

Abstract
T helper 1-driven immune responses have been implicated in protective immunity against viral infections. Interleukin (IL)-12 is a heterodimeric proinflammatory cytokine formed by a p35 and a p40 subunit that can induce differentiation of naïve T cells towards a T helper 1-response. To determine the role of IL-12 in respiratory tract infection with influenza, p35 gene deficient (p35−/−) and normal wild type mice were intranasally infected with influenza A virus. IL-12 p35−/− mice displayed a transiently enhanced rather than an impaired viral clearance, as indicated by a 10-fold reduction in viral loads on day 8 after infection. Although interferon-γ levels were significantly lower in the lungs of IL-12 p35−/− mice, their cellular immune responses were not altered, as reflected by similar T cell CD69 expression and influenza-specific T cell recruitment. Our data indicate that endogenous IL-12 impairs viral clearance during the late phase of influenza A virus infection in mice.
Keywords: Influenza, Pneumonia, Interleukin-12, Innate response
1. Introduction
Influenza A virus usually causes upper respiratory tract infection associated with fever, chills, cough, soar throat, headache, general malaise and sometimes nausea and myalgia. In addition, primary influenza infection may lead to pneumonia (Treanor, 2000, Murphy and Webster, 1996). Host defense against influenza virus is accomplished by an interplay between innate and adaptive immune responses. Adaptive immunity against respiratory tract infection with influenza virus is mediated through antigen presentation by macrophages and dendritic cells (Braciale et al., 1987). Virus-infected cells are able to produce several cytokines and chemokines that facilitate the adaptive immune response against influenza virus (Julkunen et al., 2001).
IL-12, is a heterodimeric cytokine, which consists of a p35 subunit and a p40 subunit (IL-12 p70). The expression of both subunits is independently regulated, whereby the p40 subunit is expressed in excess to the p35 subunit (Gubler et al., 1991). Therefore, most of the p40 subunit is present as monomer or homodimer (D’Andrea et al., 1992, Ling et al., 1995). The p40 subunit can also dimerize with a p19 subunit to form IL-23 (Oppmann et al., 2000). IL-12 has been shown to induce proliferation and differentiation of T cells towards interferon (IFN-γ) producing T helper (Th) 1 cells. IL-12 is also able to induce interferon (IFN)-γ production in natural killer (NK) cells. Like IL-12, IL-23 has been shown to induce proliferation of naive T cells and the production of IFN-γ (Trinchieri, 2003). IFN-γ is considered to be the most important antiviral mediator during influenza infection and induces several antiviral mechanisms, including inhibition of viral replication in virus-infected cells (Katze et al., 2002).
Although T-cell mediated immune responses have been shown to be important in protective immunity against influenza virus (Doherty et al., 1997), the role of IL-12 herein is less clear. Administration of recombinant IL-12 to influenza virus-infected mice has been reported to enhance (Tsurita et al., 2001) or to delay (Kostense et al., 1998) the clearance of influenza A. In many viral infections, including influenza, the endogenous production of IL-12 seems limited (Sareneva et al., 1998, Monteiro et al., 1998, Biron, 1999, Kurokawa et al., 2002, Pirhonen et al., 2002), and treatment with an anti-IL-12 antibody only modestly and transiently impairs the clearance of influenza virus from the lungs of mice (Monteiro et al., 1998). Of note, in this sole study in which the role of endogenous IL-12 in host defense against influenza A was investigated an antibody raised against the p40 subunit was used to inhibit IL-12p70 activity (Monteiro et al., 1998). In that investigation the extent of IL-12 neutralization was not evaluated. Moreover, since the p40 component of IL-12p70 is also part of IL-23, it is likely that this antibody also influenced the activity of endogenous IL-23 (Lankford and Frucht, 2003). In the present study we sought to determine the role of endogenous IL-12 in the host response to influenza A virus by using p35 gene deficient mice, animals in which IL-12 is the only cytokine that cannot be produced.
2. Materials and methods
2.1. Animals
All experiments were approved by the Institutional Animal Care and Use Committee of the Academic Medical Center of the University of Amsterdam. IL-12 p35−/− mice on a C57Bl/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in the animal facility of the Academic Medical Center. Normal wildtype C57BL/6 mice were obtained from Harlan Sprague–Dawley (Horst, The Netherlands). Sex- and age-matched (8-weeks old) mice were used in all experiments.
2.2. Experimental virus infection
Influenza infection was induced as described previously (van der Sluijs et al., 2003). In brief, influenza A/PR/8/34 (ATCC No. VR-95; Rockville, MD), a mouse-adapted influenza strain, was grown on LLC-MK2 cells (RIVM, Bilthoven, Netherlands). Virus was harvested by a freeze/thaw cycle, followed by centrifugation at 680 × g for 10 min. Supernatants were stored in aliquots at −80 °C. Titration was performed in LLC-MK2 cells to calculate the median cell culture infective dose (CCID50) of the viral stock (Reed and Muench, 1938). A non-infected cell culture was used for preparation of the control inoculum. None of the stocks were contaminated by other respiratory viruses, i.e. influenza B, human parainfluenza types 1, 2, 3, 4A and 4B, RSV A and B, rhinovirus, enterovirus, corona virus and adenovirus, as determined by PCR or cell culture. Viral stock was diluted just before use in phosphate-buffered saline (PBS, pH 7.4). Mice were anesthetized by inhalation of isoflurane (Abbott Laboratories, Kent, UK) and intranasally inoculated with 10 CCID50 influenza (1400 viral copies) or control in a final volume of 50 μl PBS.
2.3. Determination of viral outgrowth
Viral load was determined on days 4, 8, and 12 after viral infection using real-time quantitative PCR as described (van Elden et al., 2001, van der Sluijs et al., 2003). On days 2, 4, 8 and 12, mice (6–8 mice per group) were anesthesized using 0.3 ml FFM (fentanyl citrate 0.079 mg/ml, fluanisone 2.5 mg/ml, midazolam 1.25 mg/ml in H2O; of this mixture 7.0 ml/kg intraperitoneally) and sacrificed by bleeding out the vena cava inferior. Lungs were harvested and homogenized at 4 °C in four volumes of sterile saline using a tissue homogenizer (Biospec Products, Bartlesville, OK). A 100 μl of lung-homogenates were treated with 1 ml Trizol reagent to extract RNA. RNA was resuspended in 10 μl DEPC-treated water. cDNA synthesis was performed using 1 μl of the RNA-suspension and a random hexamer cDNA synthesis kit (Applera, Foster City, CA). A 5 μl out of 25 μl cDNA-suspension was used for amplification in a quantitative real-time PCR reaction (ABI PRISM 7700 Sequence Detector System). The viral load present in a sample was calculated using a standard curve of particle counted influenza virus included in every assay run. The following primers were used: 5′-GGACTGCAGCGTAGACGCTT-3′ (forward); 5′-CATCCTGTTGTATATGAGGCCCAT-3′ (reverse), 5′-CTCAGTTATTCTGCTGGTGCACTTGCC-3′ (5′-FAM labeled probe).
2.4. Cytokine assays
Lung homogenates were lysed with an equal volume of lysisbuffer (300 mM NaCl, 30 mM Tris, 2 mM MgCl2, 2 mM CaCl2, 1% (v/v) Triton X-100, 20 ng/ml Pepstatin A, 20 ng/ml Leupeptin, 20 ng/ml Aprotinin, pH 7.4) and incubated for 30 min on ice, followed by centrifugation at 680 × g for 10 min. Supernatants were stored at −80 °C until further use. Cytokine levels in total-lung-lysates were measured by enzyme-linked immunosorbent assays according to the Manufacturer's instructions interleukin(IL)-6 from R&D Systems, Minneapolis, MN; interferon (IFN)-γ, IL-12 p40 and IL-12 p70 from Pharmingen, San Diego, CA). In addition, IL-12 p70 were measured by Cytometric Bead Assay (CBA) (BD Biosciences, San José, CA). The detection limits of the IL-12 p70 assays were 32 pg/ml (ELISA) and 20 pg/ml (CBA) resulting in detection limits of 320 and 200 pg/g lung tissue, respectively.
2.5. Flow cytometry
Pulmonary cell suspensions were obtained by grinding the lung tissue through nylon sieves. Cells (106) were collected in 96-well U-bottomed plates in FACS staining buffer (PBS with 0.5% (w/v) bovine serum albumin). All samples were stained for 15 min at 4 °C with anti-CD4-PE (clone RM4-5, PharMingen, San Diego, CA), anti-CD8-APC (clone 53-6.7, PharMingen, San Diego, CA) and anti-CD69-FITC monoclonal antibodies (clone H1.2F3, PharMingen, San Diego, CA). Alternatively, cells were stained with CD8-APC, and a PE-labeled tetrameric complex of H-2Db molecules with the PR/8 influenza virus nucleoprotein epitope ASNENMETM (NP366–374; tetramers were kindly provided by T. Schumacher, Netherlands Cancer Institute, Amsterdam, The Netherlands). Splenic cell suspensions were obtained on day 21 after viral infection and stained with CD8-FITC (clone 53-6.7, PharMingen, San Diego, CA), CD44-PE (clone IM7, PharMingen, San Diego, CA) and APC-labeled PR-tetramers. FACS analysis was done on a FACSCalibur with Cell Quest software (BD Biosciences, San Jose, CA).
2.6. Influenza-specific immunoglobulin assays
Virus-specific Ig levels were determined, as described previously (Xiao et al., 2004). In brief, blood was collected by heart puncture in EDTA-treated tubes. Density gradient-purified influenza virus was coated onto 96-well flat-bottom microtiter plates (Nunc). Wells were incubated with serially diluted sera, followed by biotinylated goat anti-mouse IgM, IgG1, IgG2a or IgG2b mAb (Southern Biotechnology Associates) and streptavidin-conjugated HRP (Sigma–Aldrich). Substrate 3,3′,5,5′ tetramethylbenzidine (Merck) was used to develop the reaction.
2.7. Bronchoalveolar lavage (BAL)
The trachea was exposed through a midline incision and cannulated with a sterile 22-gauge Abbocath-T catheter (Abbott, Sligo, Ireland). BAL was performed by instillation of two 0.5-ml aliquots of sterile saline into the right lung. The retrieved BAL fluid (BALF, approximately 0.8 ml) was spun at 260 g for 10 min at 4 °C and the pellet was resuspended in 0.5 ml sterile PBS. Total cell numbers were counted using a Z2 Coulter Particle Count and Size Analyzer (Beckman-Coulter Inc., Miami, FL). Differential cell counts were done on cytospin preparations stained with modified Giemsa stain (Diff-Quick; Baxter, UK).
2.8. Histological examination
The left lung was harvested on days 4 or 8 after viral infection, fixed in 10% formaline and embedded in paraffin. Then, 4 μm sections were stained with hematoxylin and eosin (H/E), and were analyzed by a pathologist.
2.9. Statistical analysis
Data are expressed as mean ± S.E. Comparison between the groups was conducted by using the Mann–Whitney U-test. p < 0.05 was considered to represent a statistically significant difference.
3. Results
3.1. Expression of IL-12 p40 and IL-12 p70 during influenza virus infection
Pulmonary levels of IL-12 p40 and IL-12 p70 were measured in total lung homogenates at several time points to determine the expression of these mediators after influenza virus infection. Pulmonary levels of IL-12 p40 were elevated on days 8 and 12 after viral infection and had returned to control levels on day 21 postinfection (Fig. 1 ). Using two different assays (ELISA and CBA), IL-12 p70 could not be detected in total lung lysates of influenza virus-infected mice, indicating that IL-12 p70 concentrations did not exceed 200 pg/g lung tissue.
Fig. 1.
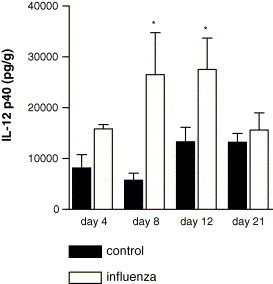
IL-12 p40 expression after influenza virus infection. Pulmonary IL-12 p40 levels in total-lung homogenates were measured in influenza-virus-infected mice (open bars) and control mice (filled bars) on days 4, 8, 12 and 21 after intranasal inoculation (6–8 mice per group). Data are expressed in pg/g lung tissue (mean ± S.E.). *p < 0.05 vs. control mice.
3.2. Viral load
To investigate the role of IL-12 on viral titers after influenza virus infection, we inoculated IL-12 p35−/− and wildtype mice and measured the viral load in the lungs over time. Viral load was 10-fold lower in IL-12 p35−/− mice than in wildtype mice on day 8 after infection (p = 0.05). Similar viral titers in wildtype mice and IL-12 p35−/− mice were found on days 2, 4 and 12 after infection. Hence, these data indicate that IL-12 transiently impairs clearance of influenza A in vivo (Fig. 2 ). Despite the fact that viral load was significantly lower in IL-12 p35−/− mice on day 8 after viral infection, a similar decline in bodyweight was observed for IL-12 p35−/− and wildtype mice (data not shown).
Fig. 2.
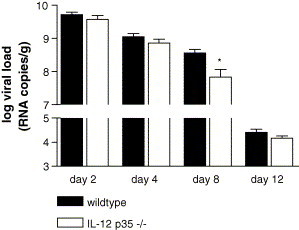
Viral load in the lungs of IL-12 p35−/− mice. Viral load was determined on days 2, 4, 8 and 12 after influenza virus infection (seven mice per time-point) in IL-12 p35−/− mice (open bars) and wildtype mice (filled bars). Viral load is expressed as viral RNA copies per lung. In control mice influenza could not be detected either (four mice per time-point, data not shown).
3.3. Survival
To further investigate the role of IL-12 during influenza virus infection, we monitored lethality at least twice a day after influenza infection in wildtype mice (n = 12) and IL-12 p35−/− mice (n = 13). Of note, we inoculated mice with a higher dose of influenza virus (200 TCID50), since a low dose of influenza virus (10 CCID50) appeared to be non-lethal for both wildtype mice and IL-12 p35−/− mice. No significant differences were observed between wildtype mice and IL-12 p35−/− mice (Fig. 3 ). These data indicate that, despite enhanced viral clearance, a lack of IL-12 does not improve survival after infection with influenza virus.
Fig. 3.
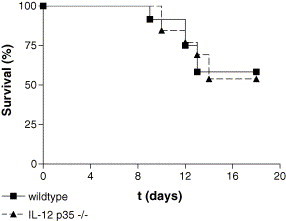
Survival after influenza virus infection in wildtype mice and IL-12 p35−/− mice. Survival after influenza infection in wildtype mice (squares) and IL-12 p35−/− mice (triangles). All mice received 200 CCID50 influenza virus intranasally on day 0. Mice were monitored at least twice a day after infection.
3.4. Pulmonary cytokine levels
To obtain insight in the role of IL-12 in cytokine release during influenza infection, IL-6, IFN-γ and IL-12 p40 concentrations were measured in total lung lysates of IL-12 p35−/− and wildtype mice, 2, 4, 8 and 12 days after infection. IL-6 levels were similar in both mouse strains at all time-points (Fig. 4 A). IFN-γ levels were significantly lower in IL-12 p35−/− mice than in wildtype mice on day 8 (p = 0.0058) and day 12 (p = 0.0064) after infection (Fig. 4B). IL-12 p40 levels were significantly higher in IL-12 p35−/− mice than in wildtype mice on day 8 (p = 0.02) and day 12 (p = 0.002) after infection (Fig. 4C).
Fig. 4.
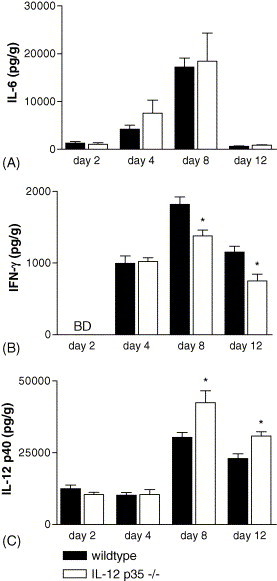
Cytokine expression in the lungs of IL-12 p35−/− mice. IL-6 (A), IFN-γ (B) and IL-12 p40 (C) levels in total-lung homogenates were measured in IL-12 p35−/− mice (open bars) and wildtype mice (filled bars) on days 2, 4, 8 and 12 after influenza virus infection (7–8 mice per group). Data are expressed in pg/g lung tissue (mean ± S.E.). *p < 0.05 vs. wildtype mice. BD = below detection level.
3.5. Cellular immune response to influenza virus infection
To investigate the role of IL-12 in the cellular immune response to influenza A we determined expression of CD69, an early activation-marker, on CD4+ and CD8+ T cells isolated from the lungs on day 8 after viral infection. Cell-surface expression of CD69 was similar on CD4+ and CD8+ T cells of IL-12 p35−/− mice and wildtype mice (Fig. 5 A and B), indicating that activation of T cells is not affected by IL-12 deficiency. We also investigated whether IL-12 regulates the recruitment of influenza-specific T cells into the lungs by using tetramer-staining. No differences were observed between IL-12 p35−/− mice and wildtype mice, suggesting that IL-12 does not play a role in the cellular immune response to influenza virus (Fig. 5C). To exclude that IL-12 plays a regulatory role in memory T cell formation, we determined the total number of influenza-specific T cells in the spleen on day 21 after influenza infection. Again, no difference was observed in IL-12 p35−/− mice and wildtype mice (Fig. 6 A). To confirm a memory phenotype of these influenza-specific T cells in the spleen, we determined CD44 expression on these T cells. In both wildtype and IL-12 p35−/− mice, more than 95% of the influenza-specific T cells expressed CD44 on the cell-surface (Fig. 6B). These data indicate that IL-12 does not regulate T cell activation or T cell memory formation during influenza virus infection in vivo.
Fig. 5.
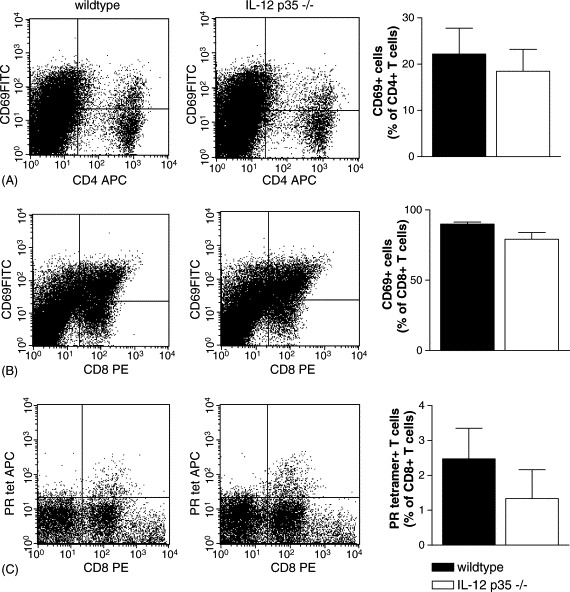
CD69 expression and influenza specific T-cell recruitment in IL-12 p35−/− mice. Representative dotplots for wildtype and IL-12 p35−/− mice are shown. CD69 expression on lung-derived CD4+ (A) and CD8+ (B) T cells in IL-12 p35−/− mice (open bars) and wildtype mice (filled bars) on day 8 after influenza virus infection (six mice per group). Influenza-specific T cell recruitment was determined by tetramer-staining (C) in IL-12 p35−/− mice (open bars) and wildtype mice (filled bars) on day 8 after viral infection (six mice per group). All data are expressed as percentage of total number of CD4+ or CD8+ T cells in the lungs. Total lymphocyte numbers in the lungs on day 8 after viral infection were similar in IL-12 p35−/− mice and wildtype mice (data not shown).
Fig. 6.
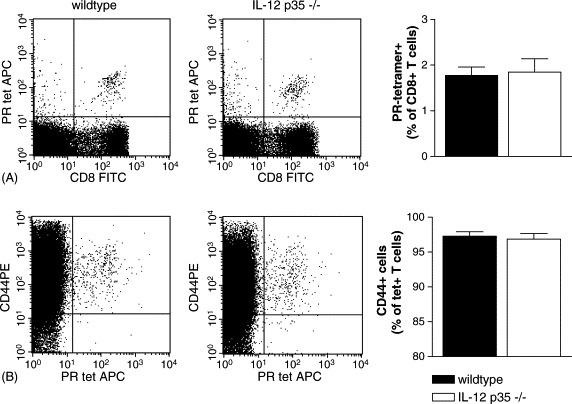
Memory T cell formation in the spleen of IL-12 p35−/− mice. Representative dotplots for wildtype and IL-12 p35−/− mice are shown. Influenza-specific T cell recruitment was determined by tetramer-staining (A) in IL-12 p35−/− mice (open bars) and wildtype mice (filled bars) on day 21 after viral infection (six mice per group). CD44 expression on spleen-derived CD8+ (B) T cells in IL-12 p35−/− mice (open bars) and wildtype mice (filled bars) on day 21 after influenza virus infection (six mice). Data are expressed as percentage of total number of CD8+ (lower graph) T cells in the lungs. Total lymphocyte numbers in the spleen on day 21 after viral infection were similar in IL-12 p35−/− mice and wildtype mice (data not shown).
3.6. Serum levels of influenza-specific immunoglobulins
Immunoglobulins have been shown to play an important role in the clearance of influenza virus. IgM, IgG1, IgG2a and IgG2b levels were measured on day 12 after viral infection. Influenza virus infection resulted in an increase of IgM and IgG2b in both mouse strains (Fig. 7 ), whereas serum titers of IgG1 and IgG2a were hardly detectable. The IgM titers were higher in wildtype mice than in IL-12 p35−/− mice, while IgG2b titers tended to be higher in IL-12 p35−/− mice.
Fig. 7.
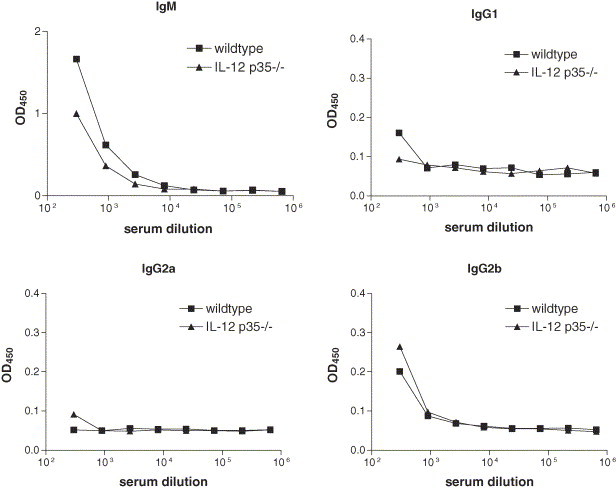
Ig responses on day 12 after influenza infection in IL-12 p35−/− mice. IgM, IgG1, IgG2a and IgG2b levels were measured in pooled sera of wildtype mice (squares) and IL-12 p35−/− mice (triangles). Each data point represents the OD450 at the specified dilution of the sera.
3.7. Cells in BALF
To evaluate the role of IL-12 in cell recruitment to the lungs during influenza infection, BALF was obtained on day 4 and day 8 after infection. On day 4, total cell counts and differentials were similar in IL-12 p35−/− mice and wildtype mice (Table 1 ). On day 8, the total number of recruited leukocytes in BALF was significantly lower in IL-12 p35−/− mice than in wildtype mice (p < 0.05, Table 1). The total number of macrophages, neutrophils and lymphocytes were also lower in BALF of IL-12 p35−/− mice, but these differences were not statistically significant (p = 0.10, 0.06 and 0.15, respectively).
Table 1.
Cells in BAL fluid
| Cells (×103) |
t = 4 |
t = 8 |
||
|---|---|---|---|---|
| Wildtype | IL-12 p35−/− | Wildtype | IL-12 p35−/− | |
| Total cell count | 348 ± 51 | 478 ± 67 | 302 ± 23 | 160 ± 33* |
| Macrophages | 180 ± 27 | 218 ± 38 | 220 ± 25 | 135 ± 29 |
| Polymorphonuclear cells | 167 ± 28 | 258 ± 36 | 73 ± 21 | 22 ± 12 |
| Lymphocytes | ND | ND | 8.3 ± 2.1 | 2.6 ± 1.0 |
Mice (n = 6 per group) received influenza A i.n. on day 0. Bronchoalveolar lavage fluid was obtained on days 4 and 8 after viral infection. All data are mean ± S.E. ND, not detectable, i.e.<0.5% of total cell counts.
p < 0.05 compared to wildtype mice.
3.8. Histopathology
To identify differences in inflammation between wildtype mice and IL-12 p35−/− mice, histopathological analysis of H/E stained lung-slides from days 4 and 8 after infection was performed. Influenza virus infection was associated with mild interstitial inflammation, endothelitis and bronchitis on day 4 after infection (Fig. 8 ). The interstitial inflammation, endothelitis and bronchitis were more pronounced on day 8 after viral infection and accompanied by pleuritis, focal necrosis together with apoptotic granulocytes (Fig. 8). Differences between wildtype mice and IL-12 p35−/− mice were not observed.
Fig. 8.
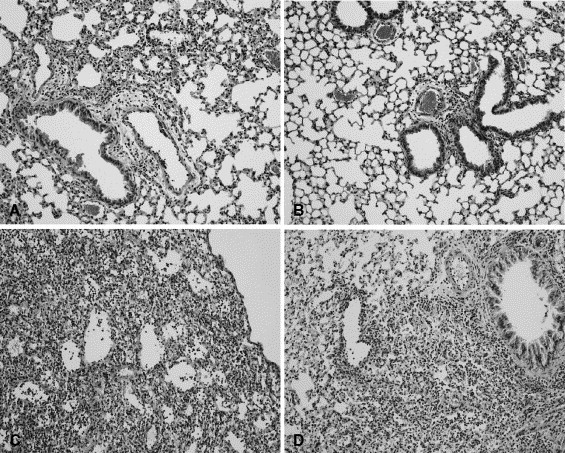
Histopathology of the lungs of IL-12 p35−/− mice. Increased lung inflammation during influenza pneumonia. Histopathological analysis of the lungs of IL-12 p35−/− mice (B and D) and wildtype mice (A and C). Lungs were isolated on day 4 (A and B) and day 8 (C and D) after viral infection and prepared for histopathological analysis. Original magnification: 100×. Slides are representative for six mice per group.
4. Discussion
Cellular immunity plays an important role in viral clearance. Host defense against viral infections are predominantly mediated by Th1 immune responses (Julkunen et al., 2001, Doherty et al., 1997). Antigen-presentation by dendritic cells leads to the activation of naive T cells to proliferate and differentiate into T helper cells (Hamilton-Easton and Eichelberger, 1995). Cytokines play a decisive role in the formation of either Th1 or Th2 responses. IL-12 has been implicated in the formation of Th1 lymphocytes and may play an important role in host defense against viral infections (Julkunen et al., 2001, Trinchieri, 2003). In the present study we investigated the role of IL-12 during influenza virus infection by using IL-12 p35−/− mice, in which the production of IL-12 is selectively eliminated. Our data demonstrate that IL-12 does not markedly contribute to innate and adaptive immunity against influenza A. If anything, IL-12 deficiency was associated with lower rather than higher viral loads at 8 days after infection. Of note, we determined viral load by real-time quantitative PCR instead of a standard plaque assay. The major differences between these two methods is that a standard plaque assay only detects viable (live) virus, i.e. virus that has been shed by infected cells, whereas molecular techniques detect all viral particles, including remnant viral genomes in dead cells.
Although the lung levels of IL-12p70 did not exceed 200 pg/g lung tissue, IL-12 p35−/− mice temporarily had lower viral loads and displayed significantly lower IFN-γ concentrations than wildtype mice after influenza A infection. Previous studies support our findings of marginal IL-12 production during influenza. Indeed, influenza-infected macrophages did not produce significant amounts of IL-12 (Sareneva et al., 1998, Pirhonen et al., 2002) and influenza infection in mice resulted in IL-12 concentrations in lungs that were barely higher than in lungs of control mice (Monteiro et al., 1998, Kurokawa et al., 2002). Our data expand the results of a previous study in which administration with an anti-p40 antibody (in theory inhibiting both IL-12 and IL-23 activity) modestly reduced local IFN-γ release during airway infection with influenza A (Monteiro et al., 1998). The fact that very low concentrations of IL-12 p70 can contribute to IFN-γ production has also been demonstrated in other experimental conditions such as whole blood cultures stimulated with endotoxin or bacteria (Lauw et al., 1999, Lauw et al., 2002).
In light of the important role for IL-12 in IFN-γ production, a logic assumption would have been that IL-12 p35−/− mice display a reduced resistance against influenza A. However, the evidence for this assumption is not readily available in the literature. To the best of our knowledge only one study directly evaluated the role of endogenous IL-12 in host defense against influenza. In this investigation, Monteiro et al. (1998) used an antibody directed against the p40 subunit to inhibit IL-12 activity in influenza infected mice; the authors did not report to what extent IL-12 activity was inhibited by the antibody. In addition, it is quite likely that the antibody also interfered with the activity of IL-23, a novel cytokine that shares the p40 subunit with IL-12 p70 and further consists of a p19 subunit (Oppmann et al., 2000); this cytokine had yet to be discovered at the time Monteiro et al. published their study. Importantly, IL-23 can also direct Th1 responses and has been claimed to be more important in cellular immunity than IL-12 (Ha et al., 2004). Anti-p40 treatment was associated with modestly elevated influenza titers in lungs at 3 days after infection when compared with mice treated with rat immunogobulin, whereas viral loads were similar in both experimental groups at days 5 and 7 after infection (Monteiro et al., 1998). Our current finding that IL-12 p35−/− mice had lower rather than higher influenza titers at day 8 after infection strongly argues against a dominant role for IL-12 in the protective immune response against this common virus. This notion is supported by an earlier study showing that administration of recombinant IL-12 to influenza virus-infected mice delayed the clearance of influenza A (Kostense et al., 1998), although this was not confirmed in another investigation (Tsurita et al., 2001). Nonetheless, our data together with the results obtained by Monteiro et al. (1998) indicate that further research is required to identify the role of IL-23 during influenza A infection.
The present finding of enhanced viral clearance in IL-12 p35−/− mice was also observed for influenza virus-infected IL-18−/− mice (van der Sluijs et al., 2005). Although IL-12 and IL-18 are not structurally related, both cytokines are known to induce a Th1 response. The enhanced viral clearance in IL-18−/− mice correlated with enhanced CD69 expression CD4+ T cells. In contrast, wildtype mice and IL-12 p35−/− mice displayed a similar expression of CD69 and influenza-specific T cell receptors on the surface of recruited lymphocytes when compared with wildtype mice. These data suggest that the cellular immune response to influenza virus is not significantly altered in IL-12 p35−/− mice. The humoral immune response appeared to be slightly altered in IL-12 p35−/− mice. The IgM response appeared to be lower in IL-12 p35−/−, which may have been caused by a lower viral load in the lungs of these mice. However, a tendency towards increased IgG2b formation in IL-12 p35−/− was observed as well, which might indicate an early class-switch from IgM to IgG. The net effect of reduced IgM and increased IgG2b titers on day 12 after infection seems to be of minor importance since both IgM and IgG have been shown to be capable of neutralizing influenza virus (Kopf et al., 2002, Renegar et al., 2004). This suggestion is in line with our findings that wildtype mice and IL-12 p35−/− mice show similar viral loads on day 12 after infection.
Cell counts in BALF indicate that the recruitment of neutrophils and to a lesser extent macrophages was reduced in IL-12 p35−/− mice. These reduced cell numbers on day 8 after infection could be the consequence of IL-12 deficiency, since IL-12 has been implicated in recruitment of macrophages (Ha et al., 1998, Liu et al., 2001, Kawakami et al., 1996, Pearlman et al., 1997) and neutrophils (Papp et al., 2000), via induction of other inflammatory mediators. Alternatively, diminished cell numbers in IL-12 p35−/− mice could have been caused by reduced viral load, providing a less potent proinflammatory stimulus. Homodimeric p40 can function as a chemotactic factor for macrophages (Heinzel et al., 1997, Ha et al., 1999), which has been shown to play an important role during respiratory tract infection with Sendai virus in mice (Russell et al., 2003). Here we found elevated p40 concentrations in lungs of IL-12 p35−/− mice on days 8 and 12 after infection. Although we did not measure the amount of monomeric and homodimeric p40, previous studies indicate that excess amounts of p40 consist for up to one-third of homodimeric p40–p40. The excess amount of p40 produced in IL-12 p35−/− mice may be, in part, present in a homodimeric form. Our study indicates a reduced number of macrophages in BAL fluid of IL-12 p35−/− mice (135 ± 29 versus 220 ± 25 in wildtype mice) on day 8 after infection, which contradicts a possible chemotactic role of homodimeric IL-12 p40. An alternative explanation would be that the excess of IL-12 p40 forms heterodimeric IL-23. This latter explanation would be in line with the hypothesis that IL-23 plays a more prominent role than IL-12 in host defense against influenza virus infection. Additional research is warranted to determine the contribution of homodimeric p40 and/or IL-23 in host defense against influenza A.
Apoptosis of virus-infected cells has been shown to be an effective mechanism for viral clearance (Brydon et al., 2003, Wark et al., 2005). IL-12 has been shown to inhibit apoptosis of T cells (Palmer et al., 2001) and dendritic cells (Pirtskhalaishvili et al., 2000). Since alveolar macrophages have a functional IL-12R (Grohmann et al., 2001), virus-infected macrophages might be protected from apoptosis, thereby limiting viral clearance. It seems unlikely that a similar mechanism applies for airway epithelial cells, since epithelial cells have not been described to express IL-12R. However, the possibility that IL-12 may have an indirect effect on apoptosis of airway epithelial cells cannot be excluded.
In conclusion, we have demonstrated that the host response to influenza A is modestly altered during the late phase of viral infection in IL-12 p35−/− mice, animals that are totally and selectively deficient for IL-12. This finding contrasts with numerous reports on strongly enhanced susceptibility of IL-12 deficient mice to infections by a variety of other pathogens, in particular mycobacteria and parasites (Romani et al., 1997). Our data strongly suggest that endogenous IL-12 is not important for protective immunity against respiratory tract infection with influenza A.
Acknowledgments
We thank Joost Daalhuisen and Ingvild Kop for technical assistance during the animal experiments and Teus van den Ham for assistance during the preparation and titration of the viral stocks. We also thank Dr. T.N. Schumacher for kindly providing PE and APC labeled tetramers.
References
- Biron C.A. Initial and innate responses to viral infections—pattern setting in immunity or disease. Curr. Opin. Microbiol. 1999;2:374–381. doi: 10.1016/s1369-5274(99)80066-6. [DOI] [PubMed] [Google Scholar]
- Braciale T.J., Morrison L.A., Sweetser M.T., Sambrook J., Gething M.J., Braciale V.L. Antigen presentation pathways to class I and class II MHC-restricted T lymphocytes. Immunol. Rev. 1987;98:95–114. doi: 10.1111/j.1600-065x.1987.tb00521.x. [DOI] [PubMed] [Google Scholar]
- Brydon E.W., Smith H., Sweet C. Influenza A virus-induced apoptosis in bronchiolar epithelial (NCI-H292) cells limits pro-inflammatory cytokine release. J Gen. Virol. 2003;84:2389–2400. doi: 10.1099/vir.0.18913-0. [DOI] [PubMed] [Google Scholar]
- D’Andrea A., Rengaraju M., Valiante N.M., Chehimi J., Kubin M., Aste M., Chan S.H., Kobayashi M., Young D., Nickbarg E. Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J. Exp. Med. 1992;176:1387–1398. doi: 10.1084/jem.176.5.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty P.C., Topham D.J., Tripp R.A., Cardin R.D., Brooks J.W., Stevenson P.G. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol. Rev. 1997;159:105–117. doi: 10.1111/j.1600-065x.1997.tb01010.x. [DOI] [PubMed] [Google Scholar]
- Grohmann U., Belladonna M.L., Vacca C., Bianchi R., Fallarino F., Orabona C., Fioretti M.C., Puccetti P. Positive regulatory role of IL-12 in macrophages and modulation by IFN-gamma. J. Immunol. 2001;167:221–227. doi: 10.4049/jimmunol.167.1.221. [DOI] [PubMed] [Google Scholar]
- Gubler U., Chua A.O., Schoenhaut D.S., Dwyer C.M., McComas W., Motyka R., Nabavi N., Wolitzky A.G., Quinn P.M., Familletti P.C., Gately M.K. Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proc. Natl. Acad. Sci. USA. 1991;88:4143–4147. doi: 10.1073/pnas.88.10.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S.J., Lee S.B., Kim C.M., Shin H.S., Sung Y.C. Rapid recruitment of macrophages in interleukin-12-mediated tumour regression. Immunology. 1998;95:156–163. doi: 10.1046/j.1365-2567.1998.00579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S.J., Lee C.H., Lee S.B., Kim C.M., Jang K.L., Shin H.S., Sung Y.C. A novel function of IL-12p40 as a chemotactic molecule for macrophages. J. Immunol. 1999;163:2902–2908. [PubMed] [Google Scholar]
- Ha S., Kim D.J., Baek K.H., Yun Y.D., Sung Y.C. IL-23 induces stronger sustained CTL and Th1 immune responses than IL-12 in hepatitis C virus envelope protein 2 DNA immunization. J. Immunol. 2004;172:525–532. doi: 10.4049/jimmunol.172.1.525. [DOI] [PubMed] [Google Scholar]
- Hamilton-Easton A., Eichelberger M. Virus-specific antigen presentation by different subsets of cells from lung and mediastinal lymph node tissues of influenza virus-infected mice. J. Virol. 1995;69:6359–6366. doi: 10.1128/jvi.69.10.6359-6366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel F.P., Hujer A.M., Ahmed F.N., Rerko R.M. In vivo production and function of IL-12 p40 homodimers. J. Immunol. 1997;158:4381–4388. [PubMed] [Google Scholar]
- Julkunen I., Sareneva T., Pirhonen J., Ronni T., Melen K., Matikainen S. Molecular pathogenesis of influenza A virus infection and virus-induced regulation of cytokine gene expression. Cytokine Growth Factor Rev. 2001;12:171–180. doi: 10.1016/s1359-6101(00)00026-5. [DOI] [PubMed] [Google Scholar]
- Katze M.G., He Y., Gale M., Jr. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- Kawakami K., Tohyama M., Xie Q., Saito A. IL-12 protects mice against pulmonary and disseminated infection caused by Cryptococcus neoformans. Clin. Exp. Immunol. 1996;104:208–214. doi: 10.1046/j.1365-2249.1996.14723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf M., Brombacher F., Bachmann M.F. Role of IgM antibodies versus B cells in influenza virus-specific immunity. Eur. J. Immunol. 2002;32:2229–2236. doi: 10.1002/1521-4141(200208)32:8<2229::AID-IMMU2229>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Kostense S., Sun W.H., Cottey R., Taylor S.F., Harmeling S., Zander D., Small P.A., Jr., Bender B.S. Interleukin 12 administration enhances Th1 activity but delays recovery from influenza A virus infection in mice. Antiviral Res. 1998;38:117–130. doi: 10.1016/s0166-3542(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Kurokawa M., Tsurita M., Brown J., Fukuda Y., Shiraki K. Effect of interleukin-12 level augmented by Kakkon-to, a herbal medicine, on the early stage of influenza infection in mice. Antiviral Res. 2002;56:183–188. doi: 10.1016/s0166-3542(02)00104-3. [DOI] [PubMed] [Google Scholar]
- Lankford C.S., Frucht D.M. A unique role for IL-23 in promoting cellular immunity. J. Leukoc. Biol. 2003;73:49–56. doi: 10.1189/jlb.0602326. [DOI] [PubMed] [Google Scholar]
- Lauw F.N., Dekkers P.E., te Velde A.A., Speelman P., Levi M., Kurimoto M., Hack C.E., van Deventer S.J., van der Poll T. Interleukin-12 induces sustained activation of multiple host inflammatory mediator systems in chimpanzees. J. Infect. Dis. 1999;179:646–652. doi: 10.1086/314636. [DOI] [PubMed] [Google Scholar]
- Lauw F.N., Branger J., Florquin S., Speelman P., van Deventer S.J., Akira S., van der Poll T. IL-18 improves the early antimicrobial host response to pneumococcal pneumonia. J. Immunol. 168. 2002:372–379. doi: 10.4049/jimmunol.168.1.372. [DOI] [PubMed] [Google Scholar]
- Ling P., Gately M.K., Gubler U., Stern A.S., Lin P., Hollfelder K., Su C., Pan Y.C., Hakimi J. Human IL-12 p40 homodimer binds to the IL-12 receptor but does not mediate biologic activity. J. Immunol. 1995;154:116–127. [PubMed] [Google Scholar]
- Liu Z., Geboes K., Heremans H., Overbergh L., Mathieu C., Rutgeerts P., Ceuppens J.L. Role of interleukin-12 in the induction of mucosal inflammation and abrogation of regulatory T cell function in chronic experimental colitis. Eur. J. Immunol. 2001;31:1550–1560. doi: 10.1002/1521-4141(200105)31:5<1550::AID-IMMU1550>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Monteiro J.M., Harvey C., Trinchieri G. Role of interleukin-12 in primary influenza virus infection. J. Virol. 1998;72:4825–4831. doi: 10.1128/jvi.72.6.4825-4831.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B.R., Webster R.G. Orthomyxoviruses. In: Fields B.N., Knipe D.M., Howley P.M., editors. Fields Virology. 3rd ed. Lippincott-Raven publishers; Philadelphia: 1996. p. 1407. [Google Scholar]
- Oppmann B., Lesley R., Blom B., Timans J.C., Xu Y., Hunte B., Vega F., Yu N., Wang J., Singh K., Zonin F., Vaisberg E., Churakova T., Liu M., Gorman D., Wagner J., Zurawski S., Liu Y., Abrams J.S., Moore K.W., Rennick D., de Waal-Malefyt R., Hannum C., Bazan J.F., Kastelein R.A. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- Palmer E.M., Farrokh-Siar L., Maguire van Seventer J., van Seventer G.A. IL-12 decreases activation-induced cell death in human naive Th cells costimulated by intercellular adhesion molecule-1. I. IL-12 alters caspase processing and inhibits enzyme function. J. Immunol. 2001;167:749–758. doi: 10.4049/jimmunol.167.2.749. [DOI] [PubMed] [Google Scholar]
- Papp Z., Middleton D.M., Rontved C.M., Foldvari M., Gordon J.R., Baca-Estrada M.E. Transtracheal administration of interleukin-12 induces neutrophil responses in the murine lung. J. Interferon Cytokine Res. 2000;20:191–195. doi: 10.1089/107999000312603. [DOI] [PubMed] [Google Scholar]
- Pearlman E., Lass J.H., Bardenstein D.S., Diaconu E., Hazlett F.E., Jr., Albright J., Higgins A.W., Kazura J.W. IL-12 exacerbates helminth-mediated corneal pathology by augmenting inflammatory cell recruitment and chemokine expression. J. Immunol. 1997;158:827–833. [PubMed] [Google Scholar]
- Pirhonen J., Matikainen S., Julkunen I. Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J. Immunol. 2002;169:5673–5678. doi: 10.4049/jimmunol.169.10.5673. [DOI] [PubMed] [Google Scholar]
- Pirtskhalaishvili G., Shurin G.V., Esche C., Cai Q., Salup R.R., Bykovskaia S.N., Lotze M.T., Shurin M.R. Cytokine-mediated protection of human dendritic cells from prostate cancer-induced apoptosis is regulated by the Bcl-2 family of proteins. Br. J. Cancer. 2000;83:506–513. doi: 10.1054/bjoc.2000.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 1938;27:s493–s497. [Google Scholar]
- Renegar K.B., Small P.A., Jr., Boykins L.G., Wright P.F. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J. Immunol. 2004;173:1978–1986. doi: 10.4049/jimmunol.173.3.1978. [DOI] [PubMed] [Google Scholar]
- Romani L., Puccetti P., Bistoni F. Interleukin-12 in infectious diseases. Clin. Microbiol. Rev. 1997;10:611–636. doi: 10.1128/cmr.10.4.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell T.D., Yan Q., Fan G., Khalifah A.P., Bishop D.K., Brody S.L., Walter M.J. IL-12 p40 homodimer-dependent macrophage chemotaxis and respiratory viral inflammation are mediated through IL-12 receptor beta1. J. Immunol. 2003;171:6866–6874. doi: 10.4049/jimmunol.171.12.6866. [DOI] [PubMed] [Google Scholar]
- Sareneva T., Matikainen S., Kurimoto M., Julkunen I. Influenza A virus-induced IFN-alpha/beta and IL-18 synergistically enhance IFN-gamma gene expression in human T cells. J. Immunol. 1998;160:6032–6038. [PubMed] [Google Scholar]
- Treanor J.J. Orthomyxoviridae: influenza virus. In: Mandell G.L., Douglas D.R., Bennett E., Dolin R., editors. Principles and Practice of Infectious Diseases. 5th ed. Churchill Livingston; New York: 2000. pp. 1834–1835. [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Tsurita M., Kurokawa M., Imakita M., Fukuda Y., Watanabe Y., Shiraki K. Early augmentation of interleukin (IL)-12 level in the airway of mice administered orally with clarithromycin or intranasally with IL-12 results in alleviation of influenza infection. J. Pharmacol. Exp. Ther. 2001;298:362–368. [PubMed] [Google Scholar]
- van Elden L.J.R., Nijhuis M., Schipper P., Schuurman R., van Loon A.M. Simultaneous detection of influenza virus A and B using real-time quantitative PCR. J. Clin. Microbiol. 2001;39:196–200. doi: 10.1128/JCM.39.1.196-200.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs K.F., van Elden L., Nijhuis M., Schuurman R., Florquin S., Jansen H.M., Lutter R., van der Poll T. Toll-like receptor 4 is not involved in host defense against respiratory tract infection with Sendai virus. Immunol. Lett. 2003;89:201–206. doi: 10.1016/S0165-2478(03)00138-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs K.F., van Elden L.J.R., Arens R., Nijhuis M., Schuurman R., Kwakkel G.J., Florquin S., Akira S., Jansen H.M., Lutter R., van der Poll T. Enhanced viral clearance in IL-18 deficient mice after pulmonary infection with influenza A virus. Immunology. 2005;114:112–120. doi: 10.1111/j.1365-2567.2004.02000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wark P.A., Johnston S.L., Bucchieri F., Powell R., Puddicombe S., Laza-Stanca V., Holgate S.T., Davies D.E. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y., Hendriks J., Langerak P., Jacobs H., Borst J. CD27 is acquired by primed B cells at the centroblast stage and promotes germinal center formation. J. Immunol. 2004;172:7432–7441. doi: 10.4049/jimmunol.172.12.7432. [DOI] [PubMed] [Google Scholar]