

Abstract
Latex agglutination of the glucose-modified latex, which were synthesized by emulsion copolymerization of allyl-modified glucose and styrene monomers by specific binding interactions between Concanavalin A (Con A) and allyl-α-d-glucopyranose, were investigated. The surface of the glucose-modified latex was characterized by dye-partition method and the number of glucose was 1517 per latex particle. The average particle size and the polydispersity index of the latex were 78.3 and 1.005, respectively. Time-evolution adsorption behavior of various concentrations of the Con A and the consequent latex agglutination were studied by UV spectrophotometer at 540 nm and ζ-potential analyzer at the fixed latex concentration of 0.02 wt%. Specific binding between Con A and allyl-α-d-glucopyranose leaded the latex particles to coagulate by decreasing the electrostatic repulsion between the particles and mobility.
Keywords: Polymer colloid, Biosensor, Glucose, Concanavalin A, Binding, Agglutination
1. Introduction
The role of carbohydrate ligands, including immune response, signal transduction, inflammatory processes, and metastasis, has been thought to be restricted to the proteins that are confined to cell surface or within lumen of intracellular organelles [1], [2], [3], [4], [5]. Therefore, development of synthetic biomaterials has aimed to control the interactions with bio-molecules and living cells by mimicking biological surface [6], [7]. Among the biomaterials, membranes have been a center of the various functions delimiting living cells. The membranes play a role in the interactions involving cell-surface glycoprotein, which mediate many biological communications and recognition processes [8].
Recently, there have been various works related to the applications aforementioned by using polymer colloids, since they possess extremely large surface area to guarantee high sensitivity. Colloid-based separation and sensor engineering seems to be very effective, for example, purification of specific protein from a crude biological mixture by either electrostatic interaction or specific ligand–protein interactions, antigen–antibody, DNA–complimentary DNA, and carbohydrate–carbohydrate binding protein [9], [10], [11], [12], [13], [14], [15], [16]. Chern et al. reported adsorption of Concanavalin A (Con A) onto dextran (glucose)-modified PMMA latex particles and charge neutralization due to Con A adsorption [17], [18]; however, the interaction between Con A and the glucose-modified PMMA latex is believed to be complicated and unclear due to the immobilization of glucose done by mere physical absorption. On the other hand, Koch and Yaacoub studied kinetics of emulsion polymerization of sugar-modified monomer, 3-O-methacryloyl-1,2:5,6-di-O-isopropylidene-α-d-glucofuranose, and properties of the sugar latex [19] where the sugar could be chemically adsorbed on to the latex surface and avoid any complex effect of dissolved glucose solution to Con A. As mentioned above, carbohydrate-based biosensors can be utilized to detect severe acute respiratory syndrome (SARS) viruses [20]. Avian influenza viruses can bind to cell-surface of the glycoproteins or glycolipds containing carbohydrate, which can be similarly recognized by detection of specific proteins with sugar-modified latex.
In this work, glucose-modified monomer was synthesized with allyl alcohol and the glucose-modified latex was prepared by emulsion copolymerization of styrene with glucose-modified monomer and small amount of sodium styrene sulfonate (NaSS). For individual control of particle size and glucose concentration on the latex surface, two-stage shot-growth method was introduced. Adsorption of the Con A onto the glucose-modified latex was observed in the phosphate buffered saline (PBS). For the better understanding of adsorption kinetics and latex agglutination induced by the interaction between allyl-α-d-glucopyranose and Con A, time-evolution adsorption isotherm and Fuchs stability ratio (W) were measured with varying Con A concentration.
2. Experimentals
2.1. Materials
Glucose (Duksan, Korea), acetic anhydride (Duksan, Korea), boron trifluoride diethyl etherate (Aldrich, USA), and sodium methoxide powder (Aldrich, USA) were used as received. Styrene (Junsei, Japan) was purified by distillation under reduced pressure and kept at −5 °C. Sodium styrene sulfonate (TCI, Japan), potassium persulfate (KPS, Junsei, Japan), and sodium lauryl sulfate (SLS, Ducksan, Korea) were used as received. Concanavalin A (Sigma, USA), calcium chloride (Duksan, Korea), manganous chloride (Duksan, Korea), sodium phosphate, and monobasic monohydrate (J.T. Baker, USA) were used as received. Distilled and deionized (DI) water was used all through the reaction.
2.2. Preparation of allyl-α-d-glucopyranose monomer
For the glucose-modified latex, allyl-α-d-glucopyranose monomer was synthesized by using stepwise reactions as given in Scheme 1 [21]. The detailed procedure is as follows: 2 g glucose was dissolved in a mixture of 17.4 g pyridine and 11.3 g acetic anhydride. The mixture was stirred at room temperature for 6 h to obtain a clear solution. Afterwards, the reaction mixture was poured into the iced water, extracted with ethyl acetate, and washed with a mixture solution of 1N hydrochloric acid, aqueous sodium chloride, and water, and then dried over magnesium sulfate anhydrous. After washing, it was filtered to remove magnesium sulfate and volatile solvent was evaporated under reduced pressure. As a result, α,β-mixture of peracetylated glucose was quantitative. Afterwards, 1 g peracetylated glucose was dissolved in 4 mL dry dichloromethane with 1.48 g allyl alcohol, and then 0.72 g boron trifluoride etherate was added to the round flask. The reaction was continued at 40 °C until the reaction completed, which was confirmed by thin layer chromatography (hexane:ethyl acetate = 5:4). The collected mixture was poured into the water, and extracted with dichloromethane. The collected dichloromethane solutions were washed several times with aqueous sodium hydrogen carbonate, dried, filtered, and then evaporated under reduced pressure. The product was fractionated by using chromatographic method (SiO2; hexane:ethyl acetate = 5:4) to obtain allyl 2,3,4,6-tetra-O-acetyl-α-d-glucopyranose as a syrup. This product was dissolved in methanol with sodium methoxide, and stirred, until the starting material had been consumed (t.l.c; ethyl acetate). Finally, the product was neutralized with Amberlite IR-H+ resin, filtered, and evaporated under reduced pressure.
Scheme 1.
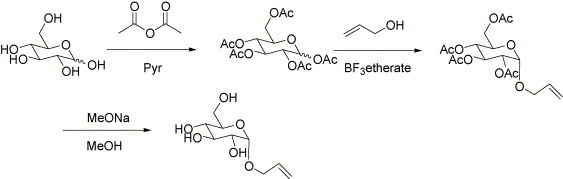
Synthetic procedure for glucose-modified monomer.
2.3. Preparation of poly(allyl-α-d-glucopyranose/styrene) latex
Poly(allyl-α-d-glucopyranose/styrene) latex was prepared by using emulsion polymerization. For the introduction of allyl-α-d-glucopyranose onto the latex surface, two-stage shot-growth method was applied [22], [23]. A 100-mL glass reactor was charged with 24 g of degassed DI water. A required amount of SLS was added with stirring and the solution was purged with nitrogen gas for 1 h. The mixture was heated to 75 °C. Two grams of styrene and 0.02 g NaSS were added under constant stirring. Then 0.04 g of KPS solution was injected for the initiation. At 90–95% monomer conversion, a calculated amount of allyl-α-d-glucopyranose solution (0.03 g) was injected. After 6 h polymerization, poly(allyl-α-d-glucopyranose/styrene) latex with 6.83 wt% solid was obtained. Final monomer conversion was 93%.
2.4. Binding reaction of poly(allyl-α-d-glucopyranose/styrene) latex with Con A
The poly(allyl-α-d-glucopyranose/styrene) latex was cleaned using dialysis tubing (Cellu Sep T2, membrane filtration products, USA) for 1 week and diluted with DI water to fix the solid content at 0.5 wt%. Then, the diluted latex was poured into the phosphate buffer solution (8 g NaCl, 0.2 g KCl, and 1.44 g Na2HPO4 are dissolved in 1 L water; pH 7.1) and the final solid content of the latex became 0.02 wt%. Afterwards, 200 μL of 1 mM CaCl2 and MnCl2 solution were added to the 2 mL latex. The prepared latex was mixed with the Con A solution of various concentrations from 1 to 20 μg/mL. The binding between Con A and the latex was determined quantitatively by using UV-spectrophotometer (UV-1601PC, Japan, 540 nm) and ζ-potential analyzer (Zeta-sizer 3000HSA, USA).
3. Results and discussion
3.1. Surface analysis of the poly(allyl-α-d-glucopyranose/styrene) latex
The glucose content of the poly(allyl-α-d-glucopyranose/styrene) latex was quantitatively determined by measuring UV absorbance at 485 nm after color development with phenol–sulfuric acid reaction (see Scheme 2 ). This reaction manifests the amount of glucose immobilized the surface of colloids, so-called “dye-partition” introduced by Palit and co-workers [24], [25], [26], which was used for end-group determination [27]. Calibration function for the UV absorption was expressed as follows:
| (1) |
The extraction process involving polymers is represented by the equilibrium. The equation that quantitatively describes dye extraction is as follows:
| (2) |
where A o is the absorbance of the organic phase in a cell of optical path length l, C the polymer concentration (the molar absorbance of the dye–polymer pair). The equilibrium constants, K E, for dye extraction were calculated from the slope and intercepts of the plot of 1/A o versus 1/[phenol]w. The total mole of hydroxyl group was calculated by Eq. (2). The molar absorbance of dye–polymer pair was 15,635 L mol−1 cm−1. The glucose-modified latexes were calculated concentration of hydroxyl group (2.55 × 10−7 mol) by slope (see Fig. 1 ). The attached glucose on the surface a colloid based on the particle number density (unit/mL) [28]. The average particle sizes and particle size distributions were measured by both scanning electron microscopy (SEM) and dynamic light scattering (DLS). The particle number (N P) was calculated from the following equation:
| (3) |
where m is the weight of glucose-modified colloids per unit volume of the latex, D n the number-average particle size, and ρ P is the density of the polymer. The values were calculated by Eq. (3) and given in Table 1 . The glucose-modified latexes were compared with the control latex that was polymerized in the absence of allyl-α-d-glucopyranose at the same condition. The UV absorbance value of the glucose-modified latex was corrected with respect to that of the control latex, since the sulfate (SO4 −) group onto the latex surface also participates in the reaction with phenol–sulfuric acid.
Scheme 2.

Color development with phenol–sulfuric acid reaction.
Fig. 1.
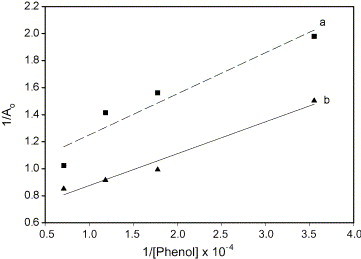
Concentration of phenol vs. absorbance at 0.5% solid contents: (a) poly(allyl-α-d-glucopyranose-styrene) latex and (b) control.
Table 1.
Properties of control and poly(allyl-α-d-glucopyranose/styrene) latexes
| Sample | a (nm) | PDIb | Glucose density (unit particle) | Particle number (mL−1) |
|---|---|---|---|---|
| Control latex | 94.4 | 1.003 | – | 1.09 × 1013 |
| Glucose-modified latex | 78.3 | 1.005 | 1517 | 2.13 × 1013 |
The number-average particle sizes were determined by SEM.
PDI: polydisperse index = .
3.2. Colloidal stability of the poly(allyl-α-d-glucopyranose/styrene) latex
The colloidal stability of the poly(allyl-α-d-glucopyranose/styrene) latex was observed and compared with control latex. Colloidal instability of the latex was observed when the latex was diluted with PBS solution due to the presence of salts. As seen in Fig. 2(b), the SEM photo shows the coagulation of colloids as compared with Fig. 2(a). When Con A concentration was 0.1 and 0.4 μg/mL Con A (50 μL) to the above 0.02 wt% (0.5 mL) mixture, the mixtures increased the degree of coagulation. Scheme 3 shows the schematic agglutination process due to the interaction between the glucose-modified latex and Con A. As seen in Scheme 3, Con A molecules in the PBS solution are forming tetramer structure at pH 7.2. Therefore, this Con A aggregate has at least four binding sites to coagulate the glucose-modified latex. Fig. 2 shows the effect of Con A concentration on the colloid stability of the latex, after incubating and evaporating for 6 h. There is a stability on the latex with various concentrations of the Con A which was directly observed by ζ-potential values. The Con A concentration decreased the stability of colloids because of the attachment of the Con A onto the surface of colloids. Decrease in the stability of colloids may be due to the charge neutralization of these negatively charged colloids induced by adsorption of the charged Con A. At pH 7.2, however, Con A is the isoelectric point (pI), i.e. electrically neutral. Therefore, this suggests that the coagulation of the latex particle should be achieved not by charge neutralization but by binding of Con A among the latex particles containing allyl-α-d-glucopyranose. As can be seen in Fig. 3 , ζ-potential of the latex decreased as the concentration of Con A increased. This can be explained by the decrease in mobility due to the attachment of Con A, which shows electrically neutral and large size, ca. 3–6 nm [29]. When the glucose-modified latex particle meets Con A, Con A binds with allyl-α-d-glucopyranose and the latex would be covered with Con A, so particle becomes bigger and charge density decreased consequently. Aforementioned, these ζ data imply that the coagulation kinetics is predominantly governed by the decrease in the electrical repulsion due to binding of Con A and followed by the agglutination of Con A-binded latex particles, whose behavior is totally different with the case of salt addition, as seen in Fig. 2(b and c).
Fig. 2.

SEM images of poly(allyl-α-d-glucopyranose-styrene) latex after incubating and evaporating room temperature for 6 h: (a) 0.02% solid content, (b) 0.02% solid content with PBS and metal salts, (c) added 0.1 μg/mL Con A, and (d) added 0.4 μg/mL Con A.
Scheme 3.
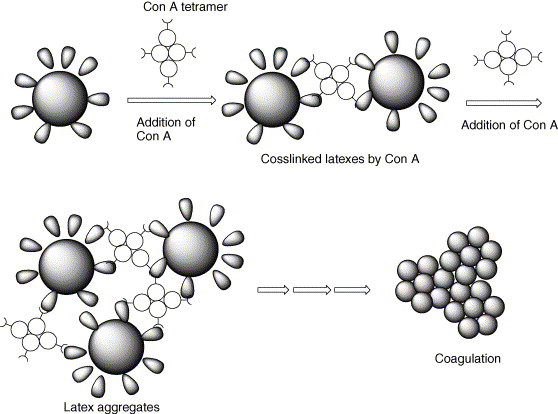
Coagulation mechanism of glucose-modified latexes in the presence of Con A tetramer at pH 7.2.
Fig. 3.
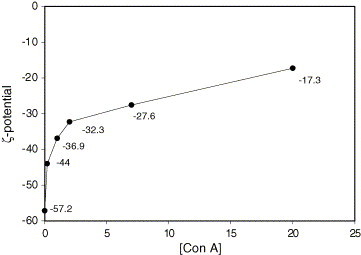
ζ-Potential data for 0.02% solid content colloids with various concentrations of Con A, pH 7.2 phosphate solution (0.1 mM Ca2+ and Mn2+) and deionized water.
3.3. Detection of Con A by the immobilized probe on the surface of colloids
UV-spectrophotometer is a powerful tool for qualitative and quantitative analysis. Analysis of the above stability is the best method for detecting the attached Con A. The stability of colloid was influenced by synergetic effects of electrostatic mechanisms. Con A diffused among of latex was observed by absorbance at 540 nm. The diffusion rate of Con A was measured by UV-intensity as a function of time. Fig. 4 shows that Con A was dispersed in 0.02 wt% solid contents of the colloid with various concentrations. Its data was a general feature of the kinetic of coagulation. This result indicates that the dimension of the coagulated colloids continues to build up as the precipitation process proceeds. According to Fuchs, the stability ratio (W) is defined as the ratio K R/K S, where K R and K S are the fast coagulation and slow coagulation rate constants, respectively. As the method of Chern's analysis [17], the value of W for the colloids sample at concentration of Con A = 1 μg/mL can be estimated as the ratio of the asymptotic slope of the absorbance versus time curve at t = 0 (initial slope as concentration of Con A → 20 μg/mL) to the slope of the absorbance versus time curve at t = 0 (initial slope at concentration of Con A → 1 μg/mL). This is because K R or K S is proportional to the initial slope of the absorbance versus time curve. The log W versus log C (concentration of Con A) diagram for the modified colloids is shown in Fig. 5 . The inflection point in the log W versus log C diagram is identified as the critical coagulation concentration of glucose-modified latex with addition of Con A. The formation of a cross-linked network structure by coagulation between the tetra-Con A and glucose active sites absorbed latex enhances the coagulation rate [30], [31].
Fig. 4.
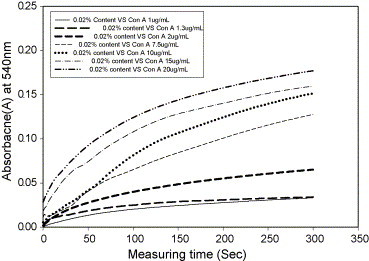
Time evolution of UV-absorbance intensity with different concentrations of Con A at 540 nm.
Fig. 5.
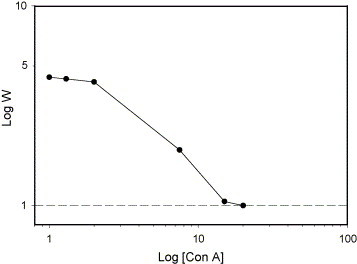
Stability ratio vs. concentration of Con A.
4. Conclusions
Glucose-modified latex was polymerized with the modified monomer and styrene by two-shot growth methods. Interactions between Con A and glucose-modified latex were determined by the stability. Binding process was determined by the kinetic of coagulation. In the work, experimental data showed that the colloids stability decreased with increasing concentration of Con A. This was explained by decreasing charge of the negatively charged colloids by binding of the Con A molecules of isoelectric point (net charge = 0). Formation of a cross-linked network structure through the affinity interactions between the tetra-Con A molecules and poly(allyl-α-d-glucopyranose/styrene) latex plays an important role in the kinetic of coagulation.
References
- 1.Varki A. Glycobiology. 1993;3:97. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dwek R.A. Chem. Rev. (Washington, DC) 1996;96:683. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 3.Lis H., Sharon N. Chem. Rev. (Washington, DC) 1998;98:637. doi: 10.1021/cr940413g. [DOI] [PubMed] [Google Scholar]
- 4.Simanek E.E., McGarvey G.J., Jablonowski J.A., Wong C.-H. Chem. Rev. (Washington, DC) 1998;98:833. doi: 10.1021/cr940226i. [DOI] [PubMed] [Google Scholar]
- 5.Weis W.I., Drickamer K. Annu. Rev. Biochem. 1996;65:441. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- 6.Ratner B.D. J. Biomed. Mater. Res. 1993;27:837. doi: 10.1002/jbm.820270702. [DOI] [PubMed] [Google Scholar]
- 7.Ruiz L., Hilborn J.G., Leonard D., Mathieu H.J. Biomaterials. 1998;19:987. doi: 10.1016/s0142-9612(97)00197-x. [DOI] [PubMed] [Google Scholar]
- 8.Kieburg C., Sadalapure K., Lindhorst T.K. Eur. J. Org. Chem. 2000:2035. [Google Scholar]
- 9.Norde W., Lyklema J. J. Colloid Interface Sci. 1978;66:277. doi: 10.1006/jcis.1996.4615. [DOI] [PubMed] [Google Scholar]
- 10.Shirahama H., Takeda K., Suzawa T. J. Colloid Interface Sci. 1986;109:552. [Google Scholar]
- 11.Tamai H., Fujii A., Suzawa T. J. Colloid Interface Sci. 1987;118:176. [Google Scholar]
- 12.Kim C.W., Rha C. Biotechnol. Bioeng. 1989;33:1205. doi: 10.1002/bit.260330917. [DOI] [PubMed] [Google Scholar]
- 13.Kondo A., Kaneko T., Higashitani K. Appl. Microbiol. Biotechnol. 1993;40:365. doi: 10.1007/BF00170394. [DOI] [PubMed] [Google Scholar]
- 14.Kondo A., Kamura H., Higashitani K. Appl. Microbiol. Biotechnol. 1994;41:99. doi: 10.1007/BF00166089. [DOI] [PubMed] [Google Scholar]
- 15.Sumi Y., Shiroya T., Fujimoto K., Wada T., Handa H., Kawaguchi H. Colloids Surf. B: Biointerfaces. 1994;2:419. [Google Scholar]
- 16.Chern C.S., Lee C.K., Ho C.C. Colloid Polym. Sci. 1999;277:507. [Google Scholar]
- 17.Chern C.S., Lee C.K., Tsai Y.J., Ho C.C. Colloid Polym. Sci. 1998;276:427. [Google Scholar]
- 18.Chern C.S., Lee C.K., Tsai Y.J. Colloid Polym. Sci. 2001;279:420. [Google Scholar]
- 19.Koch U., Yaacoub E.-J. Macromol. Chem. Phys. 2003;204:803. [Google Scholar]
- 20.Matrosovich M.N., Matrosovich T.Y., Gray T., Roberts N.A., Klenk H.-D. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4620. doi: 10.1073/pnas.0308001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu M.-Z., Fan H.-N., Guo Z.-W., Hui Y.-Z. Carbohydr. Res. 1996;290:233. [Google Scholar]
- 22.Cheong I.W., Kim J.H. Colloid Polym. Sci. 1997;275:736. [Google Scholar]
- 23.Oh J.T., Kim J.H. Enzyme Microbial Technol. 2000;27:356. doi: 10.1016/s0141-0229(00)00232-5. [DOI] [PubMed] [Google Scholar]
- 24.Mandal B.M., Palit S.R. J. Polym. Sci., Polym. Chem. Ed. 1971;9:3301. [Google Scholar]
- 25.Banthia A.K., Mandal B.M., Palit S.R. J. Polym. Sci., Polym. Chem. Ed. 1977;15:945. [Google Scholar]
- 26.Nakamura S., Kato A., Kobayashi K. J. Agric. Food Chem. 1991;39:647. [Google Scholar]
- 27.Rizzardo E., Solomon D.H. J. Macromol. Sci., Chem. 1979;A13:997. [Google Scholar]
- 28.Cheong I.W., Nomura M., Kim J.H. Macromol. Chem. Phys. 2000;201:2221. [Google Scholar]
- 29.Kanai M., Mortell K.H., Kiessling L.L. J. Am. Chem. Soc. 1997;119:9931. [Google Scholar]
- 30.Agrawal B.B.L., Goldstein I.J. Can. J. Biochem. 1968;46:1147. doi: 10.1139/o68-170. [DOI] [PubMed] [Google Scholar]
- 31.Shubin V., Samoshina Y., Menshikova A., Evseeva T. Colloid Polym. Sci. 1997;275:655. [Google Scholar]