks.
Introduction
Lead generation is a critical first step in the drug discovery process. Over the past decade, high-throughput screening (HTS) of corporate compound decks has emerged as the primary paradigm for hit or lead discovery. Typically, targets are interrogated with 1–3 million discrete compounds in parallel. The HTS era has thus far delivered mixed results. Hit rates for targets tend to be extremely low or very high. Two key factors are likely to be responsible for this. First, most compound decks in the industry are skewed in compound distribution favoring specific, well-studied protein subfamilies. Second, and most importantly for novel targets, sampling of drug-like chemistry space by a few million compounds is inadequate to consistently deliver several diverse lead classes [1]. Challenges facing HTS technologies include high false-positive and false-negative rates, the need for reporter assays, and the limitation in throughput imposed by testing compounds individually [2]. To address these issues, affinity or bio-affinity screening approaches have emerged as orthogonal methods for early lead discovery. For affinity-based techniques, the readout is typically a qualitative or quantitative signal based on the physical interaction between macromolecule (RNA, DNA or protein) and the small-molecule partner. This approach has several advantages over biochemical assays for both drug-like and fragment-like methods (see Glossary). First, it has the capability to sample mixtures (including natural products) rather than discrete entities, thus enabling the exploration of larger chemical spaces without a concomitant increase in the number of samples. Second, it has the ability to investigate multiple forms of macromolecules, such as various ternary complexes, activated or inactivated forms and forms with or without cofactor. Third, it is possible to carry out the assays at small-molecule concentrations much higher or lower than that of the corresponding biochemical assays. Last, this approach eliminates many assay artifacts that arise from nonspecific aggregation, fluorescence absorption or quenching. On the downside, affinity-based technologies do not deliver a functional readout. Thus, affinity hits require biochemical, cellular or in vivo validation. Nevertheless, the advantages of affinity techniques have extended their use into follow-up or secondary screens, especially in the field of ATP-dependent enzymes.
In this article, we review a subset of affinity techniques that has been shown to be valuable for lead discovery (Table 1 ). We provide several examples of demonstrated success, considering achievements from the perspectives of the technology and drug discovery.
Table 1.
Affinity-based technologies in primary screening applications.
| Detection | Kineticsa | Binding assay | Main applications | Pros | Cons |
|---|---|---|---|---|---|
| MS | Koff-dependent | Homogeneous | Ultra-HTS of large libraries | Improved sampling of drug-like chemistry space (higher throughput and ability to analyze mixtures), preference for slower Koff | Koff-dependent signal |
| Kd-dependent | Homogeneous reversible | Ultra-HTS of large libraries | Improved sampling of drug-like chemistry space (higher throughput and ability to analyze mixtures), suitable for weak compounds | Harder to automate and miniaturize | |
| Homogeneous, covalent | Fragment-based lead discovery | Smaller space to sample, lead-likeness | Mutated proteins might be required | ||
| Heterogeneous: immobilized protein | Ultra-HTS of large libraries, mixture capability | Improved sampling of drug-like chemistry space, enabling natural product screens | Immobilization of target | ||
| NMR | Kd-dependent | Homogeneous | Fragment-based lead discovery | Smaller space to sample, lead-likeness | Target size limits, soluble proteins only, lower throughput, labeled protein may be required |
| Heterogeneous: immobilized protein | Fragment-based lead discovery | Smaller space to sample, enables membrane proteins to be used, lead-likeness | Target size limits, labeled protein required | ||
| X-ray | Fragment-based lead discovery | Smaller space to sample, lead-likeness | Long validation for new proteins, difficulties associated with crystallization | ||
| PCR | Kd-dependent | Heterogeneous: immobilized protein | Ultra-HTS of vast DNA-templated libraries | Improved sampling of drug-like chemistry space (higher throughput and ability to analyze mixtures) | Limited chemistry scope, immobilization of target |
| SPR | Kd-dependent | Heterogeneous: immobilized compounds | Fragment-based lead discovery and traditional libraries | Applicable for both fragment-like and drug-like compounds | Immobilization of compounds |
Koff, off-rate (dissociation rate constant); Kd, dissociation constant.
Affinity selection in screens of drug-like libraries
Affinity selection techniques can be classified as homogeneous or heterogeneous. A heterogeneous screening environment is created by immobilization of either the macromolecule of interest or of the small molecule(s) on a reactive surface followed by a static or flow-based analysis of binding. By contrast, a homogeneous environment leaves both macromolecular and small-molecule species to interact in their native states. Following the binding event, a qualitative or quantitative assessment of the interaction can be made using one of many available detectors that have been used for affinity screening, such as nuclear magnetic resonance (NMR), mass spectroscopy (MS), X-ray crystallography or surface plasmon resonance (SPR). A subset of these techniques employs a filtration step before detection. Specific details of some affinity screening techniques have been reviewed [3••]. Below we consider some recently developed platforms for the affinity selection of drug-like compounds.
Glossary.
Drug-like molecules: Typical molecules are in the molecular weight range 350–550 Da and either pass or do not violate more than one component of the ‘rule-of-five’ (Lipinski rules for oral bioavailability) [48].
Lead-like molecules: Typical molecules are in the molecular weight range 300–450 Da to enable lead growth during medicinal chemistry optimization towards clinical candidates that still pass the rule-of-five.
Fragment-like molecules: Typical molecules are in the molecular weight range 120–300 Da and have higher solubility than drug-like molecules to enable screens at high micromolar or millimolar small-molecule concentrations.
Fragment-based drug or lead discovery: A lead generation technique that calls for primary screens of weak-binding (IC50 5 mM–50 μM) fragment-like molecules followed by optimization towards leads, rather than for primary screens of more potent (IC50 25 μM–50 nM) drug-like molecules via HTS. The advantage of the former is the better sampling of chemistry space, because the number of compounds relevant for drug discovery grows exponentially with molecular weight.
Fragment evolution: A fragment optimization technique that typically involves the systematic exploration of structure-activity relationships along various substitution vectors of the fragment hits.
Site-directed fragment screening: A fragment optimization technique that typically involves screening fragment libraries in the presence of another fragment added at saturation concentrations. The binding mode of the latter fragment is typically known from previous studies. Any new hits from the second screen would target new binding sites and such hits could later be the subject of ‘fragment linking’ with the previously identified fragment.
The automated ligand identification system (ALIS) screening platform (NeoGenesis; http://www.neogenesis.com/) was one of the earliest affinity selection technologies capable of screening very large numbers of compounds. The key step to ALIS is the separation of the desired protein–ligand complex from non-binding components and dissociation of the complex by reverse phase chromatography with mass detection. As part of a lead discovery effort against the anti-infective target Escherichia coli dihydrofolate reductase, a total of 3 750 000 compounds (in mixtures of 2000), were screened to identify compound 1 (Figure 1 ) [4]. The advantages of improved sampling of chemistry space were demonstrated by the discovery of a single compound (2) from a multimillion member library for β-secretase (BACE1), an enzyme that is crucial in Alzheimer's disease [5•]. The compound was selective, possessed a unique binding mode and could be optimized to form a potent inhibitor (3) of the enzyme [6]. The generality of the ALIS platform has been demonstrated against a diverse set of targets [7].
Figure 1.
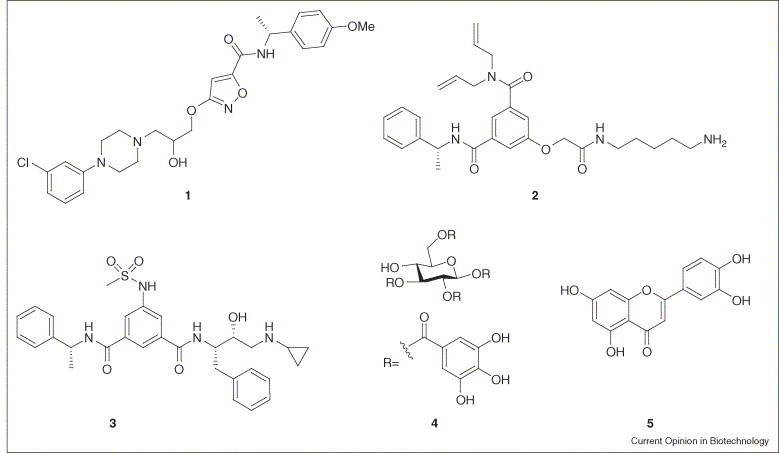
Compounds derived from affinity screens of drug-like molecules. See text for details.
A related technique, termed Speedscreen (Novartis; http://www.novartis.com/), has also been disclosed. In practice, this approach includes the same sequence of events as ALIS, but each step is carried out completely decoupled from the last. Unlike ALIS, Speedscreen takes advantage of the parallel capabilities of using a 96-well format [8]. A typical screening campaign is comprised of approximately 600 000 compounds in mixtures of 400 tested against soluble and globular proteins [9].
Researchers at Abbott (http://www.abbott.com/) have used ultrafiltration to separate binders for the oncology target Bcl-XL from non-binders, in mixtures of 2400 compounds, as part of a study comparing results from a fluorescence polarization assay and an affinity selection/mass spectrometry (ASMS) platform. From the 263 382 compounds that were screened in duplicate, 29 binders with affinities below 100 μM were identified by ASMS [10]. The authors suggested that it is valuable to apply different screening methods for lowering the false-negative rate. Similar conclusions can be made from the screening statistics for far upstream element binding protein (FBP) [11].
Capillary electrophoresis (CE) has also been shown to be a reliable tool for studying molecular interactions [12, 13]. CE has been used to interrogate crude natural product broths for potential leads [14]. A binding event can be discerned by monitoring the migration times of the protein–ligand complex versus that of the protein alone as a reference point. Upon complex formation conformational changes of the protein expose different residues to the surface, an event that can change the mobility of the complex in relation to its charged environment.
Frontal affinity chromatography coupled to a mass spectrometer (FAC-MS) is based on the immobilization of a target onto a solid support [15, 16]. Small molecules are infused continuously over the immobilized target and are monitored by a mass spectrometer. Depending on their affinity strength, the extracted ion currents for the binders are observed at later elution times when compared to a non-interacting control compound. Furthermore, the shape of the individual extracted ion currents can provide crude approximations of relative affinities under competitive conditions. In a study to discover molecules that bind to the surface spike protein of severe acute respiratory syndrome coronavirus (SARS-CoV), two small molecules have been reported. Compounds 4 and 5 were identified from the extracts of more than 121 Chinese herbs using the FAC-MS approach [17].
By using the resolving power of Fourier transform ion cyclotron resonance mass spectrometry, researchers have been able to study non-covalent interactions of RNA–ligand complexes without the need for a separation step before detection [18, 19]. The platform, termed multitarget affinity/specificity screening, allows for the multiplexing of RNA constructs and small molecules; however, the generality of this technique for other targets remains to be demonstrated.
As part of the quest to screen increasing numbers of small molecules, DNA-tagged libraries have been devised and affinity screened against immobilized targets. Binders are then amplified by PCR to reveal the chemical identity. This technology is rather new and its limitations (synthetic, false-positive or false-negative rates) are not yet clear (more details can be found at http://www.nuevolution.com or http://www.praecis.com).
Affinity detection of fragments for lead discovery
As the cost, efficiency, scalability and lead delivery-rate issues with HTS became apparent over the past decade, new terms such as lead-likeness, fragment-likeness and fragment-based drug discovery have surfaced (see Glossary). Significant resources and research have been dedicated to identifying techniques that can measure the binding of very small (molecular weight ∼120–300 Da) molecules (fragments) to macromolecules. The inherent difficulties of using biochemical tools to detect weak ligands, often at millimolar concentrations, necessitated the search for affinity-type readouts. Over the years, NMR (Abbott, Vertex; http://www.vpharm.com/) and X-ray crystallography (Abbott, Astex http://www.astex-therapeutics.com/ and Plexxikon http://www.plexxikon.com/) have emerged as the methods of choice to conduct fragment-based lead discovery. An intriguing alternative for the covalent trapping of fragments in a K d-dependent manner has also been developed by Sunesis (thiol-reactive probes; http://www.sunesis.com/).
The fragment-based approach is ideal for finding new ‘warheads’, small functional groups or ensembles of functional groups that bind to crucial residues conserved within protein families. These advantages have been demonstrated with protein tyrosine phosphatase 1B (PTP1B), a validated but historically challenging target implicated in type II diabetes. A protein-based NMR fragment screen (which detects changes in the NMR spectra of a protein on ligand binding) has been used to identify several phosphate mimics: oxalylarylaminobenzoic acids (6; Figure 2 ) [20], which proved not to be cell permeable, and isoxazolecarboxylic acids (7) [21•]. The latter was linked with a noncompetitive fragment identified by another NMR screen, which resulted in the development of a monocharged PTP1B inhibitor (8) with cellular activity. In a search for aminoglycoside mimetics against the E. coli A-site RNA, an NMR-based screen of a 10 000 member library was used to identify several different chemical series, two of which (2-aminoquinolines and aminopyridines) could rapidly be advanced to more potent derivatives in the low micromolar range [22]. Structure activity relationships determined by NMR (SAR by NMR) have been instrumental in the development of Abbott's matrix metalloproteinase program, identifying the key biphenyl group [23]. Fragment linking with hydroxamic acids followed by medicinal chemistry optimization ultimately resulted in the development candidate ABT-518 (9) for treating cancer. Site-directed fragment screening and fragment evolution (see Glossary) was also used to discover and optimize leads for the anti-apoptotic Bcl-2 family, ultimately leading to ABT-737 (10). Compound 10 was well tolerated and was shown to cause tumor regression in multiple cancer cell lines either as a single agent or in combination with chemotherapeutics [24]. Protein-based NMR fragment screens have also recently been extended to insoluble proteins [25].
Figure 2.
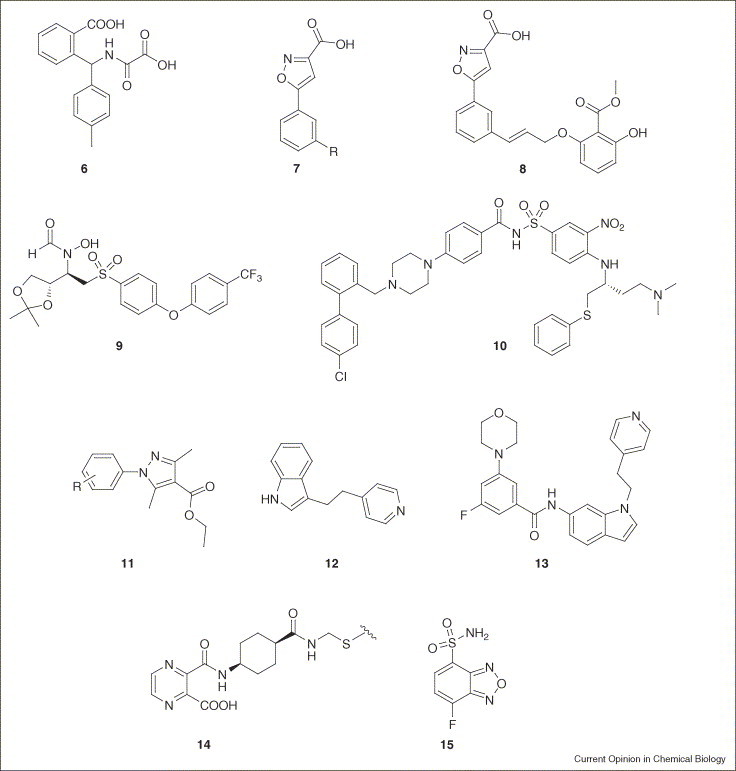
Compounds derived from fragment-based techniques. See text for details.
Unlike protein-based NMR, the SHAPES technique relies on the detection of changes in NMR spectra of the small molecules themselves. Advantages over the protein-based method are that no limits are imposed on the size of the protein, there is no requirement for labeled protein, and lower reagent consumption. On the downside, no structural information on the protein's binding site or on the relative binding mode of different fragments can be discerned. The SHAPES method has been applied pre-HTS, owing to minimal development time, or post-HTS for failed targets, as exemplified by Jnk3 MAP kinase and adipocyte lipid-binding protein [26]. Close integration with X-ray crystallography and molecular modeling was reported to be essential in optimization of the initial fragment hits.
Over the past decade, data analysis, crystal soaking technology and throughput have matured to the level where X-ray crystallography can be applied to the parallel detection of weakly binding fragments. Although drawbacks such as false-negatives [27], long development times for novel unknown proteins, and protein consumption still remain, structure-guided fragment evolution makes this technology an attractive option. Fragments can simply be screened in small mixtures (<10 members) or can be pre-filtered by biochemical assays to limit the set of scaffolds for crystallization. A proof-of-principle for the latter approach has recently been disclosed for phosphodiesterases. In that study, 1-phenyl-4-pyrazolecarboxylic esters (11) were developed using a 20 000 member scaffold library and structure-based optimization [28]. A more comprehensive study of five targets (p38, CDK2, thrombin, PTP1B and RNase A) demonstrated that weak fragments can indeed be identified using high-throughput crystallography [29••]. Chemical series evolution from fragments (12) to leads (13) was illustrated by the p38 program [30]. Although these studies clearly show the potential of the underlying technology in lead generation, the strong reliance on structure-based design for optimization makes the discovery of truly novel molecular frameworks unlikely.
Using thiol-reactive probes, researchers at Sunesis were able to screen for new chemical motifs that were active against PTP1B with the aid of ‘breakaway tethering’ [31•]. A cysteine residue was introduced outside of the enzyme active site and covalently modified with a flexible linker holding a thiol end group. The protein was then interrogated with 15 000 fragments and the screen unveiled the 1,2-pyrazinedicarboxylic acid monoamide moiety (14), which was an equipotent and competitive inhibitor of phosphotyrosine in biochemical assays. A different study in which electrophiles were screened against PTP1B found that 15 selectively reacts with a cysteine 8 Å away from the catalytic site [32•]. This covalent modification reduces the enzymatic activity of PTP1B via an allosteric perturbation of the neighboring phosphotyrosine-binding site.
Competitive affinity screens for cytokine hormone interleukin-2 (IL-2) using fragments in the presence of a previously known inhibitor revealed a series of compounds that show cooperative binding with a previously known molecule [33]. Fragment decomposition of the same known inhibitor and additional competitive fragment screens were invaluable for the development of nanomolar inhibitors with cellular activity [34]. Fragment libraries with exchangeable thiols unveiled an allosteric binding site for caspases [35] as well as agonist and antagonist fragments for the G-protein-coupled receptor C5a [36]. These results elegantly demonstrate one of the key advantages of affinity-related techniques: the ability to study or screen different enzyme forms and to find compounds with affinity for allosteric or adjacent binding sites.
Fragment-based lead discovery has been shown to be successful over the years, delivering ‘efficient’ leads (relatively high free-energy of binding per heavy atom [37••]). As the approach is relatively new, it remains to be seen whether the smaller and more hydrophilic lead entry points for medicinal chemistry actually yield smaller drug candidates that fail less often in the clinic [38].
Surface plasmon resonance in lead generation
The focus of the affinity techniques discussed so far has been either to increase the throughput for traditional libraries or to enable the detection of weakly binding fragments with low throughput. Both help sampling of discovery space but represent opposite extremes. The immobilization of small organic molecules on microarrays enables the use of SPR to detect weak interactions. The method is practically an extension of the established BIAcoreTM technology to fragments, and may hold promise for HTS of lead-like small molecules. A model study using factor VIIa and a focused set of immobilized compounds confirmed binding of the positive control (p-aminobenzamidine, ∼70 μM) and identified several new fragment ligands [39]. Follow-up biochemical activities or the tendency to form crystals, however, had no correlation with the observed SPR signal. This developing approach may mature into a semi-quantitative screening technique or a qualitative filter.
Secondary assay applications
High-throughput affinity methods other than NMR or X-ray crystallography have found their way into many applications. These include mechanistic and competition studies, the determination of binding kinetics, and compound profiling or selectivity screens.
The use of homogenous affinity selection has recently been shown to distinguish direct or allosteric competitive behavior. It could also establish compound ranking in mixture format through titration experiments of unknown compounds with a ligand [40]. The same method can also be used for ranking new derivatives in optimization studies. SPR analysis of binding kinetics with BIAcore™ has been the standard for the determination of association and dissociation rate constants and K ds [41, 42]. The major drawback — the requirement for immobilizing one of the binding partners, typically the protein — is compensated for by the lack of need for radiolabeled materials.
Selectivity is a particularly crucial factor in the development of drug candidates for ATP-dependent enzymes. There has been a significant effort to express and develop assays for a growing number of kinases and their mutants for counter-screening studies, owing to a few early reports on finding off-target liabilities (potential adverse effects) in distant sections of the kinase dendogram. Immobilized inhibitors can be used to map ATP-binding site interactions with many targets. Conceivably, each inhibitor might have to be linked via multiple orientations to avoid false-negatives arising from linker interference. Nevertheless, several surprisingly tight off-target interactions were identified for the aryl-imidazole and pyrido[2,3-d]pyrimidine kinase inhibitor classes [43, 44, 45]. A semi-homogenous variation of this technique has also been developed, which requires the immobilization of only a small number of compounds to study all ATP-competitive small molecules with any enzymes [46••]. In this approach, highly promiscuous (inhibitory constant (IC50) < 1 μM) ‘bait’ molecules are immobilized on the solid support via a biotinylated linker. The T7 phage tagged protein is introduced along with the ‘study compound’ free in solution. If the free compound does not bind to the protein, elution of the macromolecule is slower because it can extensively interact with the bait compounds on the solid support. The amount of protein bound to the support is quantified by either phage plaque assays or quantitative PCR for high sensitivity and can be plotted as a function of the test compound concentration. An entire enzyme panel can be run in a single experiment and, unlike biochemical assays, the result is not dependent on ATP concentration. Of course, when interesting novel interactions of known compounds with new targets are found, these profiling techniques can also serve as lead discovery tools to initiate chemistry efforts to derive new therapeutics.
Conclusions
Today's high-throughput screens, although frequently effective, might not be sufficient for the pharmaceutical industry's future lead discovery needs. Affinity methods aim at easing this problem in various ways and new advances in these technologies will surely follow. Better success rates will not only improve the lead discovery process, but will also enhance our understanding of small-molecule binding to proteins. To this end, a protein ‘druggability’ index has been proposed based on NMR fragment screening data [47•]. The predictive classification of proteins via small-molecule interactions is a highly intriguing concept, but further theoretical and experimental studies on the required sampling rate of the different discovery libraries (drug-like or fragments) might be necessary before this major milestone can be reached.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
We thank Kevin Chapman and Joseph Becker for their suggestions and for their careful review of the manuscript.
References
- 1.Harper G., Pickett S.D., Green D.V.S. Design of a compound screening collection for use in high throughput screening. Comb Chem High Throughput Screen. 2004;7:63–70. doi: 10.2174/138620704772884832. [DOI] [PubMed] [Google Scholar]
- 2.Hann M.M., Oprea T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr Opin Chem Biol. 2004;8:255–263. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 3••.Comess K.M., Schurdak M.E. Affinity-based screening techniques for enhancing lead discovery. Curr Opin Drug Discov Devel. 2004;7:411–416. [PubMed] [Google Scholar]; Detailed technological summary of the state-of-the-art in affinity-based screening.
- 4.Annis A.D., Athanasopoulos J., Curran P.J., Felsch J.S., Kalghatgi K., Lee W.H., Nash H.M., Orminati J.-P.A., Rosner K.E., Shipps G.W., Jr. An affinity selection-mass spectrometry method for the identification of small molecule ligands from self-encoded combinatorial libraries: discovery of a novel antagonist of E. coli dihydrofolate reductase. Int J Mass Spec. 2005;238:77–78. [Google Scholar]
- 5•.Coburn C.A., Stachel S.J., Li Y.-M., Rush D.M., Steele T.G., Chen-Dodson E., Holloway M.K., Xu M., Huang Q., Lai M.-T. Identification of a small molecule nonpeptide active site β-secretase inhibitor that displays a nontraditional binding mode for aspartyl proteases. J Med Chem. 2004;47:6117–6119. doi: 10.1021/jm049388p. [DOI] [PubMed] [Google Scholar]; Discovery of a novel and selective BACE1 inhibitor via affinity screening. Analysis of the X-ray structure of the bound inhibitor is included.
- 6.Stachel S.J., Coburn C.A., Steele T.J., Jones C.J., Loutzenhiser E.F., Gregro A.R., Rajapakse H.A., Lai M.-T., Crouthamel M.-C., Xu M. Structure-based design of potent and selective cell-permeable inhibitors of human, β-secretase (BACE-1) J Med Chem. 2004;47:6447–6450. doi: 10.1021/jm049379g. [DOI] [PubMed] [Google Scholar]
- 7.Makara G.M., Nash H., Zheng Z., Orminati J.-P.A., Wintner E.W. A reagent-based strategy for the design of large combinatorial libraries: a preliminary experimental validation. Mol Divers. 2003;7:3–14. doi: 10.1023/b:modi.0000006537.06541.8a. [DOI] [PubMed] [Google Scholar]
- 8.Muckenschnabel I., Falchetto R., Mayr L.M., Filipuzzi I. SpeedScreen: label-free liquid chromatography-mass spectrometry-based high-throughput screening for the discovery of orphan protein ligands. Anal Biochem. 2004;324:241–249. doi: 10.1016/j.ab.2003.09.040. [DOI] [PubMed] [Google Scholar]
- 9.Zehender H., Le Goff F., Lehmann N., Filipuzzi I., Mayr L.M. SpeedScreen: the ‘missing link’ between genomics and lead discovery. J Biomol Screen. 2004;9:498–505. doi: 10.1177/1087057104267605. [DOI] [PubMed] [Google Scholar]
- 10.Qian J., Voorbach M.J., Huth J.R., Coen M.L., Zhang H., Ng S.-C., Comess K.M., Petros A.M., Rosenberg S.H., Warrior U., Burns D.J. Discovery of novel inhibitors of Bcl-xL using multiple high-throughput screening platforms. Anal Biochem. 2004;328:131–138. doi: 10.1016/j.ab.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 11.Huth J.R., Yu L., Collins I., Mack J., Mendoza R., Isaac B., Braddock D.T., Muchmore S.W., Comess K.M., Fesik S.W. NMR-driven discovery of benzoylanthranilic acid inhibitors of far upstream element binding protein binding to the human oncogene c-myc promoter. J Med Chem. 2004;47:4851–4857. doi: 10.1021/jm0497803. [DOI] [PubMed] [Google Scholar]
- 12.Gayton-Ely M., Pappas T.J., Holland L.A. Probing affinity via capillary electrophoresis: advances in 2003-2004. Anal Bioanal Chem. 2005;382:570–580. doi: 10.1007/s00216-004-3033-z. [DOI] [PubMed] [Google Scholar]
- 13.Heegaard N.H. Applications of affinity interactions in capillary electrophoresis. Electrophoresis. 2003;24:3879–3891. doi: 10.1002/elps.200305668. [DOI] [PubMed] [Google Scholar]
- 14.Belenky A., Hughes D., Korneev A., Dunayevskiy Y. Capillary electrophoretic approach to screen for enzyme inhibitors in natural extracts. J Chromatogr A. 2004;1053:247–251. [PubMed] [Google Scholar]
- 15.Slon-Usakiewicz J.J., Ng W., Dai J.R., Pasternak A., Redden P.R. Frontal affinity chromatography with MS detection (FAC-MS) in drug discovery. Drug Discov Today. 2005;10:409–416. doi: 10.1016/S1359-6446(04)03360-4. [DOI] [PubMed] [Google Scholar]
- 16.Chan N.W., Lewis D.F., Rosner P.J., Kelly M.A., Schriemer D.C. Frontal affinity chromatography-mass spectrometry assay technology for multiple stages of drug discovery: applications of a chromatographic biosensor. Anal Biochem. 2003;319:1–12. doi: 10.1016/s0003-2697(03)00193-3. [DOI] [PubMed] [Google Scholar]
- 17.Yi L., Li Z., Yuan K., Qu X., Chen J., Wang G., Zhang H., Luo H., Zhu L., Jiang P. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. J Virol. 2004;78:11334–11339. doi: 10.1128/JVI.78.20.11334-11339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sannes-Lowery K.A., Drader J.J., Griffey R.H., Hofstadler S.A. Fourier transform ion cyclotron resonance mass spectrometry as a high throughput affinity screen to identify RNA binding ligands. Trends Anal Chem. 2000;19:481–491. [Google Scholar]
- 19.Cummins L.L., Chen S., Blyn L.B., Sannes-Lowery K.A., Drader J.J., Griffey R.H., Hofstadler S.A. Multitarget affinity/specificity screening of natural products: finding and characterizing high-affinity ligands from complex mixtures by using high-performance mass spectrometry. J Nat Prod. 2003;66:1186–1190. doi: 10.1021/np0301137. [DOI] [PubMed] [Google Scholar]
- 20.Liu G., Szczepankiewicz B.G., Pei Z., Janowick D.A., Xin Z., Hajduk P.J., Abad-Zapatero C., Liang H., Hutchins C.W., Fesik S.W. Discovery and structure-activity relationship of oxalylarylaminobenzoic acids as inhibitors of protein tyrosine phosphatase 1B. J Med Chem. 2003;46:2093–2103. doi: 10.1021/jm0205696. [DOI] [PubMed] [Google Scholar]
- 21•.Liu G., Xin Z., Pei Z., Hajduk P.J., Abad-Zapatero C., Hutchins C.W., Zhao H., Lubben T.H., Ballaron S.J., Haasch D.L. Fragment screening and assembly: a highly efficient approach to a selective and cell active protein tyrosine phosphatase 1B inhibitor. J Med Chem. 2003;46:4232–4235. doi: 10.1021/jm034122o. [DOI] [PubMed] [Google Scholar]; A good demonstration of the potentials offered by fragment-based screenings.
- 22.Yu L., Oost T.K., Schkeryantz J.M., Yang J., Janowick D., Fesik S.W. Discovery of aminoglycoside mimetics by NMR-based screening of Escherichia coli A-site RNA. J Am Chem Soc. 2003;125:4444–4450. doi: 10.1021/ja021354o. [DOI] [PubMed] [Google Scholar]
- 23.Wada C.K. The evolution of the matrix metalloproteinase inhibitor drug discovery program at Abbott Laboratories. Curr Top Med Chem. 2004;4:1255–1267. doi: 10.2174/1568026043388015. [DOI] [PubMed] [Google Scholar]
- 24.Oltersdorf T., Elmore S.W., Shoemaker A.R., Armstrong R.C., Augeri D.J., Belli B.A., Bruncko M., Deckwerth T.L., Dinges J., Hajduk P.J. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 25.Vanwetswinkel S., Heetebrij R.J., van Duynhoven J., Hollander J.G., Filippov D.V., Hajduk P.J., Siegal G. Trends neurosci, target immobilized NMR screening: an efficient and sensitive method for ligand discovery. Chem Biol. 2005;12:207–216. doi: 10.1016/j.chembiol.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Moore J., Abdul-Manan N., Fejzo J., Jacobs M., Lepre C., Peng J., Xie X. Leveraging structural approaches: applications of NMR-based screening and X-ray crystallography for inhibitor design. J Synchrotron Radiat. 2004;11:97–100. doi: 10.1107/s0909049503023975. [DOI] [PubMed] [Google Scholar]
- 27.Sanders W.J., Nienaber V.L., Lerner C.G., McCall J.O., Merrick S.M., Swanson S.J., Harlan J.E., Stoll V.S., Stamper G.F., Betz S.F. Discovery of potent inhibitors of dihydroneopterin aldolase using CrystaLEAD high-throughput X-ray crystallographic screening and structure-directed lead optimization. J Med Chem. 2004;47:1709–1718. doi: 10.1021/jm030497y. [DOI] [PubMed] [Google Scholar]
- 28.Card G.L., Blasdel L., England B.P., Zhang C., Suzuki Y., Gillette S., Fong D., Ibrahim P.N., Artis D.R., Bollag G. A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design. Nat Biotechnol. 2005;23:201–207. doi: 10.1038/nbt1059. [DOI] [PubMed] [Google Scholar]
- 29••.Hartshorn M.J., Murray C.W., Cleasby A., Frederickson M., Tickle I.J., Jhoti H. Fragment-based lead discovery using X-ray crystallography. J Med Chem. 2005;48:403–413. doi: 10.1021/jm0495778. [DOI] [PubMed] [Google Scholar]; The most comprehensive summary on the discovery of fragments by crystallography exemplified with five different therapeutic targets including p38, CDK2, thrombin, PTP1B and RNase A. Hit to lead evolution is demonstrated for two of the five targets.
- 30.Gill A.L., Frederickson M., Cleasby A., Woodhead S.J., Carr M.G., Woodhead A.J., Walker M.T., Congreve M.S., Devine L.A., Tisi D. Identification of novel p38α MAP kinase inhibitors using fragment-based lead generation. J Med Chem. 2005;48:414–426. doi: 10.1021/jm049575n. [DOI] [PubMed] [Google Scholar]
- 31•.Erlanson D.A., McDowell R.S., He M.M., Randal M., Simmons R.L., Kung J., Waight A., Hansen S.K. Discovery of a new phosphotyrosine mimetic for PTP1B using breakaway tethering. J Am Chem Soc. 2003;125:5602–5603. doi: 10.1021/ja034440c. [DOI] [PubMed] [Google Scholar]; Reports the discovery of a novel phosphate-mimicking group via disulfide-mediated fragment exchange. A good illustration of the tethering technology.
- 32•.Hansen S.K., Cancilla M.T., Shiau T.P., Kung J., Chen T., Erlanson D.A. Allosteric inhibition of PTP1B activity by selective modification of a non-active site cysteine residue. Biochemistry. 2005;44:7704–7712. doi: 10.1021/bi047417s. [DOI] [PubMed] [Google Scholar]; A novel approach for the inhibition of phosphatases discovered using covalent tethering.
- 33.Hyde J., Braisted A.C., Randal M., Arkin M.R. Discovery and characterization of cooperative ligand binding in the adaptive region of interleukin-2. Biochemistry. 2003;42:6475–6483. doi: 10.1021/bi034138g. [DOI] [PubMed] [Google Scholar]
- 34.Raimundo B.C., Oslob J.D., Braisted A.C., Hyde J., McDowell R.S., Randal M., Waal N.D., Wilkinson J., Yu C.H., Arkin M.R. Integrating fragment assembly and biophysical methods in the chemical advancement of small-molecule antagonists of IL-2: an approach for Inhibiting protein-protein interactions. J Med Chem. 2004;47:3111–3130. doi: 10.1021/jm049967u. [DOI] [PubMed] [Google Scholar]
- 35.Hardy J.A., Lam J., Nguyen J.T., O’Brien T., Wells J.A. Discovery of an allosteric site in the caspases. Proc Natl Acad Sci USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buck E., Bourne H., Wells J.A. Site-specific disulfide capture of agonist and antagonist peptides on the C5a receptor. J Biol Chem. 2005;280:4009–4012. doi: 10.1074/jbc.C400500200. [DOI] [PubMed] [Google Scholar]
- 37••.Hopkins A.L., Groom C.R., Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]; A new metric for the qualification of lead molecules has the potential to become a major concept or filter for hit-to-lead groups.
- 38.Vieth M., Siegel M.G., Higgs R.E., Watson I.A., Robertson D.H., Savin K.A., Durst G.L., Hipskind P.A. Characteristic physical properties and structural fragments of marketed oral drugs. J Med Chem. 2004;47:224–232. doi: 10.1021/jm030267j. [DOI] [PubMed] [Google Scholar]
- 39.Dickopfa S., Franka M., Junkera H.-D., Maiera S., Metza G., Ottlebena H., Raua H., Schellhaasa N., Schmidta K., Sekula R. Custom chemical microarray production and affinity fingerprinting for the S1 pocket of factor VIIa. Anal Biochem. 2004;335:50–57. doi: 10.1016/j.ab.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 40.Annis D.A., Nazef N., Chuang C.-C., Porter Scott M., Nash H.M. A general technique to rank protein-ligand binding affinities and determine allosteric versus direct binding site competition in compound mixtures. J Am Chem Soc. 2004;126:15495–15503. doi: 10.1021/ja048365x. [DOI] [PubMed] [Google Scholar]
- 41.Huxley P., Sutton D.H., Debnam P., Matthews I.R., Brewer J.E., Rose J., Trickett M., Williams D.D., Andersen T.B., Classon B.J. High-affinity small molecule inhibitors of T cell costimulation: compounds for immunotherapy. Chem Biol. 2004;11:1651–1658. doi: 10.1016/j.chembiol.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 42.Huber W. A new strategy for improved secondary screening and lead optimization using high-resolution SPR characterization of compound-target interactions. J Mol Recognit. 2005;18:273–281. doi: 10.1002/jmr.744. [DOI] [PubMed] [Google Scholar]
- 43.Godl K., Wissing J., Kurtenbach A., Habenberger P., Blencke S., Gutbrod H., Salassidis K., Stein-Gerlach M., Missio A., Cotton M., Daub H. An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc Natl Acad Sci USA. 2003;100:15434–15439. doi: 10.1073/pnas.2535024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daub H., Godl K., Brehmer D., Klebl B., Müller G. Evaluation of kinase inhibitor selectivity by chemical proteomics. Assay Drug Dev Technol. 2004;2:215–224. doi: 10.1089/154065804323056558. [DOI] [PubMed] [Google Scholar]
- 45.Wissing J., Godl K., Brehmer D., Blencke S., Weber M., Habenberger P., Stein-Gerlach M., Missio A., Cotton M., Muller S., Daub H. Chemical proteomic analysis reveals alternative modes of action for pyrido[2,3-d]pyrimidine kinase inhibitors. Mol Cell Proteomics. 2004;3:1181–1193. doi: 10.1074/mcp.M400124-MCP200. [DOI] [PubMed] [Google Scholar]
- 46••.Fabian M.A., Biggs W.H., Treiber D.K., Atteridge C.E., Azimioara M.D., Benedetti M.G., Carter T.A., Ciceri P., Edeen P.T., Floyd M. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]; A high-throughput technology for the profiling of kinase inhibitors is described, including several examples of marketed and developmental compounds.
- 47•.Hajduk P., Huth J.R., Fesik S.W. Druggability indices for protein targets derived from NMR-based screening data. J Med Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]; A potentially controversial analysis of hit-rate statistics obtained with fragment-based NMR screenings.
- 48.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Res. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]