

Abstract
Parasitic nematodes (roundworms) of livestock have major economic impact globally. In spite of the diseases caused by these nematodes and some advances in the design of new therapeutic agents (anthelmintics) and attempts to develop vaccines against some of them, there has been limited progress in the establishment of practical diagnostic techniques. The specific and sensitive diagnosis of gastrointestinal nematode infections of livestock underpins effective disease control, which is highly relevant now that anthelmintic resistance (AR) is a major problem. Traditional diagnostic techniques have major constraints, in terms of sensitivity and specificity. The purpose of this article is to provide a brief background on gastrointestinal nematodes (Strongylida) of livestock and their control; to summarize conventional methods used for the diagnosis and discuss their constraints; to review key molecular-diagnostic methods and recent progress in the development of advanced amplification-based and sequencing technologies, and their implications for epidemiological investigations and the control of parasitic diseases.
Keywords: Biotechnology, Specific diagnosis, Parasites, Livestock
1. Introduction
The phylum Nematoda (roundworms) includes many parasites that are of major socio-economic importance. For instance, grazing ruminants are usually parasitized by one or more nematodes (order Strongylida) which can cause parasitic gastroenteritis (PGE) (Taylor et al., 2007). Various species of strongylid nematodes can vary considerably in their pathogenicity, geographical distribution and susceptibility to anthelmintic drugs (Dobson et al., 1996). Mixed infections involving multiple genera and species are common, and usually have a greater impact on the host than monospecific infections. In addition, the species composition of the parasites present in a host animal can have an important relationship with the severity of infection (Wimmer et al., 2004). Depending on the number, species and burden of parasitic nematodes, common signs of PGE include reduced weight gain or weight loss, anorexia, diarrhea, reduced production and, in the case of blood-feeding species, anemia and edema, due to the loss of blood and/or plasma proteins (Kassai, 1999, Taylor et al., 2007). Therefore, the knowledge of the nematode species present in a particular geographical area, and their biology and epidemiology, have important implications for the control of PGE, particularly given the increasing problems of anthelmintic resistance (AR) in strongylid nematodes of livestock (Kaplan, 2004, Wolstenholme et al., 2004).
The accurate diagnosis of parasitic diseases and AR is central to these areas and the control of parasites. Traditional methods of diagnosis can be time-consuming to perform and have limitations, in terms of their specificity and sensitivity (Gasser, 2006). In particular, in the case of mixed infections, diagnosis can be laborious and time-consuming using techniques such as fecal egg counts (FEC) and larval culture and differentiation (MAFF, 1986). DNA techniques that rely on the amplification of nucleic acids, particularly those coupled to the polymerase chain reaction (PCR) (Saiki et al., 1988), are effective for the specific identification of parasites, and aid the diagnosis of infections from minute amounts of target template, if suitable genetic markers are employed. Such methods are likely to provide powerful alternative tools to traditional approaches, to underpin fundamental research into parasite epidemiology and to improve the control of parasitic diseases (Gasser, 2006). The purpose of this article was to: (i) concisely review the biology and significance of gastrointestinal strongylid nematodes of small ruminants; (ii) discuss salient aspects of parasite control and AR; (iii) review traditional methods for the diagnosis of strongylid infections and discuss their limitations; and (iv) summarize nucleic acid-based diagnostic techniques, emphasizing recent advances in the establishment of robotic PCR-based technology and its implications.
2. Gastrointestinal strongylid nematodes of small ruminants
2.1. Strongylids and their biology
The order Strongylida includes five superfamilies: the Diaphanocephaloidea, Ancylostomatoidea, Strongyloidea, Trichostrongyloidea and Metastrongyloidea. The Strongylida are characterized by the presence of a copulatory bursa in the male and are thus called bursate nematodes (Anderson, 2000). The first four of these superfamilies are monoxenous and predominantly live in the gastrointestinal tract of their vertebrate hosts (Fig. 1 ). Adult strongylid nematodes exist as females and males; the females produce relatively large numbers (depending on the species) of typically ovoid, strongylid eggs (70–150 μm), which are excreted in the feces into the external environment. The first-stage larva (L1) develops inside the egg to then hatch (within 1–2 days, depending on environmental conditions) and develops through to the second-stage larva (L2). Both the L1s and L2s feed on bacteria and other microorganisms in the external environment (feces). After the molts, the ensheathed third-stage larva (L3) develops (usually within 1–2 weeks, depending on species, temperature, humidity, pH and/or other factors). The cuticular sheath around the L3 prevents it from feeding but protects it from relatively harsh environmental conditions. After the L3 is ingested by the animal and passes through the stomach(s), it exsheaths (xL3) and (after a tissue phase) develops through to the fourth-stage larva (L4) and subsequently the adult at the predilection site in the alimentary tract. The time from the xL3 to the production of eggs by the adult female is usually 3–4 weeks.
Fig. 1.
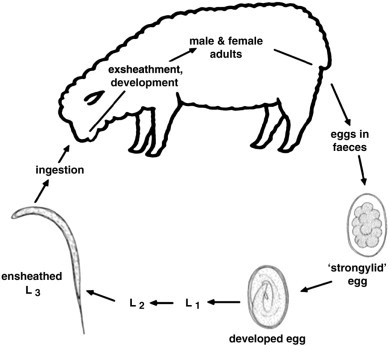
Generalized life cycle representing key gastrointestinal strongylid nematodes of small ruminants (adapted from Demeler, 2005). First-, second- and third-stage larvae (L1, L2 and L3, respectively) are ‘free-living’ in the environment. The fourth larval (L4) and adult stages (dioecious) are ‘parasitic’ in the gastrointestinal tract of the host. Disease in the host animal is caused by the adult and/or L4 stages, and depends on the species of nematode; intensity of infection; species, age and immunological/health status of the host; host response against the parasite; stress and other environmental and management factors (Kassai, 1999, Taylor et al., 2007).
2.2. Key nematodes and aspects of disease
Important gastrointestinal strongylid nematodes that infect small ruminants are listed in Table 1 . Key nematodes responsible for disease in grazing sheep include Haemonchus contortus, Teladorsagia circumcincta and intestinal species of Trichostrongylus (Besier and Love, 2003). Sheep are usually infected with one or more nematodes, but the severity of disease can vary considerably (e.g., Donald et al., 1978). Disease is predominantly linked to factors, such as the species and number of worms infecting the host, the immunological and health status of the host, environmental factors, such as climate and pasture type, stress, stocking rate, management and/or diet (Kassai, 1999, Taylor et al., 2007). Three main groups of animals are susceptible to high intensity infections: (i) young, non-immune animals, (ii) adult, immuno-compromised animals, and (iii) animals exposed to large numbers of L3s from the environment (Zajac, 2006). Nematode populations in sheep are usually over-dispersed, with the majority of sheep having low and only few sheep with high intensities of infection, respectively (Barger, 1985).
Table 1.
The key morphological characteristics, pre-patent periods and locations in the host of the most important genera and species of gastrointestinal nematodes infecting sheep in Australasia (based on Anderson, 2000, Besier and Love, 2003, Gibbons, 2010, Levine, 1968, Taylor, 2007).
| Family | Species | Morphometrics/morphology |
Prepatent period (days) |
Location in the host | |
|---|---|---|---|---|---|
| Length (mm) | Features | ||||
| Trichostrongylidae | Haemonchus contortus | ♂ 10-20 | Red pseudocoelomic fluid and white, coiled uterus, giving a barber’s pole appearance. Presence of vulvar flap depends on strain. | 18-21 | Abomasum |
| ♀ 18-30 | |||||
| Teladorsagia circumcincta | ♂ 7-8 | Small head and buccal cavity. | 15-21 | Abomasum | |
| ♀ 10-12 | In females a vulvar flap can be present. | ||||
| Trichostrongylus axei | ♂ 2-6 | Dissimilar spicules of unequal length. | 15-23 | Abomasum or stomach | |
| ♀ 3-8 | |||||
| T. colubriformis | ♂ 4-8 | Equal length spicules with triangular tip. | 15-23 | Anterior small intestine | |
| ♀ 5-9 | |||||
| T. vitrinus | ♂ 4-7 | Equal length spicules with sharp tips. | 15-23 | Anterior small intestine | |
| ♀ 5-8 | |||||
| T. rugatus | ♂ 4-7 | Dissimilar spicules with foot-like appearance | 15-23 | Small intestine | |
| ♀ 6-7 | |||||
| Cooperia curticei | ♂ 4-5 | Transverse striation of cuticle in all species. | 14-15 | Small intestine | |
| ♀ 5-6 | Watch-spring-like body posture and presence a small cephalic vesicle are characteristic. | ||||
| Molineidae | Nematodirus spathiger | ♂ 10-19 | Small but distinct cephalic vesicle. | 18 | Small intestine |
| ♀ 15-29 | Very long spicules ending in a spoon-shaped terminal piece. | ||||
| N. filicollis | ♂ 10-15 | Small but distinct cephalic vesicle. | 18 | Small intestine | |
| ♀ 15-20 | Long and slender spicules with a narrow lanceolate membrane. | ||||
| Ancylostomatidae | Bunostomum trigonocephalum | ♂ 12-17 | Anterior end is bend dorsally, | 40-70 | Small intestine |
| ♀ 19-26 | Buccal capsule with is equipped with two cutting plates. | ||||
| Chabertiidae | Oesophagostomum columbianum | ♂ 12-16 | Have two leaf crowns and a shallow buccal capsule. Position of cervical papillae used fro species differentiation. | 40‐45 | Large intestine |
| ♀ 14-18 | |||||
| Oe. venulosum | ♂ 11-16 | Cervical papillae are situated posterior to the oesophagus. | 40-45 | Large intestine | |
| ♀ 13-24 | |||||
| Chabertia ovina | ♂ 13-14 | Mouth is directed antero-ventrally. | 42-50 | Large intestine | |
| ♀ 17-20 | Buccal capsule is subglobular without teeth. | ||||
Haemonchus contortus is one of the most pathogenic and fecund strongylid nematodes of small ruminants. Adult females are capable of producing thousands of eggs per day, which can lead to rapid larval pasture contamination and associated outbreaks of haemonchosis (Levine, 1968). In sheep, the pre-patent period of H. contortus is 18–21 days. Worms are short-lived, surviving in their hosts for only a few months. The main pathogenic effects are caused by their blood feeding activity, resulting in anemia which usually becomes apparent after ~ 2 weeks of infection (Baker et al., 1959). Acute disease is usually intensity-dependent and is associated with dark-stained feces, edema, weakness, reduced production of wool and muscle mass, or sometimes sudden death. In chronic disease, decreased food intake, weight loss and anemia are most commonly observed (Kassai, 1999). Unlike many other gastrointestinal nematodes, H. contortus is not a primary cause of diarrhea, and its effects on a flock are not always detected by routine observation (Zajac, 2006).
Teladorsagia circumcincta does not feed on blood, and the main pathogenic effect is caused by its larval stages. Larval development takes place in the gastric glands, leading to nodule formation in abomasal mucosa and extensive damage to parietal cells, in turn causing a decrease in hypochloric acid production (Levine, 1968). Subsequently, the increase in abomasal pH causes a failure of pepsinogen to convert to the active form, pepsin, which results in elevated plasma pepsinogen levels and reduced protein digestion. The severity of the infection depends on concurrent infections, nutritional state of the host and also the ability to develop an immunogenic response (Stear et al., 2003). Commonly, moderate infections occur, causing diarrhea, poor weight gain, weight loss and reduced wool production (Zajac, 2006).
Species of Trichostrongylus are important parasites of grazing small ruminants. Most species occur in the small intestine and mainly exert their pathogenic effects in lambs and weaners, but have also been reported to cause significant depression of wool growth in old animals (Donald et al., 1978). In Australasia, the three most common species are T. colubriformis, T. vitrinus, and T. rugatus (Beveridge et al., 1989a). Significant pathology is caused by the exsheathed L3s of T. vitrinus, which burrow between the intestinal villi and live in sub-epithelial tunnels (Beveridge et al., 1989b). Immature nematodes developing in these tunnels are released 10–12 days following infection. The liberation of young adults is associated with extensive damage to the epithelium, with signs of generalized enteritis, including hemorrhage, edema and protein loss into the intestinal lumen, and subsequent hypoalbuminaemia and hypoproteinaemia (Taylor et al., 2007). Low intensity infections with Trichostrongylus can be difficult to distinguish from malnutrition (Taylor et al., 2007) but, if present at high intensity, infections usually cause “black scours” (watery diarrhea which stains the fleece on the hindquarters) (Levine, 1968). Trichostrongylus axei, which inhabits the abomasum, appears to be less common and occurs usually in low intensity (Donald et al., 1978).
Cooperia curticei, Nematodirus spathiger, N. filicollis and Oesophagostomum venulosum are common nematodes of the small and/or large intestine, whilst Chabertia ovina and Bunostomum trigonocephalum (hookworm), are less common (Zajac, 2006). Each of these species has relatively low pathogenicity alone, but can contribute to PGE in grazing small ruminants. Nematodirus battus is of particular disease significance in some areas, such as the British Isles, where the mass-hatching of infective L3s occurs during spring, causing disease of young lambs (Taylor and Thomas, 1986); however, this latter species has not yet been reported in Australasia.
3. Anthelmintic resistance (AR)
The control of gastrointestinal nematodes relies largely on the use anthelmintics representing three main chemical groups: the benzimidazoles (BZ), the macrocyclic lactones (ML) and the imidazothiazoles/tetrahydropyrimidines (LV) (Besier and Love, 2003, Hoste and Torres-Acosta, 2011). While there has been a recent advance with the development of a new compound, monepantel, from an alternative drug class (amino-acetonitrile derivatives, AADs) (Kaminsky et al., 2008), success in the discovery of new anthelmintics has been extremely limited over the last decades (Kaplan, 2004).
The often excessive and frequent use of these drugs has led to a widespread problem with AR in livestock parasites (Taylor et al., 2009). AR has emerged as a major bionomic and economic problem globally, being currently most pronounced in nematodes of small ruminants (von Samson-Himmelstjerna, 2006, Waller, 1994, Waller, 1997). For instance, in Australia, it has been proposed that the prevalence and extent of resistance to all major classes of broad-spectrum anthelmintics is so widespread that it threatens the profitability of the whole sheep industry (Besier and Love, 2003). Therefore, monitoring the AR status of strongylid nematode populations in livestock must be a high priority, and should be an integral part of sustainable parasite control. Various methods, such as fecal egg count reduction test (FECRT), and egg hatch- and larval development assays, have been used for estimating levels of AR in strongylid nematodes of small ruminants, cattle and horses (Coles et al., 1992).
Advances in the diagnosis of AR have focused on the implementation of a standardized protocol for the egg hatch test (von Samson-Himmelstjerna et al., 2009) and a larval migration inhibition test (Demeler et al., 2010a). However, many of these assays are quite time consuming to conduct and appear to suffer from a lack of reliability, sensitivity and reproducibility of test results (Taylor et al., 2002). Therefore, novel approaches of AR diagnosis are required.
Molecular methods have been proposed to provide new alternatives to commonly applied in vivo and in vitro techniques for the diagnosis of AR, and might be able to overcome some of their limitations (Beech et al., 2011, Demeler et al., 2010a, Demeler et al., 2010b, von Samson-Himmelstjerna, 2006). Crucial to the development of molecular diagnostic assays for AR is an in-depth knowledge of the mode of action of these chemicals, their target sites and mechanisms linked to reduced susceptibility to drugs in parasites (Beech et al., 2011, von Samson-Himmelstjerna, 2006, Wolstenholme et al., 2004).
At this stage, the BZ resistance in nematodes seems to be best understood at the molecular level, whilst much less is known about resistances against other classes of anthelmintics (Taylor et al., 2002). A single nucleotide polymorphism (SNP) at codon 200 of the beta-tubulin isotype 1 was believed to be linked to BZ resistance (Kwa et al., 1994, Wolstenholme et al., 2004) and has been demonstrated in resistant strains of H. contortus (see Geary et al., 1992), T. colubriformis (see Silvestre and Humbert, 2002) and Te. circumcincta (see Elard and Humbert, 1999) in sheep. At least two more SNPs at position 167 and 198 have been detected, but appear to be less common in different species of trichostrongylid nematodes (Beech et al., 2011, Wolstenholme et al., 2004). Besides the sequence changes in beta-tubulin, which are believed to be the major cause of BZ resistance, recent investigations have suggested a link to the drug transporter P-glycoprotein, hypothesized to play a role in the transport of the anthelmintic away from its site of action and may also select for resistance to MLs (Beech et al., 2011).
Based on current knowledge of the genetic basis of BZ resistance, allele-specific PCRs were developed to determine the genotype of adult worms of H. contortus (see Kwa et al., 1994) and Te. circumcincta (see Elard and Humbert, 1999). This work was extended by Silvestre and Humbert (2000) by combining the previously described PCR assays with a RFLP procedure, which allowed the phenetic characterization and identification of L3s of H. contortus, T. colubriformis and Te. circumcincta. Alvarez-Sanchez et al. (2005) designed a real-time PCR (RT-PCR) assay to assess the frequency of the beta-tubulin isotype 1 allele (linked to codon 200) in nematode samples. As stated by the authors, the diagnosis of BZ resistance using this assay showed an agreement with phenotypic tests, including the egg hatch test and the fecal egg count reduction test (von Samson-Himmelstjerna, 2006).
In spite of these developments, there has been no detailed evaluation of the suitability of these assays using field samples containing mixed species of gastrointestinal parasites, which limits their practical utility at this stage. In addition, all currently employed molecular assays used adult nematodes (only available through necropsy of the host) or infective L3s (requires the culturing of eggs for 1–2 weeks), but none of them has yet been assessed for the detection of AR directly from (mixed populations of) eggs, which would significantly reduce the time required for diagnosis. In contrast to the BZs, the molecular mechanisms associated with resistance to LV and ML anthelmintics are not yet deeply understood, and recent research has suggested that, in both cases, multiple genes (Beech et al., 2011) are involved in resistance and that resistance is often the result of changes in the parasite other than the immediate drug target, such as transporters and metabolism (Cvilink et al., 2009). Consequently, the multigenic nature of AR and the lack of reliable and universal markers represent a major obstacle to the development of molecular diagnostic tools for AR. No molecular test is yet available for these two groups of broad-spectrum anthelmintics.
4. Conventional diagnostic techniques and their limitations
Disease caused by gastrointestinal nematodes manifests itself in a range of clinical signs, including scouring, anemia, loss of body-condition, and in severe cases death (Hungerford, 1990). The nature and extent of clinical manifestation is also influenced by factors, such as the species and number of worms present, the plane of nutrition and immunological/reproductive status of the host (Hungerford, 1990, Levine, 1968). A number of approaches have been developed for the interpretation of clinical signs linked to PGE; these include body condition- (Russel et al., 1969), “dag”- (Larsen et al., 1994) or anemia-scoring (van Wyk and Bath, 2002). Although useful as indicators, these clinical approaches are subjective and lack specificity (van Wyk and Bath, 2002).
4.1. Fecal egg counts (FEC)
The counting of nematode eggs from feces is the commonest method for the diagnosis of gastrointestinal nematode infections. This method is inexpensive, easy to perform and does not require specialized instrumentation, making it suitable for use in most diagnostic settings. Important applications of this technique include estimating infection intensity (McKenna, 1987, McKenna and Simpson, 1987), estimating levels of contamination with helminth eggs (Gordon, 1967), evaluating the effectiveness of anthelmintics (Waller et al., 1989), determining the breeding value of an animal when selecting for worm resistance (Woolaston, 1992), and guiding decisions regarding treatment and control (Brightling, 1988).
This method involves mixing feces with a saturated salt or sugar solution (e.g., sodium nitrate or sucrose; specific gravity: 1.1–1.3) to float parasite eggs (with the exception of trematode eggs) on the surface of the suspension. An aliquot of this suspension is aspirated and eggs counted, and the number transformed into eggs per gram (EPG). Various methods have been developed, including the direct centrifugal flotation method (Lane, 1922), the Stoll dilution technique (Stoll, 1923), the McMaster method (Gordon and Whitlock, 1939) and the Wisconsin flotation method (Cox and Todd, 1962), of which the McMaster method appears to be most widely used (Nicholls and Obendorf, 1994). In the last decades, numerous modifications of these methods have been described (Levine et al., 1960, Raynaud, 1970, Roberts and O'Sullivan, 1950, Whitlock, 1948), and most teaching and research institutions use their own modifications of original protocols (Kassai, 1999). Modifications include the use of different flotation solutions (and specific gravities), sample dilutions and counting procedures, which achieve varying sensitivities and may complicate the comparisons of FEC results among different laboratories. In addition to these issues, factors, such as variation in biotic potential of different nematode species (e.g., Gordon, 1981, Le Jambre et al., 1971, Martin et al., 1985, McKenna, 1981, Roberts and Swan, 1981, Rowe et al., 2008, Stear and Bishop, 1999), water content (Gordon, 1953, Gordon, 1981, Le Jambre et al., 2007) and storage/preservation conditions (Nielsen et al., 2010, Rinaldi et al., 2011, Whitlock, 1943) of feces can each affect the interpretation of test results. Other considerations are that FECs (i) relate to patent but not pre-patent infections (Thienpont et al., 1986), (ii) do not provide any information on male or immature worms that may be present (McKenna, 1981), and (iii) can be influenced by variation in the excretion of eggs by adult worms (Villanua et al., 2006), age of the worm population, and/or the immunity, sex and age of the host (Thienpont et al., 1986). While there are some differences in morphology of eggs between some socioeconomically important nematodes (Georgi and McCulloch, 1989), specific identification is not reliable by routine microscopy (with the exception of, for example, Nematodirus spp.) (Lichtenfels et al., 1997).
FECs alone should not be used to guide treatment decisions, but should be interpreted in conjunction with information about the nutritional status, age and management of sheep in a flock (McKenna, 2002). However, according to common practice, a FEC of ≥ 200 EPG is regarded to indicate a “significant” intensity of infection (www.wormboss.com.au). The value of FEC results also depends on the parasite and host species involved. For example, FEC results for adult cattle are of limited diagnostic value, as they do not usually relate to worm burden (McKenna, 1981); FECs in cattle are usually low and require more sensitive flotation methods than for small ruminants (Mes et al., 2001); for species of Nematodirus, FECs are also regarded to be of limited value, as most pathological damage during infection is caused by the immature stages prior to egg-laying (McKenna, 1981). In addition, the detection limit of some flotation techniques is in the order 10–50 EPG (depending on protocol), which can represent a constraint for the diagnosis of AR by FECRT (Levecke et al., 2012).
Nonetheless, attempts have been made by the World Association for the Advancement of Veterinary Parasitology (WAAVP) to improve and implement FEC protocols for the assessment of AR in different species of animals (Coles et al., 2006). In addition, lectin staining for the identification of H. contortus eggs (Palmer and McCombe, 1996), computerized image recognition of strongylid eggs (Sommer, 1996) and automated egg enumeration (Mes et al., 2007) are interesting developments toward improved species identification and differentiation. However, the suitability of the latter two approaches requires rigorous evaluation for routine applications. With the development of FECPAK, a diagnostic test-kit for coproscopic examination (www.techiongroup.co.nz), efforts have been made to provide sheep farmers with a field-based FEC method. However, the implementation of such a method requires the cooperation by farmers, adequate training and quality assurance to ensure that diagnoses are accurate (McCoy et al., 2005). Also FLOTAC (Cringoli et al., 2010) seems to be a promising FEC method. Once validated for different host and parasite species, this method might deliver FECs at increased sensitivity (i.e., 1 EPG) and could represent an alternative to current flotation techniques.
4.2. Larval culture (LC)
Larval culture involves incubating fecal samples containing eggs of strongylid nematodes to allow L1s to hatch and then develop through to L3s; the latter are examined microscopically and differentiated morphologically/morphometrically. A number of protocols have been published which differ in the temperatures, times and media used for culture, and the approach of larval recovery (Dinaburg, 1942, Hubert and Kerboeuf, 1984, MAFF, 1986, Roberts and O'Sullivan, 1950, Whitlock, 1956). The most widely employed protocol includes an incubation at 27 °C for 7 days (MAFF, 1986).
Investigations of the ecology and developmental requirements of various species of gastrointestinal nematodes of livestock (Beveridge et al., 1989a, O'Connor et al., 2006) have shown that different species of strongylid nematodes require distinct conditions, such as environmental temperature and relative humidity, to develop adequately. This aspect is particularly important to consider when larval culture results are used to estimate the contribution of different species to mixed infections. It has been demonstrated that one culture protocol can favor the development of one species over others (Dobson et al., 1992). For instance, Whitlock (1956) observed that the usual culture condition (27 °C for 7 days) is suitable for most species, but that the free-living stages of Teladorsagia species develop better at somewhat lower temperatures. This statement was supported by the findings of Dobson et al. (1992), who demonstrated that the developmental success of the infective larvae in fecal cultures was lower for Te. circumcincta than for T. colubriformis when cultured alone or concurrently, indicating that LC was unreliable for estimating the contribution of individual species in mixed infections. Berrie et al. (1988) reported similar findings for the bovine parasites H. placei, Oe. radiatum and Cooperia pectinata. In this study, the recovery of larvae of H. placei was significantly lower compared with the other two species under the same LC conditions. Based on their results, the authors stated that LC and subsequent larval differentiation are unsuitable for an accurate estimation of the proportions of individual species in animals with mixed infections and only provide an indication of the species present (Berrie et al., 1988).
Further variability in LC results has been attributed to differences in the composition of the culture medium, pH, humidity and oxygen (Hubert and Kerboeuf, 1984, Roberts and O'Sullivan, 1950). Therefore, it had been proposed that a defined medium might help to obtain more consistent results (Hubert and Kerboeuf, 1984). To test this proposal, Hubert and Kerboeuf (1984) established a modified method of LC using an “on-agar” approach to provide standardized conditions. LC on agar medium led to higher recoveries of larvae compared with traditional fecal cultures, but lengthy preparation times and increased laboratory requirements appeared to limit the routine application of this method.
In addition to variation in results relating to LC conditions, the differentiation of cultured L3s provides challenges. Differentiation relies on morphological and morphometric parameters, such as the length of the tail sheath extension and total body length of L3s (Dikmans and Andrews, 1933, Gordon, 1933, MAFF, 1986, McMurtry et al., 2000, van Wyk et al., 2004). Various keys for the identification of L3s have been published (Dikmans and Andrews, 1933, Gordon, 1933, MAFF, 1986), but there is an overlap in the body length measurements between some species, and substantial variation in the length of L3s has been reported (McMurtry et al., 2000).
Van Wyk et al. (2004) used the mean length of the tail sheath extension to differentiate L3s of Teladorsagia and/or Trichostrongylus from the larvae of Haemonchus, and Chabertia and Oesophagostomum. However, although useful to differentiate genera, without the requirement to measure every single larva (thus being more time efficient), this approach has the disadvantage that it does not allow the unequivocal differentiation of all genera. For instance, Teladorsagia and Trichostrongylus (being the most common genera in winter rainfall areas) cannot be differentiated based on sheath extension length. To further refine the differentiation of these two genera, other morphological features are required. Lancaster and Hong (1987) suggested that the presence of an inflexion (“shoulder”) at the cranial extremity of Teladorsagia larvae was an informative morphological feature. However, this feature is subtle and its detection is subjective. Gordon (1933) proposed the differentiation of Teladorsagia and Trichostrongylus L3s based on the body length. Based on the measurements of 1,000 larvae of each genus, a body length of > 720 μm was used to infer Teladorsagia and ≤ 720 μm for Trichostrongylus. Although practical, this approach requires individual larvae to be measured and does not take into account variability in the length of developing larvae (as a consequence of culture conditions, climate/season, food source for developing larvae and/or immune status of the host) (McMurtry et al., 2000).
McMurtry et al. (2000) described an approach for the differentiation of Teladorsagia from Trichostrongylus L3s, which involves the exsheathment of cultured larvae with sodium hypochlorite and counting of tubercles at the posterior end of the exsheathed L3 using a microscope. As claimed by the authors, this approach allows the differentiation among populations of T. axei, T. colubriformis, T. vitrinus and Te. circumcincta. However, the authors acknowledged that there is a degree of variability in the number of tubercles and that the tails of Te. circumcincta and T. axei lack these structures (McMurtry et al., 2000).
Interestingly, L3s of the large intestinal nematodes Oesophagostomum and Chabertia cannot be differentiated morphologically/morphometrically under a light microscope, which has prevented epidemiological studies of the distribution and prevalence of these genera (and species). A less commonly used method for larval differentiation involves culture and morphological identification of L1s (Whitlock, 1959). This technique has the advantage of being rapid, since the time required for the development of the L1 stage is short; however, the same issues in relation to culture conditions and identification apply to L1s and L2s as to L3s (Lichtenfels et al., 1997). Although routinely used in most parasitology diagnostic laboratories, the technique of LC coupled to larval differentiation by microscopy is time-consuming, laborious to perform, suffers from inaccuracy (see Johnson et al., 1996, Lichtenfels et al., 1997) and cannot readily be automated.
4.3. Immunological and biochemical methods
In addition to conventional copro-diagnostic methods, various immunological and biochemical methods have been assessed or established, aimed at the specific diagnosis of infection. These methods rely mainly on the detection and measurement of parameters, such as pepsinogen, gastrin or specific antibody in serum, which might be indicative of parasite infections.
4.3.1. Immunological detection
A number of immunological methods, including those that are based on the detection of an immune response in an infected animal, and those for the detection of parasite antigens, have been developed for the specific diagnosis of parasitic infections (e.g., Engvall and Ruitenberg, 1974, Fletcher, 1965, Ogunremi et al., 2008). Based on the target molecule (antigen or antibody), such methods can be classified as either “direct” or “indirect”.
Direct immunological methods provide direct evidence of an infection and can be based on the detection of parasite antigens present in the circulation of and/or excreta from infected hosts. Parasitic extracts have a complex composition and contain molecules that are sometimes shared by other parasites (i.e., are cross-reactive) (Cohen and Sadun, 1976). Shared antigenic composition of closely related parasite species represents a challenge, particularly for nematodes, and often leads to cross-reactivity in immunological tests (Eysker and Ploeger, 2000, Noordin et al., 2005). Also the presence of host materials associated with the parasite can complicate antigen purification and can sometimes interfere with the specificity of a diagnostic assay. Furthermore, the stage of a parasite, used as an antigen source, can influence immuno-diagnostic results, as parasites undergo significant structural and biochemical changes during their development (Cohen and Sadun, 1976). As an example, the antigenic composition of larval stages differs from that of adults (Williams and Soulsby, 1970) and can give rise to variation in diagnostic specificity and sensitivity (McLaren et al., 1978).
Johnson et al. (1996) described an immunodiagnostic assay for the quantitative detection of excretory/secretory parasite antigens in host feces (coproantigens).These authors evaluated the usefulness of this approach in a murine model system using Heligmosomoides polygyrus, a trichostrongyloid gastrointestinal nematode related to the common species infecting ruminants. The authors also suggested that the enzyme-linked immunosorbent assay (ELISA) was useful for the detection of parasite antigens in host feces and might have potential for the detection of pre-patent infections. The diagnostic performance of this assay was promising under experimental conditions, but cross-reactivity, fecal components interfering with the reactivity and the loss of antigens in feces were reported (Johnson et al., 1996).
Indirect immunological methods are usually based on the detection of anti-parasite antibodies or cell-mediated immune responses in infected hosts. A variety of methods has been developed and applied to the diagnosis of nematode infections, such as the complement fixation test, indirect immunofluorescence, indirect haemagglutination and ELISA, of which the latter has been most commonly used (Doenhoff et al., 2004). However, parasitic helminths possess a huge variety of antigens, and there is limited information on which stages and antigens are actually responsible for eliciting immune responses (Berghen et al., 1993). Antibody detection from serum has several disadvantages, including that it cannot distinguish between current and past infection, which is a major challenge when evaluating the effects of chemotherapy, does often not reflect infection intensity and sometimes achieves poor specificity, particularly in disease-endemic areas (Doenhoff et al., 2004).
The detection of anti-Ostertagia antibodies in the serum of cattle has been found to be useful for epidemiological and cross-sectional studies, but is only of limited utility for diagnosis on an individual animal basis (Berghen et al., 1993). Although anti-Ostertagia antibodies are detectable in milk samples by ELISA, there are also some limitations to this approach (Charlier et al., 2010). The response to parasitic infections is variable among host individuals, and it has been shown that serum antibody levels can be influenced by factors, such as milk yield, season, mastitis, the number of pregnancies of a cow, stage of lactation and genetic constitution (Gasbarre et al., 1993, Kloosterman et al., 1993, Sanchez et al., 2004). Also the use of bulk milk samples has been investigated, which has the advantage of being an inexpensive and user-friendly approach (Charlier et al., 2010). However, bulk milk samples taken only a few weeks apart can show significant variation in test results, depending on calving patterns, number of cows contributing to the milk in a tank (i.e., dilution effect) and their relative milk yields (Pritchard, 2001).
4.3.2. Gastrin or pepsinogen detection
Gastrin is a hormone produced by G-cells in the stomach. Gastrin stimulates parietal cells to secrete acid, and also stimulates pepsinogen secretion, stomach motility and blood circulation in gastric vessels. It was proposed that strongylid nematodes can directly stimulate G-cells, causing an increased gastrin production (Berghen et al., 1993). However, as shown for pepsinogen, the specificity of this approach was questioned (Berghen et al., 1993), because other parasites or factors, such as diet, lactation and/or abomasal lesions, can also effect gastrin levels. Furthermore, in an experimental context, it has been shown that high infective doses need to be administered to parasite-naïve calves to induce a significant rise in blood gastrin (Berghen et al., 1993).
Pepsinogen is a pro-enzyme produced by chief cells of the gastric fundus. It is converted to its active form by acid produced by parietal cells. When parasitized glands of the gastric mucosa are destroyed, the hydrochloric acid production of parietal cells decreases, causing a rise in abomasal pH and resulting in less conversion of pepsinogen to active pepsin (Levine, 1968). Accumulating pepsinogen can escape between disrupted cell junctions into the blood. Therefore, an increase in serum pepsinogen concentration has been regarded to relate to mucosal damage by developing larval stages of Ostertagia (Levine, 1968). Berghen et al. (1993) reviewed the value and application of pepsinogen, gastrin and antibody responses as diagnostic indicators for ostertagiasis and identified a number of potentially limiting factors. The authors suggested that other parasitic or non-parasitic diseases can be responsible for a moderate rise in pepsinogen concentrations in blood, thus limiting the specificity of this approach.
4.4. Post mortem diagnosis
The post mortem diagnosis of infection is usually employed in parasitology to determine the number of nematodes present in the gastrointestinal tract (= intensity of infection), for epidemiological studies or to assess anthelmintic efficacy. These techniques involve the opening and washing of respective parts of the gastrointestinal tract and the examination of subsamples to estimate infection intensity. Various techniques have been described (Eysker and Kooyman, 1993, MAFF, 1986, Robertson and Elliott, 1966); the main differences among them are in the counting of nematodes, the soaking or not of the organ in water or saline (mainly used to recover immature stages), and the proportion of the total volume and the number of aliquots examined (Gaba et al., 2006). Other differences are in the length of the intestinal section examined (proximal 10 m of small intestine versus the entire length) and the mesh size of the sieve employed to remove plant debris from the washes (McKenna, 2008).
The common practice of examining the proximal 10 m of the small intestine is based on the observation that most intestinal Trichostrongylus spp. occur within the first 6 m of the small intestine (Beveridge and Barker, 1983). McKenna (2008) stated that processing only the first 10 m of the small intestine led to a recovery of < 50% of the worms located in the entire length, resulting in serious underestimates of the total number of worms present. However, the results reported were based on the necropsy of only 15 sheep, and a recovery of less than half of the total number was observed only in a few individual sheep, whereas in most infected sheep trichostrongylid nematodes were located in the proximal 10 m of the small intestine (cf. McKenna, 2008). Therefore, it can be concluded that the improvement of accuracy achieved by processing the entire small intestine is marginal and involves a significant increase in the amount of labor and time required for processing.
Eysker and Kooyman (1993) described a method that involves three parts (contents, immediate water wash of the organ and the saline wash after 5 h of soaking the organ). The disadvantage of this method is that it involves more labor at necropsy, but it has the distinct advantage that worms are separated from the bulk of the gut contents, allowing a rapid worm count. Gaba et al. (2006) assessed their approach for H. contortus and Te. circumcincta and suggested that the estimation of infection intensity, based on gut washes alone, is reliable. However, a prerequisite is that gut sections (e.g., abomasum) are processed rapidly (within 15 min) after the death of the sheep, as some worms progressively start migrating into the contents (Gaba et al., 2006). Gaba et al. (2006) also stated that immediate washing of the gut is insufficient for extracting T. axei or larvae from the mucosa.
Similarly, the selection of the mesh size of the sieves used is dictated by the purpose of the counting procedure. The use of a smaller mesh size enables a higher recovery of early L4s, but has the disadvantage that more debris remains in the subsample examined, resulting in a prolonged time for counting (McKenna, 2008). Therefore, a small sieve size (e.g., 38 μm aperture) is only required if L4s are counted. If studies are conducted to confirm AR (reflected in a reduced efficacy against adult worms), larger mesh sizes (e.g., 250 μm aperture) can be used (McKenna, 2008).
4.5. The need for standardization
Surprisingly, the performance of most diagnostic tests used routinely for the diagnosis or parasitic infections or disease have not been validated against standards of the Office International des Epizooties (Conraths and Schares, 2006, Office International des Epizooties, 2004). The validation of the performance of any diagnostic test (cf. Table 2, Table 3 ) is critical, and involves the characterization of basic parameters and can be achieved in number of steps (OIE, 2004). As a first step, a test suitable for a particular use has to be selected, developed and optimized. Subsequently, validation parameters have to be determined, such as analytical sensitivity and analytical specificity (Conraths and Schares, 2006). Following this initial assessment, the diagnostic sensitivity and specificity are determined by examining a larger number of samples for which the true disease or infection status of the animals being tested is known (determined by a “gold standard”). After a test has been evaluated, it may be considered validated (Conraths and Schares, 2006), but a continuous monitoring of test performance during routine application is also advisable in both commercial and research settings.
Table 2.
Key validation parameters employed for the assessment of a diagnostic test (based on Conraths and Schares, 2006, Pfeiffer, 2010, Thrusfield, 2005).
| Term | Definition | Method of assessment |
|---|---|---|
| Sensitivity | The proportion of animals with the disease and are test-negative. | Assessment of these two parameters requires an independent, valid criterion termed a “gold standard” used to define the true disease status of an animal. |
| Specificity | The proportion of animals without the disease and are test-negative. | |
| Agreement | The agreement in results between two diagnostic tests, with one of the tests being a generally accepted diagnostic method. | Frequently assessed by Kappa test, which measures the proportion of agreement beyond that to be expected by chance. |
| Accuracy | Refers to the concordance between test results and the ‘true’ clinical state. | Depends on the number of ‘false positives’ and ‘false negatives’, in comparison with the true infection state as determined by the “gold standard”. |
| Reliability | The extent to which test results are consistent in repeat experiments. | This includes the assessment of repeatability, reproducibility, inter- and intra-assay variability. Repeatability assessment can be done by running the test two or more times on the same samples in the same laboratory under the same conditions. Additionally the intraassay variability (between replicates within the same run) and interassay variability (replicates between different runs) can be assessed. Reproducibility can be assessed in the same manner as described before, and performed between different laboratories. |
Table 3.
Stages of validation of a diagnostic test (adapted from Conraths and Schares, 2006).
| Stages of test validation |
|---|
| 1. Feasibility studies |
| 2. Assay development and standardization |
| - Optimization of reagents, protocols and equipment |
| - Preliminary estimate of repeatability |
| - Determination of analytical sensitivity and specificity |
| 3. Determination of assay performance characteristics |
| - Diagnostic sensitivity and specificity |
| - Repeatability and reproducibility |
| 4. Monitoring the validity of assay performance |
| 5. Maintenance and enhancement of validation criteria |
5. Nucleic acid-based methods for diagnosis
Clearly, conventional methods of diagnosis (reviewed in Section 4) have some limitations, in terms of sensitivity and/or specificity. In addition, they can be time consuming and costly to perform. DNA technologies have enabled the development of new, sensitive and specific diagnostic methods that have found applications in parasitology. The ability to specifically identify and study parasites (irrespective of life-cycle stage) using DNA methods has provided new insights into parasite biology, epidemiology and ecology, and has important implications for the specific diagnosis, treatment and control of parasitic diseases (Gasser, 2006). In particular, methods that rely on the enzymatic amplification of nucleic acids can overcome some of the limitations of traditional approaches (Gasser, 2006). Methods that employ the polymerase chain reaction (PCR) (Mullis et al., 1986, Saiki et al., 1988) can selectively amplify in vitro target DNA sequences from complex genomes or matrices, and have led to advances in many areas of the biological sciences.
PCR involves the heat denaturation of double-stranded DNA, followed by a decrease in temperature to allow oligonucleotide primers to bind (= anneal) to their complementary sequence on sense and antisense strands of the target template. Then, the temperature is increased again to enhance the enzymatic activity of a thermostable DNA polymerase, which extends the complementary strands from the primer sites. These synthesis steps are usually repeated 20–40 times in an automated thermal cycler, resulting in an exponential increase in target DNA copies. The major advantage of this methodology is that it enables the study of parasite DNA from minute amounts of template, which would otherwise be insufficient for conventional analysis. The value of this technology in the field of diagnostic veterinary parasitology lies in its ability to specifically identify parasites, detect infection and analyze genetic variation, which are particularly important, given the increasing problems of AR in parasitic nematodes (cf. Gasser, 2006, Gasser et al., 2008).
5.1. Sample processing and PCR inhibition
The selection of the most suitable sample preparation method depends on the type of sample and the purpose of the PCR analysis, as there is no universal method that suits all sample matrices and/or applications (Hoorfar et al., 2004). The main goals of sample preparation are to (i) concentrate the target organisms and the template for subsequent PCR, (ii) eliminate possible PCR-inhibitors, and (iii) produce a homogenous sample for specific and sensitive enzymatic amplification (Rådström et al., 2004). Complex biological (e.g., fecal) samples can contain a wide range of inhibitory substances (e.g., bile salts, collagen, haeme, humic acids and polysaccharides), which are capable of reducing or preventing PCR amplification (Rådström et al., 2004, Wilson, 1997). Different samples can have very different compositions, and the presence of substances potentially inhibitory to the PCR often varies depending on the sample type and composition (Hoorfar et al., 2004, Wilson, 1997). ‘Spike-controls’ (natural or synthetic nucleotide sequences-/fragments introduced into samples) can be used to assess the presence of inhibitory substances in the amplification mixture and the efficiency of the DNA isolation and/or PCR reaction (Ninove et al., 2011). Therefore, the selection and evaluation of the sample preparation approach and a suitable reaction mixture, including polymerases and primers, are critical to obtain PCR-compatible samples of comparable composition, irrespective of the variation in the original matrix (e.g., batch-to-batch variation) (Hoorfar et al., 2004).
5.2. Genetic markers for specific identification or detection
The key to developing a reliable PCR method for the specific diagnosis of infection is the definition of one or more suitable DNA targets (genetic marker or locus) based on DNA sequencing. Since different genes evolve at different rates, the DNA region selected should be sufficiently variable in sequence to allow the identification of parasites to the taxonomic level required. For specific identification, the target DNA should display no or minor sequence variation within a species and differ sufficiently in sequence to consistently allow the delineation among species. In contrast, for the purpose of identifying population variants (subspecies, genotypes or “strains”), a considerable degree of variation in the sequence should exist within a species. A range of target regions in the nuclear and mitochondrial genomes have been employed to achieve the identification of parasites to species or sub-specific genotypes (Anderson et al., 1998, Blouin, 2002, Chilton, 2004, Gasser, 2006). In nuclear ribosomal genes and spacers, there is often less sequence variation among individuals within a population and between populations, which makes them suitable as specific markers. Hence, in the case of genetic markers for the specific identification of strongylid nematodes of livestock, most of the focus has been on employing nuclear ribosomal DNA (rDNA).
Although some success was achieved with other DNA targets (e.g., Callaghan and Beh, 1994, Callaghan and Beh, 1996, Christensen et al., 1994, Roos and Grant, 1993, Zarlenga et al., 1994), most studies have consistently shown that the first (ITS-1) and second (ITS-2) internal transcribed spacers (ITS) of nuclear rDNA provide reliable genetic markers for the specific identification of a range of strongylid nematodes of livestock, including species of Haemonchus, Teladorsagia and Ostertagia (abomasum); Trichostrongylus (abomasum or small intestine), Cooperia, Nematodirus, Bunostomum (small intestine); Oesophagostomum and Chabertia (large intestine); Dictyocaulus, Protostrongylus and Metastrongylus (lung) (reviewed by Gasser, 2006, Gasser et al., 2008).
A comparison of the ITS sequences from a range of strongylid nematodes has shown that the ITS-1 (364–522 bp) is usually larger in size than the ITS-2 (215–484 bp) (see Chilton, 2004). For instance, the ITS-1 region of Ostertagia ostertagi and O. lyrata (801 bp) (Zarlenga et al., 1998b) is longer than that of other trichostrongylids, including congeners, due to the presence of an internal 204 bp region, which is repeated twice (Zarlenga and Higgins, 2001, Zarlenga et al., 1998a, Zarlenga et al., 1998b). No major differences have been detected among species of Teladorsagia/Ostertagia in the lengths of their ITS-2 sequence (Chilton et al., 2001, Stevenson et al., 1996). The G + C content (39–50%) of the ITS-1 sequence of species studied is usually greater than of their ITS-2 (29–45%). The ITS-2 sequences of some species can be relatively A + T-rich (60–70%), which may relate to structural aspects of the precursor rRNA molecule. In addition, studies to date, show that the magnitude of sequence variation in both the ITS-1 and ITS-2 within a species is less (usually < 1.5%) than the levels of sequence differences among species (Gasser, 2006), providing the basis for the specific identification of strongylids and diagnosis of infections.
6. Conventional PCR tools
ITS-1 and/or ITS-2 provide useful genetic markers for the development of diagnostic PCR-based tools for strongylid nematodes (Gasser et al., 2006, Gasser et al., 2008); in addition to being species-specific in sequence, they are short (usually ≤ 800 bp), repetitive and undergo homogenisation (Elder and Turner, 1995, Gasser, 2006), the latter factors ultimately determining the efficiency, “sensitivity” and specificity of any PCR amplification procedure.
PCR-based SSCP analysis has provided a useful approach for the specific identification of strongylid nematodes using markers ITS-1 and/or ITS-2 and for detecting cryptic (= morphologically similar but genetically distinct) species at any stage of development (Gasser and Chilton, 2001, Gasser et al., 2006). Although there has been a considerable focus on nematodes of humans, there have been some applications to strongylids of livestock (reviewed by Gasser et al., 2006, Gasser et al., 2008).
Oligonucleotide primers have been designed to specific regions flanking and/or within the ITS-1 or ITS-2 for diagnostic applications (reviewed by Chilton, 2004, Gasser, 2006, Gasser et al., 2008). Using rDNA targets, this strategy has also been employed for the development of PCR assays for the genus- or species-specific identification of different developmental stages of strongylid nematodes. For instance, Zarlenga et al. (1998a) described the development of a semi-quantitative PCR assay for the diagnosis of patent Ostertagia ostertagi infection in cattle. Conserved oligonucleotide primers were used in PCR to amplify a ~ 1 kb rDNA region, spanning the ITS-1 and part of the 5.8S rRNA gene, from O. ostertagi, whereas amplicons of ~ 600 bp were amplified from H. contortus, Co. oncophora and Oe. radiatum. When DNA samples derived from adult nematodes of the different genera were mixed and amplified simultaneously, there was no evidence of inhibition in PCR, and O. ostertagi-specific amplicons were readily detected electrophoretically. There was a correlation between the intensity of the ~ 1 kb and 600 bp amplicons on gels and the percentage of O. ostertagi DNA within the mix of heterologous species. There was also a strong correlation between the percentage of O. ostertagi DNA and percentage of O. ostertagi eggs in the feces. Effective amplification was achieved from 5% of the genomic DNA isolated from a single O. ostertagi egg. Hence, the establishment of this PCR assay had major implications for diagnosis of patent O. ostertagi infection in cattle as well as for studying the epidemiology of this parasite. Other studies (reviewed by Gasser, 2006, Gasser et al., 2008) have demonstrated the diagnostic utility of PCR assays using species-specific ITS oligonucleotide primers or probes, even when the sequences of related species differ by a single nucleotide (Hung et al., 1999). For instance, Zarlenga et al. (2001) extended previous work to develop a multiplex PCR assay for the specific detection and differentiation of economically important gastrointestinal strongylid nematodes (including H. placei, O. ostertagi, Trichostrongylus spp., Co. oncophora/Co. surnabada and Oe. radiatum) of cattle.
7. Real-time PCR (RT-PCR)
7.1. Principle
RT-PCR was developed in the early 1990s (Higuchi et al., 1992) and allows enzymatic amplification to be monitored in real time in vitro. All current RT-PCR systems detect the amplification using fluorescent dyes or probes. The predominant advantages of real-time PCR over conventional PCR are that it allows high throughput analysis in a “closed-tube” format, not requiring handling or electrophoresis following amplification, that it can be employed for quantitation over a wide “dynamic range” and that it can be used to differentiate amplicons of varying sequence(s) by melting-curve analysis.
The principle of the original method was to incorporate a specific, intercalating dye (e.g., ethidium bromide) into the PCR to measure the change in fluorescence after each cycle using a digital camera and a fluorometer coupled to the reaction tube (Higuchi et al., 1993). The technique has been modified to include other (non-carcinogenic) dyes, such as SYBR Green I (Becker et al., 1996), LCGreen (Wittwer et al., 2003), SYTO9 (Monis et al., 2005a) and EvaGreen (Wang et al., 2006). Real-time PCR assays using such dyes enable the relative or absolute quantitation of amplicons by allowing the identification of the cycle (Ct) at which the amplification commences. One or more DNA standards (of differing concentrations) and test samples are subjected to cycling at the same time and their Ct values established and compared. Standard curves can be constructed based on the use of reference samples, and the relative amounts of template in test samples are calculated in relation to these curves.
Intercalating dyes, such as SYBR Green I, detect any double-stranded DNA, which is advantageous because they can be incorporated into any assay. However, a disadvantage is that the dye binds to all double-stranded DNA in a reaction, which includes primer dimers and non-specific products. This limitation can be overcome by acquiring fluorescence data at a temperature that denatures the non-specific products and leaves the specific products intact. The melting point of an amplicon is linked to the composition and length of the nucleotide sequence(s) within it, which means that a melting-curve analysis can be used to detect and/or characterize sequence variation within and among samples. Other recent advances include the complementary use of high resolution melting-curve (HRM) analysis following RT-PCR (e.g., Jeffery et al., 2007). Melting analysis using the dye LCGreen or SYTO9 has been reported to achieve acceptable levels of reproducibility, attaining better mutation detection capacity than SYBR Green I (Monis et al., 2005a, Wittwer et al., 2003). Alternative, more expensive detection systems (other than intercalating dyes) include Taqman probes (Heid et al., 1996), minor groove binder (MGB) Eclipse probes (Afonina et al., 2002), molecular beacons (Piatek et al., 1998) and fluorescence resonance energy transfer (FRET) (Chen and Kwok, 1999), ensuring specificity in the PCR through exclusive binding to the target sequence (Monis et al., 2005b).
7.2. RT-PCR assays for the diagnosis of strongylid nematode infections
In spite of promising results of RT-PCR for the diagnosis and quantification of selected protozoan and metazoan parasites (Bell and Ranford-Cartwright, 2002, Monis et al., 2005b, van Lieshout and Verweij, 2010, Zarlenga and Higgins, 2001), to date, relatively little research has focused on its use for the diagnosis of strongylid infections of livestock (cf. Gasser, 2006, Gasser et al., 2008). There have been attempts to use RT-PCR for the specific diagnosis and/or quantification of helminth eggs or larvae from the feces from infected hosts. First efforts were made by von Samson-Himmelstjerna et al., 2002, von Samson-Himmelstjerna et al., 2003, who developed RT-PCR assays for the diagnosis and quantification of ovine gastrointestinal nematodes, including H. contortus, O. leptospicularis, T. colubriformis, Co. curticei and for small strongyles (cyathostomins) of horses. However, these assays were used for the identification of larval or adult nematodes only, which limited their utility for routine diagnostic applications.
Harmon et al. (2007) evaluated the use of RT-PCR to quantify eggs of H. contortus from sheep feces and examined various aspects, such as the influence of fecal inhibitors on PCR, the effects of competing and non-competing DNA in multiplex reactions and the impact of embryonic development within the egg on the PCR result. The assay developed showed linear quantifiable amplification of DNA obtained from egg quantities ranging from five to 75 eggs, whereas DNA from higher egg numbers of 75–1000 eggs did not show significant differences in Ct, limiting the quantitative capacity of the assay to a narrow detection range (Harmon et al., 2007). During this study an impact of egg embryonic development on Ct values has only been observed between 0 and 6 h of development at 21 °C, whilst later time periods at 6, 12, and 30 h. did not show statistical differences in Ct when compared to each other (Harmon et al., 2007). Non-competitive DNA, derived from environmental sources, did not appear to have a negative impact on amplification, but in multiplex reactions, the presence of large amounts of competing trichostrongyle DNA hindered the amplification of a different target species whose DNA is present at much smaller amounts (Harmon et al., 2007).
The storage of fecal samples is often necessary in a practical context, but the possible impact of egg embryonation during prolonged storage is known to be a critical factor relating to the accuracy of quantifying egg numbers by RT-PCR (Bott et al., 2009, Harmon et al., 2007). Therefore, approaches to circumvent this issue should be developed, which could possibly involve allowing maximum development to occur prior to DNA isolation (Harmon et al., 2007). It has been proposed that the method used for DNA extraction and the presence of PCR inhibitors might be responsible for discrepancies in the linear correlation between DNA amount and number of nematode eggs (Harmon et al., 2007). Harmon et al. (2007) suggested to account for the variability arising from DNA extraction and the presence of fecal inhibitors through the use of multiplex PCR systems that quantify, in relative terms, egg numbers using Ct values, and include an exogenous DNA template to standardize Ct values and assess every sample individually for fecal inhibitors (Harmon et al., 2007). Additional work in this area had been undertaken by two other research teams, who developed RT-PCR assays for the diagnosis of infections with the human hookworms Ancylostoma duodenale, Necator americanus, and the nodule worm Oe. bifurcum (Verweij et al., 2007) and the equine parasite Strongylus vulgaris (see Nielsen et al., 2008). These assays employed specific primers and TaqMan probes to target the ITS-2 region of nuclear ribosomal DNA. Verweij et al. (2007) suggested that false-negative RT-PCR results (n = 2) in relation to LC might be explained by the amount of feces used for DNA isolation being 30-times less than that used to set up LC. However, both assays were reported by the authors to have a high specificity and a sensitivity superior to that of LC. Inhibition by fecal components was not evident. A potential limitation of these studies was that the specificity of these assays was based exclusively on the design and use of TaqMan probes. However, HRM or sequencing was not used to verify the identity or specificity of the amplicons produced.
Bott et al. (2009) established a combined microscopic/RT-PCR method that allows the semi-quantification of strongylid infections in sheep. In this study, oligonucleotide primers were designed to ITS-2 and 28S rDNA regions of seven key genera or species of strongylids of sheep, including H. contortus, Te. circumcincta, Trichostrongylus spp., Co. oncophora, C. ovina, Oe. columbianum and Oe. venulosum, and used in separate PCR reactions. To determine relative proportions of species/genera contributing to a FEC, standard curves were prepared for the RT-PCRs for individual species and demonstrated a log-linear relationship over four orders of magnitude. The Ct values obtained from species-specific PCR reactions showed a linear correlation to the numbers of eggs present per gram of feces and demonstrated the applicability of this PCR approach for semi-quantification of target species. For some of the primer pairs used in this study, as little as 0.1–2 pg of DNA was sufficient to achieve specific amplification from the respective species, which equates to a proportion of genomic DNA which can be isolated from a single egg (Bott et al., 2009). In the evaluation of this PCR assay, all amplicons generated from specific primer pairs were examined by HRM and sequence analysis to confirm their identity. Designed primers were critically assessed for their specificity (i.e., against a broad range of parasites that are known to be detectable from the feces of infected sheep, including lungworms), and there was no evidence of non-specific amplification. However, Bott et al. (2009) stated that, due to possible sequence polymorphism or heterogeneity of ITS-2 among or within individuals from different geographical locations, the performance of the PCR platform might need additional assessment, if applied in different countries or regions.
Bott et al. (2009) also discussed issues that needed consideration for future applications, such as the effect of fecal consistency on FEC and PCR results. The testing of loose/diarrhoeic and desiccated fecal samples can lead to an under- and over-estimation, respectively, of FECs (Le Jambre et al., 2007) and likely variability in semi-quantitative PCR results. Furthermore, Bott et al. (2009) discussed the need for rapid DNA isolation from fecal samples following their collection. The results of previous studies (Harmon et al., 2007) indicated that mitosis during the larval development leads to an increase of ITS-2 copy number and results in enhanced amplification during the RT-PCR. Because the storage and transport of samples at ambient temperatures is often necessary for practical reasons, approaches for the preservation of fecal material, for example, ethanol fixation might be applied. In addition, the direct extraction of DNA from feces, using commercially available kits, has been proposed as an alternative to methods that involve the concentration of eggs by fecal flotation prior to DNA isolation (cf. Bott et al., 2009, Nielsen et al., 2008). However, such direct extraction methods need to be critically assessed for their ability to remove potential inhibitors (e.g., humic acids, phenolic compounds or polysaccharides) from feces. A fecal flotation and egg isolation approach has been shown to remove such inhibitors (Bott et al., 2009), and has the advantage that it provides a FEC, which can be compared with a PCR result but which is not possible employing a direct DNA isolation-coupled PCR method. Furthermore, flotation allows for a concentration of eggs from multiple grams of feces prior to DNA isolation and PCR, thus increasing the likelihood of amplifying DNA from very small numbers of nematode eggs (Bott et al., 2009, Nielsen et al., 2008). By contrast, only small amounts of feces (e.g., ~ 0.25 g) are used in most commercially-available, direct fecal DNA isolation methods, limiting the ‘sensitivity’ of subsequent PCR. Noting these limitations, the combined microscopic-molecular method (Bott et al., 2009) was established for the specific diagnosis of patent strongylid infections in sheep, and future work is required to evaluate the performance of this method for the specific diagnosis of infections with immature (pre-patent) or hypobiotic stages (e.g., Te. circumcincta or H. contortus) of nematodes and to compare it with direct amplification from DNA isolated directly from fecal samples.
7.3. Critical evaluation and application of RT-PCR to assess the composition of strongylid nematode populations in sheep
Roeber et al. (2011) critically evaluated the performance of the RT-PCR method (Bott et al., 2009) for the diagnosis of naturally acquired strongylid nematode infections in sheep (n = 470; in a temperate climatic zone of south-eastern Australia), using a panel of 100 ‘negative control’ samples from sheep known not to harbor parasitic helminths. The authors compared the diagnostic sensitivity and specificity of this RT-PCR assay with a conventional fecal flotation method. They also established a system to rank the contribution of particular strongylid nematodes to the fecal egg counts (FECs) from ‘mixed infections’ in individual sheep. The testing of fecal samples revealed that Te. circumcincta (80%) and Trichostrongylus spp. (66%) were most prevalent, followed by C. ovina (33%), Oe. venulosum (28%) and H. contortus (1%). For most sheep tested in this study, Te. circumcincta and Trichostrongylus spp. represented the largest proportion of strongylid eggs in fecal samples from individual sheep. This was the first large-scale prevalence survey of gastrointestinal nematodes in live sheep utilizing a molecular tool. The ability to rapidly rank strongylid nematodes according to their contribution to mixed infections represents a major advantage over routine flotation methods. The conclusion from this study was that this RT-PCR tool might be able to replace the conventional technique of larval culture.
This assessment showed that the RT-PCR assay achieved high diagnostic sensitivity (98%) and specificity (100%), and the test results were in ‘good’ agreement (i.e., Kappa: 0.95) with those achieved using conventional fecal flotation. In addition, of 53 field samples which were test-negative based on coproscopy, 23 tested positive by PCR for one or more target nematodes (a result which was confirmed by sequencing), demonstrating better sensitivity of the molecular approach compared with coproscopic examination, and reinforcing that microscopy is not an adequate reference technique [i.e., is an imperfect gold standard (cf. Conraths and Schares, 2006)] for the detailed assessment of the sensitivity and specificity of a diagnostic assay. The results achieved demonstrated that the prevalences of key genera/species, such as Te. circumcincta and Trichostrongylus spp., known to be the dominant species infecting sheep in the winter rainfall environment of Victoria, Australia, were largely in accordance with information available in the published literature (e.g., Anderson, 1972, Anderson, 1973). The application of an ordinal ranking system to estimate the contribution of individual parasites to observed FEC results showed that these genera/species were also responsible for the largest proportion of strongylid eggs in the fecal samples tested. Although known to be abundant in winter rainfall environments, C. ovina and Oe. venulosum were found at high prevalence (33.6% and 28.7%, respectively). An unexpected finding was that Oe. venulosum was the main contributor to the observed FEC results for one of nine farms tested, which has important implications for the interpretation of FECs and anthelmintic control.
According to common practice (Brightling, 1988), FEC results of ≥ 200 eggs per gram (EPG) are considered to relate to a ‘significant’ worm burden, and without further considerations of the species present and their reproductive capacity, give an indication for anthelmintic treatment. This common practice, which involves frequent and, in many cases, unnecessary or excessive administration of anthelmintic drugs can promote AR development in gastrointestinal nematodes of sheep and other hosts, as recent evidence has shown (Kaplan, 2004, Wolstenholme et al., 2004). Given that Oe. venulosum is recognized to be less pathogenic than most strongylids of the upper alimentary tract (Donald et al., 1978) but has high fecundity (Gordon, 1981), FEC results (e.g., > 200 EPG) in which Oe. venulosum is the sole or main contributor would be misinterpreted, and sheep harboring this relatively non-pathogenic would be treated unnecessarily. Therefore, the specific/generic identification of infecting nematodes assists the interpretation of FEC results and, subsequently, treatment decisions, thus, directly contributing to efforts of preserving the efficacy of currently available anthelminthics.
The RT-PCR assay (Roeber et al., 2011), coupled to conventional coproscopy, and the microscopic detection of Nematodirus, Trichuris and Moniezia eggs in fecal samples revealed that the majority of sheep investigated were parasitized by two to four helminth taxa per animal. Data from this study provided new and important insights into the composition and distribution of nematodes, which would not have been achievable in such detail using any of the currently used coprological methods. The results indicated that this tool should be applicable in other climatic regions and/or major sheep producing countries in the world. Depending on the nematode species infecting sheep in a particular climatic zone, minor modifications could be made to the molecular assay to adapt it for the diagnosis of infections with other important parasites (e.g., hookworm or lungworm) and to provide opportunities to study their biology, prevalence and distributions. For instance, as the life-cycles of some lungworms, such as Muellerius capillaris and Protostrongylus rufescens, involve invertebrate intermediate hosts, such as snails and slugs, an adapted RT-PCR assay could be used to examine the prevalence and relative intensity of these parasites in their intermediate hosts and to study their ecology.
The ability to identify helminth species and to rank them according to their contribution to FEC results (Roeber et al., 2011) represents a novel approach that is time- and cost-efficient compared with classical diagnostic techniques, and enables a better interpretation of FEC results, particularly in relation to the anthelmintic treatment of infected sheep (cf. McKenna, 1996, McKenna, 1997). This advance in the diagnosis of gastrointestinal nematode infections could directly and significant contribute to enhanced parasite control.
7.4. Evaluation of RT-PCR to replace larval culture (LC) and support fecal egg count reduction testing (FECRT)
Roeber et al. (2012a) assessed the RT-PCR assay to support the diagnosis of AR in nematodes, in conjunction with conventional FECRT; in addition, a direct comparison of PCR results with those of larval culture (LC) and selective total worm counts (TWC; considered a “gold standard”) was undertaken. In this assessment, the molecular assay achieved a diagnostic sensitivity of 100% and specificity of 87.5% in relation to TWC. These percentages where similar to those achieved previously (Roeber et al., 2011) (diagnostic sensitivity 98% and specificity 100% in relation to FEC), demonstrating that the RT-PCR assay consistently achieved a high diagnostic performance. DNA sequencing results also demonstrated that this molecular assay had a better sensitivity than the routinely used TWC method and, together with FECRT, was of practical value for the detection of albendazole resistance in Te. circumcincta and T. colubriformis populations. However, although the PCR test results were in accordance with TWC, the direct comparison of molecular and LC results showed markedly different findings, depending on the recommended measurements used for larval differentiation (Dikmans and Andrews, 1933, Gordon, 1933, McMurtry et al., 2000). Using the morphometric criteria given by Dikmans and Andrews (1933), the majority of L3s from the LC of the albendazole treated group of sheep were identified as Trichostrongylus, whereas, using the measurements recommended by Gordon (1933) and McMurtry (2000), the same larvae were identified as Teladorsagia. This discrepancy emphasizes the complications and errors associated with the use of LC, which can readily lead to misinterpretations as to which nematodes are resistant to a particular drug. Overall, this study (Roeber et al., 2012a) demonstrated clearly that the molecular assay coupled to FECRT provides a rapid, efficient and universally applicable tool for the diagnosis of AR and the early detection of residual populations of worms in sheep following treatment. Future studies should be conducted to test sheep on different farms and the response of gastrointestinal nematodes to the treatment with various anthelmintics, such as imidazothiazoles/tetrahydropyrimidines, macrocyclic lactones and monepantel (Zolvix, Novartis) (Kaminsky et al., 2008).
The movement of sheep and their gastrointestinal parasites between or among farms favors the spread of drug resistance (Blouin et al., 1995). Therefore, the routine use of the RT-PCR assay, in conjunction with FECRT, could provide a universally applicable method to test sheep before transport to and/or introduction on to a new farm, in order to reduce the spread of drug resistant populations of nematodes. In addition, the assay allows the identification of sheep shedding large numbers of parasite eggs in feces and, in conjunction with information about the infecting helminth species, can be used to support “targeted, selective treatment” approaches (Kenyon et al., 2009). Such a strategy focuses on treating only sheep that will benefit most from an anthelmintic treatment, whereas sheep with low-intensity infections remain untreated to provide refugia for the dilution of resistance genes within a parasite population. Furthermore, the present RT-PCR assay had a similar or improved sensitivity compared with post mortem diagnosis, thus being able to replace the latter. In practice, this means that the presence of particular species/genera and their prevalence can be assessed reliably without the need to kill sheep.
8. Multiplexed-tandem PCR (MT-PCR) for specific diagnosis
8.1. Rationale and establishment of MT-PCR
A limitation of the RT-PCR assay developed (Roeber et al., 2011, Roeber et al., 2012a) was that it employs individual primer pairs for individual specific or generic amplification and requires numerous, manual handling steps throughout the entire procedure. Therefore, the goal was to develop a user-friendly and practical platform that would allow the rapid testing of large numbers of samples at relatively low cost with limited manual intervention, and that could be introduced into a routine testing laboratory. Although conventional multiplex RT-PCR (e.g., Chamberlain et al., 1988) seemed to be a promising prospect at first, preliminary work conducted (Bott and Gasser, unpublished) showed that primer sets used in individual PCRs (cf. Bott et al., 2009) could not be incorporated into a single reaction to achieve specific amplifications. Other restrictions of a conventional multiplex PCR approach are that each target sequence requires a probe for fluorescence-based detection at a particular wave-length and that such probes are relatively costly and require the use of multi-channel RT-PCR thermocyclers — which usually have only four to six distinct wave-length channels, thus limiting the number of species/genera that can be detected.
To circumvent these issues, Roeber et al. (2012b) explored the use of multiplexed-tandem PCR (MT-PCR) (Stanley and Szewczuk, 2005). MT-PCR consists of two amplification phases: (i) a primary ‘target enrichment’ phase (through a small number of PCR cycles) conducted using multiplexed primer sets, and (ii) a subsequent analytical amplification phase (utilizing a diluted product from the primary amplification as a template), consisting of the targeted amplification, in tandem rather than by multiplex, of each genetic locus using specific, nested primers. Because the initial amplification phase is limited to 10–15 cycles, interactions between or among multiplexed primer sets are minimized, reducing competition or the generation of artefactual products and limiting amplification bias, which would otherwise prevent downstream quantification (Stanley and Szewczuk, 2005). Because the primary amplicons are diluted (e.g., 100-fold) prior to use as templates in the secondary phase, primer carry-over and PCR inhibition are substantially reduced. By conducting the secondary (analytical) amplification phase in tandem, the method can be coupled to a single-channel, RT-PCR thermocycler, allowing rapid screening and quantification employing one fluorogenic dye (e.g., SYTO-9), thus reducing the cost associated with detection.
Roeber et al. (2012b) established a high throughput MT-PCR assay for the diagnosis of nematode infections in sheep, and critically assessed its diagnostic sensitivity and specificity relative to RT-PCR as well as conventional LC using fecal samples from different flocks of sheep from a broad geographical range in Australia. The MT-PCR achieved high diagnostic specificity (87.5%) and sensitivity (100%) based on the testing of a panel of 100 fecal DNA samples from helminth-free sheep and 30 samples from sheep with infections confirmed by necropsy. This MT-PCR assay was then used to test 219 fecal samples from sheep with naturally acquired infections from various geographical localities within Australia, and results were compared with those of LC, using 139 of the 219 samples. The MT-PCR and LC results correlated significantly for most nematodes examined, but parasites of the large intestine were significantly under-represented in the LC results. The findings showed that Trichostrongylus spp. (87%), Te. circumcincta (80%) and H. contortus (67%) had the highest prevalences, followed by Oe. venulosum (51%) and C. ovina (12%). Importantly, this MT-PCR allowed a species- or genus-specific diagnosis of patent nematode infections to be made within 24 h (compared with 7–10 days for LC).
The evaluation of two different pre-PCR genomic DNA isolation methods showed that a combined egg flotation and column-purification approach achieved better sensitivity overall compared with direct fecal DNA isolation (Roeber et al., 2012b). The testing of samples from sheep from different geographical locations in Australia showed that the prevalence of the key nematodes investigated were consistent with their presumed distribution, based on historical data and many years of studies conducted using routine diagnostic procedures (Donald et al., 1978). Epidemiological information had not been provided in such detail prior to the use of a molecular diagnostic tool, and the results achieved by routine LC and the molecular diagnostic platform showed significant correlations (rs = 0.69–0.83) for most nematodes. However, C. ovina and Oe. venulosum were both much less frequently detected in LC than by the molecular assays. Thus, the evidence showed that LC was unreliable for the diagnosis of infections with these two nematodes, and did not allow their prevalence and distribution to be studied with any confidence. Importantly, the inability of LC to detect the latter two species subsequently led to an overestimation of other nematodes, namely H. contortus, Trichostrongylus and Teladorsagia, present in the LCs examined, thus providing a biased result. This finding also highlights the utility of the present molecular-diagnostic platform for epidemiological studies and suggests that results obtained previously using traditional diagnostic techniques might have led to inaccurate information on the prevalence and distribution of particular species of gastrointestinal nematodes. The findings of Roeber et al., 2012a, Roeber et al., 2012b showed that C. ovina and Oe. venulosum may contribute much more significantly to infections in sheep than currently acknowledged. Given that the pathogenicity of these parasites is considered low (Donald et al., 1978), it is likely that the inability of classical techniques to reliably identify and quantify these parasites has led to unnecessary anthelmintic treatments at increased cost to the farmer and an increased risk of AR in nematode populations.
8.2. Applications and implications
An important future application of the MT-PCR platform could be the testing of pasture samples for infective larvae of strongylid nematodes. This approach has significant implications for studying the epidemiology and ecology (i.e., the seasonal occurrence of species and length of larval survival) of gastrointestinal nematodes in livestock, and enables assessments of the extent of contamination and risk of infection. For this purpose, pasture foliage could be collected, washed and concentrated; DNA could then be isolated from the larval suspension and molecular testing conducted. However, as the number of cells increases during the mitotic division in developing eggs and larvae, the number of ITS-2 copies also increases, which means that the MT-PCR platform will need to be calibrated to accommodate this change in copy number. This PCR platform permits different sensitivity settings (based on the number of amplification cycles in the primary PCR) and, whilst a medium sensitivity (15 cycles of primary amplification) was shown to produce the best results for the detection of eggs from feces, this setting could be adjusted for L3s from pasture samples.
There are numerous other possible applications of a high throughput MT-PCR assay. For instance, such an assay could also be used to assist in evaluating the efficacy of vaccines by monitoring FEC, either at the stage of initial vaccine development or, later, in field trials, to predict levels of protection against particular parasite species in individual host animals of different genetic backgrounds (i.e., different breeds of sheep). Immunological methods could also be used in concert with FECRT to measure protective effects of anti-nematode vaccines for sheep following challenge infection/s (with reference to well-defined controls). Moreover, to further refine the diagnosis of AR, genomic DNA regions linked to resistance (e.g., beta-tubulin gene) could be integrated as targets into the assay. However, AR is frequently polygenic (Beech et al., 2011) and/or can also be associated with drug transport mechanisms (Cvilink et al., 2009). Most mechanisms of resistance are only partially understood for many drug classes (cf. Taylor et al., 2002), and more research needs to focus on identifying reliable DNA markers for the specific detection of AR.
There is now major potential to extend the use of MT-PCR assay to other socioeconomically important pathogens, including helminths (e.g., flukes and lungworms), protists (e.g., Cryptosporidium, Eimeria and Giardia), bacteria (e.g., Campylobacter, Escherichia, Salmonella and Yersinia) and viruses (e.g., Rota- and Corona-viruses). Furthermore, such a platform could also be adapted, with modifications being made to the DNA isolation procedure and primers used, for the diagnosis of infections with blood pathogens (e.g., Babesia and Theileria) or the detection of pathogens in environmental samples. In addition, although we have focused on the development of this platform for the detection and identification of pathogens of socioeconomic importance in sheep (Roeber et al., 2012b), a similar approach would be of major benefit in assessing parasites in other hosts, including other small ruminants, cattle, horses and even humans. For example, estimating the number of nematode eggs in the feces from adult cattle is challenging, as eggs are generally present in low numbers and, thus, require more sensitive techniques of diagnosis (Agneessens et al., 2000, Mes et al., 2001). Therefore, the MT-PCR-coupled diagnostic platform could be further enhanced by automating the counting of eggs in fecal samples prior to DNA isolation. Approaches that involve the recovery of nematode eggs from feces and the automated counting of all eggs present in a sample (as opposed to the counting of eggs from sub-aliquots of a sample and their extrapolation to the total sample volume) by image recognition have been developed (Mes et al., 2001, Mes et al., 2007). Consequently, with only minor modifications, the MT-PCR-based platform could provide a major advance in the diagnosis of nematode infections in cattle.
The MT-PCR platform (Roeber et al., 2012b) could also be further enhanced through the use a robotic DNA isolation procedure. Automated DNA isolation platforms are readily available from different manufacturers and include easyMAG (BioMerieux, Mary l'Etoile, France), m2000sp (Abbott, Abbott Park, IL, USA), MagNA Pure LC 2.0 (Roche) and QiaSymphony (Qiagen, Hilden, Germany), but their suitability for the isolation of DNA from nematode eggs from fecal samples would need to be critically assessed before they could be incorporated into the present MT-PCR system. Further advantages of this approach include the possibility for large-scale, targeted collections and long-term storage of DNA samples. Unlike traditional copro-diagnostic testing, in which samples are usually examined only once to enumerate parasite eggs or larvae, and are then discarded, the use of DNA samples has the advantage that the sample material collected can be stored (at − 80 °C) in a clean and space efficient manner (i.e., either frozen or in dehydrated form), allowing the testing of the samples many years or even decades later. This provides the opportunity of carrying out (even retrospectively) studies of “new” or “emerging” pathogens. These are important applications in times of changing environmental conditions (e.g., climate change, urbanization and associated changes in pathogen transmission, distribution and epidemiology), increased problems with AR as well as the increased mobility of humans and/or animals between/among different regions or countries.
9. Prospects for field-based assays
PCR-based technologies have become central to the diagnosis of parasitic and other infectious diseases. However, most PCR methods are laboratory based and often require relatively expensive and specialized instrumentation and reagents, a clean laboratory to avoid contamination during processing and testing and skilled operators with some technical knowledge in molecular technologies. Therefore, most PCR assays are not applicable to use in the field, which represents a limitation for application in resource-poor countries or in situations where a rapid ‘on-the-spot’ diagnosis is required. Recent developments provide possibilities for the miniaturization and automation of devices for diagnostic testing.
The time required to perform PCR (typically 90–120 min) depends, to a large extent, on the ability of the instrument to cycle rapidly through the denaturation, annealing and extension steps (deMello, 2003). By reducing the thermal mass of the instrument, the total reaction times can be significantly shortened. Two different parameters can be altered to achieve this: (i) the physical dimensions of a system can be reduced to reduce the thermal mass of the instrument, and (ii) a sample can be moved through multiple reaction zones which are held at specific temperatures required for the in vitro amplification of nucleic acids (deMello, 2003). The latter approach allows the heating or cooling of small fluid elements which move through different temperature zones within 100 ms, thus allowing ultra-rapid reaction times and making the functional integration of PCR into micro-chips possible (deMello, 2003). Obeid et al. (2003) developed such a microchip that demonstrated efficient amplification of DNA within 5 min and, additionally, provides a separate channel for reverse transcription, in which RNA samples can be transcribed into DNA before entering the PCR zone of the chip for amplification. Furthermore, the continuous injection of small sample volumes (1–2 μl), separated by water and air plugs, allows the simultaneous amplification of multiple samples without cross-contamination (deMello, 2003).
Isothermal amplification methods provide prospects for the design of simple, portable and low-energy consuming operating systems (Asiello and Baeumner, 2011). Some of these methods include nucleic acid sequence-based amplification (NASBA) (Compton, 1991), loop-mediated isothermal amplification (LAMP) (Notomi et al., 2000), helicase-dependent amplification (HDA) (Vincent et al., 2004), rolling circle amplification (RCA) (Liu et al., 1996) and strand displacement amplification (SDA) (Walker et al., 1992a, Walker et al., 1992b). Advantages of such methods seem to be that they reduce costs, simplify the use of amplification reactions by eliminating laboratory equipment, prevent contamination and provide the potential to run several reactions in parallel. Furthermore, miniaturized test systems can be combined with integrated steps of sample preparation and detection of amplification products, thus providing scope for the design of monolithic diagnostic systems, also referred to as micro-total analysis systems (μTAS) or labs-on-a-chip (LOC) (Asiello and Baeumner, 2011). Although, these technologies appear to be in their early phase of development and require further research to translate into practical diagnostics, there are some reports demonstrating their utility. For example, Liu et al., 2011 developed a point-of-care diagnostic system, which is equipped with an integrated membrane for isolation, concentration and purification of nucleic acids. In this system, the amplification process is carried out using a LAMP procedure coupled to real-time detection of amplicons using a fluorescence reader. The authors assessed their system for the detection of human immunodeficiency virus (HIV-1) from oral fluids and demonstrated a detection limit of less than 10 HIV particles (Liu et al., 2011).
A number of LAMP assays have already been reported for a range of metazoan parasites, including taeniid cestodes (Nkouawa et al., 2010), Fasciola spp., Opisthorchis viverrini, Paragonimus westermani (see Ai et al., 2010, Arimatsu et al., 2012, Chen et al., 2011a) and Angiostrongylus cantonensis and Wuchereria bancrofti (see Chen et al., 2011b, Takagi et al., 2011). There are reports of LAMP assays for, for example, Toxoplasma (see Hu et al., 2012), Plasmodium (see Sirichaisinthop et al., 2011), Theileria annulata (Liu et al., 2012) or Eimeria (Barkway et al., 2011). However, these assays are still laboratory-based and, to date, there is no point-of-care diagnostic system for routine use. Although a portable system for the detection of animal diseases has been developed and was assessed for foot-and-mouth disease (FMD) (Seise et al., 2011), the steps of RNA isolation from the virus responsible for this disease still require laboratory work. These recent achievements indicate some potential for novel diagnostic systems with prospects for rapid, field-based diagnosis.
10. Future diagnostic applications of advanced sequencing technologies
Further advances in molecular diagnostics are expected from the rapidly developing sequencing technologies. In the past, studies that investigated the diversity of microorganisms in a natural sample involved the cloning and subsequent Sanger sequencing of selected genes (commonly 16S rDNA) to produce profiles of diversity in environmental samples (Tyson et al., 2004). These so-called metagenomic studies (i.e., the study of genetic material recovered directly from environmental samples) showed that cultivation-based methods are unable to detect the vast majority of microorganisms and have allowed novel insights into a previously hidden diversity of microbial life (Hugenholtz et al., 1998). Following the rapid reduction in cost of DNA sequencing (Pushkarev et al., 2009) and the development of high-throughput sequencing technologies (e.g., 454 pyrosequencing, Illumina- and SOLID sequencing platforms) (Bentley et al., 2008, Harris et al., 2008, Mardis, 2008), these technologies have become accessible to many research groups and have enabled the direct sequencing of microbe and parasite genomes from environmental samples. However, these novel approaches of DNA sequencing create read lengths which are significantly shorter than those produced by Sanger sequencing and a much larger number of sequence reads, ultimately leading to the generation of large amounts of data for analyses (Desai et al., 2012). Analyses of such data sets to obtain biologically meaningful information and the need for enhanced computing power can represent a bottleneck to this approach (Desai et al., 2012). Nonetheless, a number of studies show the value of metagenomic approaches for assessing the diversity of microbial communities in the gut of humans and other mammals (e.g., Hess et al., 2011, Lamendella et al., 2011, Lepage et al., 2013, Qin et al., 2010) or in the marine environment (e.g., Breitbart et al., 2002, Venter et al., 2004, Yooseph et al., 2010) and have led to the discovery of large numbers of previously unknown microorganisms.
Another technological advance is DNA sequencing using nano-pores. This type of analysis is emerging and involves the use of voltage to drive molecules through a nano-scale pore in a membrane between two electrolytes. This allows the analysis of charged polymers (single-stranded DNA, double-stranded DNA or RNA) by monitoring the change of the ionic current as single molecules pass through it (Schneider and Dekker, 2012). Nano-pore sequencing has the advantages that it does not require the labeling of nucleotides or amplification prior to sequencing, can be applied to single molecules and is capable of high throughput DNA analysis (Venkatesan and Bashir, 2011). Further benefits are that it is low cost, requires small reagent volumes and generates long reads, which appears to make it suitable for de novo sequencing (Venkatesan and Bashir, 2011). It has been proposed that nano-pore-based diagnostic tools could detect target molecules at extremely low concentrations and from minute sample volumes, detect simultaneously multiple biomarkers or genes, eliminate the need for time-consuming amplification and conversion steps, thus providing a rapid analysis at low cost (Venkatesan and Bashir, 2011). In addition to the sequencing through nano-pores, with the aim of rapid and affordable DNA sequencing, a number of other approaches have been developed, including the single-molecule evanescent field detection of sequencing-by-synthesis in arrays of nano-chambers (Pacific Biosciences) (Eid et al., 2009), sequencing by ligation on self-assembled DNA nano-arrays (Complete Genomics) (Drmanac et al., 2010), and the detection of H + ions released during sequencing-by-synthesis on silicon field-effect transistors from multiple polymerase template reactions (Ion Torrent) (Rothberg et al., 2011). Clearly, the rapid emergence of a range of exciting sequencing technologies provides new prospects for diagnostic applications. Current evidence suggests that some of these advanced technologies will change the face of molecular diagnostics in the near future.
11. Conclusions
The accurate diagnosis of gastrointestinal nematode infections of livestock underpins investigations of the biology, ecology and epidemiology of parasites and supports the monitoring of emerging problems with anthelmintic resistance (AR). Current, routinely used methods of diagnosis rely on the detection or morphological identification of the infective stages (eggs and/or larvae) of these nematodes in host feces. Until recently, these traditional techniques, which can be time-consuming and laborious, have not undergone any substantial technological advancement. As eggs and larvae of numerous genera and species of nematodes infecting livestock lack distinctive morphological features, traditional approaches are not usually able to achieve a species- or even genus-specific diagnosis in the live animal, making it challenging to conduct reliable studies of the biology, epidemiology and ecology of parasites, unless expensive and laborious post mortem investigations are carried out. This situation has also hampered investigations of the occurrence and distribution of AR in strongylid nematodes of livestock, which represents a global problem.
Advances in PCR-based methods and the availability of specific genetic markers in the internal transcribed spacers of nuclear rDNA have provided the opportunity of developing enhanced PCR-based tools for diagnosis (reviewed by Gasser, 2006, Gasser et al., 2008). Recent studies (Roeber et al., 2011, Roeber et al., 2012a, Roeber et al., 2012b) showed that RT-PCR and MT-PCR assays can replace the inaccurate and time-consuming method of LC. This high throughput MT-PCR (Roeber et al., 2012b) takes < 1 day to perform, compared with at least a week for LC, thus reducing the time that the farmer has to wait for a diagnosis. From a service provision perspective, this platform does not require detailed technical expertise of the operator, has high sensitivity and specificity, and has broad applicability, in that it can be used to carry out large-scale epidemiological studies, to support the diagnosis of drug resistance, and will be applicable or adaptable to other parasites and/or hosts. In addition, the MT-PCR platform delivers, rapidly, objective and detailed results to a genus or species level, which is of major value for enhanced control. Overall, the MT-PCR assay established (Roeber et al., 2012b) essentially meets the international standards (Conraths and Schares, 2006, Office International des Epizooties, 2004) required for use in a laboratory setting for research or routine diagnostic purposes and has significant advantages over classical methods, particularly in relation to the interpretation of FEC results and recommendations about anthelmintic treatment. This test improves the diagnosis of infections with nematode species which are problematic to detect or identify by traditional coprological techniques, either because of their morphological/morphometric similarity with other species/genera (i.e., Teladorsagia and Trichostrongylus, C. ovina and Oe. venulosum) or their unfavorable development under standard culture conditions. Current evidence indicates that this MT-PCR assay is highly adaptable, allowing the development of a wide range of next-generation diagnostic tools to underpin the control of socioeconomically important infectious diseases of animals and the detection and monitoring of drug resistance.
Clearly, a number of nucleic acid-based methods of diagnosis have significant advantages, particularly in terms of sensitivity and specificity, reproducibility and repeatability. These features make them suitable to be incorporated into surveillance systems, which are based on a “from pasture/stable-to-table” approach, such as the Hazard Analysis Critical Control Point (HACCP). The routine application of advanced diagnostic platforms, including practical and high performance field-based assays and, particularly, high throughput sequencing technologies as well as, now provide major scope for better disease surveillance and detailed epidemiological investigations, shorter response times to tackle and control disease outbreaks (as the diagnosis using molecular tools usually takes less time compared with culture- or microscopy-based approaches) and, ultimately, greater protection for the consumer of animal products. Epidemiological data obtained using such methods would help government authorities, such as OIE, the Food and Agricultural Organization (FAO) and/or the World Health Organization (WHO) in the detailed tracking and mapping of disease outbreaks or spreads, to make forecasts about their occurrence and to implement appropriate contingency plans and guidelines for the effective and sustainable control of parasitic and other infectious diseases. This focus will be accompanied by an expansion in revolutionary, new diagnostic technologies and important biotechnological outcomes.
Acknowledgments
This research has been supported through funds from the Australian Research Council (ARC) and the National Health and Medical Research Council (NHMRC). Other support from Melbourne Water Corporation is gratefully acknowledged. Some of our research was supported by a Victorian Life Sciences Computation Initiative (VLSCI) grant on its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government, Australia. FR is the grateful recipient of scholarships from the University of Melbourne.
References
- Afonina I.A., Reed M.W., Lusby E., Shishkina I.G., Belousov Y.S. Minor groove binder-conjugated DNA probes for quantitative DNA detection by hybridization-triggered fluorescence. Biotechniques. 2002;32:940–949. doi: 10.2144/02324pf01. [DOI] [PubMed] [Google Scholar]
- Agneessens J., Claerebout E., Dorny P., Borgsteede F.H.M., Vercruysse J. Nematode parasitism in adult dairy cows in Belgium. Vet Parasitol. 2000;90:83–92. doi: 10.1016/s0304-4017(00)00232-6. [DOI] [PubMed] [Google Scholar]
- Ai L., Li C., Elsheikha H.M., Hong S.J., Chen J.X., Chen S.H. Rapid identification and differentiation of Fasciola hepatica and Fasciola gigantica by a loop-mediated isothermal amplification (LAMP) assay. Vet Parasitol. 2010;174:228–233. doi: 10.1016/j.vetpar.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Alvarez-Sanchez M.A., Perez-Garcia J., Cruz-Rojo M.A., Rojo-Vazquez F.A. Real time PCR for the diagnosis of benzimidazole resistance in trichostrongylids of sheep. Vet Parasitol. 2005;129:291–298. doi: 10.1016/j.vetpar.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Anderson N. Trichostrongylid infections of sheep in a winter rainfall region. I. Epizootiological studies in the western district of Victoria, 1966–67. Aust J Agric Res. 1972;23:1113–1129. [Google Scholar]
- Anderson N. Trichostrongylid infections of sheep in a winter rainfall region. II. Epizootiological studies in the western district of Victoria, 1967–68. Aust J Agric Res. 1973;24:599–611. [Google Scholar]
- Anderson C.R. CAB International; 2000. Nematode parasites of vertebrates. Their development and transmission. [Google Scholar]
- Anderson T.J., Blouin M.S., Beech R.N. Population biology of parasitic nematodes: applications of genetic markers. Adv Parasitol. 1998;41:219–283. doi: 10.1016/s0065-308x(08)60425-x. [DOI] [PubMed] [Google Scholar]
- Arimatsu Y., Kaewkes S., Laha T., Hong S.-J., Sripa B. Rapid detection of Opisthorchis viverrini copro-DNA using loop-mediated isothermal amplification (LAMP) Parasitol Int. 2012;61:178–182. doi: 10.1016/j.parint.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asiello P., Baeumner A. Miniaturized isothermal nucleic acid amplification, a review. Lab Chip. 2011;11:1420–1430. doi: 10.1039/c0lc00666a. [DOI] [PubMed] [Google Scholar]
- Baker N.F., Cook E.F., Douglas J.R., Cornelius C.E. The pathogenesis of trichostrongyloid parasites. III. Some physiological observations in lambs suffering from acute parasitic gastroenteritis. J Parasitol. 1959;45:643–651. [PubMed] [Google Scholar]
- Barger I.A. The statistical distribution of trichostrongylid nematodes in grazing lambs. Int J Parasitol. 1985;15:645–649. doi: 10.1016/0020-7519(85)90010-4. [DOI] [PubMed] [Google Scholar]
- Barkway C., Pocock R., Vrba V., Blake D. Loop-mediated isothermal amplification (LAMP) assays for the species-specific detection of Eimeria that infect chickens. BMC Vet Res. 2011;7:67-. doi: 10.1186/1746-6148-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A., Reith A., Napiwotzki J., Kadenbach B. A quantitative method of determining initial amounts of DNA by polymerase chain reaction cycle titration using digital imaging and a novel DNA stain. Anal Biochem. 1996;237:204–207. doi: 10.1006/abio.1996.0230. [DOI] [PubMed] [Google Scholar]
- Beech R.N., Skuce P., Bartley D.J., Martin R.J., Prichard R.K., Gilleard J.S. Anthelmintic resistance: markers for resistance, or susceptibility? Parasitology. 2011;138:160–174. doi: 10.1017/S0031182010001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A.S., Ranford-Cartwright L.C. Real-time quantitative PCR in parasitology. Trends Parasitol. 2002;18:338–342. [PubMed] [Google Scholar]
- Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghen P., Hilderson H., Vercruysse J., Dorny P. Evaluation of pepsinogen, gastrin and antibody response in diagnosing ostertagiasis. Vet Parasitol. 1993;46:175–195. doi: 10.1016/0304-4017(93)90057-t. [DOI] [PubMed] [Google Scholar]
- Berrie D.A., East I.J., Bourne A.S., Bremner K.C. Differential recoveries from faecal cultures of larvae of some gastro-intestinal nematodes of cattle. J Helminthol. 1988;62:110–114. doi: 10.1017/s0022149x00011330. [DOI] [PubMed] [Google Scholar]
- Besier R.B., Love S.C.J. Anthelmintic resistance in sheep nematodes in Australia: the need for new approaches. Aust J Exp Agric. 2003;43:1383–1391. [Google Scholar]
- Beveridge I., Barker I.K. Morphogenesis of Trichostrongylus rugatus and distribution during development in sheep. Vet Parasitol. 1983;13:55–65. doi: 10.1016/0304-4017(83)90020-1. [DOI] [PubMed] [Google Scholar]
- Beveridge I., Pullman A.L., Martin R.R., Barelds A. Effects of temperature and relative humidity on development and survival of the free-living stages of Trichostrongylus colubriformis, T. rugatus and T. vitrinus. Vet Parasitol. 1989;33:143–153. doi: 10.1016/0304-4017(89)90062-9. [DOI] [PubMed] [Google Scholar]
- Beveridge I., Pullman A.L., Phillips P.H., Martin R.R., Barelds A., Grimson R. Comparison of the effects of infection with Trichostrongylus colubriformis, T. vitrinus and T. rugatus in Merino lambs. Vet Parasitol. 1989;32:229–245. doi: 10.1016/0304-4017(89)90123-4. [DOI] [PubMed] [Google Scholar]
- Blouin M. Molecular prospecting for cryptic species of nematodes: mitochondrial DNA versus internal transcribed spacer. Int J Parasitol. 2002;32:527–531. doi: 10.1016/s0020-7519(01)00357-5. [DOI] [PubMed] [Google Scholar]
- Blouin M.S., Yowell C.A., Courtney C.H., Dame J.B. Host movement and the genetic structure of populations of parasitic nematodes. Genetics. 1995;141:1007–1014. doi: 10.1093/genetics/141.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott N.J., Campbell B.E., Beveridge I., Chilton N.B., Rees D., Hunt P.W. A combined microscopic-molecular method for the diagnosis of strongylid infections in sheep. Int J Parasitol. 2009;39:1277–1287. doi: 10.1016/j.ijpara.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Breitbart M., Salamon P., Andresen B., Mahaffy J., Segall A., Mead D. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci U S A. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightling A. Inkata Press; Melbourne: 1988. Sheep diseases. [Google Scholar]
- Callaghan M.J., Beh K.J. A middle-repetitive DNA sequence element in the sheep parasitic nematode, Trichostrongylus colubriformis. Parasitology. 1994;109:345–350. doi: 10.1017/s0031182000078379. [DOI] [PubMed] [Google Scholar]
- Callaghan M.J., Beh K.J. A tandemly repetitive DNA sequence is present at diverse locations in the genome of Ostertagia circumcincta. Gene. 1996;174:273–279. doi: 10.1016/0378-1119(96)00093-5. [DOI] [PubMed] [Google Scholar]
- Chamberlain J.S., Gibbs R.A., Ranier J.E., Nguyen P.N., Caskey C.T. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988;16:11141–11156. doi: 10.1093/nar/16.23.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier J., Vercruysse J., Smith J., Vanderstichel R., Stryhn H., Claerebout E. Evaluation of anti-Ostertagia ostertagi antibodies in individual milk samples as decision parameter for selective anthelmintic treatment in dairy cows. Prev Vet Med. 2010;93:147–152. doi: 10.1016/j.prevetmed.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Chen X., Kwok P.Y. Homogeneous genotyping assays for single nucleotide polymorphisms with fluorescence resonance energy transfer detection. Genet Anal. 1999;14:157–163. doi: 10.1016/s1050-3862(98)00016-3. [DOI] [PubMed] [Google Scholar]
- Chen M.X., Ai L., Zhang R.L., Xia J.J., Wang K., Chen S.H. Sensitive and rapid detection of Paragonimus westermani infection in humans and animals by loop-mediated isothermal amplification (LAMP) Parasitol Res. 2011;108:1193–1198. doi: 10.1007/s00436-010-2162-x. [DOI] [PubMed] [Google Scholar]
- Chen R., Tong Q., Zhang Y., Lou D., Kong Q., Lv S. Loop-mediated isothermal amplification: rapid detection of Angiostrongylus cantonensis infection in Pomacea canaliculata. Parasitol Vectors. 2011;4:204-. doi: 10.1186/1756-3305-4-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilton N.B. The use of nuclear ribosomal DNA markers for the identification of bursate nematodes (order Strongylida) and for the diagnosis of infections. Anim Health Res Rev. 2004;5:173–187. doi: 10.1079/ahr200497. [DOI] [PubMed] [Google Scholar]
- Chilton N.B., Newton L.A., Beveridge I., Gasser R.B. Evolutionary relationships of trichostrongyloid nematodes (Strongylida) inferred from ribosomal DNA sequence data. Mol Phylogenet Evol. 2001;19:367–386. doi: 10.1006/mpev.2001.0938. [DOI] [PubMed] [Google Scholar]
- Christensen C.M., Zarlenga D.S., Gasbarre L.C. Ostertagia, Haemonchus, Cooperia, and Oesophagostomum: construction and characterization of genus-specific DNA probes to differentiate important parasites of cattle. Exp Parasitol. 1994;78:93–100. doi: 10.1006/expr.1994.1009. [DOI] [PubMed] [Google Scholar]
- Cohen S., Sadun E.H. Blackwell Scientific Publications; Oxford: 1976. Immunology of Parasitic Infections. [Google Scholar]
- Coles G.C., Bauer C., Borgsteede F.H.M., Geerts S., Klei T.R., Taylor M.A. World Association for the Advancement of Veterinary Parasitology (W.A.A.V.P.). Methods for the detection of anthelmintic resistance in nematodes of veterinary importance. Vet Parasitol. 1992;44:35–44. doi: 10.1016/0304-4017(92)90141-u. [DOI] [PubMed] [Google Scholar]
- Coles G.C., Jackson F., Pomroy W.E., Prichard R.K., von Samson-Himmelstjerna G., Silvestre A. The detection of anthelmintic resistance in nematodes of veterinary importance. Vet Parasitol. 2006;136:167–185. doi: 10.1016/j.vetpar.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Compton J. Nucleic acid sequence-based amplification. Nature. 1991;350:91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- Conraths F.J., Schares G. Validation of molecular-diagnostic techniques in the parasitological laboratory. Vet Parasitol. 2006;136:91–98. doi: 10.1016/j.vetpar.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Cox D.D., Todd A.C. Survey of gastrointestinal parasitism in Wisconsin dairy cattle. J Am Vet Med Assoc. 1962;141:706–709. [PubMed] [Google Scholar]
- Cringoli G., Rinaldi L., Maurelli M.P., Utzinger J. FLOTAC: new multivalent techniques for qualitative and quantitative copromicroscopic diagnosis of parasites in animals and humans. Nat Protoc. 2010;5:503–515. doi: 10.1038/nprot.2009.235. [DOI] [PubMed] [Google Scholar]
- Cvilink V., Lamka J., Sklov L. Xenobiotic metabolizing enzymes and metabolism of anthelminthics in helminths. Drug Metab Rev. 2009;41:8–26. doi: 10.1080/03602530802602880. [DOI] [PubMed] [Google Scholar]
- Demeler J. The physiological site of action and the site of resistance to the macrocyclic lactone anthelmintics in sheep parasitic trichostrongyloid nematodes. Doctoral thesis, Tierärztliche Hochschule Hannover; 2005.
- Demeler J., Küttler U., El-Abdellati A., Stafford K., Rydzik A., Varady M. Standardization of the larval migration inhibition test for the detection of resistance to ivermectin in gastro intestinal nematodes of ruminants. Vet Parasitol. 2010;174:58–64. doi: 10.1016/j.vetpar.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Demeler J., Küttler U., von Samson-Himmelstjerna G. Adaptation and evaluation of three different in vitro tests for the detection of resistance to anthelmintics in gastro intestinal nematodes of cattle. Vet Parasitol. 2010;170:61–70. doi: 10.1016/j.vetpar.2010.01.032. [DOI] [PubMed] [Google Scholar]
- deMello A. Microfluidics: DNA amplification moves on. Nature. 2003;422:28–29. doi: 10.1038/422028a. [DOI] [PubMed] [Google Scholar]
- Desai N., Antonopoulos D., Gilbert J., Glass E., Meyer F. From genomics to metagenomics. Curr Opin Biotechnol. 2012;23:72–76. doi: 10.1016/j.copbio.2011.12.017. [DOI] [PubMed] [Google Scholar]
- Dikmans G., Andrews J.S. A comparative morphological study of the infective larvae of the common nematodes parasitic in the alimentary tract of sheep. Trans Am Microsc Soc. 1933;52:1–25. [Google Scholar]
- Dinaburg A.G. The efficiency of the Baermann apparatus in the recovery of larvae of Haemonchus contortus. J Parasitol. 1942;28:433–440. [Google Scholar]
- Dobson R.J., Barnes E.H., Birclijin S.D., Gill J.H. The survival of Ostertagia circumcincta and Trichostrongylus colubriformis in faecal culture as a source of bias in apportioning egg counts to worm species. Int J Parasitol. 1992;22:1005–1008. doi: 10.1016/0020-7519(92)90060-x. [DOI] [PubMed] [Google Scholar]
- Dobson R.J., LeJambre L., Gill J.H. Management of anthelmintic resistance: inheritance of resistance and selection with persistent drugs. Int J Parasitol. 1996;26:993–1000. doi: 10.1016/s0020-7519(96)80078-6. [DOI] [PubMed] [Google Scholar]
- Doenhoff M., Chiodini P., Hamilton J. Specific and sensitive diagnosis of schistosome infection: can it be done with antibodies? Trends Parasitol. 2004;20:35–39. doi: 10.1016/j.pt.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Donald A.D., Southcott W.H., Dineen J.K. Commonwealth Scientific and Industrial Research Organisation (CSIRO); Melbourne: 1978. The epidemiology and control of gastrointestinal parasites of sheep in Australia. [p. xii + 153 pp.] [Google Scholar]
- Drmanac R., Sparks A., Callow M., Halpern A., Burns N., Kermani B. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Elard L., Humbert J.F. Importance of the mutation of amino acid 200 of the isotype 1 β-tubulin gene in the benzimidazole resistance of the small-ruminant parasite Teladorsagia circumcincta. Parasitol Res. 1999;85:452–456. doi: 10.1007/s004360050577. [DOI] [PubMed] [Google Scholar]
- Elder J.F., Turner B.J. Concerted evolution of repetitive DNA sequences in eukaryotes. Q Rev Biol. 1995;70:297–320. doi: 10.1086/419073. [DOI] [PubMed] [Google Scholar]
- Engvall E., Ruitenberg E.J. ELISA, enzyme linked immunosorbent assay — a new technique for sero-diagnosis of trichinosis. Parasitology. 1974;69:xxiv. [PubMed] [Google Scholar]
- Eysker M., Kooyman F.N. Notes on necropsy and herbage processing techniques for gastrointestinal nematodes of ruminants. Vet Parasitol. 1993;46:205–213. doi: 10.1016/0304-4017(93)90059-v. [DOI] [PubMed] [Google Scholar]
- Eysker M., Ploeger H.W. Value of present diagnostic methods for gastrointestinal nematode infections in ruminants. Parasitology. 2000;120:S109–S119. doi: 10.1017/s0031182099005752. [DOI] [PubMed] [Google Scholar]
- Fletcher S. Indirect fluorescent antibody technique in the serology of Toxoplasma gondii. J Clin Pathol. 1965;18:193–199. doi: 10.1136/jcp.18.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaba S., Chadoeuf J., Monestiez P., Sauve C., Cortet J., Cabaret J. Estimation of abomasum strongyle nematode infections in sheep at necropsy: tentative proposals for a simplified technique. Vet Parasitol. 2006;140:105–113. doi: 10.1016/j.vetpar.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Gasbarre L.C., Leighton E.A., Davies C.J. Influence of host genetics upon antibody responses against gastrointestinal nematode infections in cattle. Vet Parasitol. 1993;46:81–91. doi: 10.1016/0304-4017(93)90049-s. [DOI] [PubMed] [Google Scholar]
- Gasser R.B. Molecular tools—advances, opportunities and prospects. Vet Parasitol. 2006;136:69–89. doi: 10.1016/j.vetpar.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Gasser R.B., Chilton N.B. Applications of single-strand conformation polymorphism (SSCP) to taxonomy, diagnosis, population genetics and molecular evolution of parasitic nematodes. Vet Parasitol. 2001;101:201–213. doi: 10.1016/s0304-4017(01)00567-2. [DOI] [PubMed] [Google Scholar]
- Gasser R., Hu M., Chilton N., Campbell B., Jex A., Otranto D. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2006;1:3121–3128. doi: 10.1038/nprot.2006.485. [DOI] [PubMed] [Google Scholar]
- Gasser R.B., Bott N.J., Chilton N.B., Hunt P., Beveridge I. Toward practical, DNA-based diagnostic methods for parasitic nematodes of livestock — bionomic and biotechnological implications. Biotechnol Adv. 2008;26:325–334. doi: 10.1016/j.biotechadv.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Geary T.G., Nulf S.C., Favreau M.A., Tang L., Prichard R.K., Hatzenbuhler N.T. Three [beta]-tubulin cDNAs from the parasitic nematode Haemonchus contortus. Mol Biochem Parasitol. 1992;50:295–306. doi: 10.1016/0166-6851(92)90227-b. [DOI] [PubMed] [Google Scholar]
- Georgi J.R., McCulloch C.E. Diagnostic morphometry: identification of helminth eggs by discriminant analysis of morphometric data. Proc Helminthol Soc Wash. 1989;56:44–57. [Google Scholar]
- Gibbons L.M. CAB International; Wallingford: 2010. Keys to the Nematode Parasites of Vertebrates; pp. 83–102. [Google Scholar]
- Gordon H.M. Differential diagnosis of the larvæ of Ostertagia spp. and Trichostrongylus spp. of sheep. Aust Vet J. 1933;9:223–227. [Google Scholar]
- Gordon H.M. The epidemiology of helminthosis in sheep in winter-rainfall regions of Australia. I. Preliminary observations. Aust Vet J. 1953;29:337–348. [Google Scholar]
- Gordon H.M. Some aspects of the control of helminthosis in sheep. Vet Inspector NSW. 1967;31:88–99. [Google Scholar]
- Gordon H.M. Refresher Course for Veterinarians Proceedings No58: Refresher Course on sheep, 10–14 August 1981. University of Sydney: Post-Graduate Committee in Veterinary Science, University of Sydney; 1981. Epidemiology of helminthosis in sheep; pp. 551–566. [Google Scholar]
- Gordon H.M., Whitlock H.V. A new technique for counting nematode eggs in sheep faeces. J CSIR. 1939;12:50–52. [Google Scholar]
- Harmon F.R., Williams Z.B., Zarlenga D.S., Hildreth M.B. Real-time PCR for quantifying Haemonchus contortus eggs and potential limiting factors. Parasitol Res. 2007;101:71–76. doi: 10.1007/s00436-006-0428-0. [DOI] [PubMed] [Google Scholar]
- Harris T., Buzby P., Babcock H., Beer E., Bowers J., Braslavsky I. Single-molecule DNA sequencing of a viral genome. Science. 2008;320:106–109. doi: 10.1126/science.1150427. [DOI] [PubMed] [Google Scholar]
- Heid C.A., Stevens J., Livak K.J., Williams P.M. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Hess M., Sczyrba A., Egan R., Kim T.-W., Chokhawala H., Schroth G. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- Higuchi R., Dollinger G., Walsh P.S., Griffith R. Simultaneous amplification and detection of specific DNA sequences. Biotechnology. 1992;10:413–417. doi: 10.1038/nbt0492-413. [DOI] [PubMed] [Google Scholar]
- Higuchi R., Fockler C., Dollinger G., Watson R. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology. 1993;11:1026–1030. doi: 10.1038/nbt0993-1026. [DOI] [PubMed] [Google Scholar]
- Hoorfar J., Wolffs P., Radström P. Diagnostic PCR: validation and sample preparation are two sides of the same coin. APMIS. 2004;112:808–814. doi: 10.1111/j.1600-0463.2004.apm11211-1207.x. [DOI] [PubMed] [Google Scholar]
- Hoste H., Torres-Acosta J.F. Non chemical control of helminths in ruminants: adapting solutions for changing worms in a changing world. Vet Parasitol. 2011;180:144–154. doi: 10.1016/j.vetpar.2011.05.035. [DOI] [PubMed] [Google Scholar]
- Hu X., Pan C.-W., Li Y.-F., Wang H., Tan F. Urine sample used for detection of Toxoplasma gondii infection by loop-mediated isothermal amplification (LAMP) Folia Parasitol. 2012;59:21–26. doi: 10.14411/fp.2012.004. [DOI] [PubMed] [Google Scholar]
- Hubert J., Kerboeuf D. A new method for culture of larvae used in diagnosis of ruminant gastrointestinal strongylosis: comparison with fecal cultures. Can J Comp Med. 1984;48:63–71. [PMC free article] [PubMed] [Google Scholar]
- Hugenholtz P., Goebel B.M., Pace N.R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung G.C., Gasser R.B., Beveridge I., Chilton N.B. Species-specific amplification by PCR of ribosomal DNA from some equine strongyles. Parasitology. 1999;119:69–80. doi: 10.1017/s0031182099004497. [DOI] [PubMed] [Google Scholar]
- Hungerford T.G. Nineth ed. MacGraw-Hill Medical; Sydney: 1990. Diseases of livestock. [Google Scholar]
- Jeffery N., Gasser R.B., Steer P., Noormohammadi A. Classification of Mycoplasma synoviae strains using single-strand conformation polymorphism and high-resolution melting-curve analysis of the vlhA gene single-copy region. Microbiology. 2007;153:2679–2688. doi: 10.1099/mic.0.2006/005140-0. [DOI] [PubMed] [Google Scholar]
- Johnson M.J., Behnke J.M., Coles G.C. Detection of gastrointestinal nematodes by a coproantigen capture ELISA. Res Vet Sci. 1996;60:7–12. doi: 10.1016/s0034-5288(96)90122-8. [DOI] [PubMed] [Google Scholar]
- Kaminsky R., Ducray P., Jung M., Clover R., Rufener L., Bouvier J. A new class of anthelmintics effective against drug-resistant nematodes. Nature. 2008;452:176–180. doi: 10.1038/nature06722. [DOI] [PubMed] [Google Scholar]
- Kaplan R.M. Drug resistance in nematodes of veterinary importance: a status report. Trends Parasitol. 2004;20:477–481. doi: 10.1016/j.pt.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Kassai T. Butterworth Heinemann; Oxford UK: 1999. Veterinary Helminthology. [Google Scholar]
- Kenyon F., Greer A.W., Coles G.C., Cringoli G., Papadopoulos E., Cabaret J. The role of targeted selective treatments in the development of refugia-based approaches to the control of gastrointestinal nematodes of small ruminants. Vet Parasitol. 2009;164:3–11. doi: 10.1016/j.vetpar.2009.04.015. [DOI] [PubMed] [Google Scholar]
- Kloosterman A., Verhoeff J., Ploeger H.W., Lam T.J.G.M. Antibodies against nematodes in serum, milk and bulk milk samples as possible estimators of infection in dairy cows. Vet Parasitol. 1993;47:267–278. doi: 10.1016/0304-4017(93)90028-l. [DOI] [PubMed] [Google Scholar]
- Kwa M.S.G., Veenstra J.G., Roos M.H. Benzimidazole resistance in Haemonchus contortus is correlated with a conserved mutation at amino acid 200 in beta-tubulin isotype 1. Mol Biochem Parasitol. 1994;63:299–303. doi: 10.1016/0166-6851(94)90066-3. [DOI] [PubMed] [Google Scholar]
- Lamendella R., Domingo J.W.S., Ghosh S., Martinson J., Oerther D. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011;11:103-. doi: 10.1186/1471-2180-11-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster M.B., Hong C. Differentiation of third stage larvae of ‘ovine Ostertagia’ type and Trichostrongylus species. Vet Rec. 1987;120:503. doi: 10.1136/vr.120.21.503. [DOI] [PubMed] [Google Scholar]
- Lane C. The mass diagnosis of ankylostome infestation. (Part I) Trans R Soc Trop Med Hyg. 1922;16:274–315. [Google Scholar]
- Larsen J.W.A., Anderson N., Vizard A.L., Anderson G.A., Hoste H. Diarrhoea in Merino ewes during winter: association with trichostrongylid larvae. Aust Vet J. 1994;71:365–372. doi: 10.1111/j.1751-0813.1994.tb00930.x. [DOI] [PubMed] [Google Scholar]
- Le Jambre L.F., Ractliffe L.H., Uhazy L.S., Whitlock J.H. Fecal egg output of lambs in relationship to Haemonchus contortus burden. Int J Parasitol. 1971;1:157–160. doi: 10.1016/0020-7519(71)90010-5. [DOI] [PubMed] [Google Scholar]
- Le Jambre L.F., Dominik S., Eady S.J., Henshall J.M., Colditz I.G. Adjusting worm egg counts for faecal moisture in sheep. Vet Parasitol. 2007;145:108–115. doi: 10.1016/j.vetpar.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Lepage P., Leclerc M., Joossens M., Mondot S., Blottire H., Raes J. A metagenomic insight into our gut's microbiome. Gut. 2013;62:146–158. doi: 10.1136/gutjnl-2011-301805. [DOI] [PubMed] [Google Scholar]
- Levecke B., Rinaldi L., Charlier J., Maurelli M.P., Bosco A., Vercruysse J. The bias, accuracy and precision of faecal egg count reduction test results in cattle using McMaster, Cornell-Wisconsin and FLOTAC egg counting methods. Vet Parasitol. 2012;188:194–199. doi: 10.1016/j.vetpar.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Levine N.D. Burgess Publishing Company; Minneapolis: 1968. Nematode parasites of domestic animals and of man. [Google Scholar]
- Levine N.D., Mehra K.N., Clark D.T., Aves I.J. A comparison of nematode egg counting techniques for cattle and sheep feces. Am J Vet Res. 1960;21:511–515. [PubMed] [Google Scholar]
- Lichtenfels J.R., Hoberg E.P., Zarlenga D.S. Systematics of gastrointestinal nematodes of domestic ruminants: advances between 1992 and 1995 and proposals for future research. Vet Parasitol. 1997;72:225–245. doi: 10.1016/s0304-4017(97)00099-x. [DOI] [PubMed] [Google Scholar]
- Liu D., Daubendiek S., Zillman M., Ryan K., Kool E. Rolling circle DNA synthesis: small circular oligonucleotides as efficient templates for DNA polymerases. J Am Chem Soc. 1996;118:1587–1594. doi: 10.1021/ja952786k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Geva E., Mauk M., Qiu X., Abrams W., Malamud D. An isothermal amplification reactor with an integrated isolation membrane for point-of-care detection of infectious diseases. Analyst. 2011;136:2069–2076. doi: 10.1039/c1an00007a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A., Guan G., Du P., Liu Z., Gou H., Liu J. Loop-mediated isothermal amplification (LAMP) assays for the detection of Theileria annulata infection in China targeting the 18S rRNA and ITS sequences. Exp Parasitol. 2012;131:125–129. doi: 10.1016/j.exppara.2012.02.012. [DOI] [PubMed] [Google Scholar]
- MAFF . Manual of Veterinary Parasitological Laboratory Techniques. Her Majesty's Stationary Office; London, UK: 1986. pp. 20–27. [Google Scholar]
- Mardis E. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Martin P.J., Anderson N., Jarrett R.G. Resistance to benzimidazole anthelmintics in field strains of Ostertagia and Nematodirus in sheep. Aust Vet J. 1985;62:38–43. doi: 10.1111/j.1751-0813.1985.tb14230.x. [DOI] [PubMed] [Google Scholar]
- McCoy M.A., Edgar H.W., Kenny J., Gordon A.W., Dawson L.E., Carson A.F. Evaluation of on-farm faecal egg counting in sheep. Vet Rec. 2005:21–23. doi: 10.1136/vr.156.1.21. [DOI] [PubMed] [Google Scholar]
- McKenna P.B. The diagnostic value and interpretation of faecal egg counts in sheep. N Z Vet J. 1981;29:129–132. doi: 10.1080/00480169.1981.34821. [DOI] [PubMed] [Google Scholar]
- McKenna P.B. The estimation of gastrointestinal strongyle worm burdens in young sheep flocks: a new approach to the interpretation of faecal egg counts. I. Development. N Z Vet J. 1987;35:94–97. doi: 10.1080/00480169.1987.35396. [DOI] [PubMed] [Google Scholar]
- McKenna P.B. Potential limitations of the undifferentiated faecal egg count reduction test for the detection of anthelmintic resistance in sheep. N Z Vet J. 1996;44:73–75. doi: 10.1080/00480169.1996.35938. [DOI] [PubMed] [Google Scholar]
- McKenna P.B. Further potential limitations of the undifferentiated faecal egg count reduction test for the detection of anthelmintic resistance in sheep. N Z Vet J. 1997;45:244–246. doi: 10.1080/00480169.1997.36038. [DOI] [PubMed] [Google Scholar]
- McKenna P.B. Faecal egg counts as a guide for drench use. N Z Vet J. 2002;50:123–124. doi: 10.1080/00480169.2002.36295. [DOI] [PubMed] [Google Scholar]
- McKenna P.B. Comparison of two worm counting procedures for the enumeration of abomasal and small intestinal nematode parasites of sheep. Vet Parasitol. 2008;157:254–259. doi: 10.1016/j.vetpar.2008.08.003. [DOI] [PubMed] [Google Scholar]
- McKenna P.B., Simpson B.H. The estimation of gastrointestinal strongyle worm burdens in young sheep flocks: a new approach to the interpretation of faecal egg counts. II. Evaluation. N Z Vet J. 1987;35:98–100. doi: 10.1080/00480169.1987.35397. [DOI] [PubMed] [Google Scholar]
- McLaren M.L., Draper C.C., Roberts E., Minter-Goedbloed E., Lighthart G.S., Teesdale C.H. Studies on the enzyme linked immunosorbent assay (ELISA) for Schistosoma mansoni infection. Ann Trop Med Parasitol. 1978:243–253. doi: 10.1080/00034983.1978.11719312. [DOI] [PubMed] [Google Scholar]
- McMurtry L.W., Donaghy M.J., Vlassoff A., Douch P.G.C. Distinguishing morphological features of the third larval stage of ovine Trichostrongylus spp. Vet Parasitol. 2000;90:73–81. doi: 10.1016/s0304-4017(00)00230-2. [DOI] [PubMed] [Google Scholar]
- Mes T.H.M., Ploeger H.W., Terlou M., Kooyman F.N., Van der Ploeg M.P., Eysker M. A novel method for the isolation of gastro-intestinal nematode eggs that allows automated analysis of digital images of egg preparations and high throughput screening. Parasitology. 2001;123:309–314. doi: 10.1017/s0031182001008496. [DOI] [PubMed] [Google Scholar]
- Mes T.H.M., Eysker M., Ploeger H.W. A simple, robust and semi-automated parasite egg isolation protocol. Nat Protoc. 2007;2:486–489. doi: 10.1038/nprot.2007.56. [DOI] [PubMed] [Google Scholar]
- Monis P.T., Giglio S., Keegan A.R., Thompson R.C.A. Emerging technologies for the detection and genetic characterization of protozoan parasites. Trends Parasitol. 2005;21:340–346. doi: 10.1016/j.pt.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Monis P.T., Giglio S., Saint C.P. Comparison of SYTO9 and SYBR Green I for real-time polymerase chain reaction and investigation of the effect of dye concentration on amplification and DNA melting curve analysis. Anal Biochem. 2005;340:24–34. doi: 10.1016/j.ab.2005.01.046. [DOI] [PubMed] [Google Scholar]
- Mullis K., Faloona F., Scharf S., Saiki R., Horn G., Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol. 1986;51:263–273. doi: 10.1101/sqb.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- Nicholls J., Obendorf D.L. Application of a composite faecal egg count procedure in diagnostic parasitology. Vet Parasitol. 1994;52:337–342. doi: 10.1016/0304-4017(94)90125-2. [DOI] [PubMed] [Google Scholar]
- Nielsen M.K., Peterson D.S., Monrad J., Thamsborg S.M., Olsen S.N., Kaplan R.M. Detection and semi-quantification of Strongylus vulgaris DNA in equine faeces by real-time quantitative PCR. Int J Parasitol. 2008;38:443–453. doi: 10.1016/j.ijpara.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Nielsen M.K., Vidyashankar A.N., Andersen U.V., DeLisi K., Pilegaard K., Kaplan R.M. Effects of fecal collection and storage factors on strongylid egg counts in horses. Vet Parasitol. 2010;167:55–61. doi: 10.1016/j.vetpar.2009.09.043. [DOI] [PubMed] [Google Scholar]
- Ninove L., Nougairede A., Gazin C., Thirion L., Delogu I., Zandotti C. RNA and DNA bacteriophages as molecular diagnosis controls in clinical virology: a comprehensive study of more than 45,000 routine PCR tests. PLoS One. 2011;6:e16142. doi: 10.1371/journal.pone.0016142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkouawa A., Sako Y., Li T., Chen X., Wandra T., Swastika I.K. Evaluation of a loop-mediated isothermal amplification method using fecal specimens for differential detection of Taenia species from humans. J Clin Microbiol. 2010;48:3350–3352. doi: 10.1128/JCM.00697-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordin R., Smith H.V., Mohamad S., Maizels R.M., Fong M.Y. Comparison of IgG-ELISA and IgG4-ELISA for Toxocara serodiagnosis. Acta Trop. 2005;93:57–62. doi: 10.1016/j.actatropica.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Notomi T., Okayama H., Masubuchi H., Yonekawa T., Watanabe K., Amino N. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:E63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid P., Christopoulos T., Crabtree H.J., Backhouse C. Microfabricated device for DNA and RNA amplification by continuous-flow polymerase chain reaction and reverse transcription-polymerase chain reaction with cycle number selection. Anal Chem. 2003;75:288–295. doi: 10.1021/ac0260239. [DOI] [PubMed] [Google Scholar]
- O'Connor L.J., Walkden-Brown S.W., Kahn L.P. Ecology of the free-living stages of major trichostrongylid parasites of sheep. Vet Parasitol. 2006;142:1–15. doi: 10.1016/j.vetpar.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Office International des Epizooties (OIE) OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. 2004. Quality Management in Veterinary Testing Laboratories. Paris, France. [Google Scholar]
- Ogunremi O., Halbert G., Mainar Jaime R., Benjamin J., Pfister K. Accuracy of an indirect fluorescent-antibody test and of a complement-fixation test for the diagnosis of Babesia caballi in field samples from horses. Prev Vet Med. 2008;83:41–51. doi: 10.1016/j.prevetmed.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Palmer D.G., McCombe I.L. Lectin staining of trichostrongylid nematode eggs of sheep: rapid identification of Haemonchus contortus eggs with peanut agglutinin. Int J Parasitol. 1996;26:447–450. doi: 10.1016/0020-7519(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Pfeiffer D.U. Wiley-Blackwell; Oxford, UK: 2010. Veterinary Epidemiology: An Introduction. [Google Scholar]
- Piatek A.S., Tyagi S., Pol A.C., Telenti A., Miller L.P., Kramer F.R. Molecular beacon sequence analysis for detecting drug resistance in Mycobacterium tuberculosis. Nat Biotechnol. 1998;16:359–363. doi: 10.1038/nbt0498-359. [DOI] [PubMed] [Google Scholar]
- Pritchard G. Milk antibody testing in cattle. In Pract. 2001;23:542–549. [Google Scholar]
- Pushkarev D., Neff N., Quake S. Single-molecule sequencing of an individual human genome. Nat Biotechnol. 2009;27:847–850. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J., Li R., Raes J., Arumugam M., Burgdorf K., Manichanh C. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rådström P., Knutsson R., Wolffs P., Lövenklev M., Löfström C. Pre-PCR processing: strategies to generate PCR-compatible samples. Mol Biotechnol. 2004;26:133–146. doi: 10.1385/MB:26:2:133. [DOI] [PubMed] [Google Scholar]
- Raynaud J.P. Etude de l'efficacité d'une technique de coproscopie quantitative pour le diagnostic de routine et le controle des infestations parasitaires des bovins, ovins, equins et porcins. Ann Parasitol Hum Comp. 1970;45:321–342. [PubMed] [Google Scholar]
- Rinaldi L., Coles G.C., Maurelli M.P., Musella V., Cringoli G. Calibration and diagnostic accuracy of simple flotation, McMaster and FLOTAC for parasite egg counts in sheep. Vet Parasitol. 2011;177:345–352. doi: 10.1016/j.vetpar.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Roberts F.H.S., O'Sullivan P.J. Methods for egg counts and larval cultures for strongyles infesting the gastro-intestinal tract of cattle. Aust J Agric Res. 1950;1:99–102. [Google Scholar]
- Roberts J.L., Swan R.A. Quantitative studies of ovine haemonchosis. I. Relationship between faecal egg counts and total worm counts. Vet Parasitol. 1981;8:165–171. [PubMed] [Google Scholar]
- Robertson T.G., Elliott D.C. The laboratory assessment of worm parasite populations in sheep. N Z J Agric Res. 1966;9:350–358. [Google Scholar]
- Roeber F., Jex A.R., Campbell A.J.D., Campbell B.E., Anderson G.A., Gasser R.B. Evaluation and application of a molecular method to assess the composition of strongylid nematode populations in sheep with naturally acquired infections. Infect Genet Evol. 2011;11:849–854. doi: 10.1016/j.meegid.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Roeber F., Jex A., Campbell A.J.D., Nielsen R., Anderson G., Stanley K. Establishment of a robotic, high-throughput platform for the specific diagnosis of gastrointestinal nematode infections in sheep. Int J Parasitol. 2012;42:1151–1158. doi: 10.1016/j.ijpara.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Roeber F., Larsen J.W.A., Anderson N., Campbell A.J.D., Anderson G.A., Gasser R.B. A molecular diagnostic tool to replace larval culture in conventional faecal egg count reduction testing in sheep. PLoS One. 2012;7:e37327. doi: 10.1371/journal.pone.0037327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos M.H., Grant W.N. Species-specific PCR for the parasitic nematodes Haemonchus contortus and Trichostrongylus colubriformis. Int J Parasitol. 1993;23:419–421. doi: 10.1016/0020-7519(93)90021-p. [DOI] [PubMed] [Google Scholar]
- Rothberg J., Hinz W., Rearick T., Schultz J., Mileski W., Davey M. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- Rowe A., McMaster K., Emery D., Sangster N. Haemonchus contortus infection in sheep: parasite fecundity correlates with worm size and host lymphocyte counts. Vet Parasitol. 2008;153:285–293. doi: 10.1016/j.vetpar.2008.01.040. [DOI] [PubMed] [Google Scholar]
- Russel A.J.F., Doney J.M., Gunn R.G. Subjective assessment of body fat in live sheep. J Agric Sci. 1969;72:451–454. [Google Scholar]
- Saiki R.K., Gyllensten U.B., Erlich H.A. IRL Press; Oxford UK: 1988. The Polymerase Chain Reaction. [Google Scholar]
- Sanchez J., Markham F., Dohoo I., Sheppard J., Keefe G., Leslie K. Milk antibodies against Ostertagia ostertagi: Relationships with milk IgG and production parameters in lactating dairy cattle. Vet Parasitol. 2004;120:319–330. doi: 10.1016/j.vetpar.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Schneider G., Dekker C. DNA sequencing with nanopores. Nat Biotechnol. 2012;30:326–328. doi: 10.1038/nbt.2181. [DOI] [PubMed] [Google Scholar]
- Seise B., Brinker A., Kretschmer R., Schwarz M., Rudolph B., Kaulfuß T. Chip-based detection system for the on-site analysis of animal diseases. Eng Life Sci. 2011;11:148–156. [Google Scholar]
- Silvestre A., Humbert J.F. A molecular tool for species identification and benzimidazole resistance diagnosis in larval communities of small ruminant parasites. Exp Parasitol. 2000;95:271–276. doi: 10.1006/expr.2000.4542. [DOI] [PubMed] [Google Scholar]
- Silvestre A., Humbert J.F. Diversity of benzimidazole-resistance alleles in populations of small ruminant parasites. Int J Parasitol. 2002;32:921–928. doi: 10.1016/s0020-7519(02)00032-2. [DOI] [PubMed] [Google Scholar]
- Sirichaisinthop J., Buates S., Watanabe R., Han E.-T., Suktawonjaroenpon W., Krasaesub S. Evaluation of loop-mediated isothermal amplification (LAMP) for malaria diagnosis in a field setting. Am J Trop Med Hyg. 2011;85:594–596. doi: 10.4269/ajtmh.2011.10-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C. Digital image analysis and identification of eggs from bovine parasitic nematodes. J Helminthol. 1996;70:143–151. doi: 10.1017/s0022149x00015303. [DOI] [PubMed] [Google Scholar]
- Stanley K., Szewczuk E. Multiplexed tandem PCR: gene profiling from small amounts of RNA using SYBR Green detection. Nucleic Acids Res. 2005;33:e180-e. doi: 10.1093/nar/gni182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stear M.J., Bishop S.C. The curvilinear relationship between worm length and fecundity of Teladorsagia circumcincta. Int J Parasitol. 1999;29:777–780. doi: 10.1016/s0020-7519(99)00019-3. [DOI] [PubMed] [Google Scholar]
- Stear M.J., Bishop S.C., Henderson N.G., Scott I. A key mechanism of pathogenesis in sheep infected with the nematode Teladorsagia circumcincta. Anim Health Res Rev. 2003;4:45–52. doi: 10.1079/ahrr200351. [DOI] [PubMed] [Google Scholar]
- Stevenson L.A., Gasser R.B., Chilton N.B. The ITS-2 rDNA of Teladorsagia circumcincta, T. trifurcata and T. davtiani (Nematoda: Trichostrongylidae) indicates that these taxa are one species. Int J Parasitol. 1996;26:1123–1126. [PubMed] [Google Scholar]
- Stoll N.R. Investigations on the control of hookworm disease. XV. An effective method of counting hookworm eggs in faeces. Am J Epidemiol. 1923;3:59–70. [Google Scholar]
- Takagi H., Itoh M., Kasai S., Yahathugoda T., Weerasooriya M., Kimura E. Development of loop-mediated isothermal amplification method for detecting Wuchereria bancrofti DNA in human blood and vector mosquitoes. Parasitol Int. 2011;60:493–497. doi: 10.1016/j.parint.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Taylor D.M., Thomas R.J. The development of immunity to Nematodirus battus in lambs. Int J Parasitol. 1986;16:43–46. doi: 10.1016/0020-7519(86)90063-9. [DOI] [PubMed] [Google Scholar]
- Taylor M.A., Hunt K.R., Goodyear K.L. Anthelmintic resistance detection methods. Vet Parasitol. 2002;103:183–194. doi: 10.1016/s0304-4017(01)00604-5. [DOI] [PubMed] [Google Scholar]
- Taylor M.A., Coop R.L., Wall R.L. 3rd ed. Blackwell Publishing; Oxford: 2007. Veterinary Parasitology. [Google Scholar]
- Taylor M.A., Learmount J., Lunn E., Morgan C., Craig B.H. Multiple resistance to anthelmintics in sheep nematodes and comparison of methods used for their detection. Small Rumin Res. 2009;86:67–70. [Google Scholar]
- Thienpont D., Rochette F., Vanparijs O.F.J. 2nd ed. Janssen Research Foundation; Beerse, Belgium: 1986. Diagnosing Helminthiasis by Coprological Examination. [Google Scholar]
- Thrusfield M.V. 3rd ed. Blackwell Science Ltd.; Oxford UK: 2005. Veterinary epidemiology. [Google Scholar]
- Tyson G., Chapman J., Hugenholtz P., Allen E., Ram R., Richardson P. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- van Lieshout L., Verweij J. Newer diagnostic approaches to intestinal protozoa. Curr Opin Infect Dis. 2010;23:488–493. doi: 10.1097/QCO.0b013e32833de0eb. [DOI] [PubMed] [Google Scholar]
- van Wyk J.A., Bath G.F. The FAMACHA system for managing haemonchosis in sheep and goats by clinically identifying individual animals for treatment. Vet Res. 2002;33:509–529. doi: 10.1051/vetres:2002036. [DOI] [PubMed] [Google Scholar]
- van Wyk J.A., Cabaret J., Michael L.M. Morphological identification of nematode larvae of small ruminants and cattle simplified. Vet Parasitol. 2004;119:277–306. doi: 10.1016/j.vetpar.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Venkatesan B., Bashir R. Nanopore sensors for nucleic acid analysis. Nat Nanotechnol. 2011;6:615–624. doi: 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- Venter J.C., Remington K., Heidelberg J., Halpern A., Rusch D., Eisen J. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Verweij J.J., Brienen E.A.T., Ziem J., Yelifari L., Polderman A.M., Van Lieshout L. Simultaneous detection and quantification of Ancylostoma duodenale, Necator americanus, and Oesophagostomum bifurcum in fecal samples using multiplex real-time PCR. Am J Trop Med Hyg. 2007;77:685–690. [PubMed] [Google Scholar]
- Villanua D., Perez-Rodriguez L., Gortazar C., Hofle U., Vinuela J. Avoiding bias in parasite excretion estimates: the effect of sampling time and type of faeces. Parasitology. 2006;133:251–259. doi: 10.1017/S003118200600031X. [DOI] [PubMed] [Google Scholar]
- Vincent M., Xu Y., Kong H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004;5:795–800. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Samson-Himmelstjerna G. Molecular diagnosis of anthelmintic resistance. Vet Parasitol. 2006;136:99–107. doi: 10.1016/j.vetpar.2005.12.005. [DOI] [PubMed] [Google Scholar]
- von Samson-Himmelstjerna G., Harder A., Schnieder T. Quantitative analysis of ITS2 sequences in trichostrongyle parasites. Int J Parasitol. 2002;32:1529–1535. doi: 10.1016/s0020-7519(02)00163-7. [DOI] [PubMed] [Google Scholar]
- von Samson-Himmelstjerna G., Buschbaum S., Wirtherle N., Pape M., Schnieder T. TaqMan minor groove binder real-time PCR analysis of beta-tubulin codon 200 polymorphism in small strongyles (Cyathostomin) indicates that the TAC allele is only moderately selected in benzimidazole-resistant populations. Parasitology. 2003;127:489–496. doi: 10.1017/s0031182003003974. [DOI] [PubMed] [Google Scholar]
- von Samson-Himmelstjerna G., Coles G., Jackson F., Bauer C., Borgsteede F., Cirak V. Standardization of the egg hatch test for the detection of benzimidazole resistance in parasitic nematodes. Parasitol Res. 2009;105:825–834. doi: 10.1007/s00436-009-1466-1. [DOI] [PubMed] [Google Scholar]
- Walker G.T., Fraiser M.S., Schram J.L., Little M.C., Nadeau J.G., Malinowski D.P. Strand displacement amplification — an isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992;20:1691–1696. doi: 10.1093/nar/20.7.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker G.T., Little M.C., Nadeau J.G., Shank D.D. Isothermal in vitro amplification of DNA by a restriction enzyme/DNA polymerase system. Proc Natl Acad Sci U S A. 1992;89:392–396. doi: 10.1073/pnas.89.1.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller P.J. The development of anthelmintic resistance in ruminant livestock. Acta Trop. 1994;56:233–243. doi: 10.1016/0001-706x(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Waller P.J. Anthelmintic resistance. Vet Parasitol. 1997;72:391–412. doi: 10.1016/s0304-4017(97)00107-6. [DOI] [PubMed] [Google Scholar]
- Waller P.J., Donald A.D., Dobson R.J., Lacey E., Hennessy D.R., Allerton G.R. Changes in anthelmintic resistance status of Haemonchus contortus and Trichostrongylus colubriformis exposed to different anthelmintic selection pressures in grazing sheep. Int J Parasitol. 1989;19:99–110. doi: 10.1016/0020-7519(89)90027-1. [DOI] [PubMed] [Google Scholar]
- Wang W., Chen K., Xu C. DNA quantification using EvaGreen and a real-time PCR instrument. Anal Biochem. 2006;356:303–305. doi: 10.1016/j.ab.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Whitlock H.V. A method for preventing the development of strongylid eggs in sheep faeces during transport and storage. J CSIR. 1943;16:215–216. [Google Scholar]
- Whitlock H.V. Some modifications of the McMaster helminth egg-counting technique and apparatus. J CSIR. 1948;21:177–180. [Google Scholar]
- Whitlock H.V. An improved method for the culture of nematode larvae in sheep faeces. Aust Vet J. 1956;32:141–143. [Google Scholar]
- Whitlock H.V. The recovery and identification of the first stage larvae of sheep nematodes. Aust Vet J. 1959;35:310–316. [Google Scholar]
- Williams J.F., Soulsby E.J.L. Antigenic analysis of developmental stages of Ascaris suum. I. Comparison of eggs, larvae and adults. Exp Parasitol. 1970;27:150–162. doi: 10.1016/s0014-4894(70)80019-4. [DOI] [PubMed] [Google Scholar]
- Wilson I.G. Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol. 1997;63:3741–3751. doi: 10.1128/aem.63.10.3741-3751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer B., Craig B.H., Pilkington J.G., Pemberton J.M. Non-invasive assessment of parasitic nematode species diversity in wild Soay sheep using molecular markers. Int J Parasitol. 2004;34:625–631. doi: 10.1016/j.ijpara.2003.11.022. [DOI] [PubMed] [Google Scholar]
- Wittwer C.T., Reed G.H., Gundry C.N., Vandersteen J.G., Pryor R.J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- Wolstenholme A.J., Fairweather I., Prichard R., von Samson-Himmelstjerna G., Sangster N.C. Drug resistance in veterinary helminths. Trends Parasitol. 2004;20:469–476. doi: 10.1016/j.pt.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Woolaston R.R. Selection of Merino sheep for increased and decreased resistance to Haemonchus contortus: peri-parturient effects on faecal egg counts. Int J Parasitol. 1992;22:947–953. doi: 10.1016/0020-7519(92)90052-m. [DOI] [PubMed] [Google Scholar]
- Yooseph S., Nealson K., Rusch D., McCrow J., Dupont C., Kim M. Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature. 2010;468:60–66. doi: 10.1038/nature09530. [DOI] [PubMed] [Google Scholar]
- Zajac A.M. Gastrointestinal nematodes of small ruminants: life cycle, anthelmintics, and diagnosis. Vet Clin N Am Food Anim Pract. 2006;22:529–541. doi: 10.1016/j.cvfa.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Zarlenga D.S., Higgins J. PCR as a diagnostic and quantitative technique in veterinary parasitology. Vet Parasitol. 2001;101:215–230. doi: 10.1016/s0304-4017(01)00568-4. [DOI] [PubMed] [Google Scholar]
- Zarlenga D.S., Stringfellow F., Nobary M., Lichtenfels J.R. Cloning and characterization of ribosomal RNA genes from three species of Haemonchus (Nematoda: Trichostrongyloidea) and identification of PCR primers for rapid differentiation. Exp Parasitol. 1994;78:28–36. doi: 10.1006/expr.1994.1003. [DOI] [PubMed] [Google Scholar]
- Zarlenga D.S., Gasbarre L.C., Boyd P., Leighton E., Lichtenfels J.R. Identification and semi-quantitation of Ostertagia ostertagi eggs by enzymatic amplification of ITS-1 sequences. Vet Parasitol. 1998;77:245–257. doi: 10.1016/s0304-4017(98)00114-9. [DOI] [PubMed] [Google Scholar]
- Zarlenga D.S., Hoberg E.P., Stringfellow F., Lichtenfels J.R. Comparisons of two polymorphic species of Ostertagia and phylogenetic relationships within the Ostertagiinae (Nematoda: Trichostrongyloidea) inferred from ribosomal DNA repeat and mitochondrial DNA sequences. J Parasitol. 1998;84:806–812. [PubMed] [Google Scholar]