

Abstract
Reovirus is a gastroenteric virus with a genome that consists of ten segments of double-stranded RNA. The segmented nature of the genome allows for genetic mixing when cells are simultaneously infected with two different viral serotypes. The ability of viral reassortment to take place in asynchronous infections has not previously been investigated with mammalian reoviruses. In this study, five different cell lines, representing mouse, monkey, and human, were infected synchronously or asynchronously with various sets of two different temperature-sensitive (ts) reovirus mutants in order to study the genetic interactions which occur. Recombinant viruses were detected at high frequency when infection by the two different ts mutants was separated by as much as 24 h, suggesting that superinfection exclusion does not play a role in reovirus mixed infections. The apparent lack of superinfection exclusion in reovirus infections may have important implications in its evolution.
Keywords: Double-stranded RNA, Human, Monkey, Mouse, Reovirus, Temperature-sensitive
Mammalian reoviruses can infect a variety of host species but cause significant pathology only in a subset of these hosts, most notably in neonatal mice (Fields and Greene, 1982; Bass et al., 1988; Ramig et al., 1989). The inability for reovirus to cause substantial pathogenesis in the human tissues they infect (primarily lung and gut) resulted in the acronym reo, for `respiratory enteric orphan'. Despite the agent's apparent inability to cause significant human disease, reoviruses have been extensively studied through the years due to a unique property of their genome; the reovirus genome is composed of ten discrete segments of dsRNA which are assembled into complete virions with extremely high efficiency (for review see Joklik and Roner, 1996). The mechanism which enables such efficient virus assembly is unknown, but the ability of the individual genome segments to reassort among different reovirus serotypes has been well studied and exploited experimentally (Fields and Joklik, 1969; Fields, 1971; Chakraborty et al., 1979; Ahmed and Fields, 1981; Ramig and Fields, 1983; Antczak and Joklik, 1992).
Reassortment can be easily observed when using sets of two different temperature-sensitive (ts) mutants of reovirus in mixed infections. If used individually, neither of the two parental ts mutants can form viable progeny virions capable of replication at 39°C since each contains a mutation in one of the ten gene segments, resulting in decreased functional efficiency at higher temperatures. However, when cells are simultaneously co-infected with two ts viral mutants whose lesions reside on different gene segments, reassortment can be detected as recombinant wild-type-like (ts+) progeny capable of replication at the conditionally-lethal (nonpermissive) temperature of 39°C because a portion of virions being produced will package the wild-type genes of both parents, omiting the two mutated genes from each respective parent. Reassortment can be calculated by the formula:
| (AB)NP−(A+B)NP(AB)P × 100 |
where A and B are the viral titers of the two different temperature sensitive mutants; NP=nonpermissive temperature of 39°C and P=permissive temperature of 32°C. Although synchronous co-infections between two different virus serotypes or ts mutants consistently result in reassortment amongst all investigated genera of the family Reoviridae, the effects of asynchronous infections have yielded opposing results when different genera were studied.
A wide variety of cell types, when infected with a virus, are unable to host a successful secondary infection of the same or a closely-related virus; this phenomenon is called superinfection exclusion with the second, or superinfecting, virus being prevented in some way from successfully replicating in the infected cells. Superinfection exclusion is not well understood and a number of different mechanisms have been suggested to explain it. For example, competition for host factors required for entry (e.g. surface glycoproteins) or for viral replication and transcription (e.g. various host enzymes, or chaperones) may hinder the progress of a superinfecting virus. Superinfection exclusion was first described in bacteriophages (Dulbecco, 1952; Visconti, 1953) but many viruses which infect eukaryotic cells also display an exclusion phenotype. The first animal virus shown to undergo superinfection exclusion was vesicular stomatitis virus (VSV) (Whitaker-Dowling et al., 1983). Similarly, vaccinia virus-infected BSC40 cells did not permit the replication of superinfecting vaccinia virus (Christen et al., 1990). Although the methodology of superinfection exclusion has not been elucidated, adsorption of the superinfecting virus occurred for both VSV and vaccinia, and the exclusion was thought to target an early replicative stage of the second virus. In both vertebrate and invertebrate cells infected with Sindbis virus, superinfection exclusion occurred at an intracellular stage since transfected viral RNA was also inhibited from replicating in infected mosquito cells (Karpf et al., 1997).
In other virus systems, interference with entry or penetration of the superinfecting virus has been observed. Influenza virus synthesizes a neuraminidase which destroys the cell surface receptor initially required for infection (Palese et al., 1974), thereby preventing progeny virions from re-infecting the same host cell. This destruction of the cell surface receptor for influenza viruses may also prevent the re-infection of a host cell by a different influenzavirus serotype; coronavirus expresses an esterase which may have similar functions (Vlasak et al., 1988). For herpesvirus, the cell surface expression of glycoprotein D inhibits superinfection of both HSV-1 or HSV-2 serotypes by preventing virus penetration (Dean et al., 1995).
Superinfection exclusion occurs in some members of the Reoviridae family as well. In Bluetongue virus (BTV), a prototype of the orbivirus genus, reassortment (or recombination) failed to occur when the second virus followed the first infecting virus by >4 h in vitro, or 5 days in vivo (el-Hussein et al., 1989; Ramig et al., 1989). The mechanism for this exclusion in BTV is not yet known. On the other hand, rotaviruses (another member of Reoviridae) do not undergo superinfection exclusion (Ramig, 1990). Recombination was still detected between two rotavirus ts mutants even if inoculation with the second virus occurred 24 h after the initial infection. With regard to reassortment of genome segments and protein function, the members of the Reoviridae family function in a very similar fashion. It is reasonable to assume that all viral genera in a family would share such a notable characteristic as superinfection exclusion. To attempt to resolve the conflicting results thus observed, the possibility of superinfection exclusion occurring in mammalian reoviruses is examined.
For the present study, four temperature-sensitive mutants were used in the superinfection experiments. These mutants were chosen for analysis because they have been well characterized by our lab (Coombs et al., 1994; Coombs, 1996; Shing and Coombs, 1996). tsC447, tsD357, tsG453 and tsH11.2 all map to independent genome segments, and the locations of the lesions are known. At permissive temperature, the growth rates of these mutants were comparable to that of wild-type reovirus, thus allowing the detection of wild-type-like recombinant progeny from mixed infections between the ts mutants when subsequently assayed at nonpermissive temperature. The L929 fibroblast cell line was grown in MEM medium, supplemented with 2.5% FCS/2.5% VSP and antibiotics as described (Coombs et al., 1994). Infections were carried out in 2-dm vials containing approximately 8×105 cells. To begin the infection, the medium was removed from the cell line and the appropriate virus mutant was added at a multiplicity of infection (MOI) of 5 PFU per cell and allowed to adsorb for 1 h at room temperature. After adsorption, 500 μl of fresh medium was added to the cells, which were then incubated at 32°C. At the appropriate time post-infection, the medium was removed and the superinfecting virus was adsorbed at an MOI of 5 PFU for 1 h. Fresh medium (500 μl) was again added after virus adsorption, and all samples were incubated at 32°C. All infections were stopped 72 h after adsorption of the first virus. The infected cell monolayers were disrupted by three cycles of freeze/thawing followed by sonication in order to efficiently release the virus. Virus replication was determined by plaque assay titrations at 32°C and 39°C as described (Coombs, 1996) and recombination frequencies determined by the formula provided earlier. Recombination ratios also were standardized to values obtained at Time 0, when the virus mutants were added synchronously to the cells. In this way, a decline in recombination efficiency could be easily detected.
Self crosses failed to recombine and form wild-type-like progeny since the mutational lesion that creates the temperature-sensitive phenotype is on the same genome segment. In contrast, virtually all possible pairs of synchronous infections in L929 cells between the four different groups of ts mutants (i.e. tsC×tsD, tsH×tsD,... tsG×tsH) resulted in wild-type-like recombinant progeny, as illustrated by recombination values above zero (Table 1 , 0 h points), and as previously reported (Fields and Joklik, 1969; Ramig and Fields, 1979; Coombs, 1996). One cross, tsC×tsG, failed to recombine at 39°C (see Table 1). Both tsC447 and tsG453 were characterized by our lab using extensive reassortment analysis (Coombs et al., 1994; Shing and Coombs, 1996); thus, the likelihood that these two mutants are from the same recombination group is unlikely. It has been previously noted that recombination values obtained by the standard mathematical equation often result in wide variations upon repeat of the same virus co-infections (Ramig, 1983). Chakraborty and coworkers also observed this peculiar anomoly (Chakraborty et al., 1979). They discovered that certain ts mutants interfered with the growth of ts+ virus at 39°C. The tsC x tsG cross was specifically noted for the low levels of both reassortment and growth of progeny virions.
Table 1.
Recombination between selected reovirus temperature-sensitive mutants resulting from synchronous and asynchronous co-infections
| MUTANT | EOPa | Percentage ts+ recombinantsb when crossed with |
|||
| tsC447 | tsD357 | tsG453 | tsH11.2 | ||
| 0 hc | |||||
| tsC447 | 9.1x10−6 | 0.000 | 2.06±0.86 | 0.000 | 0.190±0.06 |
| tsD357 | 8.49x10−4 | 0.000 | 2.75±0.74 | 0.925±0.405 | |
| tsG453 | 2.8x10−5 | 0.000 | 1.11±0.62 | ||
| tsH11.2 | 1.5x10−4 | 0.000 | |||
| 4 hc | |||||
| tsC447 | 0.000 | 1.075±0.005 | 0.000 | 0.195±0.055 | |
| tsD357 | 0.000 | 7.53±6.57 | 1.765±0.975 | ||
| tsG453 | 0.000 | 0.60±0.10 | |||
| tsH11.2 | 0.000 | ||||
| 24 hc | |||||
| tsC447 | 0.000 | 1.85±0.62 | 0.000 | 0.26±0.01 | |
| tsD357 | 0.000 | 1.42±1.18 | 0.54±0.22 | ||
| tsG453 | 0.000 | 1.025±0.115 | |||
| tsH11.2 | 0.000 | ||||
Efficiency of plating=titer A39÷titer A32.
bInfections incubated at permissive temperature of 32°C for 72 h, freeze/thawed, and titered at both 32°C and 39°C as detailed in the text. Percent ts+ recombinants={[(titer AB39)−(titer A39+titer B39)]÷titer AB32}×100.
cTime (in hours) when virus 2 was added to a culture infected with virus 1.
It is generally accepted that recombination is an `all or none' phenomenon, and what is important is whether recombination occurs at all. If it does, then the dysfunctional genome segment of one ts mutant can be replaced by the corresponding wild-type genome segment of another ts mutant; obviously these two viruses would then be classified as two separate groups of ts mutants. For example, the wild-type M1 gene of tsC mutants (whose ts lesion resides in the S2 gene) can replace the mutated M1 gene segment of tsH viral serotypes, thus resulting in progeny that no longer display a temperature-sensitive phenotype. If two tsH mutants were crossed, there would be no wild-type M1 gene from either parent, and therefore production of wild-type-like recombinant progeny would not occur. Recombination experiments such as these were initially used to identify what gene mutation within the viral genome resulted in the temperature-sensitive phenotype, and to classify the virus mutants into groups A through J, depending on which gene segment contained the temperature-sensitive mutation (reviewed in Ramig and Fields, 1983; Ramig and Ward, 1991; Coombs, 1998).
To determine the time frame during which reovirus superinfection exclusion might or might not occur, L929 cells were mixedly infected under asynchronous conditions. Therefore, at the appropriate time after the initial infection, superinfection was carried out with either the same virus (as a control) or with a virus from a different group of ts mutants. Table 2 and Fig. 1 show representative results for synchronous and asynchronous infection of L929s with representative pairs of reovirus ts mutants. Recombination still occurred between the two virus mutants (either tsC and tsH or tsG and tsD), even when the superinfecting virus was added 24 h after the first virus. Later times for superinfection were attempted but the cells displayed cytopathic effects, primarily loss of adhesion from the substrate, by 36 h post-infection with virus 1. A gradual decline in the recombination rate was observed, as easily visualized in Fig. 1, and probably relates to the diminished cell viability upon infection with the second virus, since the first virus had already finished a complete round of replication. In cases where superinfection exclusion has been documented, addition of the superinfecting virus at relatively early time points of 4–6 h after initial virus infection consistently resulted in a dramatic decline in recombination efficiency up to 100-fold (for examples see Whitaker-Dowling et al., 1983; el-Hussein et al., 1989; Ramig et al., 1989; Christen et al., 1990). Comparison of these abrupt and dramatic declines in recombination in cases of superinfection exclusion with our gradual and minor declines indicates that superinfection exclusion did not play a role in asynchronous infections of L929 cells. The order by which each mutant was adsorbed (first or second infecting virus) was also monitored to determine its significance. As shown in Fig. 1, the recombination ratio, which compares the percentage of wild-type-like recombinant progeny obtained from synchronous and asynchronous infections, did not significantly vary depending on which ts mutant was adsorbed first. More importantly, there did not appear to be any exclusion of the second infecting virus, even 24 h after infection with the first virus. L929 cells that were infected with a wider variety of reovirus mutants at key time points of 4- and 24 h also suggest that superinfection exclusion did not occur since recombination was still evident when the second virus was adsorbed 24 h after the initial infection (Table 1). This suggests a general phenomenon that is not specific for various reovirus serotypes. For example, tsH11.2 carries a defective reovirus serotype T1L-derived M1 gene (Coombs et al., 1994; Coombs, 1996), and the other ts mutants used were derived from serotype T3D, yet superinfection exclusion did not occur with any of the crosses.
Table 2.
Recombination between reovirus temperature-sensitive mutants in asynchronous infections of L929 cells
| First virus | Second virus | % ts+ recombinantsa from indicated crosses with the second virus added at the indicated time (hours after first virus) |
||||||
| 0 | 2 | 4 | 8 | 12 | 18 | 24 | ||
| tsH11.2 | tsC447 | 1.14 | 3.02 | 1.30 | 0.60 | 0.10 | 0.12 | 0.10 |
| (1.00)b | (2.65) | (1.14) | (0.526) | (0.100) | (0.110) | (0.100) | ||
| tsC447 | tsH11.2 | 2.03 | 2.19 | 2.53 | 2.94 | 0.87 | 1.17 | 0.10 |
| (1.00) | (1.08) | (1.25) | (1.45) | (0.429) | (0.576) | (0.0500) | ||
| tsD357 | tsG453 | 9.94 | 10.9 | 5.9 | 8.9 | 1.58 | 5.8 | 2.6 |
| (1.00) | (1.10) | (0.594) | (0.895) | (0.159) | (0.584) | (0.262) | ||
| tsG453 | tsD357 | 8.8 | 0.78 | 12.7 | 6.5 | 3.9 | 6.6 | 0.124 |
| (1.00) | (0.089) | (1.44) | (0.739) | (0.443) | (0.750) | (0.014) | ||
Fig. 1.
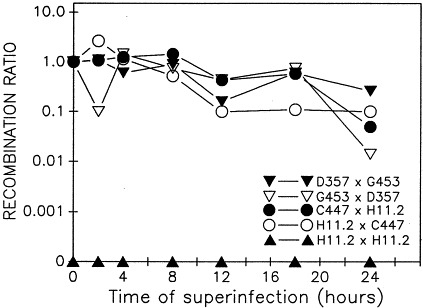
Recombination between reovirus temperature-sensitive mutants in asynchronous infections of L929 cells was investigated by infecting the cells at various times and incubating the mixed infections at 32°C for 72 h. Recombination ratio values for various ts mutant crosses were calculated and standardized to the 0 time point (set as a recombination ratio of 1.0), a time when reassortment readily occurs. The recombination ratios (values in parentheses from Table 2 above) were plotted to facilitate visualization of the relative levels of recombination that occurred when the second virus was added at the indicated time. ▾, cells already infected with tsD357 for the indicated periods of time were superinfected with tsG453 (tsD357×tsG453); ▿, cells previously infected with tsG453 superinfected with tsD357 (tsG453×tsD357); •, cells previously infected with tsC447 superinfected with tsH11.2 (tsC447×tsH11.2); ○, cells previously infected with tsH11.2 superinfected with tsC447 (tsH11.2×tsC447); ▴, cells previously infected with tsH11.2 superinfected with tsH11.2 (tsH11.2×tsH11.2).
Reoviruses produce cytopathic effects in a wide variety of cultured cells. Four additional cell lines were used to determine if the results obtained in L929 cells were cell-specific, or a characteristic attributable to reovirus. If L cell results were different from results obtained in alternate cell types, then the use of different cell lines might explain the rotavirus/BTV dichotomy. The cell lines chosen included MA104 cells (a monkey kidney cell line, kindly donated by Dr L. Babiuk, VIDO, University of Saskatchewan), which had been used to study superinfection exclusion in rotaviruses (Ramig, 1990), and three human-derived cell lines, polarized HT29 (enterocytes, a gift from Dr N. Simonsen, University of Manitoba), U373s (astrocytes), and U937s (macrophages). Both the astrocyte and macrophage cell lines were generously provided by Dr A. Nath, University of Manitoba. All cell lines used were grown in MEM medium except for the U937 cell line which was grown in RPMI medium. The tropism of reovirus for gastrointestinal tissue suggested that the human-derived HT29 cell line might provide a more representative substrate for a `normal' infection by a human reovirus than the mouse L929 cells. Astrocytes were used in our study due to the serious neuropathological effects of reovirus that have been extensively shown to occur in mice (Margolis et al., 1971; Hrdy et al., 1982; Dichter and Weiner, 1984; Cohen et al., 1990; Flamand et al., 1991), and because there is some indication that reovirus may be associated with human CNS disease (Johansson et al., 1996). Macrophages are important cells with respect to immunopathological events; theoretically, if superinfection exclusion of reovirus only occurred in U937 cells, it could suggest a cell-specific immunoevasion strategy of reovirus. As U937's have previously been utilized for reovirus studies (Farone et al., 1993), we knew that the virus could successfully replicate within this type of macrophage. For the remaining cell lines, we first confirmed that reoviruses could indeed replicate in the astrocytes, gut and monkey kidney cell lines. Two wild-type reovirus serotypes (T1L, T3D) were used to infect the cells; although viral titers obtained varied between cell types, all could accommodate replication of reovirus as seen in Fig. 2 . Total viral yields from infection with temperature-sensitive mutants in the five cell lines also demonstrated the ability of reovirus to replicate in all cell lines examined. For both synchronous and asynchronous infections, viral titers at permissive temperature ranged from 5×106–5×109 PFU/ml in the five cell lines used. This represents an increase of 0.5 to 3 orders of magnitude over input titer. Replication of the T3D reovirus serotype in HT29 cells was minimal, yet the titer for the T1L serotype increased by two orders of magnitude.
Fig. 2.
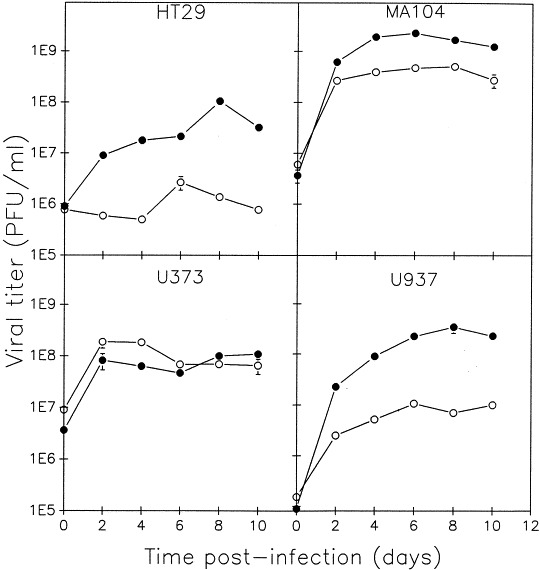
Capacity of reovirus strains to replicate in various monkey- and human-derived cell lines. Reovirus serotype 1, strain Lang (T1L, •) and serotype 3, strain Dearing (T3D, ○) were adsorbed to the indicated cells (HT29, human polarized enterocytes; MA104, monkey kidney; U373, human astrocytoma; and U937, human macrophages) at an MOI of 5 PFU/cell. After adsorption cells were cultured at 37°C. Infected cells were harvested at the indicated times post-infection, freeze-thawed, and the amount of virus produced determined by standard plaque assay. Data points represent the average of two separate experiments.
Fig. 3 shows the results of asynchronous infections of HT29, MA104, U373 and U937 cell lines with various reovirus temperature-sensitive mutants. Wild-type-like recombinant progeny were still detected at the nonpermissive temperature of 39°C even when the superinfecting virus was added 24 h after the initial infection. The self-cross of tsH×tsH mutants (which was representative of all other ts self-crosses) failed to recombine at any time point, supporting the significance of the recombination rates obtained for the various ts mutant crosses. The crosses shown in Fig. 3 are representative of all 16 ts mutant crosses that we examined, except for tsC×tsG, which, as indicated earlier, failed to significantly reassort, even under synchronous conditions. In the L929, HT29, MA104, U373 and U937 cell lines studied, there was no evidence of superinfection exclusion occurring in asynchronous infections with reovirus temperature sensitive mutants. The variability in recombination rates was most dramatic in MA104 cells; upon repeating the superinfection experiment, the recombination values varied more than one order of magnitude. The variability obtained with MA104 cells highlights the need to investigate viral mechanisms, such as superinfection exclusion, in a variety of cell types as was done in this study. In the other four cell lines used, the recombination rates obtained were significantly more constant, better illustrating the absence of superinfection exclusion with mammalian reoviruses.
Fig. 3.
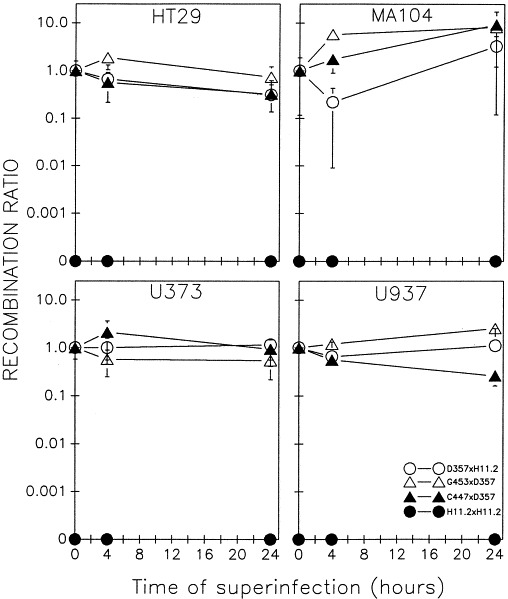
Recombination between reovirus temperature-sensitive mutants in asynchronous infections of HT29, MA104, U373 and U937 cell lines. Recombination ratio values for various ts mutant crosses in the indicated cells were determined as described in the legend to Fig. 1. ○, tsD357×tsH11.2; ▵, tsG453×tsD357; ▴, tsC447×tsD357; •, tsH11.2×tsH11.2.
From these studies, we conclude that superinfection exclusion plays no part in reovirus infections in vitro. By utilizing a number of different virus mutants and a variety of cell types derived from different species, the lack of exclusion does not appear to be mutant- or cell-specific. Although the in vivo scenario was not examined, various studies support the significance of using established cell lines to investigate the reovirus lifecycle (el-Hussein et al., 1989; Ramig et al., 1989; Farone et al., 1993).
The inability of reovirus to exclude superinfecting viruses of the same genera from genetic interactions during the infection process has significant consequences. Genome reassortment has been theorized to be a molecular mechanism for RNA virus variation (for examples see representative rotavirus (Gombold and Ramig, 1994) and influenza (Young and Palese, 1979; Palese and Young, 1982) articles). The absence of superinfection exclusion would increase the number of virus particles that could infect a single cell, thus enhancing the likelihood of reassortment occurring among the progeny virions. If the absence of superinfection exclusion is advantageous with regard to virus variation, why do many viruses, including VSV, Sindbis, herpesvirus, and vaccinia, exclude superinfecting viruses? Mechanisms thought to be involved with the reovirus life cycle suggest that it is relatively independent from host cell functions when compared to other viruses, including influenza virus (Krug et al., 1976; Plotch et al., 1979, Plotch et al., 1981) and all DNA viruses. Dependence on host cell factors (e.g. the `cap-snatching' seen in influenza) and the cell cycle (e.g. DNA viruses) would explain the need for excluding any superinfecting viruses which may outcompete the first virus. Furthermore, most viruses, especially negative-strand RNA viruses, readily generate and amplify defective interfering (DI) virus particles (McNulty et al., 1978; Lazzarini et al., 1981; Perrault, 1981; Kuge et al., 1986; Frey and Hemphill, 1988; Odagiri et al., 1994). DI particles often have smaller genomes which can be more efficiently replicated than a virus containing a complete genome. Thus, perhaps the reproductive benefit derived from preventing entry of defective virions into the cell, with subsequent enhancement of replication of the resident first virus, outweighs the advantage of increasing virus variation. Interestingly, reovirus may replicate efficiently regardless of the presence of additional defective reovirus particles, as was noted by Ahmed and Fields (Ahmed and Fields, 1981).
From a practical aspect, the lack of superinfection exclusion would allow an altering of the standard protocol for generating reovirus reassortants. Co-infection with two reovirus serotypes is routinely used to produce and later isolate reassortant progeny; however, the percentage of progeny that indeed reassort, depend on the ability of the parental viruses to replicate. For instance, T1L and T3D are significantly more efficient at replicating in L929 cells than the viral serotype T2J. If one were to infect with T2J first, and wait 12–24 h before infecting with T1L or T3D, the slower replicator would have an advantage that might result in a higher yield of reassortant progeny. This method of generating reassortants was utilized in a study with avian reoviruses (Ni and Kemp, 1990), indirectly demonstrating the absence of superinfection exclusion in avian reoviruses as well. Since reovirus strains do not require synchronous co-infection to reassort, this would greatly affect infections in vivo, where opportunities for a second reovirus infection would be notable.
Acknowledgements
We thank Drs L. Babiuk, A. Nath, and J.N. Simonsen for the monkey and human cell lines used in this study; and Mark Makarovsky and Matthew Oughton for helpful discussion. This research was supported by grant MT-11630 from the Medical Research Council of Canada. N.D. Keirstead is a recipient of a scholarship from the Natural Sciences and Engineering Research Council of Canada.
References
- Ahmed R, Fields B.N. Reassortment of genome segments between reovirus defective interfering particles and infectious virus: construction of temperature-sensitive and attenuated viruses by rescue of mutations from DI particles. Virology. 1981;111:351–363. doi: 10.1016/0042-6822(81)90339-1. [DOI] [PubMed] [Google Scholar]
- Antczak J.B, Joklik W.K. Reovirus genome segment assortment into progeny genomes studied by the use of monoclonal antibodies directed against reovirus proteins. Virology. 1992;187:760–776. doi: 10.1016/0042-6822(92)90478-8. [DOI] [PubMed] [Google Scholar]
- Bass D.M, Trier J.S, Dambrauskas R, Wolf J.L. Reovirus type I infection of small intestinal epithelium in suckling mice and its effect on M cells. Lab. Invest. 1988;58:226–235. [PubMed] [Google Scholar]
- Chakraborty P.R, Ahmed R, Fields B.N. Genetics of reovirus: the relationship of interference to complementation and reassortment of temperature-sensitive mutants at non-permissive temperatures. Virology. 1979;94:119–127. doi: 10.1016/0042-6822(79)90442-2. [DOI] [PubMed] [Google Scholar]
- Christen L, Seto J, Niles E.G. Superinfection exclusion of vaccinia virus in virus-infected cell cultures. Virology. 1990;174:35–42. doi: 10.1016/0042-6822(90)90051-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.A, Williams W.V, Weiner D.B, Geller H.M, Greene M.I. Ligand binding to the cell surface receptor for reovirus type 3 stimulates galactocerebroside expression by developing oligodendrocytes. Proc. Natl. Acad. Sci. (USA) 1990;87:4922–4926. doi: 10.1073/pnas.87.13.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs K.M. Identification and characterization of a double-stranded RNA-reovirus temperature-sensitive mutant defective in minor core protein μ2. J. Virol. 1996;70:4237–4245. doi: 10.1128/jvi.70.7.4237-4245.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs K.M. Temperature-sensitive mutants of reovirus. Curr. Top. Microbiol. Immunol. 1998;233:69–107. doi: 10.1007/978-3-642-72092-5_4. [DOI] [PubMed] [Google Scholar]
- Coombs K.M, Mak S.C, Petrycky-Cox L.D. Studies of the major reovirus core protein sigma 2: reversion of the assembly-defective mutant tsC447 is an intragenic process and involves back mutation of Asp-383 to Asn. J. Virol. 1994;68:177–186. doi: 10.1128/jvi.68.1.177-186.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean H.J, Warner M.S, Terhune S.S, Johnson R.M, Spear P.G. Viral determinants of the variable sensitivity of herpes simplex virus strains to gD-mediated interference. J. Virol. 1995;69:5171–5176. doi: 10.1128/jvi.69.8.5171-5176.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichter M.A, Weiner H.L. Infection of neuronal cell cultures with reovirus mimics in vitro patterns of neurotropism. Ann. Neurol. 1984;16:603–610. doi: 10.1002/ana.410160512. [DOI] [PubMed] [Google Scholar]
- Dulbecco R. Mutual exclusion between related phages. J. Bacteriol. 1952;63:209–217. doi: 10.1128/jb.63.2.209-217.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Hussein A, Ramig R.F, Holbrook F.R, Beaty B.J. Asynchronous mixed infection of Culicoides variipennis with bluetongue virus serotypes 10 and 17. J. Gen. Virol. 1989;70:3355–3362. doi: 10.1099/0022-1317-70-12-3355. [DOI] [PubMed] [Google Scholar]
- Farone A.L, O'Brien P.C, Cox D.C. Tumor necrosis factor-alpha induction by reovirus serotype 3. J. Leuk. Biol. 1993;53:133–137. doi: 10.1002/jlb.53.2.133. [DOI] [PubMed] [Google Scholar]
- Fields B.N. Temperature-sensitive mutants of reovirus type 3: Features of genetic recombination. Virology. 1971;46:142–148. doi: 10.1016/0042-6822(71)90013-4. [DOI] [PubMed] [Google Scholar]
- Fields B.N, Greene M.I. Genetic and molecular mechanisms of viral pathogenesis: implications for prevention and treatment. Nature. 1982;300:19–23. doi: 10.1038/300019a0. [DOI] [PubMed] [Google Scholar]
- Fields B.N, Joklik W.K. Isolation and preliminary genetic and biochemical characterization of temperature-sensitive mutants of reovirus. Virology. 1969;37:335–342. doi: 10.1016/0042-6822(69)90217-7. [DOI] [PubMed] [Google Scholar]
- Flamand A, Gagner J.P, Morrison L.A, Fields B.N. Penetration of the nervous systems of suckling mice by mammalian reoviruses. J. Virol. 1991;65:123–131. doi: 10.1128/jvi.65.1.123-131.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey T.K, Hemphill M.L. Generation of defective-interfering particles by rubella virus in Vero cells. Virology. 1988;164:22–29. doi: 10.1016/0042-6822(88)90615-0. [DOI] [PubMed] [Google Scholar]
- Gombold J.L, Ramig R.F. Genetics of the rotaviruses. Curr. Top. Microbiol. Immunol. 1994;185:129–177. doi: 10.1007/978-3-642-78256-5_6. [DOI] [PubMed] [Google Scholar]
- Hrdy D.B, Rubin D.H, Fields B.N. Molecular basis of reovirus neurovirulence: role of the M2 gene in avirulence. Proc. Natl. Acad. Sci. (USA) 1982;79:1298–1302. doi: 10.1073/pnas.79.4.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson P.J, Sveger T, Ahlfors K, Ekstrand J, Svensson L. Reovirus type 1 associated with meningitis. Scan. J. Inf. Dis. 1996;28:117–120. doi: 10.3109/00365549609049060. [DOI] [PubMed] [Google Scholar]
- Joklik W.K, Roner M.R. Molecular recognition in the assembly of the segmented reovirus genome. Prog. Nucleic Acid Res. Mol. Biol. 1996;53:249–281. doi: 10.1016/s0079-6603(08)60147-6. [DOI] [PubMed] [Google Scholar]
- Karpf A.R, Lenches E, Strauss E.G, Strauss J.H, Brown D.T. Superinfection exclusion of alphaviruses in three mosquito cell lines persistently infected with Sindbis virus. J. Virol. 1997;71:7119–7123. doi: 10.1128/jvi.71.9.7119-7123.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug R.M, Morgan M.A, Shatkin A.J. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J. Virol. 1976;20:45–53. doi: 10.1128/jvi.20.1.45-53.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge S, Saito I, Nomoto A. Primary structure of poliovirus defective-interfering particle genomes and possible generation mechanisms of the particles. J. Mol. Biol. 1986;192:473–487. doi: 10.1016/0022-2836(86)90270-6. [DOI] [PubMed] [Google Scholar]
- Lazzarini R.A, Keene J.D, Schubert M. The origins of defective interfering particles of the negative-strand RNA viruses. Cell. 1981;26:145–154. doi: 10.1016/0092-8674(81)90298-1. [DOI] [PubMed] [Google Scholar]
- Margolis G, Kilham L, Gomatos N. Reovirus type III encephalitis: Observations of virus-cell interactions in neural tissues. I. Light microscopy studies. Lab. Invest. 1971;24:91–109. [PubMed] [Google Scholar]
- McNulty M.S, Curran W.L, Allan G.M, McFerran J.B. Synthesis of coreless, probably defective virus particles in cell cultures infected with rotaviruses. Arch. Virol. 1978;58:193–202. doi: 10.1007/BF01317601. [DOI] [PubMed] [Google Scholar]
- Ni Y, Kemp M.C. Selection of genome segments following coinfection of chicken fibroblasts with avian reoviruses. Virology. 1990;177:625–633. doi: 10.1016/0042-6822(90)90528-y. [DOI] [PubMed] [Google Scholar]
- Odagiri T, Tominaga K, Tobita K, Ohta S. An amino acid change in the nonstructural NS2 protein of an influenza A virus mutant is responsible for the generation of defective interfering (DI) particles by amplifying DI RNAs and suppressing complementary RNA synthesis. J. Gen. Virol. 1994;75:43–53. doi: 10.1099/0022-1317-75-1-43. [DOI] [PubMed] [Google Scholar]
- Palese P, Young J.F. Variation of influenza A. B, and C viruses. Science. 1982;215:1468–1474. doi: 10.1126/science.7038875. [DOI] [PubMed] [Google Scholar]
- Palese P, Tobita K, Ueda M, Compans R.W. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology. 1974;61:397–410. doi: 10.1016/0042-6822(74)90276-1. [DOI] [PubMed] [Google Scholar]
- Perrault J. Origin and replication of defective interfering particles. Curr. Top. Microbiol. Immunol. 1981;93:151–207. doi: 10.1007/978-3-642-68123-3_7. [DOI] [PubMed] [Google Scholar]
- Plotch S.J, Bouloy M, Krug R.M. Transfer of 5′-terminal cap of globin mRNA to influenza viral complementary RNA during transcription in vitro. Proc. Natl. Acad. Sci. (USA) 1979;76:1618–1622. doi: 10.1073/pnas.76.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotch S.J, Bouloy M, Ulmanen I, Krug R.M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped mRNAs to generate primers that initiate viral RNA transcription. Cell. 1981;23:847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- Ramig R.F. Factors that affect genetic interactions during mixed infection with temperature- sensitive mutants of simian rotavirus SA11. Virology. 1983;127:91–99. doi: 10.1016/0042-6822(83)90374-4. [DOI] [PubMed] [Google Scholar]
- Ramig R.F. Superinfecting rotaviruses are not excluded from genetic interactions during asynchronous mixed infections in vitro. Virology. 1990;176:308–310. doi: 10.1016/0042-6822(90)90260-x. [DOI] [PubMed] [Google Scholar]
- Ramig R.F, Fields B.N. Revertants of temperature-sensitive mutants of reovirus: evidence for frequent extragenic suppression. Virology. 1979;92:155–167. doi: 10.1016/0042-6822(79)90221-6. [DOI] [PubMed] [Google Scholar]
- Ramig, R., Fields, B.N., 1983. Genetics of Reovirus. In: Joklik, W. (Ed.), The Reoviridae. Plenum Press, New York, pp. 197-228.
- Ramig R.F, Ward R.L. Genomic segment reassortment in rotaviruses and other Reoviridae. Adv. Virus Res. 1991;39:163–207. doi: 10.1016/s0065-3527(08)60795-2. [DOI] [PubMed] [Google Scholar]
- Ramig R.F, Garrison C, Chen D, Bell-Robinson D. Analysis of reassortment and superinfection during mixed infection of Vero cells with bluetongue virus serotypes 10 and 17. J. Gen. Virol. 1989;70:2595–2603. doi: 10.1099/0022-1317-70-10-2595. [DOI] [PubMed] [Google Scholar]
- Shing M, Coombs K.M. Assembly of the reovirus outer capsid requires mu1/sigma3 interactions which are prevented by misfolded sigma3 protein in reovirus temperature-sensitive mutant tsG453. Virus Res. 1996;46:19–29. doi: 10.1016/s0168-1702(96)01372-x. [DOI] [PubMed] [Google Scholar]
- Visconti D. Resistance to lysis from without in bacteria infected with T2 bacteriophage. J. Bacteriol. 1953;66:247–253. doi: 10.1128/jb.66.3.247-253.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasak R, Luytjes W, Leider J, Spaan W, Palese P. The E3 protein of bovine coronavirus is a receptor-destroying enzyme with acetylesterase activity. J. Virol. 1988;62:4686–4690. doi: 10.1128/jvi.62.12.4686-4690.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Dowling P, Youngner J.S, Widnell C.C, Wilcox D.K. Superinfection exclusion by vesicular stomatitis virus. Virology. 1983;131:137–143. doi: 10.1016/0042-6822(83)90540-8. [DOI] [PubMed] [Google Scholar]
- Young J.F, Palese P. Evolution of human influenza A viruses in nature: recombination contributes to genetic variation of H1N1 strains. Proc. Natl. Acad. Sci. (USA) 1979;76(12):6547–6551. doi: 10.1073/pnas.76.12.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]