

Abstract
A hybridisation-based genosensor was designed on a 100 nm sputtered gold film. This material worked as an immobilisation and transduction surface. A 30-mer sequence that encodes a short lysine-rich region, unique to SARS (severe acute respiratory syndrome) virus, was chosen as target. A complementary strand (probe), labelled with a thiol group at the 3′-end, was immobilised on the film. After blocking the surface, hybridisation with the biotin-conjugated SARS strand (at the 3′-end) took place. Interaction with alkaline phosphatase-labelled streptavidin permits amplified indirect electrochemical detection. The analytical signal is constituted by an electrochemical process of indigo carmine, the soluble product of the enzymatic hydrolysis of 3-indoxyl phosphate. The use of a sensitive electrochemical technique such as square wave voltammetry allowed a detection limit of 6 pM to be obtained for this DNA sequence, lower than any other found in the bibliography. The parameters affecting the methodology were studied, with special attention being placed on selectivity. Specificity was clearly enhanced when interaction time and stringency (in the form of formamide percentage) were increased. With 1 h of strand interaction and employing 50% of formamide in the hybridisation buffer, a 3-base mismatch strand was perfectly distinguished from the complementary.
Keywords: DNA, Hybridisation assay, Genosensor, Enzyme assay, Gold film, Electrochemical detection (ED), SARS
1. Introduction
Biosensors have become a very important area of Analytical Chemistry, since the majority of current and future analytical requirements are being solved by simple and sensitive devices (Nakamura and Karube, 2003). Apart from the well-established enzymatic sensors, the selectivity of affinity interactions such as those of antigen–antibody or nucleic acid hybridisation is exploited for the respective development of promising immuno and nucleic acid assays (Campas and Katakis, 2004). Nucleic acid detection is becoming relevant not only in the field of food analysis but also in clinical diagnosis. A DNA/RNA test addresses the question of whether a patient is infected with a particular pathogen or not. It is advantageous compared to immunoassay in cases of recent infection or underlying immunodeficiency. Moreover, it is very useful for treatment monitoring, since elimination of pathogen nucleic acids indicates successful handling.
Miniaturisation, on the other hand, is one of the more recent trends in Analytical Chemistry. Although it is important in all fields, it is of special relevance in the area of biosensors. The simplicity of the electrochemical instrumentation and the type of measurement that is made adapt well to miniaturisation requirements (Matisyk, 2003). Several alternative ways of electrochemically detecting DNA are possible (Wang, 2002). The intrinsic electroactivity of adenine and guanine can be the basis for direct measurement of nucleic acids (Wu et al., 2003, De-Los-Santos-Alvarez et al., 2002). The determination of electroactive indicators that intercalate or otherwise associate with double-stranded DNA (Gherghi et al., 2004, Zhu et al., 2003) is another alternative. The use of enzymes is a possibility that enhances assay sensitivity enormously due to their inherent amplification. Horseradish-peroxidase has been used as an enzyme label (Alfonta et al., 2001, Pividori et al., 2001) as well as glucose oxidase (Domínguez et al., 2004) and alkaline phosphatase (Carpini et al., 2004, Aguilar and Ritsch, 2003). Although enzymes can be directly conjugated, the use of a well-known affinity interaction such as avidin–biotin is an easy alternative. Alkaline phosphatase (AP) converts orthophosphoric monoesters into alcohols and thus various phosphate esters can act as substrates. Phenylphosphate (Jenkins et al., 1988), p-aminophenylphosphate (Meusel et al., 1995) and p-nitrophenylphosphate (Tie et al., 1992) are among the most common. α-naphthylphosphate (NP), which is hydrolysed to naphthol, has been widely used (Abad-Villar et al., 2002a, Carpini et al., 2004, Xu et al., 2001). 3-Indoxyl phosphate (3-IP) has been proposed by our research group as an adequate substrate for AP in immunoelectrochemical approaches (Fernández-Sánchez and Costa-García, 1998). Due to its satisfactory electrochemical behaviour, it has been used for the development of AP-based enzyme immunoassays (Bengoechea-Álvarez et al., 2002), this being the first time for it to be used to perform DNA assays on gold surfaces.
The surface of the genosensor is important in the performance of the analytical device because it is where immobilisation and transduction take place (Pividori et al., 2000). The use of carbon and gold electrodes as electrochemical transducers for DNA hybridisation sensors has recently been summarised (Lucarelli et al., 2004). Gold has always been an appropriate material for the electrochemical detection of substances, while also allowing different formats. Thin-film gold electrodes produced by magnetron sputtering have been voltammetrically and amperometrically characterized (Brett et al., 1997). The quartz crystal balance for piezoelectric measurements and surface plasmon resonance, likewise based on thin gold electrodes, have been respectively used for DNA analysis (Minunni et al., 2003, Jordan et al., 1997). Gold also offers the relevant possibility of generating self-assembled DNA monolayers through thiol groups (Levicky et al., 1998, Zhang et al., 2002) due to the strong binding between sulphur and gold, which may be considered almost covalent (Bain et al., 1989).
In this paper, a DNA hybridisation assay with enzymatic electrochemical detection was carried out on a 100 nm sputtered gold film that allows working with small volumes. Although thick gold substrates are reported in the literature for enzymatic DNA detection (screen-printed gold electrodes in Carpini et al., 2004, and 2 μm thick film in Xu et al., 2001), this is the first time that this type of assay has been carried out on a thin gold film. Reducing the cell volume has several advantages (Wijayawardhana et al., 1999), the first being the decrease in the diffusion distances required for analytes to reach their surface-bound receptor partners. Moreover, in the case of enzymatic detection, the product dilution, which is a critical factor in achieving low detection limits, diminishes. A simple, cheap and easy-to-handle homemade device is presented that allows the simultaneous performance of the hybridisation procedure and sequential detection of more that 20 assay sites.
The sequence chosen as target is included in the 29 751-base genome of the SARS (severe acute respiratory syndrome)-associated coronavirus (Marra et al., 2003). This is the causative agent of an outbreak of atypical pneumonia, first identified in Guangdong Province, China, that has spread to several countries. The sequence corresponds to a gene that encodes the nucleocapsid protein (422 amino acids), specifically a short lysine-rich region that appears to be unique to SARS and suggestive of a nuclear localization signal. A 30-mer oligonucleotide with bases comprised between numbers 29 218 and 29 247, both included, was chosen.
In this paper, a complementary strand to the chosen SARS sequence is labelled with a thiol group and immobilised on the gold surface. The target (30-mer oligonucleotide with a sequence included in the SARS-coronavirus) is conjugated to biotin and hybridised with the probe. Addition of AP-labelled streptavidin allows enzymatic detection via the electrochemical signal of the product. The parameters affecting all assay steps were studied and analytical characteristics, including selectivity, are discussed.
2. Experimental
2.1. Reagents
Synthetic 30-mer oligonucleotides were obtained from Eurogentec. The target sequence employed corresponds to a portion of the SARS (severe acute respiratory symptom) virus, precisely the bases comprised between 29 218 and 29 247, both included. For selectivity studies, a 3-base mismatch strand was also purchased. Mismatches are located in bases number 5, 15 and 26. Both strands are biotinylated at the 3′-end to allow hybridisation detection. Biotinylated target (193 nmol): 5′-ACA-GAG-CCT-AAA-AAG-GAC-AAA-AAG-AAA-AAG-3′-biotin. Biotinylated 3-base mismatch target (184 nmol): 5′-ACA-GCG-CCT-AAA-AAC-GAC-AAA-AAG-AGA-AAG-3′-biotin.
A complementary strand, the probe, was also obtained from Eurogentec. It was immobilised on the gold surface and, depending on the study, was labelled with biotin, a thiol group or both. The thiol group was separated from the first base by an aliphatic linker of three carbons.
Biotinylated and thiolated probe (77 nmol): Biotin-5′-CTT-TTT-CTT-TTT-GTC-CTT-TTT-AGG-CTC-TGT-3′-(CH2)3-SH. Biotinylated probe (65 nmol): Biotin-5′-CTT-TTT-CTT-TTT-GTC-CTT-TTT-AGG-CTC-TGT-3′. Thiolated probe (103 nmol): 5′-CTT-TTT-CTT-TTT-GTC-CTT-TTT-AGG-CTC-TGT-3′-(CH2)3-SH.
Oligonucleotide solutions were prepared in TE buffer pH 8 (0.1 M Tris–HCl buffer solution, 1 mM in EDTA). Aliquots were prepared and maintained at −20 °C. Working solutions were conserved at 4 °C. Hybridisation took place in a 2×SSC (saline sodium citrate) buffer, i.e. a 30 mM sodium citrate buffer with 300 mM sodium chloride and pH 7.0.
α-Naphthol and indigo carmine (IC) were purchased from Sigma. For stock solution preparation, α-naphthol was solved in methanol and the solution was made up to the desired volume with Milli-Q water. The stock solution of IC was prepared in 0.1 M H2SO4. Solutions were protected from the light and kept refrigerated at 4 °C. Dilutions were made, in both cases, with a mixture of 0.1 M Tris–HCl solution, pH 9.8 and 5.4 M H2SO4 in a 2:1 proportion. Working solutions were prepared daily.
3-Indoxyl phosphate (3-IP) was also purchased from Sigma. Solutions were prepared daily in 0.1 M Tris–HCl buffer, 10 mM MgCl2, pH 9.8. They were kept at 4 °C and protected from the light.
Bovine serum albumin (BSA, fraction V), biotin-labelled albumin (biotin content: 9 mol/mol albumin) and casein from sheep milk were obtained from Sigma. A gelatine derivative called “Perfect-Block” was obtained from MoBiTec. All solutions were prepared daily in 0.1 M Tris–HCl buffer, pH 7.2.
Alkaline phosphatase (AP)-labelled streptavidin (ST-AP) was prepared in 0.1 M Tris–HCl buffer, 1 mM MgCl2, pH 7.2. Aliquots were prepared and maintained at −20 °C. Working solutions were conserved at 4 °C.
Thioctic acid, 1-hexanethiol and 6-mercapto-1-hexanol were purchased from Aldrich and were solved in ethanol.
Methanol (99%) and absolute ethanol together with hydrochloric (37%) and sulphuric (95–97%) acids were purchased from Merck, as was magnesium chloride and sodium citrate. Trizma base and sodium chloride were supplied by Sigma. EDTA was obtained from Fluka.
Water was purified employing a Milli-Q plus 185 device from Millipore.
2.2. Instruments
The three-electrode potentiostatic system employed is schematised in Fig. 1 . Working electrodes were made on 5 cm × 5 cm supports of a 0.125 mm thick polyimide substrate called Kapton HN® (Goodfellow). They were covered with gold by a sputtering process using an Emitech sputter coater model K550. A conductor wire was attached to the centre of one of the sides by means of an epoxy resin (CW2400) obtained from RS Components, which was cured at room temperature.
Fig. 1.
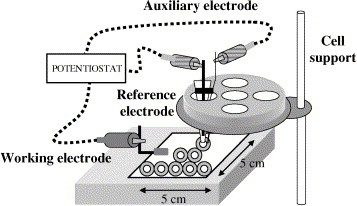
Schematic diagram of the three-electrode potentiostatic system.
The working area was limited by self-adhesive washers of 5 mm internal diameter (19.6 mm2 internal area). The total area of the gold surface allowed approximately 23 washers to be stuck. The gold film was placed on a support to which a crocodile connection was fixed.
Reference and auxiliary electrodes were coupled in a micropipette tip. The reference electrode consisted of an anodised silver wire introduced in a tip through a syringe rubber piston. The tip was filled with saturated KCl solution and contained a low resistance liquid junction. The platinum wire that acted as auxiliary electrode was fixed with insulating tape. For measurement recording, the tip was fixed on an electrochemical cell Metrohm support allowing horizontal and vertical movement.
Measurements were performed with an Autolab PGSTAT 10 (EcoChemie) potentiostat interfaced to an ADL Pentium 120 computer system and controlled by Autolab GPES software version 4.8 for Windows 98.
A Crison Micro-pH 2001 pH-meter and a Sanyo (MIR-162) incubator were also used.
2.3. Procedures
2.3.1. Gold sputtering
A kapton slide was cleaned with ethanol and, after drying, was covered with gold by a sputtering process. Gold atoms were deposited (from the cathode) over the kapton (placed on the anode) in a vacuum chamber filled with argon. The thickness of the gold layer was controlled by means of the duration and intensity of the discharge. For a 100 nm thick layer, a 35 mA discharge was applied over 220 s.
2.3.2. Hybridisation assay
An amount of 5 μL of thiolated DNA (1.02 μM) was deposited on the gold film and maintained at 37 °C for 20 min or at 4 °C for 12 h. After cleaning with 0.1 M Tris–HCl buffer pH 7.2, 15 μL of a 2% blocking agent solution was added and maintained for 10 min. Cleaning was performed with a 2×SSC buffer solution pH 7. Hybridisation took place for 60 min with the biotinylated complementary strand for 60 min at room temperature. The film was washed with 0.1 M Tris–HCl buffer, 10 mM of MgCl2, pH 7.2. A 20 μL drop of AP-labelled streptavidin at a concentration of 10−9 M was then deposited for 60 min. The film was subsequently washed with 0.1 M Tris–HCl buffer, 10 mM MgCl2, pH 9.8. Enzymatic hydrolysis took place for 10 min after adding 20 μL of a 3 mM solution of 3-IP. The reaction was stopped with 5 μL of concentrated H2SO4, and the electrochemical measurement was performed after adding 5 μL of Milli-Q water.
For immobilisation studies, the biotinylated and thiolated probe was deposited on the gold film under the same conditions. After cleaning, blocking was performed in a similar way. Washing was then carried out with the 0.1 M Tris–HCl buffer solution, 1 mM MgCl2, pH 7.2, before the addition of ST-AP. The rest of the procedure was similar to that stated above.
2.3.3. Electrochemical measurement
The tip with reference and auxiliary electrodes was introduced into the 30 μL drop (20 μL originating from the 3-IP solution plus 5 μL of concentrated H2SO4 and 5 μL of Milli-Q water) deposited on the gold film. A potential of −0.35 V was applied for 30 s before scanning the potential between −0.15 and +0.3 V following a square wave format with frequency of 50 Hz and amplitude of 50 mV.
3. Results and discussion
The main steps involved in the construction of the genosensor were immobilisation of a single strand of nucleic acids (probe), hybridisation with a complementary strand (target) and electrochemical detection. As electrochemical detection was carried out indirectly, using an enzymatic label (AP) that converts a substrate in an electroactive product, the electrochemical behaviour of several substrates in this system was studied first.
3.1. Electrochemical detection
Among all the possible AP substrates mentioned in Section 1, 3-indoxyl phosphate (IP) and α-naphthyl phosphate (NP) were the ones chosen. NP has been widely used by our research group to carry out immunoassays on gold bands in which the detection of the naphthol generated was performed by amperometry in a flow cell (Abad-Villar et al., 2002b, Abad-Villar et al., 2003). IP was proposed by our group (Fernández-Sánchez and Costa-García, 1998) as a suitable substrate based on the favourable processes that indigo carmine (IC), a soluble derivative of the product generated (indigo blue), presented. Moreover, compared with other substrates, its kinetic constants were found to be more favourable. Although the electrochemical behaviour on gold electrodes have already been studied (Castaño-Álvarez et al., 2004), the particularities of this system made it necessary to perform a comparative study between both products: α-naphthol and IC.
3.1.1. Electrochemical techniques
For these studies, 30 μL of solution was deposited on the surface limited by an adhesive washer on the gold band. This volume was chosen in order to stop the reference and auxiliary electrodes from coming into contact with the film and the drop from spilling over the washer. Measurements were recorded in a 2:1 mixture of 0.1 M Tris–HCl buffer solution pH 9.8 and 5.4 M H2SO4. Said composition was chosen as this buffer is usually employed for AP-enzymatic hydrolysis and sulphuric acid is used for stopping the reaction and solubilising the generated indigo. This solution was also employed for background measurements and for cleaning the film before a new measurement.
As the comparison has to be carried out in the best conditions for both products, the most adequate electrochemical technique was chosen in each case. When a cyclic voltammogram is recorded between −0.05 and +0.2 V with a scan rate of 50 mV s−1 in a 4 × 10−4 M solution of IC in 2 M sulphuric acid, two anodic processes appear. The first process (+0.1 V) is reversible, while the second (+0.8 V) is irreversible in nature. When cyclic voltammetry is compared with differential pulse (DPV), alternating current (ACV) and square wave voltammetry (SWV), an enhancement of the signal is observed for ACV and SWV. Intensities of 1.95, 1.84, 11.60 and 11.40 μA were respectively obtained for CV, DPV, ACV and SWV in a 6 × 10−5 M solution of IC. Of the two more sensitive techniques, SWV was chosen due to presenting the best-defined signal, with a peak potential of +0.15 V.
In the case of α-naphthol, the cyclic voltammogram recorded between 0.0 and +1.0 V presents an anodic process at +0.75 V. As this process is irreversible in nature, improvements are not expected with ACV and SWV. When cyclic and differential pulse voltammetry were compared, similar signals were obtained for a 4 × 10−4 M solution (5.57 and 4.94 μA for CV and DPV, respectively). In this case, CV was chosen for the sake of simplicity.
The parameters related to the electrochemical technique were optimised, namely frequency and amplitude for SWV and scan rate for CV. Frequency was varied between 10 and 100 Hz, obtaining higher intensities for higher frequencies. However, the widths of the peaks also increased. Peak currents were 2.18 and 4.26 μA for 50 and 100 Hz, respectively, whereas half-height widths were 90 and 140 mV. Therefore, taking into account both variables, 50 Hz was chosen as the optimum value. Employing this frequency, amplitude was varied between 10 and 100 mV. Since the maximum signal was obtained at 50 mV, this amplitude was chosen for the remainder of the work. When cyclic voltammograms were recorded at 50 and 100 mV s−1, a non-distorted higher signal was obtained for the second value, which was the one chosen for the remaining studies.
3.1.2. Repeatability
The repeatability of the measurements was then tested using SWV for IC and CV for α-naphthol. In order to obtain precise measurements, solid electrodes are commonly pre-treated (mechanically, chemically or electrochemically) (Adams, 1959). In the case of gold films, as mechanical or chemical treatments might damage the surface, electrochemical activation was tested to achieve repeatable signals in the same area of the film. Negative potentials at constant intensity and negative intensities at constant potential were tested. The best results were obtained by applying a potential of −0.35 V for 30 s before scanning the potential between −0.05 and +0.3 V for IC. In the case of α-naphtol, application of −0.5 V for 30 s gave repeatable results (Table 1 ). In both cases, cleaning with background solution was carried out between measurements.
Table 1.
Comparative electrochemical behaviour of IC and α-naphthol
| Indigo carmine | α-Naphthol | |
|---|---|---|
| Technique | SWV (50 Hz, 50 mV) | CV (100 mV s−1) |
| Potential scan | −0.05 to +0.30 V | +0.40 to +0.95 V |
| Pre-treatment | −0.35 V, 30 s | −0.5 V, 30 s |
| Precision (same area) R.S.D. (C, n) | 3.0% (6 × 10−6 M, 12) | 3.5% (3 × 10−5 M, 11) |
| Precision (different areas) R.S.D. (C, n) | 6.7% (4 × 10−5 M, 7) | 6.1% (1 × 10−4 M, 10) |
| 5.2% (6 × 10−6 M, 12) | 5.6% (3 × 10−5 M, 12) | |
| Linear dynamic range | 10−6 to 10−4 M | 10−5 to 5 × 10−4 M |
| Sensitivity | 4.7 × 105 μA L mol−1 | 1.3 × 105 μA L mol−1 |
| Detection limit | 10−6 M | 7 × 10−6 M |
The reproducibility of recorded signals following the pre-treatments stated above was tested. A total number of 23 measurements could be made in different areas of the same gold film. The effect was observed at different concentrations of IC (6 × 10−6 and 4 × 10−5 M) and α-naphthol (3 × 10−5 and 10−4 M). Values for the relative standard deviation (R.S.D.) are shown in Table 1. Differentiated zones (centre, borders, etc.) were not observed on the film.
3.1.3. Detection limit and linear dynamic range
With the optimised parameters, calibration curves were performed for IC and α-naphthol. Two different procedures were carried out, the first one recording measurements in the same area of the film and washing in-between with background solution. In the second procedure, signals were obtained in different areas of the film. They were cleaned before depositing the drop of solution. The results are compared in Table 1. Detection limits were calculated as the concentration corresponding to a signal that is three times the standard deviation of the intercept. IC presents a higher linear dynamic range, enhanced sensitivity and lower limit of detection in both cases, i.e. for calibration performed in the same area or different areas of the gold film.
The results with respect to reproducibility are similar, requiring an analogous pre-treatment in both compounds. An important difference is the peak potential, +0.15 V for IC and +0.75 V for α-naphthol. The employment of low potentials is an advantage, as there are fewer substances that present electrochemical processes, which thus avoids interferences. Since the peak potential of IC is 0.6 V lower than that of α-naphthol and as it gave better results in general, 3-IP was chosen as substrate for AP.
3.1.4. Enzymatically generated indigo carmine
When IC was enzymatically generated, similar voltammograms to those recorded in IC solutions were obtained. Bearing in mind that streptavidin adsorbs easily onto gold surfaces, ST-AP (10 μL of 7.4 × 10−8 M solution) was immobilised on the gold film for 10 min by physical adsorption. After cleaning (with 0.1 M Tris–HCl buffer, 1 mM MgCl2, pH 7.2), 20 μL of 6 mM 3-IP were added. This concentration has already been employed in previous studies (Díaz-González et al., 2002, Castaño-Álvarez et al., 2004). After 15 min, 3 μL of concentrated H2SO4, and 7 μL of Milli-Q water were added. The acid was needed not only to stop the reaction, but also to sulphonate and solubilise the indigo blue that was formed. Water was added to increase the volume until 30 μL, considered the optimum total volume for an adequate operation. The acid:water volume proportion may be changed when great quantities of indigo blue are produced, always maintaining the final volume (10 μL). No signal was obtained when the procedure was followed without immobilising AP on the working electrode. The only significant difference between signals recorded for IC solutions or enzymatically generated IC is that, in the latter case, the peak potential moved slightly to more negative potentials. This is possibly due to the in situ generation of the electroactive substance, which favoured the electrochemical process. The potential scan was consequently performed between −0.15 and +0.3 V. As the signal obtained for 10 min (29 μA) is not much lower than that obtained for 15 min (32 μA), 10 min was the one subsequently employed. The relative standard deviation of measurements carried out in different surfaces (n = 4) of a gold film was 5.3%.
3.2. Oligonucleotide immobilisation
Immobilisation of oligonucleotide strands was carried out through a thiol group attached to the 3′-end. The linker contains a three-carbon aliphatic spacer that permits higher single-strand DNA (ssDNA) flexibility. Longer hydrocarbonated chains were not chosen because electronic transference may be affected (Taft et al., 2003). In order to study immobilisation, a label was attached to the other end to generate a detectable signal. As AP is a protein of approximately 150 000 Da, labelling a strand with AP can affect its affinity. To solve this problem, the strand may be labelled at its end with a small molecule, biotin. Then the attachment of AP takes place by employing AP-conjugated streptavidin (ST-AP) through the well-known streptavidin–biotin interaction.
The procedure specified in Section 2 and schematised in Fig. 2 a includes the immobilisation of the double-labelled (thiolated and biotinylated) oligonucleotide, blocking of the surface in order to minimise non-specific adsorptions, reaction with AP-labelled streptavidin, enzymatic hydrolysis of 3-IP and recording of the analytical signal. Cleaning was performed between steps with 0.1 M Tris–HCl buffer pH 7.2 or 9.8 (the latter pH before the enzymatic hydrolysis of 3-IP).
Fig. 2.
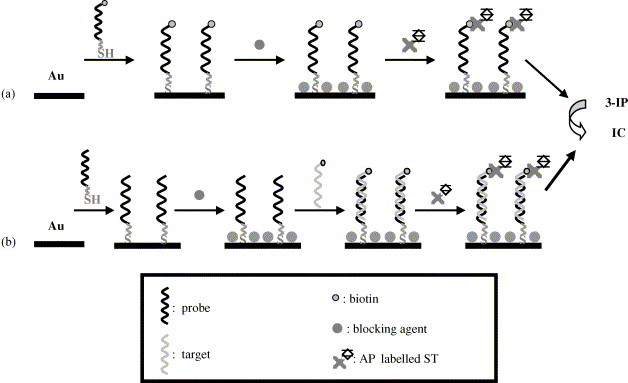
Assay procedures carried out for: (a) immobilisation and (b) hybridisation studies.
Interaction between biotin and streptavidin was first studied using biotinylated albumin, which adsorbs perfectly onto gold. In this case, 10 μL of a 2% reagent solution was employed. Albumin was employed instead of biotinylated albumin to record background signals. In this case, the signal/background ratio was not very high, indicating the appreciable level of non-specific adsorptions. In order to increase this ratio, several parameters such as interaction times and ST-AP concentration were varied. Concentrations of ST-AP ranging from 10−9 to 10−6 M (20 μL) were tested with biotinylated albumin being adsorbed for 30 min and an interaction time with ST-AP of 1 h. The best values for this ratio were obtained for a 10−7 M concentration. However, when the double-labelled oligonucleotide strand was immobilised (10 μL of 1.02 μM solution for 30 min), the amount of indigo blue produced made it necessary to decrease the concentration of 3-IP (from 6 to 3 mM) and to change the volume of acid and water added. Therefore, 5 μL of acid and 5 μL of water were subsequently added. Since the background signal was still high, the influence of ST-AP concentration was tested once more. When varying the concentration from 10−10 to 10−7 M, the best signal/background ratio was obtained for 10−9 M, which was the one employed for the remainder of the study.
When different concentrations of double-labelled DNA, ranging from 0.102 to 20.4 μM, were immobilised, signals increased up to 15.3 μM (Fig. 3 a). As regards the drop volume, 5, 10, 20 and 30 μL drops were tested for a 5.10 μM strand solution (concentration situated on the slope of Fig. 3a). The signal corresponding to 5 μL was the highest recorded. As the 5 μL-drop had evaporated for 30 min (time of DNA immobilisation), the study of DNA concentration was repeated under conditions that guaranteed drop evaporation (Fig. 3b). The highest signal now corresponded to 5.10 μM. Therefore, a DNA concentration of 1.02 μM, which is located on the steepest slope of the graph, was chosen for the rest of the study.
Fig. 3.
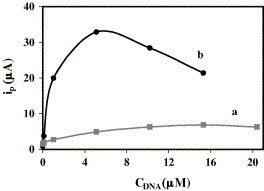
Influence of double-labelled DNA concentration on the analytical signal when: (a) 10 μL of DNA are immobilised for 30 min at room temperature and (b) 5 μL of DNA are immobilised for 20 min at 37 °C.
3.2.1. Effect of evaporation
Evaporation seems to be a critical condition in the immobilisation of SH-DNA. A more rigorous study, shown in Fig. 4 , was performed. Using a drop volume of 5 μL and a strand concentration of 1.02 μM, different immobilisation times were employed at room temperature (22 °C approximately), 37 and 47 °C. It can be observed that the enhancement of the signal with immobilisation time was greater when evaporation occurred, which corresponds to 30, 15 and 10 min for room temperature, 37 and 47 °C, respectively. When immobilisation took place at 4 °C overnight (12 h, also attaining evaporation), the signal was similar to that obtained at 37 °C (27 μA versus 29 μA, respectively). Therefore, both methodologies could be employed. The repeatability of the signals obtained for both immobilisation procedures was also similar, obtaining an R.S.D. of 9 and 8% for five measurements when immobilisation was carried out at 37 °C for 20 min and 4 °C for 12 h, respectively. Considering five different gold films, precision was analogous, with an R.S.D. of 10%.
Fig. 4.
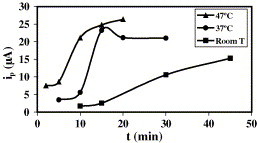
Influence of temperature on DNA immobilisation: C = 1 μM, Vdrop = 5 μL.
3.2.2. Surface blocking
Blocking the surface is one of the most important steps to minimise and control non-specific adsorptions. Two main types of agents were considered: proteins and sulphur-containing compounds. The signal/background ratio (S/B), and therefore the blocking capacity, was observed for each compound. A 15 μL drop of a 2% solution was deposited on the gold band after DNA immobilisation and cleaning. For proteins, the drop was maintained for 30 min, and in the case of sulphur-containing compounds, until its evaporation (10 min), taking into account the kinetics of alkanethiol adsorption on gold (Pan et al., 1996).
Proteins such as albumin are common blocking agents in bioanalysis and adsorb well onto gold surfaces. Aqueous solutions of bovine serum albumin (BSA), casein and a gelatine derivative called Perfect Block were tested. Of the three, albumin and casein presented similar signals and backgrounds. An analogous signal was obtained for the gelatine derivative for a higher background.
The other group of blocking agents are compounds that contain sulphur atoms and therefore present a high affinity for gold. They are usually employed for generating self-assembled monolayers (Duan and Meyerhoff, 1994, Steel et al., 1998, Bain et al., 1989). Ethanolic solutions of thioctic acid, 6-mercapto-1-hexanol and 1-hexanethiol were tested. The first one possesses a disulphide bond, whereas the other two are hydrocarbonated chains with a thiol function, one of them with a hydroxy group at the other end. Although there is a preference adsorption for thiols, disulphides give rise to similar species (Bain et al., 1989). The length of the chain was chosen so as to allow hybridisation and electrochemical processes to take place, since too dense monolayers do not allow electronic transference (Kelley et al., 1999). Shorter chains are more volatile and hence more difficult to handle. Thioctic acid and 6-mercapto-1-hexanol produced high background and the S/B ratio was not favourable (1.56 and 1.4, respectively). Although for 1-hexanethiol the background was similar to that obtained for other compounds, the signal was much higher, resulting in a more adequate ratio (4.9). 6-Mercapto-1-hexanol is normally employed for DNA assays; however, the authors have not found any reference to the use of 1-hexanethiol in this type of application despite its favourable behaviour.
A comparison between the signals obtained for background and immobilised DNA when BSA and 1-hexanethiol were employed as blocking agents is shown in Fig. 5 . It can be observed that the background with albumin is negligible and the S/B ratio is high (22.4), but the DNA signal is better defined for 1-hexanethiol where the capacitive current approaches zero.
Fig. 5.
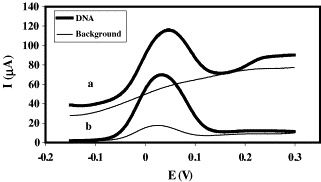
Signals and backgrounds obtained when: (a) albumin and (b) 1-hexanethiol are employed as blocking agents: C = 1 μM, Vdrop = 5 μL, Timmob = 4 °C, timmob = 12 h.
In order to decrease the background obtained for 1-hexanethiol, the concentration of this blocking agent was increased from 2 to 5 and 10%. However, the S/B ratio did not increase, but varied from 4.6 (for a 2% of blocking agent) to 3.3 and 4.1 for 5 and 10% of blocking agent, respectively.
In conclusion, the blocking agents that provided better results were albumin and casein. Nevertheless, as the signal of 1-hexanethiol is high, it could be a better blocking agent when hybridisation takes place.
3.2.3. Effect of SH group
The favourable immobilisation of DNA through its thiol group is observed in Fig. 6 . Signals were obtained for unmodified DNA and SH-DNA (both biotinylated) following similar procedures and employing 1-hexanethiol. Although single-stranded DNA adsorbs onto gold substrates, it adopts a coiled configuration and therefore cannot form an ordered structure (Zhang et al., 2002). An almost negligible adsorption of unmodified DNA is observed that is greatly favoured by the insertion of a thiol group. This was observed not only when using 1-hexanethiol, but also for albumin. Moreover, the same results were obtained when DNA immobilisation took place at 4 °C for 12 h or at 37 °C for 20 min.
Fig. 6.
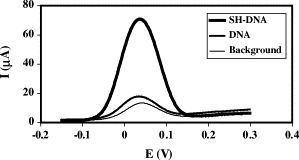
Signals recorded for biotinylated SH-DNA, biotinylated DNA and background: C = 1 μM, Vdrop = 5 μL, Timmob = 4 °C, timmob = 12 h.
3.3. Hybridisation assay
Once electrochemical detection, DNA immobilisation and blocking had been studied, the affinity interaction on which the sensor is based (hybridisation) was subsequently studied. The difference with respect to the procedure carried out for immobilisation is that the oligonucleotide fixed to the gold surface is now modified only with a thiol group at the 3′-end and the complementary strand is labelled with biotin also at the 3′-end. The main steps are shown in Fig. 2b. As cleaning is always performed with the buffer employed for the next step, the blocking agent is now cleaned with 2×SSC buffer. For hybridisation, a 20 μL drop of a complementary strand was added.
3.3.1. Blocking agent
The hybridisation signal was similar when immobilisation was performed at 4 or 37 °C, but there was a significant difference when albumin or 1-hexanethiol was employed. The S/B ratio for 1-hexanethiol was analogous to that obtained when a double-labelled (with biotin and thiol) strand was used. However, it was much lower for albumin. This might be caused by steric hindrance, due to the large size of albumin, which may hinder hybridisation. It has been reported that the creation of mixed monolayers of thiol-modified DNA probe and a spacer thiol (mercaptohexanol) in two steps makes DNA molecules stand up consistently in such a way that they become accessible for hybridisation (Herne and Tarlov, 1997). Therefore, 1-hexanethiol was employed for the remainder of the study as blocking agent.
Attempts were made to decrease the background obtained with this compound. As mentioned in the previous section, an increase in concentration was not effective. Interaction time (10 min) could not be increased because it is the time in which the ethanolic solution evaporated. Higher drop volumes could not be employed because of drop spill. Repeated additions of the agent two and three times once it had evaporated were tested without good results. Two blocking steps, before and after hybridisation, did not show better results. A decrease in the ST-AP concentration, in the range comprised between 10−9 and 10−11 M, was also tested. Although background decreased, the best S/B ratio was obtained for 10−9 M. Values of 1.5, 3.9 and 6.6 of S/B were obtained for 10−11, 10−10 and 10−9 M, respectively. Accordingly, the same methodology was employed, always recording the background signal.
3.3.2. Hybridisation time and potential
Fig. 7 shows the effect of time on hybridisation at room temperature and 37 °C. The signal increases slightly with time up to evaporation (30 min for 37 °C and more than 60 min for room temperature). This is the contrary to what occurred for immobilisation. Evaporation favours DNA adsorption and diminishes the hybridisation signal. As the drop volume cannot be increased to increase interaction time, hybridisation may be performed for 30 min at 37 °C or for 60 min at room temperature. As similar signals were obtained at 37 °C with 20 μL for 30 min and 10 μL for 15 min, the latter procedure was employed to reduce analysis time and reagent consumption.
Fig. 7.
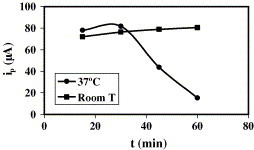
Influence of temperature on the hybridisation step: C = 1 μM, Vdrop SH-DNA = 5 μL, Vdrop DNA = 20 μL, Timmob = 4 °C, timmob = 12 h.
As DNA is a polyanionic molecule, application of potentials can affect hybridisation due to orientation of the strands. +0.3, +0.1, −0.1 and −0.3 V were applied in 0.1 M Tris–HCl pH 7.2 medium for 30 s after SH-DNA immobilisation. Signals obtained for negative potentials were higher, but so were the backgrounds. Likewise, no improvement was achieved when the potential was applied after blocking with thiol or after both steps (immobilisation and blocking). Therefore application of potential was omitted.
For the rest of the study, 5 μL of a 0.10 μM SH-DNA concentration was immobilised for 12 h at 4 °C or 20 min at 37 °C (the solution is consequently evaporated). Blocking was performed with 2% of 1-hexanethiol for 10 min. When hybridisation took place, incubation was carried out for 1 h with a 10−9 M solution of ST-AP. Indigo carmine was generated from 3-IP (20 μL of a 3 mM solution) for 10 min. Measurement was performed after adding 5 μL of concentrated H2SO4 and 5 μL of H2O.
3.4. Analytical characteristics
Once the parameters that affect the procedure had been studied, the concentration of both strands was tested. Using a 0.101 μM solution of complementary strand, SH-DNA was varied between 0.102 and 5.10 μM. The maximum value appeared at 2.04 μM, where a plateau was obtained. On the other hand, a calibration curve for the complementary strand (c-DNA) was performed under optimised conditions, employing a 1.02 μM solution of SH-DNA in order to ascertain the dynamic range of the assay. The peak intensity (i p) was linear with a concentration in the interval 0.102–5.10 nM following the equation:
The detection limit, calculated as the concentration corresponding to a signal that is three times the standard deviation of the intercept, was found to be 50 pM.
In order to test the selectivity of the assay, hybridisation was carried out with a 3-base mismatch complementary strand. When hybridisation was performed for 15 min at 37 °C with both strands (complementary and 3-base mismatch) in a concentration of 4.04 nM, there was no discrimination between the signals. No difference was observed when stringency conditions were applied: potential application, buffer composition, formamide addition, ionic strength and temperature.
Hybridisation time was then increased up to 1 h in a 2× SSC buffer with 50% formamide. It was thus necessary to increase the drop volume to 20 μL and to work at room temperature to avoid evaporation. A marked difference was observed for a 3.03 nM solution of the target strand. For 15 min of hybridisation time, signals were 35.27 and 37.07 μA for the complementary and 3-mismatch strand, respectively. These changed to 47.61 and 3.96 μA respectively when hybridisation took place for 1 h. Therefore, 100% discrimination was achieved. This makes differentiation possible, although not so marked (Xu et al., 2001), not only for a 2-base but also for 1-base mismatch.
Repeatability was tested under the following conditions: 1 h of hybridisation and a 2×SSC buffer containing 50% of formamide. The value of the R.S.D. was 11% for nine measurements.
A new calibration curve was obtained (see voltammograms in Fig. 8 ), employing the same conditions. As signals increased considerably, sensitivity and the detection limit improved. For a calibration performed between 0.01 and 1.01 nM, linearity is represented by the equation:
Fig. 8.
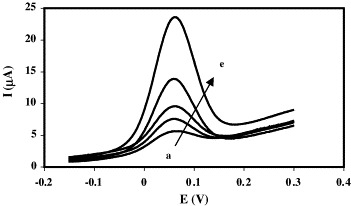
Response curves for: (a) background; (b) 0.01 nM; (c) 0.10 nM; (d) 0.51 nM; (e) 1.01 nM c-DNA concentration. Vdrop DNA = 20 μL, thybrid = 60 min, hybridisation buffer = 2× SSC + 50% formamide.
The detection limit, calculated as above, was found to be 6 pM. This means an improvement of various orders of magnitude when compared with the detection limits reported in the literature for DNA assays that detect α-naphthol on thick-film gold electrodes (0.25 nM for Carpini et al., 2004, and 0.22 nM for Xu et al., 2001). This good result is possibly due to the use of 3-IP as substrate together with a sensitive electrochemical technique and an adequate procedure. Hundred and forty six nanomolar is the detection limit obtained for an AP-based DNA assay that employs p-aminophenylphosphate as substrate (Aguilar and Ritsch, 2003). Specific quantification of 1 nM is achieved with other enzymatic schemes (Domínguez et al., 2004). Detection limits of 0.20 nM (Zhu et al., 2003) and 30 nM (Wu et al., 2003) were achieved for indirect detection using an electrochemical indicator and for direct electrochemical detection, respectively.
4. Conclusions
A sensitive, simple, cheap and miniaturised homemade device for electrochemical detection on small volumes based on thin gold films has been designed. It was successfully applied to the development of genosensors, in particular for the detection of a SARS (severe acute respiratory syndrome) virus specific sequence. Immobilisation of a DNA probe on the gold surface was carried out through a thiol group attached to the 3′-end with an aliphatic spacer. The parameters affecting immobilisation were studied employing a double-labelled (biotinylated and thiolated) DNA strand. AP-labelled streptavidin was used for enzymatic detection. Blocking with 1-hexanethiol produced well-defined signals. Evaporation of solutions was a critical point for achieving optimum results, favouring immobilisation while preventing hybridisation. The enzymatic hydrolysis of 3-IP allowed low detection limits (6 pM) to be achieved with respect to analogous schemes reported in the literature. Selectivity of the assay was demonstrated using a 3-base mismatch DNA strand. Clear discrimination was possible employing 1 h of hybridisation time and 50% of formamide in the buffer. Work is in progress to study selectivity in more depth, with the aim of also obtaining discrimination for 2-base and 1-base mismatch strands.
Acknowledgement
This work was supported by European Project UE-02-QLK-CT-70963.
Footnotes
The paper was presented at 8th World Congress on Biosensors 2004.
References
- Abad-Villar E.M., Fernández-Abedul M.T., Costa-García A. Gold bands as a suitable surface for enzyme immunoassays. Biosens. Bioelectron. 2002;17:797–802. doi: 10.1016/s0956-5663(02)00080-5. [DOI] [PubMed] [Google Scholar]
- Abad-Villar E.M., Fernández-Abedul M.T., Costa-García A. Simultaneous and sequential enzyme immunoassays on gold bands with flow electrochemical detection. Anal. Chim. Acta. 2002;453:63–69. [Google Scholar]
- Abad-Villar E.M., Fernández-Abedul M.T., Costa-García A. The use of gold bands for flow immunoelectrochemical devices. Anal. Bioanal. Chem. 2003;377:267–272. doi: 10.1007/s00216-003-2122-8. [DOI] [PubMed] [Google Scholar]
- Adams R.N. Marcel Dekker; New York: 1959. Electrochemistry at Solid Electrodes. [Google Scholar]
- Aguilar Z.P., Ritsch I. Immobilized enzyme-linked DNA-hybridization assay with electrochemical detection for Cryptosporidium parvum hsp70 mRNA. Anal. Chem. 2003;75:3890–3897. doi: 10.1021/ac026211z. [DOI] [PubMed] [Google Scholar]
- Alfonta L., Singh A.K., Willner I. Liposomes labelled with biotin and horseradish peroxidase: a probe for the enhanced amplification of antigen–antibody or oligonucleotide-DNA sensing processes by the precipitation of an insoluble product on electrodes. Anal. Chem. 2001;73:91–102. doi: 10.1021/ac000819v. [DOI] [PubMed] [Google Scholar]
- Bain C.D., Troughton E.B., Tao Y.-T., Evall J., Whitesides G.M., Nuzzo R.G. Formation of monolayer films by the spontaneous assembly of organic thiols from solution onto gold. J. Am. Chem. Soc. 1989;111:321–335. [Google Scholar]
- Bengoechea-Álvarez M.J., Fernández-Abedul M.T., Costa-García A. Flow amperometric detection of indigo for enzyme-linked immunosorbent assays with use of screen-printed electrodes. Anal. Chim. Acta. 2002;462:31–37. [Google Scholar]
- Brett A.M.O., Matysik F.-M., Vieira M.T. Thin-film gold electrodes produced by magnetron sputtering. Voltammetric characteristics and application in batch injection analysis with amperometric detection. Electroanalysis. 1997;9:209–212. [Google Scholar]
- Campas M., Katakis I. DNA biochip arraying, detection and amplification strategies. Trends Anal. Chem. 2004;23:49–62. [Google Scholar]
- Carpini G., Lucarelli F., Marrazza G., Mascini M. Oligonucleotide-modified screen-printed gold electrodes for enzyme-amplified sensing of nucleic acids. Biosens. Bioelectron. 2004;20:167–175. doi: 10.1016/j.bios.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Castaño-Álvarez M., Fernández-Abedul M.T., Costa-García A. Gold electrodes for detection of enzymes assays with 3-indoxylphosphate as substrate. Electroanalysis. 2004;16:1487–1496. [Google Scholar]
- De-Los-Santos-Alvarez P., Lobo-Castanon M.J., Miranda-Ordieres A.J., Tuñón-Blanco P. Voltammetric determination of underivatized oligonucleotides on graphite electrodes based on their oxidation products. Anal. Chem. 2002;74:3342–3347. doi: 10.1021/ac015749m. [DOI] [PubMed] [Google Scholar]
- Díaz-González M., Fernández-Sánchez C., Costa-García A. Indirect determination of alkaline phosphatase based on the amperometric detection of indigo carmine at a screen-printed electrode in a flow system. Anal. Sci. 2002;18:1209–1213. doi: 10.2116/analsci.18.1209. [DOI] [PubMed] [Google Scholar]
- Domínguez E., Rincón O., Narváez A. Electrochemical DNA sensors based on enzyme dendritic architectures: an approach for enhanced sensitivity. Anal. Chem. 2004;76:3132–3138. doi: 10.1021/ac0499672. [DOI] [PubMed] [Google Scholar]
- Duan C., Meyerhoff M.E. Separation-free sandwich enzyme immunoassays using microporous gold electrodes and self-assembled monolayer/immobilized capture antibodies. Anal. Chem. 1994;66:1369–1377. doi: 10.1021/ac00081a003. [DOI] [PubMed] [Google Scholar]
- Fernández-Sánchez C., Costa-García A. 3-Indoxyl phosphate: an alkaline phosphatase substrate for enzyme immunoassays with voltammetric detection. Electroanalysis. 1998;10:249–255. [Google Scholar]
- Gherghi I.Ch., Girousi S.Th., Voulgaropoulos A., Tzimou-Tsitouridou R. Interaction of the mutagen ethidium bromide with DNA, using a carbon paste electrode and a hanging mercury drop electrode. Anal. Chim. Acta. 2004;505:135–144. [Google Scholar]
- Herne T.M., Tarlov M.J. Characterization of DNA probes immobilized on gold surfaces. J. Am. Chem. Soc. 1997;119:8916–8920. [Google Scholar]
- Jenkins S.H., Heineman W.R., Halsall H.B. Extending the detection limit of solid-phase electrochemical enzyme-immunoassay to the attomole level. Anal. Biochem. 1988;168:292–299. doi: 10.1016/0003-2697(88)90321-1. [DOI] [PubMed] [Google Scholar]
- Jordan C.E., Frutos A.G., Thiel A.J., Corn R.M. Surface plasmon resonance imaging measurements of DNA hybridization adsorption and streptavidin/DNA multilayer formation at chemically modified gold surfaces. Anal. Chem. 1997;69:4939–4947. doi: 10.1021/ac961012z. [DOI] [PubMed] [Google Scholar]
- Kelley S.O., Boon E.M., Barton J.K., Jakson N.M., Hill M.G. Single-base mismatch detection based on charge transduction through DNA. Nucl. Acids Res. 1999;27:4830–4837. doi: 10.1093/nar/27.24.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levicky R., Herne T.M., Tarlow M.J., Satija S.K. Using self-assembly to control the structure of DNA monolayers on gold: a neutron reflectivity study. J. Am. Chem. Soc. 1998;120:9787–9792. [Google Scholar]
- Lucarelli F., Marrazza G., Turner A.P.F., Marcini M. Carbon and gold electrodes as electrochemical transducers for DNA hybridisation sensors. Biosens. Bioelectron. 2004;19:515–530. doi: 10.1016/s0956-5663(03)00256-2. [DOI] [PubMed] [Google Scholar]
- Marra M.A. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Matisyk F.-M. Miniaturization of electroanalytical systems. Anal. Bioanal. Chem. 2003;375:33–35. doi: 10.1007/s00216-002-1635-x. [DOI] [PubMed] [Google Scholar]
- Meusel M., Renneberg R., Spener F., Schmitz G. Development of a heterogeneous amperometric immunosensor for the determination of apolipoprotein-E in serum. Biosens. Bioelectron. 1995;10:577–586. doi: 10.1016/0956-5663(95)96933-p. [DOI] [PubMed] [Google Scholar]
- Minunni M., Tombelli S., Scielzi R., Mannelli I., Mascini M., Gaudiano C. Detection of β-thalassemia by a DNA piezoelectric biosensor coupled with polymerase chain reaction. Anal. Chim. Acta. 2003;481:55–64. [Google Scholar]
- Nakamura H., Karube I. Current research activity in biosensors. Anal. Bioanal. Chem. 2003;377:446–468. doi: 10.1007/s00216-003-1947-5. [DOI] [PubMed] [Google Scholar]
- Pan W., Durning C.J., Turro N.J. Kinetics of alkanethiol adsorption on gold. Langmuir. 1996;12:4469–4473. [Google Scholar]
- Pividori M.I., Merkoçi A., Alegret S. Electrochemical genosensor design: immobilisation of oligonucleotides onto transducer surfaces and detection methods. Biosens. Bioelectron. 2000;15:291–303. doi: 10.1016/s0956-5663(00)00071-3. [DOI] [PubMed] [Google Scholar]
- Pividori M.I., Merkoçi, Alegret S. Classical dot-blot format implemented as an amperometric hybridisation genosensor. Biosens. Bioelectron. 2001;16:1133–1142. doi: 10.1016/s0956-5663(01)00242-1. [DOI] [PubMed] [Google Scholar]
- Steel A.B., Herne T.M., Tarlov M.J. Electrochemical quantitation of DNA immobilised on gold. Anal. Chem. 1998;70:4670–4677. doi: 10.1021/ac980037q. [DOI] [PubMed] [Google Scholar]
- Taft B.J., O’Keefe M., Fourkas J.T., Kelley S.O. Engineering DNA-electrode connectivities: manipulation of linker length and structure. Anal. Chim. Acta. 2003;496:81–91. [Google Scholar]
- Tie F., Pan A., Ru B., Wang W., Hu Y. An improved ELISA with linear sweep voltammetry detection. J. Immunol. Meth. 1992;149:115–120. doi: 10.1016/s0022-1759(12)80055-x. [DOI] [PubMed] [Google Scholar]
- Wang J. Electrochemical nucleic acid biosensors. Anal. Chim. Acta. 2002;469:63–71. [Google Scholar]
- Wijayawardhana C.A., Halsall H.B., Heineman W.R. Microvolume rotating disk electrode (RDE) amperometric detection for a bead-based immunoassay. Anal. Chim. Acta. 1999;399:3–11. [Google Scholar]
- Wu K., Fei J., Bai J., Hu S. Direct electrochemistry of DNA, guanine and adenine at a nonstructured film-modified electrode. Anal. Bioanal. Chem. 2003;376:205–209. doi: 10.1007/s00216-003-1887-0. [DOI] [PubMed] [Google Scholar]
- Xu D., Huang K., Liu Z., Liu Y., Ma L. Microfabricated disposable DNA sensors based on enzymatic amplification electrochemical detection. Electroanalysis. 2001;13:882–887. [Google Scholar]
- Zhang, R.-Y., Pang, D.-W., Zhang, Z.-L., Yan, J.-W, Yao, J.-L, Tian, Z.-Q., Mao, B.-W., Sun, S.-G., 2002. Investigation of ordered ds-DNA monolayers on gold electrodes.
- Zhu N., Cai H., He P., Fang Y. Tris (2,2′-bipyridyl)cobalt (III)-doped silica nanoparticle DNA probe for the electrochemical detection of DNA hybridisation. Anal. Chim. Acta. 2003;481:181–189. [Google Scholar]