

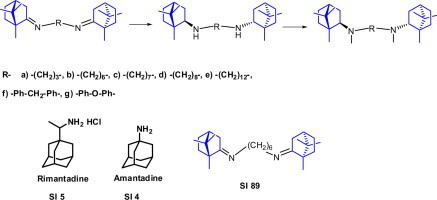
Graphical abstract
Keywords: Camphor, Diimine derivatives, Antivirals, Influenza
Abstract
Influenza is a continuing world-wide public health problem that causes significant morbidity and mortality during seasonal epidemics and sporadic pandemics. The purpose of the study was synthesis and investigation of antiviral activity of camphor-based symmetric diimines and diamines. A set of C2-symmetric nitrogen-containing camphor derivatives have been synthesized. The antiviral activity of these compounds was studied against rimantadine- and amantadine-resistant influenza virus A/California/7/09 (H1N1)pdm09 in MDCK cells. The highest efficacy in virus inhibiting was shown for compounds 2a–e with cage moieties bound by aliphatic linkers. The therapeutic index (selectivity index) for 2b exceeded that for reference compounds amantadine, deitiforin and rimantadine almost 10-fold. As shown by structure–activity analysis, the length of the linker has a dramatic effect on the toxicity of compounds. Compound 2e with –C12H24– linker exhibited the lowest toxicity (CTD50 = 2216 μM). Derivatives of camphor, therefore, can be considered as prospective antiinfluenza compounds active against influenza viruses resistant to adamantane-based drugs.
1. Introduction
Development of new drugs for treatment and prophylaxis of viral infections is one of the most important directions of modern pharmacology and medicinal chemistry. Outbreaks of avian influenza H5N1 (1997–2006) followed by pandemic of ‘swine flu’ A(H1N1)pdm09, as well as the recent introduction of avian virus of H7N9 subtype to the human population make it necessary to revise the problem of searching for and developing novel antivirals.1
As demonstrated by practice-based research, natural compounds are the most promising source for the development of drugs. Approximately half of novel drugs approved for clinical use in USA are the naturally derived compounds and their modified derivatives.2 There are currently few antivirals against influenza infection, for most of which fast emergence of drug resistance has been documented.3 For this reason, creation of novel effective antivirals with the alternative mechanism and broad spectrum of activity is one of the most important goals in the field of treatment and prophylaxis of influenza.
Due to specific genome organization (in particular, segmented nature and lack of the mechanism of correction of replication errors) and short-term life cycle, influenza virus demonstrates high genetic variability and high rate of mutations. As a result, its antigenic structure is highly variable due to selective pressure of host immune response. In addition, the presence of specific antivirals also serves as a selective factor resulting in fast emergence of drug-resistant virus strains. Taken together, these processes lead to the selection of viral variants that are capable both of escaping the immune inactivation by neutralizing antibodies and of overcoming the suppressive action of antiviral drugs.
Three chemical classes of compounds are currently used as anti-influenza drugs. They are targeted against different stages of viral life cycle in infected cells. Among them, the most commonly used compounds include neuraminidase inhibitors zanamivir (Relenza®), oseltamivir (Tamiflu®), peramivir (Rapiacta®), and laninamivir (Inavir®).4 They interfere with the activity of viral neuraminidase, which plays an essential role in the release of progeny virus particles from the surface of a host cell. Ribavirin and favipiravir are antivirals of broad range of activity, which exhibit a suppressive effect against almost all RNA-genome human viruses.5 Meanwhile, being a nucleoside analog, ribavirin possesses numerous side effects, including the reduction of haemoglobin level, neutropenia, pulmonary edema, etc.6
Adamantane derivatives, which were first discovered as specific anti-influenza drugs with the direct mechanism of activity are represented by two compounds: Rimantadine (α-methyl-1-adamantylamine hydrochloride) and amantadine (1-aminoadamantane).7 These compounds block virally encoded protein M2 that acts as a proton channel required for acidification of the virion core, and thus prevent hemagglutinin cleavage and further fusion of membranes of the viral envelope and lysosomal vacuole necessary for virus decapsidation.8, 9
Despite high initial efficacy of adamantanes as anti-influenza drugs, drug-resistant strains can be easily selected both in vitro and in patients receiving adamantane-based therapy.10 Adamantane resistance among circulating influenza A viruses increased rapidly worldwide starting from 2003 to 2004. The percentage of adamantane-resistant influenza A virus isolates increased from 0.4% in 1994–1995 to 92% in 2005–2006 influenza season.11 The resistance is mainly conferred by amino acid substitutions in M2 protein L26F, V27A, S31N and G34E.12
Being widely used cage structures, adamantanes were used for the rational design of a large class of antiviral compounds.13 In particular, attempts were undertaken to overcome the adamantane-resistance of influenza virus. Among them, aminospiroadamantanes, although inactive against the mutant S31N, were revealed to be a submicromolar inhibitor of the clinically important mutant V27A (IC50 = 0.31 μM) and also showed to be active against the mutant L26F (IC50 = 5.6 μM).14
Bananins, which have been shown to possess an inhibitory activity against SARS coronavirus, are the closest compounds to classical adamantanes.15 More widely, cage structures appeared promising core structures for the development of novel antivirals. Among them, Deitiforin (2-(1′-aminoethyl)bicyclo[2.2.1]heptane) is one of the most interesting drugs based on natural cage compounds, bornanes (Fig. 1 ).16 In this regard, it is important to note that this compound is in fact of natural origin and that borneol and isoborneol have been widely known in folk medicine as anti-inflammatory compounds.17
Figure 1.
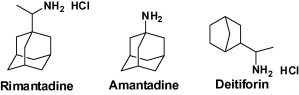
Аnti-influenza drugs.
Natural compounds, including monoterpenoids, are a promising source of new antiviral compounds.18, 19 Оxygen-containing monoterpenoid with a para-menthane framework showed a significant activity against the influenza virus,20 nitrogen containing compounds based on the hydrochlorides of 1- and 2-adamantylamines and monoterpene aldehydes demonstrated antiviral activity against the influenza virus higher than that of rimantadine and amantadine.21 Meanwhile, it is known that Schiff bases are among the most important groups of biomolecules. These compounds have been found to reveal both remarkable biological activities and a variety of valuable practical applications.22, 23 The aim of this work was to synthesize new nitrogen containing compounds, including Schiff bases and amines, based on camphor and to study their antiviral activity against rimantadine-resistant influenza virus A/California/07/09 (H1N1)pdm09.
2. Results and discussion
2.1. Chemistry
Starting from the known terpene camphor 1, we have prepared new N,N′-bis(isobornyl)diimine 2a–g and diamine 3b,c,e,f,g and 4b,c using the conventional methods of amine chemistry. Camphor is an inexpensive natural compound, one of the first plant metabolites isolated in the chemically pure form. For a long time, the technology for isolating this compound from camphor tree wood has been known in China in Japan. The analeptic properties of camphor have been known since the 18th century. Today, it is used to treat acute and chronic heart failure, respiratory depression, soporific and narcotic poisoning, as well as an agent for topical use.24 A typical feature of a camphor molecule is its cage-hydrocarbon (rigid) structure that is similar to a certain extent to that of adamantane. Having the nearly spherical structure, adamantane derivatives, amantadine and rimantadine, exhibit high penetrability through the cell membrane and, hence, high antiviral activity.
Aliphatic diimines (Schiff bases) 2a–e were obtained according to the procedure in Ref. 25 using azeotropic distillation of water and purified by column chromatography. Diimines 2f,g were obtained by the method described previously26 using tetraethylxysilane as a dehydrating agent. In the latter case, we isolated iminoamines 5f,g along with C2-symmetric camphor diimine. Aliphatic diimines 2b,c,e are reduced in a regio- and stereo selective manner,27 yielding symmetric exo-, exo-diamines 3b,c,e. N-Methylation with dimethyl sulfate of amines 3b,c give tertiary amines 4b,c.
The reduction of diimines 2f,g by NaBH4 in the presence of NiCl2 yielded the corresponding aromatic secondary amines 3f,g. However, the degree of conversion in this case is low; the yield of diamines was 34–46%. The attempts to select conditions for reducing compounds 2f,g were unsuccessful. Many well-established methods failed to give the clean reaction, including the use of complex metal hydrides (i.e. NaBH4, NaBH3CN, NaBH(OAc)3, LiAlH4, Zn(BH4)2) in a number of solvents. The stability of compounds 2f,g against reduction can probably be attributed to their rigid aromatic structure and the presence of conjugation in the molecule (Scheme 1 ).
Scheme 1.
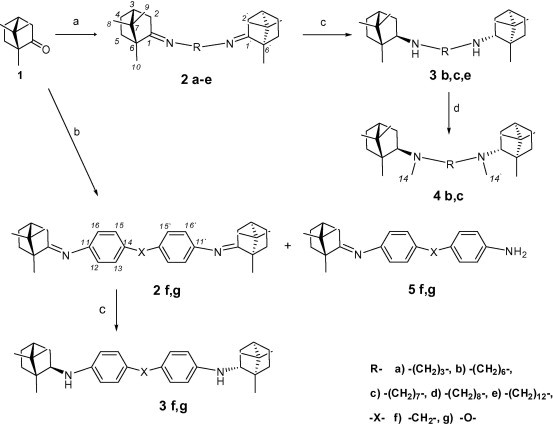
Reagents and conditions: (a) NH2-R-NH2 (0.50 equiv), BF3·Et2O (1–5 mol %), PhMe, reflux (Dean–Stark), 12–48 h; (b) NH2-Ph-X-Ph-NH2 (0.50 equiv), SiOEt4 (1 equiv), H2SO4 (5 mol %) reflux; (c) NiCl2 (4 equiv), NaBH4 (20 equiv), MeOH, –30 °C to rt; (d) (CH3O)2SO2 (10 equiv), K2CO3 (1.0 equiv), MeOH, rt, 12 h.
The endo position of H1 proton in compounds 3b,c,e,f,g is supported by the values of the constants of spin–spin coupling between it and protons H2n and Н2k (8.8 Hz and 7.5 Hz, respectively). In case of the exo position of Н1 atom, its constant of spin–spin coupling with Н2k proton would be equal to ∼12–13 Hz. The absence of long-range spin–spin proton coupling between protons Н5k and Н1 also argues in favor of the fact that the Н1 proton occupies the exo position. Compounds 2b–g, 3b,c,e,f,g, 4b,c and 5f,g are new and have not been previously described in the literature. Symmetric camphor diimine and diamine based on commercially available (1R)-camphor with ethane-1,2-diamine and propane-1,3-diamine were previously used as a chiral bidentate ligands.28
2.2. Study of biological activity. Antiviral activity
The obtained compounds 2a–g, 3b,c,g, 4b,c and 5f,g have been studied as potential antiviral agents. Adamantane- and bornane-based derivatives were used as reference compounds due to their close similarity to the compounds under investigation in having rigid cage fragments in their structures.
It should be noted that the initial camphor molecule does not possess a virus-inhibiting activity. Meanwhile, the study revealed a high inhibiting effect of symmetric compounds with aliphatic linkers between two camphor residues (compounds 2a–e) against influenza virus A/California/07/09 (H1N1)pdm09, mostly due to a decrease in toxicity. Compounds 2a,b,c,e were shown to possess the highest antiviral activity (Table 1 ). Diimine 2b has a selectivity index exceeding that for reference compounds 9-fold and more.
Table 1.
Antiviral activity of camphor-based compounds 1–5 against influenza virus A/California/7/09 (H1N1)pdm09 in MDCK cells
| Compound | CTD50a, μM | ED50b, μM | SIc |
|---|---|---|---|
| 1 | 3289.5 ± 216.0 | 1644.7 ± 144.4 | 2 |
| 2a | 283.6 ± 25.4 | 9.3 ± 0.8 | 30 |
| 2b | 1346.6 ± 126.1 | 15.1 ± 1.1 | 89 |
| 2c | 1256.3 ± 114.2 | 22.8 ± 2.3 | 55 |
| 2d | 390.1 ± 35.9 | 24.2 ± 2.1 | 16 |
| 2e | 2216.2 ± 214.8 | 45.0 ± 3.9 | 49 |
| 2f | >1072.9 | 351.4 ± 32.3 | 3 |
| 2g | >1067.0 | 373.4 ± 35.1 | 3 |
| 3b | 5.4 ± 0.4 | 0.77 ± 0.1 | 7 |
| 3c | 10.4 ± 1.0 | 3.2 ± 0.2 | 3 |
| 3e | 14.4 ± 1.5 | 4.4 ± 0.3 | 3 |
| 3f | >638.2 | 153.2 ± 11.9 | 4 |
| 3g | >635.5 | 207.6 ± 19.5 | 3 |
| 4b | 110.3 ± 9.1 | 110.1 ± 10.4 | 1 |
| 4c | 697.6 ± 72.2 | 40.4 ± 3.8 | 17 |
| 5f | 30.1 ± 2.9 | 15.0 ± 1.2 | 2 |
| 5g | 32.9 ± 2.7 | 4.2 ± 0.0 | 8 |
| Rimantadine | 335.2 ± 26.8 | 67.0 ± 4.9 | 5 |
| Amantadine | 284.1 ± 21.4 | 64.2 ± 4.7 | 4 |
| Deitiforin | 1266.2 ± 81.5 | 208.6 ± 15.4 | 6 |
| Ribavirind | >2000.0 | 24.6 | >81.0 |
CTD50—cytotoxic concentration; the concentration resulting in death of 50% of cells.
ED50—50% effective concentration; the concentration leading to 50% inhibition of virus replication.
SI—selectivity index, ratio CTD50/ED50.
Data are taken from Ref. 29.
During the course of the study, three types of modifications were applied to these compounds. As shown by structure–activity analysis, compounds bearing an amino group lost their efficacy in suppression of virus replication. With few exceptions, these compounds had much higher toxicity (low CTD50 values), resulting in lowering of their SIs. Indeed, ED50 values for diamines 3b and 3c are 0.77 and 3.2 μM, respectively, the values that could be recognized as a high antiviral activity. Nevertheless, their toxicity appeared to be high and, as a consequence, SI’s are low. Second, the introduction of aromatic moieties into linker also resulted in lack of virus suppression. Indeed, symmetrical diimines 2f,g connected by aromatic linkers, amino-imines 5f,g and diamine 3g did not exhibit high antiviral activity. The combination of these two modifications did not restore the antiviral activity (compounds 3f,g).
Based on the results of the study, two issues should be discussed. First, among the compounds 2a–2e, with one exception (2e), the strong dependence of anti-viral activity of the linker length can be observed. One can note that optimal number of carbon atoms in the linker is 6 (2b). The activity of compounds decreased when this number both increased (2c, 2d) and decreased (2a). This led to speculation that there are two sites of interaction of terminal camphor moieties with viral target, and that they demonstrate optimal activity when connected with the spacer of appropriate length.
Second, it should be noted that the described experiments were conducted using rimantadine-resistant strain of influenza virus. The resistance is due to amino acid substitution S31N (GenBank accession number AGI54797.1) typical of resistant strains. Therefore, despite structure similarity of substances under investigation with the known anti-influenza drugs amantadine and rimantadine, as well as with other M2 cage inhibitors, the mechanism of activity of symmetric camphor-based diimines should be different. Initially, adamantane derivatives were shown to interact with amino acids within the M2 proton channel.30 Recently, another adamantane-binding site was discovered in M2.31, 32 The mutations conferring amantadine- and rimantadine-resistance are located in transmembrane domain of M2 facing inside the channel. S31N is the most widely distributed mutation among drug-resistant strains. Due to spatial limitations, few modifications and side groups can be introduced into adamantane derivative without lacking its ability to fit into proton channel. Interaction with external amino acids suggests more wide range of chemical modifications resulting in high virus-inhibiting activity.
In order to address these issues, we performed a computer simulation of interaction of the most active compound 2b with M2 proton channel of influenza virus (Fig. 2 ).
Figure 2.
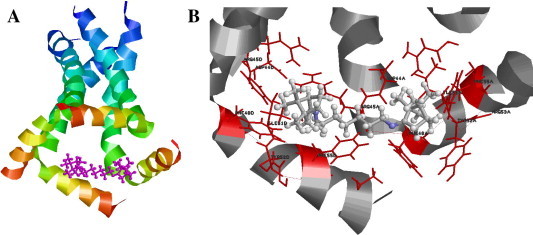
Model of interaction of 2b (marked in magenta) with M2 proton channel of influenza virus. (A) General view (outer domain of M2 is on the top and labeled blue), (B) close view from the inner side of M2.
According to the model, 2b binds to two adjacent sites in M2 in the area of amino acids 44–55. Each camphor moiety is coordinated with charged (Asp44 and Arg45) and aromatic (Phe48, Tyr52 and Phe55) amino acids. No hydrogen bonds have been established between 2b and M2. Their interaction can be provided by hydrophobic contacts with Phe48, Ile51 and Phe55.
It should be mentioned that despite docking experiment describes the interaction of 2b with specific amino acids of M2, in a framework of our study no direct evidence of such binding was obtained. For this reason, direct experiments should be performed to confirm this hypothesis using biophysical (conductance or binding testing) or virological (selection of resistant mutants and analysis of amino acid changes) methods.
As follows from the results obtained, symmetric dimeric derivatives of camphor with linker of specific length are more active than those with different spacer. This might be due to existence of two binding sites in the viral target. Combination of two camphor moieties in one molecule might increase the strength of binding of the compound with target by using two binding sites instead of one. Regarding the M2 protein, this is illustrated on the model presented, where two camphor moieties bind to two adjacent sites (amino acids 44–55) of neighboring polypeptide chains of M2. One can suppose, for instance, that such type of binding could restrict the ability of cytoplasmic domain of M2 to open the proton channel, or stabilizes its closed state. Nevertheless, once again, direct M2-inhibiting activity of 2b should be further demonstrated by specific tests.
3. Conclusion
In the present study, we have synthesized a set of C2-symmetric compounds with two camphor moieties (1,7,7-trimethylbicyclo[2.2.1]heptan) and two imine (amine) groups bound by linkers of various size and rigidity. The antiviral activity of these compounds was studied against influenza virus A/California/7/09 (H1N1)pdm09 in MDCK cells. The highest efficacy in virus inhibiting was shown for compounds 2a–e with cage moieties bound by aliphatic linkers. The therapeutic index (selectivity index) for 2b exceeded that for reference compounds amantadine, deitiforin and rimantadine almost 10-fold. As shown by structure–activity analysis, the length of the linker has a dramatic effect on the toxicity of compounds. Compound 2e with –C12H24– linker exhibited the lowest toxicity (CTD50 = 2216 μM).
The results obtained suggest that structure containing two imine groups linked by the aliphatic chain is the key element required for antiviral activity of the studied compounds. This can be illustrated by comparing of three compounds, diimines 2b,c,e and their derivatives diamines 3b,c,e. Despite low effective concentrations of 3b,c,e their toxicity is much higher than that for 2. This results in lower SI values. In summary, symmetric diimines 2a–e should be considered as a highly promising class of compounds effective against influenza.
4. Experimental section
4.1. General chemical methods
Reagents and solvents were purchased from commercial suppliers and used as received. Dry solvents were obtained according to the standard procedures. GC: 7820A gas chromatograph (Agilent Tech., USA); flame-ionization detector; HP-5 capillary column, He as carrier gas (flow rate 2 ml/min, flow division 99:1). Optical rotation: polAAr 3005 spectrometer; CHCl3 soln. 1H and 13C NMR spectra: Bruker DRX-500 apparatus at 500.13 MHz (1H) and 125.76 MHz (13C) in CDCl3; chemical shifts δ in ppm relative to residual CHCl3 [δ(H) 7.24, δ(C) 76.90 ppm], J in Hz. The structure of the products was determined by analyzing the 1H and 13C NMR spectra, assignments on a routine basis by a combination of 1D and 2D experiments (COSY, COLOC, HSCQ, HMBC). HR-MS: DFS Thermo Scientific spectrometer in a full scan mode (15–500 m/z, 70 eV electron impact ionization, direct sample administration). Spectral and analytical studies were carried out at the Collective Chemical Service Center of the Siberian Branch of the Russian Academy of Sciences. Column chromatography (CC) was performed on silica gel (60–200 l, Macherey-Nagel). The purity of the target compounds was determined by gas chromatography methods. All the target compounds reported in this paper have a purity of at least 95%.
The following reagents were used in this study: (1R)-(+) camphor (Alfa Aesar 98%, 45.5 (СHCl3, с 0.8)), ее 99, propane-1,3-diamine (Aldrich 98%), hexane-1,6-diamine (Aldrich 98%), heptane-1,7-diamine (Acros 98%), octane-1,8-diamine (Acros 98%), dodecane-1,12-diamine (ABCR 98%), 4,4′-methylenedianiline (ABCR 97%) 4,4′-oxydianiline (ABCR 97%).
The enumeration of atoms in compounds is given in order to assign the signals in the NMR spectra and does not coincide with the enumeration of atoms in the IUPAC name (see Supplementary). The specific rotation is given in (deg ml)∗(g dm)−1; concentrations of solutions, in (g) (100 ml)−1.
4.2. General synthetic procedure for diimin 2a–e (method a)
To a solution of (1R)-(+)-camphor 1 (3 g, 19.7 mmol, 1.0 equiv) and diamine (9.8 mmol, 0.5 equiv) in toluene (60 ml) 0.15 ml BF3·Et2O in 5 ml toluene were added. The solution was heated at reflux with a Deane-Stark trap condenser until no further water appeared. The combined organic layers were washed two times with brine, dried (Na2SO4) and evaporated to dryness. The reaction solution was then concentrated and crude product purified by flash silica gel column chromatography (hexane-ethyl acetate eluent) to obtain the desired diimine 2a–e.
4.2.1. N1E,N3E-N1,N3-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)propane-1,3-diamine (2a)
Yield: 74%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.68 s (C9H3, C9′H3), 0.86 s (C8H3, C8′H3), 0.90 s (C10H3, C10′H3), 1.12 ddd (H4endo, H4′endo, 2 J 12.3, J 4endo,5endo 9.3, J 4endo,5exo 4.2 Hz), 1.28 ddd (H5endo, H5′endo,2 J 13.0, J 5endo,4endo 9.3, J 5endo,4exo 4.4 Hz), 1.59 ddd (H5exo, H5′exo 2 J 13.0, J 5exo,4exo 12.2, J 5exo,4endo 4.2 Hz),), 1.77 d (H2endo, H2′endo, 2 J 16.9 Hz), 1.73–1.83 m (H4exo, H4′exo, 2H12), 1.86 dd (H3, H3′ J 3,2exo = J 3,4exo = 4.4 Hz), 2.27 ddd (H2exo, H2′exo, 2 J 16.9, J 2exo,3 4.4, J 2exo,4exo 3.3 Hz), 3.17 dt (H11а, H11′а, 2 J 12.2, J 11a,12 7.3 Hz), 3.22 dt (H11b, H11′b, 2 J 12.2, J 11b,12 7.3 Hz). 13C NMR (δ, ppm): 181.43 s (C1, C1′), 35.18 t (C2, C2′), 43.71 d (C3, C3′), 27.34 t (C4, C4′), 32.06 t (C5, C5′), 53.27 s (C6, C6′), 46.69 s (C7, C7′), 18.81 q (C8, C8′), 19.40 q (C9, C9′), 11.28 q (C10, C10′), 49.98 t (C11, C11′), 31.31 t (C12). −31 (СHCl3, c = 0.72). HRMS: calcd for C23H38N2: 342.3030, found: 342.3029. This compound was previously obtained.33 Spectral date agree with those specified in literature.
4.2.2. N1E,N6E-N1,N6-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)hexane-1,6-diamine (2b)
Yield: 70%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.70 s (C9H3, C9′H3), 0.87 s (C8H3, C8′H3), 0.91 s (C10H3, C10′H3), 1.14 ddd (H4endo, H4′endo, 2 J 12.2, J 4endo,5endo 9.3, J 4endo,5exo 4.1 Hz), 1.24–1.32 m (2H13, 2H13′, H5endo, H5′endo), 1.50–1.57 m (2H12, 2H12′), 1.60 ddd (H5exo, H5′exo, 2 J 13.0, J 5exo,4exo 12.2, J 5exo,4endo 4.1 Hz), 1.77 d (H2endo, H2′endo, 2 J 16.8 Hz), 1.79 ddddd (H4exo, H4′exo 2 J 12.2, J 4exo,5exo 12.2, J 4exo,5endo = J 4exo,3 = 4.3, J 4exo,2exo 3.2 Hz), 1.87 dd (H3, H3′, J 3,2exo = J 3,4exo = 4.3 Hz), 2.27 ddd (H2exo, H2′exo, 2 J 16.8, J 2exo,3 4.3, J 2exo,4exo 3.2 Hz), 3.12 dt (H11a, H11′a, 2 J 12.0, J 11a,12 7.3 Hz), 3.17 dt (H11b, H11′b, 2 J 12.0, J 11b,12 7.3 Hz). 13C NMR (δ, ppm): 180.99 s (C1, C1′), 35.21 t (C2, C2′), 43.71 d (C3, C3′), 27.37 t (C4, C4′), 32.09 t (C5, C5′), 53.23 s (C6, C6′), 46.66 s (C7, C7′), 18.83 q (C8, C8′), 19.40 q (C9, C9′), 11.31 q (C10, C10′), 52.19 t (C11, C11′), 30.44 t (C12, C12′), 27.31 t (C13, C13′). 18 (СHCl3, с = 1.0). HRMS: calcd for C26H44N2: 384.3503, found: 384.3499.
4.2.3. N1E,N7E-N1,N7-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)heptane-1,7-diamine (2c)
Yield: 70%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.70 s (C9H3, C9′H3), 0.87 s (C8H3, C8′H3), 0.91 s (C10H3, C10′H3), 1.14 ddd (H4endo, H4′endo, 2 J 12.2, J 4endo,5endo 9.3, J 4endo,5exo 4.2 Hz), 1.20–1.29 m (2H13, 2H13′, 2H14), 1.30 ddd (H5endo, H5′endo, 2 J 12.8, J 5endo,4endo 9.3, J 5endo,4exo 4.3 Hz), 1.49–1.56 m (2H12, 2H12′), 1.60 ddd (H5exo, H5′exo 2 J 12.8, J 5exo,4exo 12.2, J 5exo,4endo 4.2 Hz), 1.77 d (H2endo, H2′endo, 2 J 16.8 Hz), 1.79 ddddd (H4exo, H4′exo, 2 J 12.2, J 4exo,5exo 12.2, J 4exo,5endo 4.3, J 4exo,3 4.3, J 4exo,2exo 3.2 Hz), 1.87 dd (H3, H3′, J 3,4exo 4.3, J 3,2exo 4.3 Hz), 2.28 ddd (H2exo, H2′exo, 2 J 16.8, J 2exo,3 4.3, J 2exo,4exo 3.2 Hz), 3.12 dt (H11a, H11a′, 2 J 12.1, J 11a,12 7.3 Hz), 3.17 dt (H11b, H11′b, 2 J 12.1, J 11b,12 7.3 Hz). 13C NMR (δ, ppm): 180.97 s (C1, C1′), 35.21 t (C2, C2′), 43.73 d (C3, C3′), 27.38 t (C4, C4′), 32.10 t (C5, C5′), 53.23 s (C6, C6′), 46.67 s (C7, C7′), 18.84 q (C8, C8′), 19.40 q (C9, C9′), 11.32 q (C10, C10′), 52.26 t (C11, C11′), 30.39 t (C12, C12′), 27.40 t (C13, C13′), 29.28 t (C14). 20.7 (СHCl3, c = 0.66). HRMS: calcd for C27H46N2: 398.3653, found: 398.3656.
4.2.4. N1E,N8E-N1,N8-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)octane-1,8-diamine (2d)
Yield: 61%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.69 s (C9H3, C9′H3), 0.86 s (C8H3, C8′H3), 0.91 s (C10H3, C10′H3), 1.13 ddd (H4endo, H4′endo, 2 J 12.3, J 4endo,5endo 9.3, J 4endo,5exo 4.2 Hz), 1.19–1.30 m (2H13, 2H13′, 2H14, 2H14′), 1.29 ddd (H5endo, H5′endo, 2 J 13.0, J 5endo,4endo 9.3, J 5endo,4exo 4.4 Hz), 1.47–1.56 m (2H12, 2H12′), 1.59 ddd (H5exo, H5′exo 2 J 13.0, J 5exo,4exo 12.2, J 5exo,4endo 4.2 Hz), 1.77 d (H2endo, H2′endo, 2 J 16.7 Hz), 1.74-1.82 m (H4exo, H4′exo), 1.86 dd (H3, H3′, J 3,2exo 4.4, J 3,4exo 4.4 Hz), 2.27 ddd (H2exo, H2′exo, 2 J 16.7, J 2exo,3 4.4, J 2exo,4exo 3.2 Hz), 3.12 dt (H11a, H11′a, 2 J 12.1, J 11a,12 7.3 Hz), 3.16 dt (H11b, H11′b, 2 J 12.1, J 11b,12 7.3 Hz). 13C NMR (δ, ppm): 181.00 s (C1, C1′), 35.19 t (C2, C2′), 43.71 d (C3, C3′), 27.35 t (C4, C4′), 32.07 t (C5, C5′), 53.23 s (C6, C6′), 46.66 s (C7, C7′), 19.38 q (C8, C8′), 18.81 q (C9, C9′), 11.29 q (C10, C10′), 52.22 t (C11, C11′), 30.37 t (C12, C12′), 27.33 t (C13, C13′), 29.32 t (C14, C14′). −32.0 (СHCl3, c = 0.92). HRMS: calcd for C28H48N2: 412.3812, found: 412.3813.
4.2.5. N1E,N12E-N1,N12-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)dodecane-1,12-diamine (2e)
Yield: 49%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.71 s (C9H3, C9′H3), 0.88 s (C8H3, C8′H3), 0.92 s (C10H3, C10′H3), 1.15 ddd (H4endo, H4′endo, 2 J 12.2, J 4endo, 5endo 9.3, J 4endo, 5exo 4.2 Hz), 1.18–1.28 m (2H13, 2H13′, 2H14, 2H14′, 2H15, 2H15′, 2H16, 2H16′), 1.30 ddd (H5endo, H5′endo, 2 J 12.8, J 5endo, 4endo 9.3, J 5endo, 4exo 4.3 Hz), 1.49–1.57 m (2H12, 2H12′), 1.61 ddd (H5exo, H5′exo, 2 J 12.8, J 5exo,4exo 12.2, J 5exo, 4endo 4.2 Hz), 1.78 d (H2endo, H2′endo, 2 J 16.8 Hz), 1.76-1.84 m (H4exo, H4′exo), 1.88 dd (H3, H3′, J 3,2exo 4.4, J 3,4exo 4.4 Hz), 2.28 ddd (H2exo, H2′exo, 2 J 16.8, J 2exo,3 4.4, J 2exo, 4exo 3.2 Hz), 3.13 dt (H11a, H11′a, 2 J 12.1, J 11a,12 7.3 Hz), 3.17 dt (H11b, H11′b, 2 J 12.1, J 11b,12 7.3 Hz). 13C NMR (δ, ppm): 180.98 s (C1, C1′), 35.23 t (C2, C2′), 43.75 d (C3, C3′), 27.40 t (C4, C4′), 32.11 t (C5, C5′), 53.25 s (C6, C6′), 46.68 s (C7, C7′), 18.85 q (C8, C8′), 19.41 q (C9, C9′), 11.33 q (C10, C10′), 52.28 t (C11, C11′), 30.42 t (C12, C12′), 27.41 t (C13, C13′), 29.38 t, 29.46 t, 29.49 t (C14, C14′; C15, C15′; C16, C16′). −32.7 (СHCl3, c = 0.8). HRMS: calcd for C32H56N2: 468.4435, found: 468.4435.
4.3. General method for synthesis of compounds 2f-g (method b)
Camphor (15 mmol) and the diamine (7.5 mmol) were combined and treated with one drop of coned H2SO4. Si(OEt)4 (15 mmol) was added and the mixture was placed into a flask equipped with a still head. The solution was heated at 160 °C under argon. The distillate (EtOH) was discarded and the residue was dissolved in Et2O (50 mL) and washed with saturated NaHCO3 solution and H2O (25 mL each). The Et2O solution was dried (Na2SO4) and solvent removed under reduced pressure. The residue was dissolved in 10 mL of 95% EtOH and treated with 2 mL of 1 M KOH in EtOH. The solution was stirred for 15-20 min and filtered; the precipitate was washed with Et2O. The filtrate was washed with H2O (220 mL) and dried (Na2SO4). The solvent was removed under reduced pressure and purified by flash silica gel column chromatography (hexane-ethyl acetate eluent) to obtain the desired 2f,g and 5 f,g.
4.3.1. (E)-N-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)-4-(4-((E)-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)benzyl)aniline (2f)
Yield: 42%. Yellowish amorphous solid; mp 129–130 °С; 1H NMR (δ, ppm, J/Hz): 0.84 s (C9H3, C9′H3), 0.95 s (C8H3, C8′H3), 1.06 s (C10H3, C10′H3), 1.18–1.26 m (H4endo, H4′endo), 1.49 ddd (H5endo, H5′endo, 2 J 13.0, J 5endo,4endo 9.5, J 5endo,4exo 4.3 Hz), 1.73 d (H2endo, H2′endo, 2 J 17.7 Hz), 1.74 ddd (H5exo, H5′exo, 2 J 13.0, J 5exo,4exo 12.2, J 5exo,4endo 4.2 Hz), 1.81-1.89 m (H4exo, H4′exo), 1.87 dd (H3, H3′, J 3,2exo 4.3, J 3,4exo 4.3 Hz), 2.19 ddd (H2exo, H2′exo, 2 J 17.7, J 2exo,3 4.3, J 2exo,4exo 3.2 Hz), 3.88 s (2H17), 6.63 d (H12, H12′, H16, H16′, J 12,13(16,15) 8.2 Hz), 7.05 d (H13, H13′, H15, H15′, J 13,12(15,16) 8.2 Hz). 13C NMR (δ, ppm): 184.43 s (C1, C1′), 36.14 t (C2, C2′), 43.71 d (C3, C3′), 27.34 t (C4, C4′), 31.97 t (C5, C5′), 53.81 s (C6, C6′), 46.99 s (C7, C7′), 18.93 q (C8, C8′), 19.45 q (C9, C9′), 11.12 q (C10, C10′), 150.01 s (C11, C11′), 119.37 d (C12, C12′), 129.24 d (C13, C13′), 135.86 s (C14, C14′), 129.24 d (C15, C15′), 119.37 d (C16, C16′), 40.58 t (C17). 29.0 (СHCl3, с = 0.6). HRMS: calcd for C33H42N2: 466.3347, found: 466.3343.
4.3.2. (E)-4-(4-aminobenzyl)-N-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)aniline (5f)
Yield: 14%. Yellowish amorphous solid; mp 130–132 °С; 1H NMR (δ, ppm, J/Hz): 0.84 s (C9H3), 0.95 s (C8H3), 1.07 s (C10H3), 1.19–1.26 m (H4endo), 1.50 ddd (H5endo, 2 J 12.8, J 5endo,4endo 9.5, J 5endo,4exo 4.3 Hz), 1.73 d (H2endo, 2 J 17.7 Hz), 1.73–1.78 m (H5exo), 1.82–1.90 m (H4exo, H3), 2.18 ddd (H2exo, 2 J 17.7, J 2exo,3 4.3, J 2exo,4exo 3.2 Hz), 3.53 br s (NH2), 3.81 s (2H17), 6.60 d (H20, H22, J 20,19 (22,23) 8.2 Hz), 6.63 d (H12, H16, J 12,13 (16,15) 8.2 Hz), 6.95 d (H19, H23, J 19,20 8.2 Hz), 7.05 d (H13, H15, J 13,12 8.2 Hz). 13C NMR (δ, ppm): 184.40 s (C1), 36.12 t (C2), 43.68 d (C3), 27.31 t (C4), 31.94 t (C5), 53.77 s (C6), 46.96 s (C7), 18.90 q (C8), 19.42 q (C9), 11.11 q (C10), 149.91 s (C11), 119.34 d (C12, C16), 129.13 d (C13, C15), 136.22 s (C14), 40.32 t (C17), 131.48 s (C18), 129.56 d (C19, C23), 115.11 d (C20,C22), 144.25 s (C21). 18.0 (СHCl3, с = 0.6). HRMS: calcd for C23H28N2: 332.2244, found: 332.2247.
4.3.3. (E)-N-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)-4-(4-((E)-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)amino)phenoxy)aniline (2g)
Yield: 34%. White amorphous solid; mp 164–165 °С; 1H NMR (δ, ppm, J/Hz): 0.83 s (C9H3, C9′H3), 0.95 s (C8H3, C8′H3), 1.06 s (C10H3, C10′H3), 1.18–1.26 m (H4endo, H4′endo), 1.49 ddd (H5endo, H5′endo, 2 J 13.0, J 5endo,4endo 9.4, J 5endo,4exo 4.3 Hz), 1.75 ddd (H5exo, H5′exo, 2 J 13.0, J 5exo,4exo 12.2, J 5exo,4endo 4.2 Hz), 1.76 d (H2endo, H2′endo, 2 J 17.7 Hz), 1.82-1.90 m (H4exo, H4′exo, H3, H3′), 2.21 ddd (H2exo, H2′exo, 2 J 17.7, J 2exo,3 4.4, J 2exo,4exo 3.2 Hz), 6.67 d (H12, H12′, H16, H16′, J 12,13 (16,15) 8.8 Hz), 6.89 d (H13, H13′, H15, H15′, J 13,12 8.8 Hz). 13C NMR (δ, ppm): 184.94 s (C1, C1′), 36.17 t (C2, C2′), 43.70 d (C3, C3′), 27.31 t (C4, C4′), 31.93 t (C5, C5′), 53.86 s (C6, C6′), 46.98 s (C7, C7′), 18.89 q (C8, C8′), 19.43 q (C9, C9′), 11.10 q (C10, C10′), 147.29 s (C11, C11′), 120.53 d (C12, C12′), 118.98 d (C13, C13′), 153.36 s (C14, C14′), 118.98 d (C15, C15′), 120.53 d (C16, C16′). 30.3 (СHCl3, с = 0.56). HRMS: calcd for C32H40N2: 468.3135, found: 468.3138.
4.3.4. (E)-4-(4-aminophenoxy)-N-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylidene)aniline (5g)
Yield: 11%. White amorphous solid; mp 118–120 °С; 1H NMR (δ, ppm, J/Hz): 0.83 s (C9H3), 0.95 s (C8H3), 1.06 s (C10H3), 1.18–1.26 m (H4endo), 1.49 ddd (H5endo, 2 J 13.0, J 5endo,4endo 9.4, J 5endo,4exo 4.4 Hz), 1.71-1.78 m (H5exo), 1.75 d (H2endo, 2 J 17.7 Hz), 1.82-1.90 m (H4exo, H3), 2.20 ddd (H2exo, 2 J 17.7, J 2exo,3 4.4, J 2exo,4exo 3.2 Hz), 3.53 br s (NH2), 6.63 d (H19, H21, J 19,18 (21,22) 8.8 Hz), 6.65 d (H12, H16, J 12,13 (16,15) 8.8 Hz), 6.82 d (H18, H22, J 18,19 (22,21) 8.8 Hz, 6.85 d (H13, H15, J 13,12 (15,16) 8.8 Hz). 13C NMR (δ, ppm): 184.89 s (C1), 36.18 t (C2), 43.71 d (C3), 27.32 t (C4), 31.93 t (C5), 53.85 s (C6), 46.97 s (C7), 18.90 q (C8), 19.44 q (C9), 11.12 q (C10), 146.79 s (C11), 120.47 d (C12, C16), 118.18 d (C13, C15), 154.21 s (C14), 149.54 s (C17), 120.17 d (C18, C22), 116.07 d (C19, C21), 142.04 s (C20). 28.2 (СHCl3, с = 0.56). HRMS: calcd for C22H426O1N2: 334.2040, found: 334.2036.
4.4. General synthetic procedure for reduction of diimine 2a-g (method c)
Camphor diimine (10 mmol) and NiCl2·6H20 (40 mmol) were taken in methanol (100 ml) and cooled to –30 °C. NaBH4 (200 mmol) was added in portions to this mixture from a solid addition flask under stirring at −30 °C over a period of 1 h. The reaction mixture was further stirred for 1 h at −30 °C and for 4 h at room temperature. 3 N NaOH solution (15 ml) and ether (100 ml) were added, and the contents were filtered. The aqueous and organic layers of the filtrate were separated. The organic layer was washed with saturated NaCl solution (2 × 15 ml), dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure and crude product purified by flash silica gel column chromatography to obtain the desired 3b,c,e,f,g.
4.4.1. N1,N6-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)hexane-1,6-diamine (3b)
Yield: 92%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.77 s (C9H3, C9′H3), 0.83 s (C10H3, C10′H3), 0.98 s (C8H3, C8′H3), 0.99–1.07 m (H4endo, H4′endo, H5endo, H5′endo), 1.25–1.32 m (2H13, 2H13′), 1.35–1.44 m (2H12, 2H12′), 1.44–1.55 m (H5exo, H5′exo, 2H2, 2H2′), 1.60–1.69 m (H3, H3′, H4exo, H4′exo), 2.40 dt (H11a, H11′a, 2 J 11.3, J 11a,12, 7.0 Hz), 2.46 dd (H1endo, H1′endo, J 1endo,2endo 7.0, J 1endo,2exo 6.2 Hz), 2.50 dt (H11b, H11′b, 2 J 11.3, J 11b,12 7.2 Hz). 13C NMR (δ, ppm): 66.84 d (C1, C1′), 39.06 t (C2, C2′), 45.18 d (C3, C3′), 27.28 t (C4, C4′), 36.89 t (C5, C5′), 48.20 s (C6, C6′), 46.51 s (C7, C7′), 20.41 q (C8, C8′), 20.50 q (C9, C9′), 12.05 q (C10, C10′), 48.78 t (C11, C11′), 30.38 t (C12, C12′), 27.34 t (C13, C13′). 55 (СHCl3 c = 0.72). HRMS: calcd for C26H48N2: 388.3808, found: 388.3812.
4.4.2. N1,N7-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)heptane-1,7-diamine (3c)
Yield: 87%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.77 s (C9H3, C9′H3), 0.84 s (C10H3, C10′H3), 0.98 s (C8H3, C8′H3), 0.99–1.07 m (H4endo, H4′endo, H5endo, H5′endo), 1.23–1.31 m (2H13, 2H13′, 2H14), 1.35–1.43 m (2H12, 2H12′), 1.43–1.56 m (H5exo, H5′exo, 2H2, 2H2′), 1.61–1.70 m (H3, H3′, H4exo, H4′exo), 2.40 dt (H11a, H11′a, 2 J 11.3, J 11a,12 7.0 Hz), 2.47 dd (H1endo, H1′endo, J 1endo,2endo 7.0, J 1endo,2exo 6.2 Hz), 2.50 dt (H11b, H11′b, 2 J 11.3, J 11b,12 7.2 Hz). 13C NMR (δ, ppm): 66.85 d (C1, C1′), 39.07 t (C2, C2′), 45.18 d (C3, C3′), 27.28 t (C4, C4′), 36.90 t (C5, C5′), 48.21 s (C6, C6′), 46.52 s (C7, C7′), 20.41 q (C8, C8′), 20.50 q (C9, C9′), 12.06 q (C10, C10′), 48.84 t (C11, C11′), 30.37 t (C12, C12′), 27.38 t (C13, C13′), 29.42 t (C14). 75.1 (СHCl3, c = 0.66). HRMS: calcd for C27H50N2: 402.3965, found: 402.3960.
4.4.3. N1,N12-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)dodecane-1,12-diamine (3e)
Yield: 82%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.78 s (C9H3, C9′H3), 0.84 s (C10H3, C10′H3), 0.99 s (C8H3, C8′H3), 0.99-1.07 m (H4endo, H4′endo, H5endo, H5′endo), 1.21–1.30 m (2H13, 2H13′, 2H14, 2H14′, 2H15, 2H15′, 2H16, 2H16′), 1.36–1.43 m (2H12, 2H12′), 1.43–1.54 m (H5exo, H5′exo), 1.49–1.58 m (2H2, 2H2′), 1.61–1.64 m (H3, H3′), 1.63–1.71 m (H4exo, H4′exo), 2.41 dt (H11a, H11′a, 2 J 11.3, J 11a,12 7.0 Hz), 2.47 dd (H1endo, H1′endo, J 1endo,2endo 7.0, J 1endo,2exo 6.2 Hz), 2.51 dt (H11b, H11′b, 2 J 11.3, J 11b,12 7.3 Hz). 13C NMR (δ, ppm): 66.87 d (C1, C1′), 39.08 t (C2, C2′), 45.20 d (C3, C3′), 27.29 t (C4, C4′), 36.92 t (C5, C5′), 48.22 s (C6, C6′), 46.53 s (C7, C7′), 20.42 q (C8, C8′), 20.51 q (C9, C9′), 12.06 q (C10, C10′), 48.87 t (C11, C11′), 30.41 t (C12, C12′), 27.41 t (C13, C13′), 29.52 t, 29.50 t, 29.49 t (C14, C14′; C15, C15′; C16, C16′). −88.6 (СHCl3, c = 1). HRMS: calcd for C32H60N2: 472.4751, found: 472.4752.
4.4.4. (1R,2R,4R)-1,7,7-trimethyl-N-(4-(4-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylamino)benzyl)phenyl)bicyclo[2.2.1]heptan-2-amine (3f)
Yield: 46%. Yellowish amorphous solid; mp 134 °С; 1H NMR (δ, ppm, J/Hz): 0.86 s (C9H3, C9′H3), 0.92 s (C10H3, C10′H3), 1.02 s (C8H3, C8′H3), 1.13–1.30 m (H4endo, H4′endo, H5endo, H5′endo), 1.56-1.81 m (H2exo, H2′exo, H3, H3′, H4exo, H4′exo, H5exo, H5′exo), 1.87 dd (H2endo, H2′endo, 2 J 12.7, J 2endo,1endo 8.3 Hz), 3.24 dd (H1endo, H1′endo, J 1endo,2endo 8.3, J 1endo,2exo 4.6 Hz), 3.62 br s (2NH), 3.75 s (2H17), 6.49 d (H12, H12′, H16, H16′, J 12,13 (16,15) 8.2 Hz), 6.96 d (H13, H13′, H15, H15′, J 13,12 8.2 Hz). 13C NMR (δ, ppm): 61.60 d (C1, C1′), 40.69 t (C2, C2′), 45.07 d (C3, C3′), 27.28 t (C4, C4′), 36.63 t (C5, C5′), 48.58 s (C6, C6′), 47.02 s (C7, C7′), 20.35 q (C8, C8′), 20.35 q (C9, C9′), 12.15 q (C10, C10′), 146.10 s (C11, C11′), 112.54 d (C12, C12′), 129.34 d (C13, C13′), 130.00 s (C14, C14′), 129.34 d (C15, C15′), 112.54 d (C16, C16′), 39.89 t (C17). 25.0 (СHCl3, с = 0.6). HRMS: calcd for C33H46N2: 470.3649, found: 470.3656.
4.4.5. (1R,2R,4R)-1,7,7-trimethyl-N-(4-(4-((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylamino)phenoxy)phenyl)bicyclo[2.2.1]heptan-2-amine (3g)
Yield: 34%. White amorphous solid; mp 123–125 °С; 1H NMR (δ, ppm, J/Hz): 0.85 s (C9H3, C9′H3), 0.92 s (C10H3, C10′H3), 1.02 s (C8H3, C8′H3), 1.13–1.20 m (H4endo, H4′endo), 1.24 ddd (H5endo, H5′endo, 2 J 13.0, J 5endo,4endo 9.3, J 5endo,4exo 4.2 Hz), 1.61 ddd (H5exo, H5′exo, 2 J 13.0, J 5exo,4exo 12.2, J 5exo,4endo 4.2 Hz), 1.64–1.80 m (H2exo, H2′exo, H3, H3′, H4exo, H4′exo), 1.86 dd (H2endo, H2′endo, 2 J 12.8, J 2endo,1endo 8.3 Hz), 3.21 dd (H1endo, H1′endo, J 1endo,2endo 8.3, J 1endo,2exo 4.7 Hz), 3.60 br s (2NH), 6.50 d (H12, H12′, H16, H16′, J 12,13 (16,15) 8.8 Hz), 6.80 dd (H13, H13′, H15, H15′, J 13,12 8.8 Hz). 13C NMR (δ, ppm): 62.14 d (C1, C1′), 40.65 t (C2, C2′), 45.11 d (C3, C3′), 27.29 t (C4, C4′), 36.68 t (C5, C5′), 48.65 s (C6, C6′), 47.04 s (C7, C7′), 20.38 q (C8, C8′), 20.36 q (C9, C9′), 12.17 q (C10, C10′), 143.73 s (C11, C11′), 113.42 d (C12, C12′), 119.31 d (C13, C13′), 149.33 s (C14, C14′), 119.31 d (C15, C15′), 113.42 d (C16, C16′). 33.0 (СHCl3, с = 0.6). HRMS: calcd for C32H44O1N2: 472.3450, found: 472.3448.
4.5. General method for synthesis of compounds 4b,c (method d)
Camphor diamine (1.5 mmol), K2CO3 (1.5 mmol), and dimethyl sulfate (15 mmol) were mixed in methanol. Reaction mixture was stirring at room temperature during 12 h. Than 10 ml 5% NaOH and saturated NaCl solution was added. The aqueous layer was extracted with CH2Cl2 (3 × 10 ml). The combined organic phases were dried over Na2SO4. The solvent was evaporated to dryness and crude product purified by flash silica gel column chromatography to obtain the desired 4b and 4c.
4.5.1. N1,N6-dimethyl-N1,N6-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl) hexane-1,6-diamine (4b)
Yield: 52%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.77 s (C9H3, C9′H3), 0.92 s (С8H3, C8′H3), 0.95 s (C10H3, C10′H3), 0.98–1.06 m (H4endo, H4′endo, H5endo, H5′endo), 1.18–1.26 m (2H13, 2H13′), 1.37 dd (H2endo, H2′endo, 2 J 12.6, J 2endo,1endo 8.8 Hz), 1.31–1.50 m (H5exo, H5′exo, 2H12, 2H12′), 1.57 dd (H3, H3′, J 3,2exo = J 3,4exo 4.3 Hz), 1.59–1.69 m (H4exo, H4′exo), 1.85 dddd (H2exo, H2′exo, 2 J 12.6, J 2exo,1endo 6.0, J 2exo,3 4.3, J 2exo,4exo 3.2 Hz), 2.14 s (С14H3, C14′H3), 2.15 dd (H1endo, H1′endo, J 1endo,2endo 8.5, J 1endo,2exo 6.0 Hz), 2.18 ddd (H11а, Н11′а, 2 J 12.8, J 11a,12a 9.8, J 11a,12b 5.4 Hz), 2.39 ddd (Н11b , Н11′b, 2 J 12.8, J 11b,12b 10.0, J 11b,12а 5.4 Hz). 13C NMR (δ, ppm): 73.23 d (C1, C1′), 34.93 t (C2, C2′), 44.91 d (C3, C3′), 27.31 t (C4, C4′), 37.22 t (C5, C5′), 49.38 s (C6, C6′), 46.94 s (C7, C7′), 19.61 q (C8, C8′), 20.72 q (C9, C9′), 14.39 q (C10, C10′), 57.33 t (C11, C11′), 26.94 t (C12, C12′), 27.47 t (C13, C13′), 40.80 q (C14, C14′). −85.6 (СHCl3 c = 0.5). HRMS: calcd for C28H52N2: 416.4127, found: 416.4125.
4.5.2. N1,N7-dimethyl-N1,N7-bis((1R,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)heptane-1,7-diamine (4c)
Yield: 44%. Colourless oil; 1H NMR (δ, ppm, J/Hz): 0.77 s (C9H3, C9′H3 ′), 0.91 s (С8H3, C8′H3), 0.95 s (C10H3, C10′H3), 0.98–1.05 m (H4endo, H4′endo, H5endo, H5′endo), 1.17–1.31 m (2H13, 2H13′, 2H14), 1.37 dd (H2endo, H2′endo, 2 J 12.4, J 2endo,1endo 8.6 Hz), 1.32–1.44 m (H5exo, H5′exo, 2H12, 2H12′), 1.57 dd (H3, H3′, J 3,2exo = J 3,4exo = 4.3 Hz), 1.60–1.68 m (H4exo, H4′exo), 1.85 dddd (H2exo, H2′exo, 2 J 12.4, J 2exo,1endo 6.1, J 2exo,3 4.3, J 2exo,4exo 3.2 Hz), 2.14 s (С15H3, C15H3 ′), 2.15 dd (H1endo, H1′endo, J 1endo,2exo 8.6, J 1endo,2exo 6.1 Hz), 2.18 ddd (H11а, Н11′а, 2 J 12.8, J 11a,12a 9.8, J 11a,12b 5.3 Hz), 2.40 ddd (Н11b , Н11′b, 2 J 12.8, J 11b,12b 10.2, J 11b,12а 5.4 Hz). 13C NMR (δ, ppm): 73.21 d (C1, C1′), 34.92 t (C2, C2′), 44.92 d (C3, C3′), 27.32 t (C4, C4′), 37.22 t (C5, C5′), 49.37 s (C6, C6′), 46.94 s (C7, C7′), 19.61 q (C8, C8′), 20.73 q (C9, C9′), 14.37 q (C10, C10′), 57.35 t (C11, C11′), 26.86 t (C12, C12′), 27.44 t (C13, C13′), 29.66 t (C14, C14′), 40.82 q (C15, C15′). 85.6 (СHCl3, c = 0.5). HRMS: calcd for C29H54N2: 430.4283, found: 430.4282.
4.6. Viruses and cells
Influenza virus A/California/07/09 (H1N1)pdm09 from the collection of viruses of the Research Institute of Influenza (St. Petersburg, Russia) was used. The virus was cultivated in 10-12-day-old chicken embryos for 48 h at +37 °C. MDCK cells (ATCC CCL 34) in a minimal essential medium (MEM, PAA, Austria, Cat.# E15-825) were seeded on 96-well plates (Orange Scientific no. 5530100) and incubated at 37 °C in 5% CO2 until a confluent monolayer formed. To cultivate the virus, 5% albumin, trypsin (1 μg/ml), and 16 mM HEPES (pH 7.6) were added.
4.7. Toxicity studies
Microtetrazolium test (MTT) was used to study cytotoxicity of the compounds (Mossman, 1983). Briefly, series of two-fold dilutions of each compound (1000–4 μg/ml) in MEM were prepared. MDCK cells were incubated for 48 h at 37 °C in 5% CO2 in the presence of the dissolved substances. The degree of destruction of the cell monolayer was then evaluated in the microtetrazolium test (MTT). The cells were washed twice with saline, and a solution of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (ICN Biochemicals Inc., Aurora, Ohio) (0.5 μg/ml) in saline was added to the wells. After 1 h incubation, the wells were washed and the formazan residue dissolved in DMSO (0.1 ml per well). The optical density of cells was then measured on a Victor 2 1440 multifunctional reader (Perkin Elmer, Finland) at 535 nm and plotted against concentration of the compounds. Each concentration was tested in three parallels. The 50% cytotoxic dose (CTD50) of each compound (i.e., the compound concentration that causes the death of 50% cells in a culture, or decreasing the optical density twice as compared to the control wells) was calculated from the data obtained.
4.8. Determination of the antiviral activity
The compounds in appropriate concentrations were incubated with MDCK cells for 1 h at 37 °C. The cell culture was then infected with 10-fold dilutions of the virus (10−1 to 10−6). The plates were incubated for 48 h at 37 °C in the presence of 5% CO2. The infection activity of the virus was evaluated in the hemagglutination reaction with chicken erythrocytes. A virus-containing solution (100 μl) was placed in the wells of a round-bottom plate. An equal amount of a 1% suspension of chicken erythrocytes in saline was added. The reaction was evaluated after 60 min incubation at room temperature. Each concentration of the compounds was tested in three parallels. A virus titer was considered as reciprocal to decimal logarithm of maximum dilution that caused complete agglutination of erythrocytes and was expressed in logarithms of the 50% experimental infection dose (log10 EID50). The antiviral activity of the compounds was estimated by the decrease in the virus titer as compared to the control. The 50% effective dose (ED50) of the drug, i.e., the concentration at which the virus production decreased by a factor of two (a virus titer per 0.3 log10 EID50) and the selectivity index (the ratio between CTD50 and ED50) were calculated from the data obtained.
4.9. Computer modeling
The molecular docking for modeling the interaction between compounds under investigation and influenza virus M2 protein (protein database code 2LOJ) was done by Hex online server (http://hexserver.loria.fr/).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2014.02.038.
Supplementary data
This article contains supplementary informations.
References
- 1.Nicoll A., Brown C., Karcher F., Penttinen P., Hegermann-Lindencrone M., Villanueva S., Ciotti M., Jean-Gilles L., Rehmeta S., Nguyen-Van-Tamd J. Bull. World Health Organ. 2012;90:311. doi: 10.2471/BLT.11.097972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Clercq E. Vol. 5. Elsevier; 2007. (Advances in Antiviral Drug Design). [Google Scholar]
- 3.Hauge S.H., Dudman S., Borgen K., Lackenby A., Hungnes O. Infect. Dis. 2009;15:155. doi: 10.3201/eid1502.081031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ison M.G. Influenza Other Respir. Viruses. 2013;7:7. doi: 10.1111/irv.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee S.M., Yen H.L. Antiviral Res. 2012;96:391. doi: 10.1016/j.antiviral.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torriani F.J., Rodriguez-Torres M., Rockstroh J.K., Lissen E., Gonzalez-Garcia J., Lazzarin A., Carosi G., Sasadeusz J., Katlama C., Montaner J., Sette H., Passe S., De Pamphilis J., Duff F., Schrenk U.M., Dieterich D.T.N. Eng. J. Med. 2004;351:438. doi: 10.1056/NEJMoa040842. [DOI] [PubMed] [Google Scholar]
- 7.Davies W.L., Grunert R.R., Haff R.F., McGahen J.W., Neumayer E.M., Paulshock M., Watts J.C., Wood T.R., Hermann E.C., Hoffmann C.E. Science. 1964;144:862. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- 8.Scholtissek C., Quack G., Klenk H.D., Webster R.G. Antiviral Res. 1998;37:83. doi: 10.1016/s0166-3542(97)00061-2. [DOI] [PubMed] [Google Scholar]
- 9.Cady S.D., Schmidt-Rohr K., Wang J., Soto C.S., DeGrado W.F., Hong M.H. Nature. 2010;463:689. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belshe R.B., Burk B., Newman F., Cerruti R.L., Sim I.S. J. Infect. Dis. 1989;159:430. doi: 10.1093/infdis/159.3.430. [DOI] [PubMed] [Google Scholar]
- 11.Antiviral Agents for the Treatment and Chemoprophylaxis of Influenza. Recommendations and Reports2011, 60, 1. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6001a1.htm). [PubMed]
- 12.Abed Y., Goyette N., Boivin G. Antimicrob. Agents Chemother. 2005;49:556. doi: 10.1128/AAC.49.2.556-559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoidis G., Kolocouris N., Naesens L., Clercq E.D. Bioorg. Med. Chem. 2009;17:1534. doi: 10.1016/j.bmc.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 14.DeGrado, W.F.; Wang, J. (University of Pennsylvania), 2011, WO 2011/022191.
- 15.Tanner J.A., Zheng B.J., Zhou J., Watt R.M., Jiang J.Q., Wong K.L., Lin Y.P., Lu L.Y., He M.L., Kung H.F., Kesel A. J. Chem. Biol. 2005;12:303. doi: 10.1016/j.chembiol.2005.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tandura S.N., Zarubaev V.V., Anfimov P.M., Kiselev O.I. Antibiot. Khimioter. 2013;58:36. [PubMed] [Google Scholar]
- 17.Xiong Z.Y., Xiao F.M., Xu X., Wu Y.F., Jiang X.M. Zhongguo Zhong Yao Za Zhi. 2013;38:786. [PubMed] [Google Scholar]
- 18.Jassim S.A.A., Naji M.A. J. Appl. Microbiol. 2003;95:412. doi: 10.1046/j.1365-2672.2003.02026.x. [DOI] [PubMed] [Google Scholar]
- 19.Astani A., Reichling J., Schnitzler P. Phytother. Res. 2010;24(5):673–679. doi: 10.1002/ptr.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ardashov O.V., Zarubaev V.V., Shtro A.A., Korchagina D.V., Volcho K.P., Salakhutdinov N.F., Kiselev O.I. Lett. Drug Des. Discov. 2011;8:375. [Google Scholar]
- 21.Teplov G.V., Suslov E.V., Zarubaev V.V., Shtro A.A., Karpinskaya L.A., Rogachev A.D., Korchagina D.V., Volcho K.P., Salakhutdinov N.F., Kiselev O.I. Lett. Drug Des. Discov. 2013;10:477. [Google Scholar]
- 22.Sztanke K., Maziarka A., Osinka A., Sztanke M. Bioorg. Med. Chem. 2013;21:3648. doi: 10.1016/j.bmc.2013.04.037. [DOI] [PubMed] [Google Scholar]
- 23.Bhat M.A., Al-Omar M.A. Med. Chem. Res. 2013;22:4522. [Google Scholar]
- 24.Mann J.C., Hobbs J.B., Banthorpe D.V., Harborne J.B. Longman Scientific & Technical; Harlow, Essex, England: 1994. Natural Products: Their Chemistry And Biological Significance. [Google Scholar]
- 25.Caselli A., Giovenzana G.B., Palmisano G., Sisti M., Pilati T. Tetrahedron: Asymmetry. 2003;14:1451. [Google Scholar]
- 26.Love B., Ren J. J. Org. Chem. 1993;58:5556. [Google Scholar]
- 27.Periasamy M., Devasagayaraj A., Satyanarayana N., Narayana C. Synth. Commun. 1989;19:565. [Google Scholar]
- 28.Sanjeevakumar N., Periasamy M. Tetrahedron: Asymmetry. 1842;2009:20. [Google Scholar]
- 29.Sokolov D.N., Zarubaev V.V., Shtro A.A., Polovinka M.P., Luzina O.A., Komarova N.I., Salakhutdinov N.F., Kiselev O.I. Bioorg. Med. Chem. Lett. 2012;22:7060. doi: 10.1016/j.bmcl.2012.09.084. [DOI] [PubMed] [Google Scholar]
- 30.Pinto L.H., Lamb R.A. Trends Microbiol. 1995;3:271. doi: 10.1016/s0966-842x(00)88942-8. [DOI] [PubMed] [Google Scholar]
- 31.Schnell J.R., Chou J.J. Nature. 2008;451:591. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pielak R.M., Schnell J.R., Chou J.J. Proc. Natl. Acad. Sci. U.S.A. 2009;106:7379. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raza Z., Dakovic S., Vinkovic V., Dunjic V. Croat. Chem. Acta. 1996;69:1545. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This article contains supplementary informations.