

Abstract
Inhalation of Saccharopolyspora rectivirgula causes “farmer's lung” disease, a classic example of hypersensitivity pneumonitis (HP). Monocyte chemoattractant protein-1 (MCP-1) is increased in the bronchoalveolar lavage fluid of mice challenged with S rectivirgula, and S rectivirgula induces MCP-1 secretion by alveolar macrophages. We tested the hypothesis that MCP-1 and its receptor CC chemokine receptor-2 (CCR2) are essential to the development of experimental HP by treating mice with MCP-1 antibody and using CCR2−/− mice. Administration of anti–MCP-1 did not change the response to intratracheally administered S rectivirgula. CCR2−/− animals responded in a fashion similar to that of wild-type animals to intratracheally administered.S rectivirgula. To determine the influence of the MCP-1–CCR2 interaction in vitro, we transferred S rectivirgula–cultured spleen cells from S rectivirgula–sensitized mice, to naïve recipients. Later, challenge of the recipients with intratracheal S rectivirgula and examination of both lung histology and bronchoalveolar lavage fluid characteristics were used to determine whether adoptive transfer had occurred. We found that cultured cells from CCR2−/− animals were fully capable of adoptive transfer. We conclude that interaction of MCP-1 with CCR2 is not necessary for the development of pulmonary inflammation in response to intratracheally administered S rectivirgula or cells able to adoptively transfer experimental HP.
Abbreviations: ANOVA, analysis of variance; BALF, bronchoalveolar lavage fluid; CCR2, CC chemokine receptor-2; EHP, experimental hypersensitivity pneumonitis; FLD, “farmer's lung” disease; HEPA, high-efficiency particulate air; HP, hypersensitivity pneumonitis; HSD, honestly significant difference; IFN-γ, interferon-γ; IL, interleukin; MCP, monocyte chemoattractant protein; MIP-1α, macrophage inflammatory protein-1α; SEM, standard error of the mean; Th, T-helper; TNF, tumor necrosis factor
Hypersensitivity pneumonitis is a group of lung diseases that result from repeated pulmonary exposure to various organic, antigenic materials with uncertain immunopathogenetic mechanisms.1
Inhalation of Saccharopolyspora rectivirgula causes FLD, a classic example of HP. HP is characterized by neutrophilia in BALF (within 48 hours of inhalation), followed by BALF lymphocytosis.2 Chemokines that can cause chemotaxis of lymphocytes (eg, MIP-1α, MCP-1, and IL-8) are present in BALF and are released by alveolar macrophages from patients with acute HP.3, 4
We found that 1 intratracheal exposure of mice to S rectivirgula induced a remarkable increase of BALF MIP-1α and MCP-1 that preceded BALF neutrophilia, lymphocytosis, and increase of macrophages. We also noted a marked increase of BALF IL-2, IL-6, and, to a lesser extent, IL-1α and TNF. Murine macrophages (both a cell line and alveolar macrophages) produced the chemokines listed above when stimulated with S rectivirgula.5 These data suggest that local chemokine production is responsible for later influx of cells into compartments assessable to bronchoalveolar lavage and that macrophages represent a possible source of these substances.
Chemokine responses are mediated by the interaction of the soluble chemokine proteins with receptors belonging to the superfamily of 7-transmembrane G protein–coupled receptors. Eleven different CC-family chemokine receptors have been characterized. CCR2 is the major MCP-1 receptor, and MCP-1 binds to CCR2 with high affinity.6, 7, 8
In addition to chemotaxis, chemokines can influence differentiation of lymphocytes into Th1 or Th2 cells, with MCP-1 promoting Th1 cell development. Because our model of EHP appears to be mediated by Th1 cells,9 we hypothesized that absence of MCP-1 effect would decrease the expression of EHP.
We therefore tested the hypothesis that MCP-1–CCR2 interaction increases the development of EHP by treating mice with MCP-1 antibody and using mice deficient in CCR2, the major receptor for MCP-1.
Methods
Animals
CCR2−/− pairs (C57BL/6J × 129/Ola background) were a gift of Gary Huffnagle (University of Michigan, Ann Arbor, Mich). Male B6129PF2 wild-type, C57Bl/6 (Jackson Laboratory, Bar Harbor, Maine), and male CCR2−/− mice were housed under laminar-flow hoods with HEPA-filtered air in the Veterinary Medical Unit, fully accredited by the American Association for Accreditation of Laboratory Animal Care.
Antigen
S rectivirgula was prepared as previously described.9, 10, 11
Study design
Our primary outcome was measurement of the extent of pulmonary histologic abnormalities, but we also measured BALF cell types and counts. BALF cytokine and chemokine measurements were used to characterize the inflammatory milieu within the lung.
To measure the effect of anti–MCP-1, we administered 0.5 mg of polyclonal rabbit anti–MCP-1 or phosphate-buffered saline solution intraperitoneally in 0.2 mL 1 hour before and 24 and 72 hours after 7.2 μg/g of intratracheally administered S rectivirgula to C57Bl/6 mice. Animals were killed at 96 hours. This schedule was based on previous work that demonstrated effectiveness of in vivo treatments with anti-MCP.12, 13, 14 We generated polyclonal rabbit anti–MCP-1 by immunizing rabbits with murine recombinant MCP-1 (R&D Systems, Minneapolis, Minn) in multiple intradermal sites with complete Freund's adjuvant. Serum was purified in a protein A column.
To assess the direct effect of intratracheally administered S rectivirgula on CCR2−/− and wild-type mice, we anesthetized mice and injected lyophilized S rectivirgula, suspended in sterile pyrogen-free normal saline solution, into the trachea by mouth. Mice were killed 3 days (cytokine and chemokine levels) or 4 days (histologic study and BALF cells) thereafter. These times were chosen because cytokine levels peak 72 hours after intratracheal administration of S rectivirgula 5 and because histologic changes peak at 96 hours.15
We also measured the ability of cultured cells from sensitized CCR2−/− and wild-type mice to transfer EHP to mice of the same strain. Mice were immunized with 1 intratracheal injection of S rectivirgula and killed 4 days thereafter. Spleen cells were cultured with S rectivirgula (30 μg/mL) for 72 hours. The cells of RPMI-1640 media (Life Technologies, Gaithersburg, Pa) were then injected into naïve recipients, which were challenged 8 days thereafter with intratracheally administered S rectivirgula and killed 6, 24, 48, or 96 hours thereafter.
BALF
BALF cells were obtained by means of lavage with 6 washes (1 mL each) of normal saline solution. The supernatant from the first 3 combined washes was frozen at −70°C for later chemokine and cytokine analysis.
Cytokines and chemokines
Cytokines and chemokines were measured with the use of an enzyme-linked immunosorbent assay. Unknown samples were compared to a standard curve of corresponding recombinant mouse cytokine or chemokine.
Histologic study
The lungs were inflated with formalin under 20 cm of water pressure for 48 hours and embedded in a single paraffin block, after which a 5-μm section waqs cut and stained with hematoxylin and eosin. The slides were evaluated without knowledge of treatment. The area covered by an eyepiece grid (0.99 × 0.99 mm at a magnification of 100× magnification) was judged to be normal or abnormal. An abnormal field is one with increased number of cells in the interstitium or alveoli or both. An average of 300 fields was evaluated from each mouse (50% of the area under the coverslip). This method yields reproducible results (r = .89 for duplicate readings of 301 animals).16
Data analysis
We analyzed BALF cellular data, cytokines, chemokines, and the extent of histologic changes with the use of ANOVA and Tukey's conservative HSD procedure for post hoc testing. We considered P values of less than .05 significant. Post hoc tests of significance with multiple ANOVA were then applied.17
Results
Effect of anti–MCP-1
The rabbit anti–MCP-1 preparation could block at least 40 ng/mL of MCP-1 and did not cross-react with IL-4, IL-5, IL-6, IL-10, IL-12p40, IL-12p70, IL-13, IL-16, TNF, transforming growth factor-α, or MIP-1α. Treatment of mice with anti–MCP-1 did not change (t test) the extent of pulmonary histopathologic findings in response to 7.2 μg/g intratracheally administered S rectivirgula (6.9 ± 2.2, mean, sem) compared with that in animals treated with an equal volume of phosphate-buffered saline solution (8.6 – 3.4), nor did it change the number or characteristics of BALF cells (data not shown).
CCR2−/− animals
The extent of histologic abnormalities in both the CCR2−/− and wild-type mice was dependent on the amount of S rectivirgula administered but not the type of animal (P < .05, CCR2−/− vs wild-type; Fig 1, two-way ANOVA).
Fig 1.
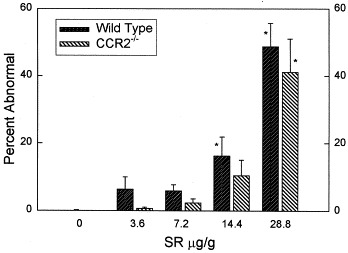
Extent of pulmonary histologic abnormalities in animals administered different amounts of S rectivirgula (SR) intratracheally. Animals were killed 96 hours after receiving the specified amount of S rectivirgula. Percent abnormal, percentage of 300 microscopic fields/animal that were abnormal. Bars represent the mean of 6 or 7 experiments; error bars denote SEM. *P < .05 vs 0 μg/g S rectivirgula (ANOVA with Tukey's HSD procedure).
We noted an increase in BALF cells in both wild-type and CCR2−/−mice after administration of every dose of S rectivirgula (Table I). The total numbers of macrophages and eosinophils were the same in the wild-type and CCR2−/− animals. We found small differences in the total number of lymphocytes (CCR2−/− < wild-type, multiple ANOVA). The numbers of cells, macrophages, lymphocytes, neutrophils, and eosinophils were related to the amount of S rectivirgula administered, and we found no differences among the strains (P < .05, multiple ANOVA).
Table I.
BALF cell characteristics 96 hours after intratracheal S rectivirgula
| S rectivirgula (μg/g body wt) | Total Cells | Macrophages | Lymphocytes | Neutrophils | Eosinophils |
|---|---|---|---|---|---|
| B6129 wild-type | |||||
| 0 | 4.6 ± 1.1 | 4.6 ± 1.0 | 0.07 ± 0.02 | .003 ± .002 | .001 ± .001 |
| 3.6 | 9.9 ± 2.1 | 7.9 ± 1.3 | 1.5 ± 0.7 | 0.53 ± 0.31 | .02 ± .01 |
| 7.2 | 16.3 ± 1.4 | 12.5 ± 0.9 | 2.3 ± 0.7 | 1.5 ± 0.5 | .04 ± .01 |
| 14.4 | 25.8 ± 3.5 | 18.3 ± 1.9 | 2.2 ± 0.6 | 5.3 ± 2.8 | .04 ± .01 |
| 28.8 | 51.4 ± 8.6* | 26.3 ± 3.3* | 2.1 ± 0.5 | 22.7 ± 5.7* | .36 ± .23 |
| CCR2−/− | |||||
| 0 | 5.5 ± 1.4 | 5.4 ± 1.3 | 0.05 ± 0.02 | 0.02 ± 0.01 | 0 ± 0 |
| 3.6 | 10.2 ± 1.1 | 9.1 ± 1.2 | 0.30 ± 0.10 | 0.8 ± 0.2 | .07 ± .03 |
| 7.2 | 18.6 ± 1.6 | 15.1 ± 1.0 | 0.57 ± 0.17 | 2.8 ± 0.9 | .08 ± .03 |
| 14.4 | 26.8 ± 3.6* | 21.0 ± 3.4* | 0.48 ± 0.16 | 5.1 ± 1.1 | .16 ± .12 |
| 28.8 | 51.9 ± 8.7* | 29.2 ± 7.2* | 1.7 ± 1.3 | 20.7 ± 7.8* | .38 ± .15 |
Data expressed as mean ± SEM of 6 or 7 experiments. All values are × 10−5.
P < .05 vs S rectivirgula.
The amount of S rectivirgula administered was related to MCP-1, MIP-1α, and IL-12p40 levels (P < .05, multiple ANOVA). We noted differences between the wild-type and CCR2−/− mice with regard to MCP-1 and MIP-1α (CCR2−/− > wild-type) and IL-12p40 (CCR2−/− < wild-type) (Table II). This increase in MCP-1 and MIP-1α and decrease in IL-12p40 in CCR2−/− compared with those in wild-type mice is similar to results reported by others.18, 19, 20, 21
Table II.
BALF cytokine and chemokine characteristics 72 hours after intratracheal S rectivirgula
| S rectivirgula (μg/g body wt) | MCP-1 | MIP-1α | IL12p40 | IL1α | TNF | IL6 | IL18 |
|---|---|---|---|---|---|---|---|
| B6129 wild-type | |||||||
| 0 | 762 ± 329 | 44 ± 7 | 58 ± 21 | 27 ± 6 | 1228 ± 149 | 516 ± 78 | 106 ± 13 |
| 3.6 | 863 ± 371 | 35 ± 8 | 581 ± 405 | 26 ± 7 | 462 ± 182 | 386 ± 98 | 110 ± 13 |
| 7.2 | 899 ± 391 | 37 ± 5 | 855 ± 313 | 29 ± 4 | 1350 ± 187 | 551 ± 43 | 136 ± 20 |
| 14.4 | 1112 ± 250 | 47 ± 8 | 3083 ± 1370*, † | 40 ± 9 | 873 ± 287 | 615 ± 123 | 123 ± 22 |
| 28.8 | 1817 ± 631*, † | 51 ± 6*, † | 2246 ± 822 | 48 ± 14 | 1146 ± 299 | 928 ± 284 | 128 ± 21 |
| CCR2−/− | |||||||
| 0 | 727 ± 297 | 39 ± 7 | 48 ± 20 | 23 ± 6 | 662 ± 208 | 335 ± 54 | 104 ± 13 |
| 3.6 | 1104 ± 433 | 38 ± 9 | 157 ± 49 | 32 ± 7 | 918 ± 180 | 598 ± 69 | 84 ± 23 |
| 7.2 | 1925 ± 599 | 41 ± 9 | 154 ± 35 | 35 ± 8 | 1040 ± 144 | 682 ± 122 | 64 ± 5 |
| 14.4 | 4230 ± 785 | 61 ± 8 | 278 ± 73 | 45 ± 5 | 973 ± 306 | 701 ± 120 | 95 ± 14 |
| 28.8 | 22,885 ± 3572* | 97 ± 11* | 241 ± 51 | 41 ± 8 | 983 ± 288 | 783 ± 82 | 113 ± 24 |
Data expressed as mean ± SEM of 6–7 experiments (picograms per milliliter).
P < .05 vs S rectivirgula (ANOVA with Tukey's HSD correction);
P < .05, wild type vs CCR2−/−.
Adoptive transfer of EHP
In both wild-type and CCR2−/− animals, transfer of cultured spleen cells conferred the ability to respond with more extensive pulmonary histologic abnormalities (compared with animals administered medium) to an intratracheal challenge with S rectivirgula (Fig 2).
Fig 2.
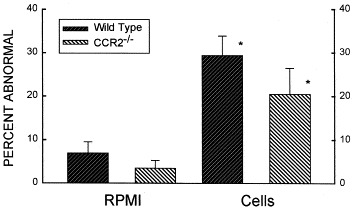
Extent of pulmonary histologic changes in recipients of cultured cells or medium. Animals were killed 96 hours after administration of an intratracheal challenge with 7.2 μg/g S rectivirgula. Cells, animals given 20 × 106 cultured (72 hours, 30 μg/mL S rectivirgula) spleen cells from sensitized animals 8 days before intratracheal S rectivirgula challenge; RPMI, animals given medium only, 8 days before intratracheal S rectivirgula challenge. Bars denote the mean value of 6 to 9 experiments; error bars indicate SEM. *P < .05 vs RPMI animals (ANOVA with Tukey's HSD procedure).
In both CCR2−/− and wild-type mice, we found extensive infiltration of the perivascular and peribronchiolar areas with a mixture of lymphocytes, macrophages, and plasma cells (Fig 3), similar to findings in C3H/HeJ, C57Bl/6, A/J, and BALB/c mice.10, 22
Fig 3.
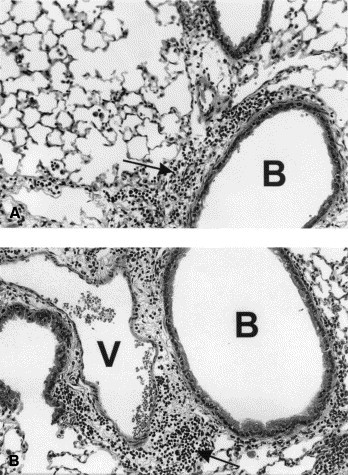
Pulmonary histologic findings in wild-type and CCR2−/− mice 96 hours after intratracheal injection of 7.2 μg/g S rectivirgula. Animals had received 20 × 106 cultured spleen cells from sensitized mice 8 days before challenge with S rectivirgula.A, Wild type. Hematoxylin and eosin stain; original magnification 400×. Note extensive perivascular, peribronchiolar, and alveolar infiltration with a mixture of neutrophils, macrophages, and lymphocytes. B, bronchiole. Arrow denotes perivascular and peribronchiolar infiltrate. B, CCR2−/−. Hematoxylin and eosin stain; original magnification 400×. Note extensive perivascular, peribronchiolar, and alveolar infiltration with a mixture of neutrophils, macrophages, and lymphocytes. B, bronchiole; V, vessel. Arrow denotes perivascular and peribronchiolar infiltrate.
We found minor differences with regard to total BALF cell number (Fig 4), with increased numbers of cells in wild-type animals that received transferred cells compared with those that received RPMI-1640. Both strains exhibited increased numbers of lymphocytes in cell recipients compared with RPMI recipients. CCR2−/− animals that had received transferred cells had increased numbers of neutrophils. Two-way ANOVA with the factors of strain and treatment (RPMI or cells) demonstrated that treatment (ie, RPMI or cells) was related (P < .05) to the number of cells and neutrophils, with no difference between the strains. The total numbers of lymphocytes were different between the strains, as well as between treatments, with more lymphocytes in the wild-type than in the CCR2−/− animals. We found few eosinophils, with no differences between groups.
Fig 4.
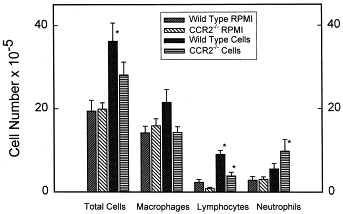
BALF cells in recipients of cultured cells or medium. Animals were killed 96 hours after being administered an intratracheal challenge with 7.2 μg/g S rectivirgula. Cells, animals given 20 × 106 cultured (72 hours, 30 μg/mL S rectivirgula) spleen cells from sensitized animals 8 days before intratracheal S rectivirgula challenge; RPMI, animals given medium only, 8 days before intratracheal S rectivirgula challenge. Bars represent mean value of 6 to 9 experiments; error bars denote SEM. *P < .05 vs RPMI animals (ANOVA with Tukey's HSD procedure).
We found an increase of IL-12p40 in wild-type animals that received cells and RPMI, with a peak at 48 hours (Fig 5; multiple ANOVA, P < .05). We also found an increase in IL-6 in wild-type animals that received cells compared with that in CCR2−/− animals that received cells (Fig 6; multiple ANOVA, P < .05).
Fig 5.
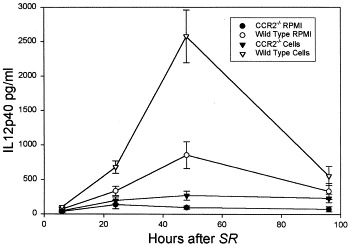
BALF IL12p40 in recipients of cultured cells or medium. Animals were killed at various times after receiving an intratracheal challenge with 7.2 μg/g S rectivirgula. Bars represent the mean value of 6 to 8 experiments; error bars denote SEM. Cells, animals administered 20 × 106 cultured (72 hours, 30 μg/mL S rectivirgula) spleen cells from sensitized animals 8 days before intratracheal S rectivirgula (SR) challenge; RPMI, animals administered medium only, 8 days before intratracheal S rectivirgula challenge. Multiple ANOVA, wild-type > CCR2−/− mice (P < .05).
Fig 6.
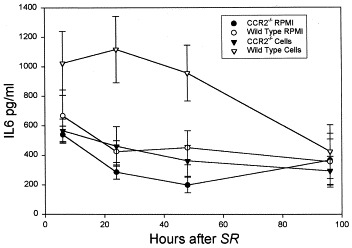
BALF IL-6 in recipients of cultured cells or medium. Animals were killed at various times after receiving an intratracheal challenge with 7.2 μg/g S rectivirgula (SR). Bars represent the mean value of 6 to 8 experiments; error bars denote SEM. Cells, animals administered 20 × 106 cultured (72 hours, 30 μg/mL S rectivirgula) spleen cells from sensitized animals 8 days before intratracheal S rectivirgula challenge; RPMI, animals administered medium only, 8 days before intratracheal S rectivirgula challenge. ANOVA, wild-type that received cells > CCR2−/− mice that received cells (P < .05).
We analyzed culture supernatants to determine the cytokine-secreting profiles of injected cells and found that the supernatants from the CCR2−/− animals produced less IFN-γ than those from the wild-type animals (Table III). IL-12p40, IL-18, IL-6, and IL-1α were detected in the supernatants, with no difference between strains.
Table III.
Culture supernatant
| Parameter | CCR2−/− | Wild-type |
|---|---|---|
| IL-4 | 0 | 10.2 ± 4.6 |
| IL-5 | 0 | 3.8 ± 3.8 |
| IFN-γ | 19.3 ± 7.7* | 340 ± 39.6 |
| IL-12p40 | 371.5 ± 31.3 | 414.8 ± 37.6 |
| IL-18 | 62.4 ± 9.6 | 60.8 ± 6.4 |
| IL-6 | 226.2 ± 44.5 | 312.8 ± 50.2 |
| IL-1α | 9.8 ± 1.9 | 5.7 ± 1.6 |
Spleen cells from CCR2−/− or wild-type S rectivirgula–sensitized animals cultured for 72 hours (30 μg/mL S rectivirgula).
Data expressed as mean ± SEM of 6 experiments (picograms per milliliter).
P < .05, CCR2−/− vs wild-type (t test).
Discussion
HP is characterized by the appearance of neutrophils (<48 hours) and then (>5 days) lymphocytes in BALF.2 The mechanisms of BALF neutrophilia and lymphocytosis may include the effects of chemokines, which have been detected in the BALF of patients with HP.3, 4 Chemokines are a family of small, discrete chemotactic proteins that are divided into different groupings depending on their structure. CC chemokines do not have another amino acid between the first 2 of 4 conserved cysteines and preferentially attract monocytes, eosinophils, basophils, and lymphocytes.23 MCP-1, known as JE in the mouse, is a prominent member of the CC-chemokine family that is apparently important in pathologic conditions associated with monocyte/macrophage infiltration such as atherosclerosis,24 rheumatoid arthritis,25 experimental allergic encephalomyelitis,26 idiopathic pulmonary fibrosis,27 acute respiratory distress syndrome,28 psoriasis,29 experimental glomerulonephritis,30 diabetes,31 and allograph rejection.32 MCP-1 is produced by many different cell types, such as types I and II alveolar cells, monocytes, endothelial cells, macrophages, fibroblasts, mast cells, keratinocytes, and smooth-muscle cells.33, 34, 35, 36
The ligand for MCP-1 is CCR2, which is expressed on monocytes, activated T-cells, B-cells, natural killer cells, fibroblasts, and mast cells.23, 37, 38 CCR2 is 1 of a family of 7 transmembrane-spanning receptors that affect cells by way of interaction with different G-proteins. Although MCP-1 is the principal agonist for CCR2, other agonists include MCP-2, MCP-3, MCP-4, MCP-5, and human immunodeficiency virus Tat.39, 40, 41, 42, 43 However, in mice, only MCP-1, MCP-3, and MCP-5 bind CCR2.
In summer-type HP, the most common cause of HP in Japan, BALF MCP-1 concentration has been show to correlate with BALF lymphocyte count.4 In rats with immune complex–mediated lung injury, there is an increased amount of BALF monocyte chemotactic activity that could be neutralized by antibody to MCP-1, markedly increased expression of alveolar macrophage MCP-1 messenger RNA, and increased in vitro secretion of MCP-1 from alveolar macrophages.44 MCP-1 levels are increased after bleomycin treatment in rats from days 3 to 21.45
We previously demonstrated that intratracheal. administration of S rectivirgula, the agent that causes FLD in human beings, caused the appearance of substantial amounts of MCP-1 in BALF, which preceded later increases of BALF neutrophil and mononuclear cell counts.5 Therefore our findings of increased S rectivirgula–induced BALF MCP-1 that preceded the influx of lymphocytes suggest that MCP-1 is responsible for cellular influx into BALF.
However, many other potential chemotactic factors are present in the lungs of subjects exposed to the agents that cause HP and in our model of EHP in mice, such as complement fragments and other chemokines. Therefore it is not clear that MCP-1 is responsible for BALF mononuclear cell influx in our model.
In addition to its chemotactic activity, MCP-1 influences the differentiation of antigen-specific CD4+ cells by promoting the development of Th1 cells.46 In our model, cells from sensitized mice cultured with an S rectivirgula extract transfer increased sensitivity to intratracheally administered S rectivirgula, as defined by the extent of pulmonary inflammation.15 The cells that effect the transfer are CD4+ with Th1 characteristics; Th1, but not Th2, cell lines are able to transfer EHP.9, 47
We tested the hypothesis that MCP-1 enhances the development of EHP. Our first approach was to block MCP-1 activity in vivo using antibody. We found no effect of anti–MCP-1 on the pulmonary histologic or BALF response using a schedule that others have demonstrated to be effective.12, 13, 14 The use of neutralizing antibodies to ablate the influence of a cytokine or chemokine may not be effective as a result of dose or schedule issues. In addition, antibody can cause the formation of antigen-antibody complexes and induce complement mediated tissue injury.
Because MCP-1–deficient mice are not readily available, we used CCR2–deficient mice to determine the influence of MCP-1 on EHP. Compared with wild-type animals, CCR2-deficient mice exhibit decreased48, 49 tissue inflammation when confronted with inflammatory stimuli or infection. They also demonstrate reduced delayed-type hypersensitivity reactions and granuloma formation49, 50 and are more susceptible to infections with Listeria 51 Leishmania,52 central nervous system coronavirus,53 and Cryptococcus 21 and exhibit decreased macrophage recruitment into the lung after infection with influenza.18 These mice also exhibit increased airway reactivity after sensitization to Aspergillus 20 and resistance to experimental allergic encephalomyelitis.54 In general, CCR2-deficient animals exhibit a decreased Th1 response, with reduced production of Th1 cytokines such as IFN-γ50, 54 and IL-12 and increased production of IL-4 and IL-5, 21
We therefore compared the histologic and BALF response to one intratracheal injection of S rectivirgulain wild-type and CCR2−/− animals. We found no difference with regard to the extent or characteristics of pulmonary histologic abnormalities in CCR2−/− mice compared with wild type animals (Fig 1). The degree of pulmonary inflammation as reflected by BALF cell characteristics was related to the amount of S rectivirgula administered and not to the mouse strain, with minor isolated differences in lymphocyte counts (Table I). The amount of MCP-1 was increased in the BALF of the CCR2−/− animals (Table II), similar to observations in other systems.18, 19, 20, 21 We could not predictably detect IFN-γ in the BALF of our animals, but the amount of BALF IL-12p40 was decreased in the CCR2−/− animals.
We did not find decreased numbers of macrophages and increased numbers of neutrophils and eosinophils in the BALF of CCR2-deficient mice compared with wild-type mice (Table I), as reported previously after intraperitoneal injection of thioglycollate.49 This may be related to differences of stimuli (thioglycollate vs S rectivirgula), anatomic site (peritoneum vs lung), or timing (36 vs 96 hours). Moore and colleagues likewise reported no difference of pulmonary inflammation in CCR2−/− and wild-type mice 1 week after intratracheal injection of fluorescein isothiocyanate, despite later differences in lung fibrosis at day 21.19 Blease found only small differences in BALF cells after intratracheal challenge with Aspergillus.20 Traynor only found differences of total cell number in the lungs of Cryptococcus-infected nimals after 2 weeks of infection.21 Therefore it is likely that the lack of differences of pulmonary inflammation that we observed in CCR2−/− and wild-type animals was related mostly to the anatomic site in that pulmonary inflammation is often relatively normal in CCR2−/− mice, as opposed to the peritoneum.
We did not find evidence of a switch of immune response from a Th1 to a Th2 profile in CCR2−/− mice described by Traynor and colleagues in experimental cryptococcal infection21 or Sato in Leishmania-infected mice.52 The reason for this difference is likely related to timing (4 days in our model vs more than 1 week in the cryptococcal model and 5 weeks in the Leishmania model) and, perhaps, the nature of the pulmonary challenge (nonreplicating bacteria vs replicating fungus). However, we did find a decrease in IFN-γ secretion in cultured cell supernatants (Table III), similar to those reported by Sato.52
We did not find increases in concentrations of the inflammatory cytokines IL-6, tumor necrosis factor, or IL-1α in BALF (Table II) in either mouse strain; this was probably the result of the time elapsed; since intratracheal challenge with S rectivirgula (72 hours). We previously demonstrated that 72 hours after S rectivirgula exposure, these cytokines had returned to baseline levels.5
We next cultured cells from both CCR2−/− and wild-type mice sensitized to S rectivirgula. Cultured cells from CCR2−/− animals secreted less IFN-γ than did cells from wild-type mice (Table III) and therefore resembled Th0 cells. Although the amount of IFN-γ was decreased, it was not absent in the CCR2−/− culture supernatants, similar to results reported by others.48, 50, 52, 55 The presence of IFN-γ in the culture supernatants from the CCR2−/− animals may have been caused by IL-18, which induces IFN-γ secretion56 and was present in supernatants at the same concentrations in cell cultures from wild-type and CCR2−/− animals.
Cultured cells from wild-type and CCR2−/− mice were equally competent in their ability to adoptively transfer EHP (defined as an increased extent of pulmonary histologic changes after intratracheal challenge with S rectivirgula; Fig 2). This is similar to findings in models of murine asthma and mycobacterial infection employing CCR2−/− animals.20, 57 The nature of the pulmonary inflammatory response was not different in the CCR2−/− recipients compared with the wild-type recipients (Fig 3, Fig 4). We did find differences in IL-12p40 and IL-6 in the BALF of the CCR2−/− mice and the control animals (Fig 4, Fig 5). Because we did not find a difference with regard to pulmonary histologic or BALF cellular response in the CCR2−/− mice (Fig 2, Fig 4), it is likely that these cytokines are not necessary for the expression of pulmonary inflammation in this adoptive-transfer model.
The lack of effect of absence of the MCP-1–CCR2 interaction on development of cells capable of transferring EHP suggests that additional pathways in this model of experimental hypersensitivity pneumonitis result in the recruitment of inflammatory cells to the lung after exposure to S rectivirgula. This is likely a consequence of the redundant nature of the chemokine-chemokine receptor system, with multiple chemokines interacting with many different receptors, and perhaps the nature of the antigen we use. S rectivirgula can activate complement,58 cause secretion of inflammatory cytokines,59 and act as an adjuvant.60 In addition, timing of exposure to MCP-1 may be important. Overexpression of MCP-1 during sensitization of mice to either PPD or Schistosoma antigen markedly altered the extent of the granulomatous response, whereas overexpression after sensitization did not change the Th1- or Th2-type granulomatous response.61
In summary, we could not demonstrate an essential role for MCP-1 and its receptor CCR2 in our model of EHP, despite considerable evidence linking EHP and this chemokine. This failure likely reflects the complex nature of the interaction between the lung and antigenic material that causes HP in human beings.
It is likely that there exist multiple redundant pathways of lung injury in HP, which implies that measures aimed at a single mechanism would not be expected to decrease the pulmonary inflammatory response in this disease. To the extent that HP resembles other inflammatory lung diseases (eg, idiopathic pulmonary fibrosis, cystic fibrosis, adult respiratory distress syndrome), similar lack of a single dominant mechanism of injury would predict the necessity of multiple interactions to reduce lung damage.
Acknowledgements
We acknowledge the excellent technical assistance of Susan Baca in maintaining the CCR2−/− colony and Kathy Kilpatrick for performing polymerase chain reaction assays.
Footnotes
Supported by the Veterans Administration Research Service and by National Heart, Lung, and Blood Institute grant HL44253.
References
- 1.Schuyler M. Hypersensitivity pneumonitis. In: Fishman A, ed. Pulmonary diseases and disorders. 3rd ed. McGraw-Hill, NY, 1997:1085-97
- 2.Drent M., van Velzen-Blad H., Diamant M., Wagenaar S.S., Hoogsteden H.C., van den Bosch J.M. Bronchoalveolar lavage in extrinsic allergic alveolitis: effect of time elapsed since antigen exposure. Eur Resp J. 1993;6:1276–1281. [PubMed] [Google Scholar]
- 3.Denis M. Proinflammatory cytokines in hypersensitivity pneumonitis. Am J Resp Crit Care Med. 1995;151:164–169. doi: 10.1164/ajrccm.151.1.7812548. [DOI] [PubMed] [Google Scholar]
- 4.Sugiyama Y., Kasahara T., Mukaida N., Matsushima K., Kitamura S. Chemokines in bronchoalveolar lavage fluid in summer-type hypersensitivity pneumonitis. Eur Resp J. 1995;8:1084–1090. doi: 10.1183/09031936.95.08071084. [DOI] [PubMed] [Google Scholar]
- 5.Schuyler M., Gott K., Cherne A. Mediators of hypersensitivity pneumonitis. J Lab Clin Med. 2000;136:29–38. doi: 10.1067/mlc.2000.107694. [DOI] [PubMed] [Google Scholar]
- 6.Kurihara T., Bravo R. Cloning and functional expression of mCCR2, a murine receptor for the C- C chemokines JE and FIC. J Biol Chem. 1996;271:11603–11607. doi: 10.1074/jbc.271.20.11603. [DOI] [PubMed] [Google Scholar]
- 7.Boring L., Gosling J., Monteclaro F.S., Lusis A.J., Tsou C.L., Charo I.F. Molecular cloning and functional expression of murine JE (monocyte chemoattractant protein 1) and murine macrophage inflammatory protein 1alpha receptors: evidence for two closely linked C-C chemokine receptors on chromosome 9. J Biol Chem. 1996;271:7551–7558. doi: 10.1074/jbc.271.13.7551. [DOI] [PubMed] [Google Scholar]
- 8.Yamagami S., Tokuda Y., Ishii K., Tanaka H., Endo N. cDNA cloning and functional expression of a human monocyte chemoattractant protein 1 receptor. Biochem Biophys Res Commun. 1994;202:1156–1162. doi: 10.1006/bbrc.1994.2049. [DOI] [PubMed] [Google Scholar]
- 9.Schuyler M., Gott K., Cherne A., Edwards B. Th1 CD4+ cells adoptively transfer experimental hypersensitivity pneumonitis. Cell Immunol. 1997;177:169–175. doi: 10.1006/cimm.1997.1107. [DOI] [PubMed] [Google Scholar]
- 10.Schuyler M., Gott K., Haley P. Experimental murine hypersensitivity pneumonitis. Cell Immunol. 1991;136:303–317. doi: 10.1016/0008-8749(91)90354-e. [DOI] [PubMed] [Google Scholar]
- 11.Schuyler M., Gott K., Shopp G., Crooks L. CD3+ and CD4+ cells adoptively transfer experimental hypersensitivity pneumonitis. Am Rev Resp Dis. 1992;146:1582–1588. doi: 10.1164/ajrccm/146.6.1582. [DOI] [PubMed] [Google Scholar]
- 12.Campbell E.M., Charo I.F., Kunkel S.L., Strieter R.M., Boring L., Gosling J. Monocyte chemoattractant protein-1 mediates cockroach allergen-induced bronchial hyperreactivity in normal but not CCR2−/− mice: the role of mast cells. J Immunol. 1999;163:2160–2167. [PubMed] [Google Scholar]
- 13.Matsukawa A., Hogaboam C.M., Lukacs N.W., Lincoln P.M., Strieter R.M., Kunkel S.L. Endogenous monocyte chemoattractant protein-1 (MCP-1) protects mice in a model of acute septic peritonitis: cross-talk between MCP-1 and leukotriene B4. J Immunol. 1999;163:6148–6154. [PubMed] [Google Scholar]
- 14.Fillion I., Ouellet N., Simard M., Bergeron Y., Sato S., Bergeron M.G. Role of chemokines and formyl peptides in pneumococcal pneumonia-induced monocyte/macrophage recruitment. J Immunol. 2001;166:7353–7361. doi: 10.4049/jimmunol.166.12.7353. [DOI] [PubMed] [Google Scholar]
- 15.Schuyler M., Gott K., Edwards B., Nikula K.J. Experimental hypersensitivity pneumonitis. Effect of CD4 cell depletion. Am J Resp Crit Care Med. 1994;149:1286–1294. doi: 10.1164/ajrccm.149.5.7909706. [DOI] [PubMed] [Google Scholar]
- 16.Schuyler M. Hypersensitivity pneumonitis. In: Fishman A, ed. Pulmonary diseases and disorders. 3rd ed. McGraw-Hill, NY, 1997:1085-97
- 17.Systat for Windows, version 8.0: Statistics. Evanston, IL: SPSS Inc; 1998
- 18.Dawson T.C., Beck M.A., Kuziel W.A., Henderson F., Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol. 2000;156:1951–1959. doi: 10.1016/S0002-9440(10)65068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore B.B., Paine R., 3rd, Christensen P.J., Moore T.A., Sitterding S., Ngan R. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol. 2001;167:4368–4377. doi: 10.4049/jimmunol.167.8.4368. [DOI] [PubMed] [Google Scholar]
- 20.Blease K., Mehrad B., Standiford T.J. Enhanced pulmonary allergic responses to Aspergillus in CCR2−/− mice. J Immunol. 2000;165:2603–2611. doi: 10.4049/jimmunol.165.5.2603. [DOI] [PubMed] [Google Scholar]
- 21.Traynor T.R., Kuziel W.A., Toews G.B., Huffnagle G.B. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J Immunol. 2000;164:2021–2027. doi: 10.4049/jimmunol.164.4.2021. [DOI] [PubMed] [Google Scholar]
- 22.Schuyler M., Gott K., Mapel V., Cherne A., Nikula K.J. Experimental hypersensitivity pneumonitis: influence of Th2 bias. Int J Exp Pathol. 1999;80:335–348. doi: 10.1046/j.1365-2613.1999.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunkel S.L., Strieter R.M., Lindley I.J., Westwick J. Chemokines: new ligands, receptors and activities. Immunol Today. 1995;16:559–561. doi: 10.1016/0167-5699(95)80076-X. [DOI] [PubMed] [Google Scholar]
- 24.Gosling J., Slaymaker S., Gu L., Tseng S., Zlot C.H., Young S.G. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest. 1999;103:773–778. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villiger P.M., Terkeltaub R., Lotz M. Production of monocyte chemoattractant protein-1 by inflamed synovial tissue and cultured synoviocytes. J Immunol. 1992;149:722–727. [PubMed] [Google Scholar]
- 26.Berman J.W., Guida M.P., Warren J., Amat J., Brosnan C.F. Localization of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J Immunol. 1996;156:3017–3023. [PubMed] [Google Scholar]
- 27.Antoniades H.N., Neville-Golden J., Galanopoulos T., Kradin R.L., Valente A.J., Graves D.T. Expression of monocyte chemoattractant protein 1 mRNA in human idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 1992;89:5371–5375. doi: 10.1073/pnas.89.12.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosseau S., Hammerl P., Maus U., Walmrath H.D., Schutte H., Grimminger F. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2000;279:L25–35. doi: 10.1152/ajplung.2000.279.1.L25. [DOI] [PubMed] [Google Scholar]
- 29.Gillitzer R., Wolff K., Tong D., Muller C., Yoshimura T., Hartmann A.A. MCP-1 mRNA expression in basal keratinocytes of psoriatic lesions. J Invest Dermatol. 1993;101:127–131. doi: 10.1111/1523-1747.ep12363613. [DOI] [PubMed] [Google Scholar]
- 30.Fujinaka H., Yamamoto T., Takeya M., Feng L., Kawasaki K., Yaoita E. Suppression of anti-glomerular basement membrane nephritis by administration of anti-monocyte chemoattractant protein-1 antibody in WKY rats. J Am Soc Nephrol. 1997;8:1174–1178. doi: 10.1681/ASN.V871174. [DOI] [PubMed] [Google Scholar]
- 31.Grewal I.S., Rutledge B.J., Fiorillo J.A., Gu L., Gladue R.P., Flavell R.A. Transgenic monocyte chemoattractant protein-1 (MCP-1) in pancreatic islets produces monocyte-rich insulitis without diabetes: abrogation by a second transgene expressing systemic MCP-1. J Immunol. 1997;159:401–408. [PubMed] [Google Scholar]
- 32.Russell M.E., Adams D.H., Wyner L.R., Yamashita Y., Halnon N.J., Karnovsky M.J. Early and persistent induction of monocyte chemoattractant protein 1 in rat cardiac allografts. Proc Natl Acad Sci U S A. 1993;90:6086–6090. doi: 10.1073/pnas.90.13.6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rollins B.J., Yoshimura T., Leonard E.J., Pober J.S. Cytokine-activated human endothelial cells synthesize and secrete a monocyte chemoattractant, MCP-1/JE. Am J Pathol. 1990;136:1229–1233. [PMC free article] [PubMed] [Google Scholar]
- 34.Paine R., 3rd, Rolfe M.W., Standiford T.J., Burdick M.D., Rollins B.J., Strieter R.M. MCP-1 expression by rat type II alveolar epithelial cells in primary culture. J Immunol. 1993;150:4561–4570. [PubMed] [Google Scholar]
- 35.Brieland JK, Flory CM, Jones ML, Miller GR, Remick DG, Warren JS, et al. Regulation of monocyte chemoattractant protein-1 gene expression and secretion in rat pulmonary alveolar macrophages by lipopolysaccharide, tumor necrosis factor-alpha, and interleukin-1 beta. Am J Respir Cell Mol Biol 1995;12:104-9 [DOI] [PubMed]
- 36.Poon M., Hsu W.C., Bogadanov V.Y., Taubman M.B. Secretion of monocyte chemotactic activity by cultured rat aortic smooth muscle cells in response to PDGF is due predominantly to the induction of JE/MCP-1. Am J Pathol. 1996;149:307–317. [PMC free article] [PubMed] [Google Scholar]
- 37.Hogaboam C.M., Bone-Larson C.L., Lipinski S., Lukacs N.W., Chensue S.W., Strieter R.M. Differential monocyte chemoattractant protein-1 and chemokine receptor 2 expression by murine lung fibroblasts derived from Th1- and Th2-type pulmonary granuloma models. J Immunol. 1999;163:2193–2201. [PubMed] [Google Scholar]
- 38.Frade J.M., Mellado M., del Real G., Gutierrez-Ramos J.C., Lind P., Martinez A.C. Characterization of the CCR2 chemokine receptor: functional CCR2 receptor expression in B cells. J Immunol. 1997;159:5576–5584. [PubMed] [Google Scholar]
- 39.Sarafi M.N., Garcia-Zepeda E.A., MacLean J.A., Charo I.F., Luster A.D. Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J Exp Med. 1997;185:99–109. doi: 10.1084/jem.185.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albini A, Ferrini S, Benelli R, Storzini S, Ginncinglio D, Aluigi MG, et al. HIV-1 Tat protein mimicry of chemokines. Proc Natl Acad Sci U S A 1998;95:13153-8 [DOI] [PMC free article] [PubMed]
- 41.Garcia-Zepeda E.A., Combadiere C., Rothenberg M.E. Human monocyte chemoattractant protein (MCP)-4 is a novel CC chemokine with activities on monocytes, eosinophils, and basophils induced in allergic and nonallergic inflammation that signals through the CC chemokine receptors (CCR)-2 and -3. J Immunol. 1996;157:5613–5626. [PubMed] [Google Scholar]
- 42.Gong X., Gong W., Kuhns D.B., Ben-Baruch A., Howard O.M., Wang J.M. Monocyte chemotactic protein-2 (MCP-2) uses CCR1 and CCR2B as its functional receptors. J Biol Chem. 1997;272:11682–11685. doi: 10.1074/jbc.272.18.11682. [DOI] [PubMed] [Google Scholar]
- 43.Combadiere C., Ahuja S.K., Van Damme J., Tiffany H.L., Gao J.L., Murphy P.M. Monocyte chemoattractant protein-3 is a functional ligand for CC chemokine receptors 1 and 2B. J Biol Chem. 1995;270:29671–29675. doi: 10.1074/jbc.270.50.29671. [DOI] [PubMed] [Google Scholar]
- 44.Brieland J.K., Jones M.L., Clarke S.J., Baker J.B., Warren J.S., Fantone J.C. Effect of acute inflammatory lung injury on the expression of monocyte chemoattractant protein-1 (MCP-1) in rat pulmonary alveolar macrophages. Am J Resp Cell Mol Biol. 1992;7:134–139. doi: 10.1165/ajrcmb/7.2.134. [DOI] [PubMed] [Google Scholar]
- 45.Zhang K., Gharaee-Kermani M., Jones M.L., Warren J.S., Phan S.H. Lung monocyte chemoattractant protein-1 gene expression in bleomycin-induced pulmonary fibrosis. J Immunol. 1994;153:4733–4741. [PubMed] [Google Scholar]
- 46.Lukacs N.W., Chensue S.W., Karpus W.J., Lincoln P., Keefer C., Strieter R.M. C-C chemokines differentially alter interleukin-4 production from lymphocytes. Am J Pathol. 1997;150:1861–1868. [PMC free article] [PubMed] [Google Scholar]
- 47.Schuyler M., Gott K., Shopp G., Crooks L. CD3+, CD4+, CD8-, Ia- T cells adoptively transfer murine experimental hypersensitivity pneumonitis. Chest. 1993;103:143S–145S. doi: 10.1378/chest.103.2_supplement.143s. [DOI] [PubMed] [Google Scholar]
- 48.Cooper A.M., Magram J., Ferrante J., Orme I.M. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J Exp Med. 1997;186:39–45. doi: 10.1084/jem.186.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, et al. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci U S A 1997;94:12053-8 [DOI] [PMC free article] [PubMed]
- 50.Boring L, Gosling J, Chensue SW, Kunkel S L, Farese RV, Jr, Broxmeyer HI, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest 1997;100:2552-61 [DOI] [PMC free article] [PubMed]
- 51.Kurihara T., Warr G., Loy J., Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sato N, Ahuja SK, Quinones M, Kosteiki V, Reddick RL, Melby PC, et al. CC chemokine receptor (CCR)2 is required for Langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, b cell outgrowth, and sustained neutrophilic inflammation. J Exp Med 2000;192:205-18 [DOI] [PMC free article] [PubMed]
- 53.Chen B.P., Kuziel W.A., Lane T.E. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol. 2001;167:4585–4592. doi: 10.4049/jimmunol.167.8.4585. [DOI] [PubMed] [Google Scholar]
- 54.Izikson L., Klein R.S., Charo I.F., Weiner H.L., Luster A.D. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Magram J, Sfarra J, Connaughton S, Faherty D, Warrier R, Carvajal D, et al. IL-12-deficient mice are defective but not devoid of type 1 cytokine responses. Ann N Y Acad Sci 1996;795:60-70 [DOI] [PubMed]
- 56.Robinson D, Shibuya K, Mui A, Zonin F, Murphy E, Sana T, et al. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity 1997;7:571-81 [DOI] [PubMed]
- 57.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1–deficient mice. J Exp Med 1998;187:601-8 [DOI] [PMC free article] [PubMed]
- 58.Smith S.M., Burrell R., Snyder I.S. Complement activation by cell wall fractions of Micropolyspora faeni. Infect Immun. 1978;22:568–574. doi: 10.1128/iai.22.2.568-574.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Denis M., Cormier Y., Tardif J., Ghadirian E., Laviolette M. Hypersensitivity pneumonitis: whole Micropolyspora faeni or antigens thereof stimulate the release of proinflammatory cytokines from macrophages. Am J Resp Cell Mol Biol. 1991;5:198–203. doi: 10.1165/ajrcmb/5.2.198. [DOI] [PubMed] [Google Scholar]
- 60.Bice D., McKarron K., Hoffman E., Salvaggio J. Adjuvant properties of Micropolyspora faeni. Int Arch Allergy Appl Immunol. 1974;55:267–273. doi: 10.1159/000231935. [DOI] [PubMed] [Google Scholar]
- 61.Matsukawa A., Lukacs N.W., Standiford T.J., Chensue S.W., Kunkel S.L. Adenoviral-mediated overexpression of monocyte chemoattractant protein-1 differentially alters the development of Th1 and Th2 type responses in vivo. J Immunol. 2000;164:1699–1704. doi: 10.4049/jimmunol.164.4.1699. [DOI] [PubMed] [Google Scholar]